

Abstract
6-Thiopurine (6-TP) prodrugs include 6-thioguanine and azathioprine. Both are widely used to treat autoimmune disorders and certain cancers. This study showed that a 6-thioguanosine triphosphate (6-TGTP), converted in T-cells from 6-TP, targets Rac1 to form a disulfide adduct between 6-TGTP and the redox-sensitive GXXXXGK(S/T)C motif of Rac1. This study also showed that, despite the conservation of the catalytic activity of RhoGAP (Rho-specific GAP) on the 6-TGTP-Rac1 adduct to produce the biologically inactive 6-thioguanosine diphosphate (6-TGDP)-Rac1 adduct, RhoGEF (Rho-specific GEF) cannot exchange the 6-TGDP adducted on Rac1 with free guanine nucleotide. The biologically inactive 6-TGDP-Rac1 adduct accumulates in cells because of the ongoing combined actions of RhoGEF and RhoGAP. Because other Rho GTPases, such as RhoA and Cdc42, also possess the GXXXXGK(S/T)C motif, the proposed mechanism for the inactivation of Rac1 also applies to RhoA and Cdc42. However, previous studies have shown that CD3/CD28-stimulated T-cells contain more activated Rac1 than other Rho GTPases such as RhoA and Cdc42. Accordingly, Rac1 is the main target of 6-TP in activated T-cells. This explains the T-cell-specific Rac1-targeting therapeutic action of 6-TP that suppresses the immune response. This proposed mechanism for the action of 6-TP on Rac1 performs a critical role in demonstrating the capability to design a Rac1-targeting chemotherapeutic agent(s) for autoimmune disorders. Nevertheless, the results also suggest that the targeting action of other Rho GTPases in other organ cells, such as RhoA in vascular cells, may be linked to cytotoxicities because RhoA plays a key role in vasculature functions.
Keywords: disulfide, immunosuppression, Ras-related C3 botulinum toxin substrate 1 (Rac1), redox regulation, T-cell, 6-TGNP-Rac1 adduct, CD4+ cells, Rac1 inactivation, redox-sensitive GXXXXGK(S/T)C motif, thiopurine prodrugs
Introduction
Purine-based antimetabolite 6-thiopurine (6-TP)3 prodrugs include 6-thioguanine (6-TG), 6-mercaptopurine, and azathioprine. They are widely used to treat autoimmune disorders such as inflammatory bowel disease and cancers such as acute lymphoblastic leukemia (1–3). In cells, enzymes convert the inactive prodrug 6-TP into pharmacologically active deoxy-6-thioguanosine phosphate (which is also called 6-thioguanine nucleotide) and 6-thioguanosine phosphate (6-TGNP), which includes 6-thioguanosine diphosphate (6-TGDP) and triphosphate (6-TGTP) (1–6). The deoxy-6-thioguanosine phosphate derived from 6-TP prodrugs can be incorporated into de novo synthesis of DNA as a form of 6-TG. Moreover, 6-TG in DNA can be recognized as a DNA lesion by the mismatch repair system. This recognition results in induction of the apoptosis of acute lymphoblastic leukemia (7–10). Recent studies show that 6-TGNP derived from 6-TP prodrugs binds to Rac1 to form the 6-TGNP·Rac1 complex (5, 11). Its formation, in turn, induces immunosuppression by blockage of the RhoGEF Vav-mediated Rac1 activation in T lymphocytes, which is where inactivation of Rac1 suppresses the function and survival of CD4+ cells (5, 11).
Rac1 is a member of the Ras superfamily of GTPases, and this superfamily includes both the Ras and Rho families of GTPases, which play key roles in various cellular signaling cascades (12, 13). The Rho family of GTPases encompasses Rac (e.g. Rac1, Rac2, and Rac3), RhoA, RhoC, and Cdc42, whereas the Ras family of GTPases includes HRas, KRas, and MRas. These proteins function by cycling between the inactive GDP-bound and active GTP-bound states (13, 14). This GDP/GTP cycling is controlled by various regulators such as guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs) (15, 16). Dysregulation of the expression and/or activity of Rho and Ras GTPases has been linked to various diseases, including autoimmune disorders, circulatory diseases, and certain cancers (5, 11, 17–21). Importantly, Rac GTPases continue to attract more and more clinical interest because of the emerging evidence of their role in such autoimmune disorders as inflammatory bowel disease (5, 11).
A precise molecularly based mechanism for the inactivation of Rac1 mediated by 6-TGNP via the blockage of the action of Vav remains unknown. The only difference between 6-TGNP and a regular guanosine phosphate (GNP) is the C-6 atom of the guanine base, where 6-TGNP and GNP, respectively, possess a sulfur and an oxygen atom at the C-6 position (10). Therefore, blockage of Vav action on the 6-TGNP-bound Rac1 in T-cells is certainly because of the presence of the C-6 sulfur atom on 6-TGNP. Accordingly, configuration of the potential chemical and physical features of the sulfur-containing thiol moiety of the Rac1-bound 6-TGNP associated with Vav action is critical because it can be a thiol-based medication platform to design more effective drugs. Furthermore, lack of the precise 6-TGNP-mediated Rac1 inactivation mechanism also potentially hinders maximization of the therapeutic effects of 6-TP prodrugs. For example, if an unknown cellular factor(s) is necessary for blockage of the Vav action on the 6-TGNP-bound Rac1, ignorance of such a potential factor limits the opportunity to maximize the therapeutic applications of 6-TP prodrugs. Perhaps the most significant disadvantage of not understanding the details of this mechanism lie in what we may consequently miss. For example, our lack of understanding may hinder recognition of any side effects of 6-TGNP that may occur through its binding to other critical Rho GTPases such as RhoA and Cdc42. This notion arises because, although the previous study shows that 6-TGNP preferentially blocks the Vav action on Rac1 but not on RhoA and Cdc42 (11), Vav has a rather broad spectrum of catalytic specificity (22). Thus, Vav is capable of activating all of these Rho proteins, including Rac1, RhoA, and Cdc42. Despite what is known, the mechanistic reason for why 6-TGNP selectively targets Rac1, rather than other Rho GTPases, remains poorly understood. Consequently, it is difficult to preclude or minimize the potential side effects of 6-TP prodrugs associated with these Rho GTPases.
We have recently identified a distinct redox-sensitive GXXXXGK(S/T)C motif that is conserved in many Rho subfamily proteins such as Rac1, RhoA, RhoC, and Cdc42 (23, 24). Notably, the thiol moiety of the Rac1 cysteine (Cys18, Rac1 numbering) in the GXXXXGK(S/T)C motif is located at the Rac1 nucleotide-binding site, but the equivalent thiol moiety of the Ras cysteine (Cys118, Harvey Ras numbering) in the NKCD motif is remote from the Ras nucleotide-binding site (13). Biologically relevant redox agents include nitric oxide (NO), nitrogen dioxide (•NO2), and dinitrogen trioxide (N2O3) as well as the superoxide anion radical (O2˙̄) and hydrogen peroxide (25–27). Of these, •NO2 and O2˙̄ have been shown to directly target and react with these redox-sensitive residues and activate Rho proteins (13).
The redox-sensitive Rac1 Cys18 residue in the GXXXXGK(S/T)C motif also is likely susceptible to reaction with another cysteine residue of proteins or molecules that contains a thiol moiety to produce protein disulfide. This susceptibility is exemplified by the formation of an intra-protein disulfide of the GXXXXGK(S/T)C motif-containing RhoA (24). Intriguingly, a disulfide cross-link between 6-TGNP and Rac1 is possible. This possibility exists because 6-TGNP, which is derived from 6-TP prodrugs (10), possesses a thiol moiety and the Rac1 Cys18 is located at the GTPase nucleotide-binding site. If this potential disulfide bond formation is real, it could cause 6-TGNP to stick on the Rac1 nucleotide-binding site, disabling its displacement with the cellularly abundant GTP to produce an active GTP-bound Rac1. Although the resultant cellular effect is different, this potential mechanistic scenario, the sticking of 6-TGNP on the Rac1 nucleotide-binding site through a disulfide bond, also is observed in another Rho protein, RhoC, which also has the GXXXXGK(S/T)C motif (28). This potential disulfide bond formation between 6-TGNP and Rac1 may explain how Rac1 is rendered inactive by blocking the Vav action on it. However, other possibilities may be at work, such as the Rac1-bound 6-TGNP altering or modifying the mechanical properties of Vav. It also is unclear what effect, if any, the presence of the thiol moiety of the Rac1-bound 6-TGNP has on the catalytic function of RhoGAPs. This information is necessary to assess the overall effects of 6-TGNP on regulation of Rac1 activity.
This study uses CD4+ cell-based redox biochemistry approaches to examine the mechanistic features of the 6-TGNP-mediated Rac1 inactivation. These features are associated with the activities of Vav and RhoGAP that detail a precise molecularly based mechanism for the 6-TGNP-mediated Rac1 inactivation. The results have the potential to aid further improvement of usage of 6-TP prodrugs and promote the development of potentially better drugs for immunosuppressive effects. Another advantage may be insight into the prevention or alleviation of the potential cytotoxicities of 6-TP prodrugs as immunosuppressive drugs.
Experimental Procedures
Chemicals and Preparation of Proteins
All chemicals used in this study were the highest grades available. Full-length Rac1 and Vav (encompassing the DH-PH-cysteine-rich domain of Vav2) constructs derived from humans were expressed in and purified from Escherichia coli as described previously (29). The GST tags of the GST-tagged p21-activated kinase 1 protein-binding domain (PAK-PBD, Cytoskeleton) and the catalytic domain of RhoGAP (Cytoskeleton) were cleaved off by incubation with HRV 3C protease (Fisher) for 12 h at 4 °C. The cleaved GST tag and uncleaved GST-tagged proteins were removed by passing through the GSTrap FF column (GE Healthcare).
Preparation and Treatments of CD4+ Cells
Human CD4+ cells used in this study were from a single individual and were obtained from Astarte Biologics. Unstimulated CD4+ cells died in 2–3 days. Therefore, all CD4+ cells were stimulated with coated antibodies to CD3 (1.0 μg/ml) and soluble CD28 (1.0 μg/ml) as well as interleukin 2 (IL-2, 20 units/ml R&D Systems) for 1 day before their use as described previously (11). When necessary, CD4+ cells (1 × 104) were repeatedly treated with 6-TG (1 μm for every 12 h) and/or diethylenetriamine/nitric oxide adduct (DETA/NO, 10 μm every 8 h) for 3 days.
Western Blot Analyses for the Activity Markers of CD4+ Cells
The CD4+ cells treated with and without 6-TG and/or DETA/NO were lysed by sonification in an extraction buffer that contains 5 mm MgCl2, 1 mm EDTA, 5% glycerol, 1% deoxycholate, and metal-free 10 mm Tris-HCl (pH 7.4). The cell extracts were subjected to Western analyses of Rac1, Vav, phosphorylated Vav (pVav), Ezrin-Radixin-Moesin (ERM), and actin, respectively, by using antibodies against Rac1 (monoclonal anti-Rac1, Cytoskeleton), Vav (monoclonal anti-Vav, Millipore), pVav (monoclonal anti-pVav, Santa Cruz Biotechnology), ERM (polyclonal anti-ERM, Cell Signaling Technology), and actin (monoclonal anti-actin, Sigma).
Quantification of Interferon-γ Secretion from CD4+ Cells
Quantity secretions of interferon-γ (IFN-γ) from CD4+ cells were determined by using a human IFN-γ ELISA kit (Abfrontier).
Quantification of Phosphorylated ERM in CD4+ Cells
To examine the level of ERM phosphorylation (pERM), CD4+ cells were stimulated in a complete RPMI 1640 medium with ∼50 μCi of 32P/ml. The cells were then treated with and without 6-TG and/or DETA/NO as indicated above. The cells were sonicated to produce cell extracts. ERM was then immunoprecipitated (IPed) from the cell extracts by using a Thermo Scientific Pierce direct IP kit and the polyclonal anti-ERM antibody (Pierce) and then analyzed by SDS-PAGE and autoradiographed.
Measurements of Rac1, RhoA, and Cdc42 Activities in CD4+ Cells
The Rac1 activity assay was performed with a cell extract by using the Rac1 G-LISA activation assay kit (Cytoskeleton). The RhoA and Cdc42 activities in the cell extracts were determined by using RhoA-GTP and a Cdc42-GTP activation assay kit (NewEast Biosciences), respectively.
Preparation of Rac1 Immunoprecipitation
Briefly, to prepare the IPed Rac1samples from CD4+ cells treated with and without 6-TG and/or DETA/NO, CD4+ cell lysates in an extraction buffer were incubated with a monoclonal anti-Rac1 antibody (Cytoskeleton) linked to glutathione-agarose beads (Pierce) for 1 h. The agarose beads were pulled down by centrifugation (3,000 × g for 10 min). The beads were washed three times with the extraction buffer. The beads were then treated with an elution buffer to release the bead-bound Rac1 proteins. The elution buffer contained 50% methanol, 0.1% formic acid, 0.1 mm MgCl2, and 1 mm EDTA. The supernatant that contained proteins (i.e. Rac1) was separated by centrifugation (3,000 × g for 5 min). Note that the entire sample preparation process(es) was done without any reducing agents such as dithiothreitol (DTT) or β-mercaptoethanol at 0 °C.
Mass Spectrometry-based Identification of the Formation of 6-TGNP-Rac1 Adduct in CD4+ Cells
Mass spectrometric analyses were performed to determine the feature state of Rac1 in CD4+ cells treated with and without 6-TG and/or DETA/NO. Analyses of electrospray-mass spectrometry (ESI-MS) and tandem mass spectrometry (MS/MS) for the IPed samples from CD4+ cells were performed as described previously (28), except that preparation of the IPed sample used a monoclonal anti-Rac1 antibody (Cytoskeleton) rather than the anti-RhoC antibody. Briefly, the IPed Rac1 samples in the elution buffer were diluted 10-fold with a buffer containing 1 mm EDTA in 50 mm Tris-HCl (pH 7.4). The IPed Rac1 samples were then digested with trypsin for 10 h. The trypsin-treated samples were then subjected to analysis with ESI-MS and MS/MS in the positive ion mode ([molecular mass + H]+).
Immunoblot-based Determination of 6-TGNP-Rac1 Adduct in CD4+ Cells
The IPed Rac1 fractions from the CD4+ cells in the elution buffer (see above) were diluted 10-fold with a sample buffer containing 2% SDS, 5% glycerol, 0.01 bromphenol blue in 50 mm Tris-HCl (pH 6.8), and denatured by heating for 1 min at 100 °C. The heat-denatured Rac1 samples were then subjected to Western analysis and probed with a monoclonal anti-6-TGNP antibody (which is an anti-6-TGNP antibody detecting both 6-TGDP and 6-TGTP, Pierce). Note that unless otherwise indicated, all potential reducing agents such as DTT or β-mercaptoethanol were excluded from all experimental processes.
Examination of the Biological Activity of 6-TGNP-Rac1 Adduct in CD4+ Cells
A fluorescence-based 6-TGNP-Rac1 adduct activity assay was performed in accordance with the previously described method (30). To label Rac1 with rhodamine B dye, Rac1 (100 μm) purified from E. coli was mixed with a 5-fold excess rhodamine B in a buffer containing 20 mm GDP, 50 mm NaCl, 5 mm MgCl2, and 1 mm EDTA in 10 mm Tris-HCl (pH 7.4). The mixture was incubated for 20 min at room temperature. The unreacted free rhodamine B was removed by applying the mixture onto a size-exclusion column (Sephadex G-25, 1.5 × 6 cm) at room temperature. The rhodamine B-attached Rac1 was loaded with 6-TGNP to produce the 6-TGNP-bound Rac1 and then further treated with NO under aerobic conditions to produce the 6-TGNP-Rac1 adduct. When necessary, the IPed Rac1 from CD4+ cells also was labeled with rhodamine B dye by using the same method established for labeling Rac1 purified from E. coli (see above). However, after production of the rhodamine B-attached IPed Rac1, the treatment step(s) of the rhodamine B-attached IPed Rac1 with the 6-TGNP and/or NO was skipped; this omission was because the feature states of the untreated IPed Rac1 with its original 6-TGNP adduct were of interest to this research.
The rhodamine B-attached 6-TGNP-Rac1 adduct or the rhodamine B-attached-IPed Rac1 (10 nm) and PAK-PBD (1 μm) were transferred into a fluorescence assay cuvette containing 5 mm MgCl2, 1 mm EDTA, 5% glycerol, 1% deoxycholate, and metal-free 10 mm Tris-HCl (pH 7.4). The assay mixture was excited at 510 nm, and the change in fluorescence emission intensity at 565 nm due to the potential binding of the rhodamine B-attached 6-TGNP-Rac1 adduct or the rhodamine B-attached IPed Rac1 to PAK-PBD was monitored with a fluorimeter (LS50B PerkinElmer Life Sciences) over a period of 0–80 s at 25 °C. The 6-TGNP-bound Rac1 that was not treated with NO was used as a control in this experiment.
Examination of the Catalytic Function of RhoGAP on the 6-TGNP-Rac1 Adduct
To examine any potential catalytic activity of RhoGAP on the 6-TGNP-Rac1 adduct or on IPed Rac1, the 6-TGNP-Rac1 adduct or IPed Rac1 (10 nm) was incubated with RhoGAP (1 μm) in a buffer containing 5 mm MgCl2, 1 mm EDTA, and metal-free 10 mm Tris-HCl (pH 7.4) for 1 h at 25 °C. The 6-TGNP-bound Rac1 not treated with NO was used as a control in this assessment. Note that the 6-TGNP-Rac1 adduct and the 6-TGNP-bound Rac1 were prepared from Rac1 purified from E. coli, yet the IPed Rac1 was directly IPed from CD4+ cells. The RhoGAP catalysis was stopped by lowering the temperature to 0 °C. The 6-TGNP was released from Rac1 by treatment with 2.5% SDS, 200 mm (NH4)2SO4, and 1 mm DTT at 0 °C. Centricon (10-kDa cutoff, Millipore) at 0 °C was used to filter out protein portions such as Rac1 and RhoGAP from the mixture of 6-TGNP, Rac1, and RhoGAP. As noted under “Preparation of Rac1 Immunoprecipitation,” the IPed Rac1 sample preparation was performed at 0 °C. Therefore, except for the RhoGAP treatment step, all steps, from the Rac1 IP from CD4+ cells to the release of 6-TGNP from the IPed Rac1, were conducted at 0 °C. If the IPed Rac1 were in the form of the 6-TGTP-Rac1 adduct, this low temperature (i.e. 0 °C) sample preparation minimized the potential nonspecific temperature-dependent hydrolysis of the γ-phosphate of the 6-TGTP-Rac1 adduct. Accordingly, the form of the identified 6-TGNP (6-TGDP or/and 6-TGTP) released from the IPed Rac1 in the absence of RhoGAP under our experimental conditions (i.e. 0 °C) likely represents the original feature status of 6-TGNP adducted to Rac1 in CD4+ cells. Recognition of the original feature status of 6-TGNP adducted to Rac1 in CD4+ cells is crucial for ensuring a correct interpretation of the features of 6-TGNP released from the IPed Rac1 after treatment of the IPed Rac1 with RhoGAP. Nevertheless, the Centricon-filtered sample containing the eluted 6-TGNP fraction was further concentrated by using a freeze dryer lyophilizer and then analyzed by using a PEI-cellulose plate thin layer chromatograph (TLC) as described previously (31). The 6-TGNP on the TLC plate was colorimetric detected by the TLC plate spraying a malachite green solution that contains 10 μm malachite, 10 mm (NH4)6Mo7O24, 0.5% (w/w) HClO4, and 0.1% Triton X-100 at 25 °C. This malachite green-based phosphate analysis was adapted from the previously reported method (32). Note that a longer exposure of the 6-TGNP on the TLC plate to the malachite green solution (e.g. more than 10 min) produces a denser band intensity of the 6-TGTP than that of the 6-TGDP. It is likely that the initial reaction to the malachite green solution involves the terminal phosphate moieties of 6-TGTP and 6-TGDP (i.e. the γ-phosphate of 6-TGTP and the β phosphate of 6-TGDP, respectively). However, a longer exposure of 6-TGNP on the TLC plate to the malachite green solution allows the solution to react with other phosphates of 6-TGNP. Because 6-TGTP possesses three phosphates but 6-TGDP possesses only two, the overall phosphate density of 6-TGTP detected by the malachite green likely exceeds that of 6-TGDP. Accordingly, the exposure of 6-TGNP on the TLC plate to the malachite green solution was limited to 5 min. Nevertheless, the phosphate bands that developed in association with the 6-TGNP at 630 nm in these 5 min were then further analyzed with densitometry. A mixture of fresh 6-TGTP and 6-TGDP (1:1, mol/mol) was used as a control for this analysis. Accordingly, the 6-GNP (6-TGTP and 6-TGDP) bands were identified by comparing them with the control bands of the control 6-TGTP and 6-TGDP.
To further link the catalytic action of RhoGAP on the biological activity of the 6-TGNP-Rac1 adduct, RhoGAP (100 nm) was added in the course of the fluorescence-based activity assay of the 6-TGNP-Rac1 adduct or the IPed Rac1 with PAK-PBD (see under “Examination of the Biological Activity of 6-TGNP-Rac1 adduct in CD4+ Cells”). As a control, RhoGAP was added over the course of the fluorescence-based activity assay of the 6-TGNP-bound Rac1 with PAK-PBD. At the time points of 20 and 60 s, an aliquot of samples was withdrawn from the solution of the fluorescence-based activity assay. Centricon (10-kDa cutoff) at 0 °C was used to remove the proteins of these aliquot samples. The free phosphate (Pi) content of the protein-excluded solution was colorimetrically determined by using the malachite green solution in which the sample solution was mixed with the malachite green solution, incubated for 5 min at 25 °C, and then the UV/visible absorbance at 630 nm was measured (32). Pi (Aldrich) was used as the standard.
Determination of the Mechanistic Features of the 6-TGNP-Rac1 Adduct in CD4+ Cells
The fractions of the IPed Rac1 samples from CD4+ cells in the elution buffer (see above) were diluted 10-fold with a buffer containing 50 mm NaCl, 5 mm MgCl2, and 1 mm EDTA in 10 mm Tris-HCl (pH 7.4). To examine the potential nucleotide exchange of Rac1 with 6-TGNP in CD4+ cells, some of the IPed Rac1 samples were treated with a 100-fold excess of 6-TGDP in the presence of Vav (10 nm) purified from E. coli for 1 h to load 6-TGDP onto Rac1; they were further treated with NO (∼5 μm) for 10 min to produce the 6-TGDP-Rac1 disulfide adduct. In addition, to examine the potential effect of a reducing agent on the nucleotide exchange of the IPed Rac1 samples with 6-TGDP, the 6-TGDP/NO-treated IPed Rac1 protein samples (above) were further treated with DTT (10 mm) for 1 h. These 6-TGDP/NO-treated IPed Rac1 as well as 6-TGDP/NO/DTT-treated IPed Rac1 samples were then subjected to Western blots and probed with an anti-6-TGNP antibody.
Measurement of Vav Activity in CD4+ Cells
Vav protein was purified directly from the CD4+ cell extract by using protein A-Sepharose beads coated with a polyclonal anti-Vav antibody (raised against amino acids 581–596 of human Vav1, Millipore) and the immunizing peptide (the Vav1 peptide 581–596) (33). A fluorescence-based Vav activity assay was performed, with a slight modification, according to the previously described method (29, 34). Briefly, Vav protein (10 nm) purified from CD4+ cells (see above) was transferred into a fluorescence assay cuvette containing a nucleotide-bound substrate Rac1 (expressed in and then purified from E. coli, 1 μm), 2′,3′-O-(N′-methylanthraniloyl) guanosine-5′-O-diphosphate-tagged GDP (mantGDP; 200 μm), 5 mm MgCl2, 1 mm EDTA, 5% glycerol, 1% deoxycholate, and metal-free 10 mm Tris-HCl (pH 7.4). The change in fluorescence intensity because of the potential replacement of the Rac1-bound nucleotide with the fluorescence mant-GDP in the presence and absence of Vav was monitored with a fluorimeter over a period of 0–800 s at 25 °C. Note that the protein concentration of each Vav (e.g. purified from CD4+ cells treated with 6-TG and DETA/NO or etc.) is essential for this comparative kinetic analysis. Hence, the concentration of all purified Vav proteins was very carefully determined through use of both the bicinchoninic acid and Bradford methods. This concentration was then carefully adjusted to 10 nm for each fluorescence assay.
Determination of the Proliferation of CD4+ Cells
Proliferations of CD4+ cells were determined by using a fluorescence-based CyQuant NF cell proliferation assay (Invitrogen) according to the manufacturer's protocol. CD4+ cells were plated at a density of ∼1000 cells per well in a 96-well plate and then treated with an NO donor DETA/NO and/or 6-TG as indicated above. As a positive control, CD4+ cells also were treated with the soluble guanylyl cyclase inhibitor, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (10 μm for every 8 h) for 3 days. Green fluorescent CyQuant GR dye (50 μl) was added to each well and incubated for 30 min at 37 °C. Then the fluorescence intensity of each sample was measured with a fluorescence microplate reader.
Determination of the Viability and Apoptotic Effects of CD4+ Cells
A colorimetric assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was used to assess the viability of CD4+ cells treated with 6-TG in the presence and absence of DETA/NO (35). The apoptotic effect of 6-TG as well as an apoptotic inducer (2-amino-N-quinolin-8-yl-benzenesulfonamide; 10 mm) on CD4+ cells in the presence and absence of DETA/NO was measured using an ApoAlert Caspase-3/8 colorimetric assay kit (Takara, Japan).
Statistical Analyses of Measurements
When necessary, statistics were obtained with a Tukey analysis of variance (ANOVA) by using GraphPad Prism software.
Results
Inhibitory Effects of 6-TPs on the Rac1 Activity of CD4+ Cells
To examine the effect of 6-TG on Rac1 activity in CD4+ cells, Rac1 activity as well as the dephosphorylation of ERM and the production of cytokine IFN-γ were monitored after the CD4+ cells were treated with 6-TG. Studies show that the levels of the ERM dephosphorylation and IFN-γ production reflect the result of Rac1 activity in CD4+ cells in which Rac1 activation stimulates not only cell motility coupled with dephosphorylation of the ERM but also the induction of IFN-γ production (11, 36).
The level of cellular expression of Rac1 and ERM in CD4+ cells in the presence or absence of 6-TG was unchanged (Fig. 1A). However, a significant portion of Rac1 was inactivated within 3 days when CD4+ cells were treated with 6-TG (Fig. 1B). A large portion of ERM in CD4+ cells was observed to have been phosphorylated to produce pERM in 3 days after treatment with 6-TG (Fig. 1B). The suppression of the secretion of IFN-γ from CD4+ cells also was coupled with the inactivation of 6-TG-mediated Rac1 (Fig. 1B). The results suggest that 6-TG inactivates Rac1, leading to suppression of T-cell ERM dephosphorylation and secretion of IFN-γ. These results are consistent with previous studies showing that 6-TP was linked to the inactivation of Rac1 and CD4+ cells (11, 37).
FIGURE 1.

6-TG and/or DETA/NO inhibits the T-cell Rac1 activity, IFN-γ secretion, and ERM dephosphorylation. A, Western blot analyses of CD4+ cells for the determination of the expression of Rac1 and ERM proteins were performed with anti-Rac1 and anti-ERM antibodies as described under “Experimental Procedures.” B, Rac1 activity, IFN-γ secretion, and pERM fraction in each treatment of CD4+ cells was measured as described under “Experimental Procedures.” The value of Rac1 activity and IFN-γ secretions determined from untreated cells was set at 100%. All pERM values were normalized against the value of pERM as determined from cells treated with both 6-TG and DETA/NO that were set at 100%. The data represented in the figure represent the mean values of triplicate measurements, and each of the vertical error bars indicates standard error (S.D.). The results of the ANOVA Tukey test were as follows: *, p < 0.01, versus the Rac1 activity of the untreated sample; **, p < 0.01, versus the IFN-γ secretion level of the untreated sample; and ***, p < 0.01, versus the quantity of pERM determined from cells treated with both 6-TG and DETA/NO.
6-TP-mediated Rac1 Inactivation by the Formation of 6-TGNP-Rac1 Adduct in CD4+ Cells
The only difference between 6-TGNP and GNP is the C6 thiol moiety on the purine base of 6-TGNP. Yet, 6-TGNP inactivates Rac1 in CD4+ cells and GNP cannot. Rac1 possesses the Cys18 residue that has the redox-sensitive thiol side chain located at the nucleotide-binding site (23). Taken together, as noted elsewhere, it can be reasoned that the 6-TGNP-mediated Rac1 inactivation in CD4+ cells is possibly because of the disulfide bond formation of the Cys18 side chain with the bound 6-TGNP. To examine this possibility, an IP-based MS approach was performed. This approach encompasses ESI as well as MS/MS analyses for the Rac1 in CD4+ cells treated with 6-TG.
The analyses (Fig. 2) suggest that Rac1 inactivation by CD4+ cells treated with 6-TG occurs because the thiol moiety of 6-TGNP (cellularly converted from the treated 6-TG) reacts with the redox-sensitive side chain Cys18 of the Rac1 GXXXXGK(S/T)C motif to produce a 6-TGNP-Rac1 disulfide adduct. Because sulfur atoms exist on each of the Cys18 side chains and on the 6-TG moiety of the bound 6-TGNP, the feature state of the 6-TGNP-Rac1 disulfide adduct is likely to be that the bound 6-TGNP is covalently linked to the side chain Cys18 of the Rac1.
FIGURE 2.

6-TGNP, derived from 6-TG, targets Rac1 to produce 6-TGNP-Rac1 disulfide adduct in CD4+ cells. A, ESI-MS analyses of CD4+ cells for the detection of the 6-TGNP-Rac1 disulfide adduct were performed as described under “Experimental Procedures.” The MS peak at 1481.6 Da was only shown when CD4+ cells were treated with 6-TG (panel b) and not shown when CD4+ cells were not treated with 6-TG (panel a). B, MS/MS analysis was performed to identify the molecule that has a mass of 1481.6 Da. Major MS/MS peaks shown were best fitted to the masses of ion fragments of the 18TCLLIVFSK-6-TGDP adduct as follows: (i) 1462.8 Da, formed upon loss of H2O from the β-phosphate of the 6-TGDP moiety of 18TCLLIVFSK-6-TGDP adduct; (ii) 1445.6 Da., formed by losing an H2O and an OH from the β- and α-phosphate of the 6-TGDP moiety of 18TCLLIVFSK-6-TGDP adduct; (iii) 1400.4 Da, formed upon loss of COOH from the C terminus of the 18TCLLIVFSK-6-TGDP adduct as well as an H2O and an OH from the β- and α-phosphate of the 6-TGDP moiety of 18TCLLIVFSK-6-TGDP adduct; (iv) 1022.8 Da, formed upon loss of the 6-TGDP moiety from the 18TCLLIVFSK-6-TGDP adduct; and (v) the 978.3 Da, formed by losing the peptide C terminus COOH and the 6-TGDP moiety from the 18TCLLIVFSK-6-TGDP adduct.
It is notable, however, that because two forms of 6-TGNP exist, 6-TGDP and 6-TGTP, two forms of the 6-TGNP-Rac1 adduct, 6-TGDP- and 6-TGTP-Rac1 adducts, respectively, are also possible. Among them, the ESI and MS/MS analyses only identified the 6-TGDP-Rac1 adduct in the 6-TG-treated CD4+ cells. However, the presence of the 6-TGTP-Rac1 adduct in the 6-TG-treated CD4+ cells cannot be ruled out. This is because even if the 6-TGTP-Rac1 adduct were present in the 6-TG-treated CD4+ cells, the labile γ-phosphate of the 6-TGTP-Rac1 disulfide adduct can be easily hydrolyzed during the room temperature preparation of the ESI and MS/MS sample that produces the 6-TGDP-Rac1 disulfide adduct.
Although the level of Rac1 expression was unchanged when CD4+ cells were treated with or without 6-TG (Fig. 3A), Western analyses using an anti-6-TGNP antibody detected the 6-TGNP-Rac1 adduct in CD4+ cells treated with 6-TG (Fig. 3B). This presence of the 6-TGNP-Rac1 adduct in the 6-TG-treated cells is consistent with the result of ESI and MS/MS analyses.
FIGURE 3.
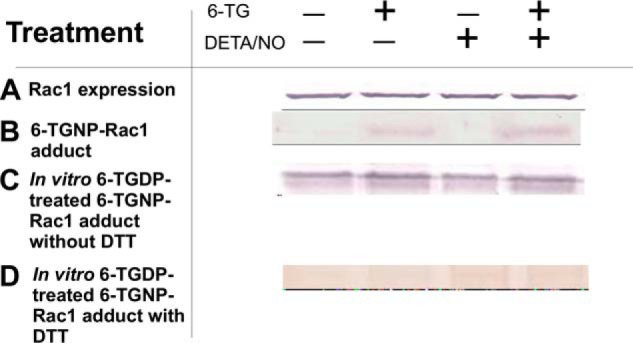
DETA/NO does not enhance formation of the 6-TGNP-Rac1 disulfide adduct in CD4+ cells. A, Western analysis of CD4+ cells for the determination of Rac1 expression was performed using an anti-Rac1 antibody as described under “Experimental Procedures.” B, presence of the 6-TGNP-Rac1 disulfide adduct in the IPed Rac1 from CD4+ cells was examined by Western analysis by using an anti-6-TGNP antibody as noted under “Experimental Procedures.” C, Vav-mediated formation of the 6-TGNP-Rac1 disulfide adduct in the IPed Rac1 from CD4+ cells in the presence of 6-TGDP and/or NO was analyzed by Western blot by using an anti-6-TGNP antibody as described under “Experimental Procedures.” D, DTT-mediated removal of the 6-TGNP-Rac1 disulfide adduct in the IPed Rac1 from CD4+ cells also was examined by Western analysis by using an anti-6-TGNP antibody as described under “Experimental Procedures.” B, C, and D, a densitometer was used to quantify band intensities. The fractions of the 6-TGNP-Rac1 adduct were then expressed as normalized values against the values of the treatment with both 6-TG and DETA/NO of sample C, in which the band intensity was set at 100%. Accordingly, the band intensities of sample B were as follows: nontreatment (0%); 6-TG treatment (29%); DETA/NO treatment (0%); and 6-TG and DETA/NO treatment (38%). The band intensities of sample C were as follows: nontreatment 95%; 6-TG treatment (100%); DETA/NO treatment (92%); and 6-TG and DETA/NO treatment (100%). The band intensities of sample D were as follows: nontreatment (3%); 6-TG treatment (8%); DETA/NO treatment (9%); and 6-TG and DETA/NO treatment (1%). Standard deviations associated with the densitometry analyses of three independent experiments were less than 20% of the values given.
Biologically Inactive Form of 6-TGDP-Rac1 Adduct in CD4+ Cells
Although the MS analyses suggest that the formation of the 6-TGNP-Rac1 adduct inactivates Rac1 in CD4+ cells, it is unclear what form of the disulfide adduct is responsible for the Rac1 inactivation. To configure the form of the disulfide adduct that is responsible for the Rac1 inactivation, a fluorescence-based in vitro binding study of Rac1 adducted with 6-TGDP and 6-TGTP with PAK-PBD was performed. These 6-TGDP- and 6-TGTP-Rac1 adducts were prepared from Rac1 that was expressed in and purified from E. coli (see “Experimental Procedures”). Note that whenever the molecular weight of the fluorescence probe-tagged molecule was increased by either a covalent bond formation or by a simple binding interaction, the intensity of the fluorescence emission was increased (34). The Rac1 proteins used in this analysis were tagged with the fluorescence rhodamine B (see “Experimental Procedures”). Accordingly, an increase in the rhodamine B fluorescence intensity by addition of PAK-PBD to the assay solution containing the rhodamine B-tagged 6-TGNP-Rac1 adduct signals the binding of 6-TGNP-Rac1 adduct with PAK-PBD.
The rhodamine B fluorescence-based binding study (Fig. 4A) shows that only the 6-TGTP-Rac1 adduct definitely has a binding affinity with PAK-PBD. Fig. 4A also shows that the binding affinities of the GTP- and GDP-bound Rac1 with PAK-PBD are, respectively, substantial and minimal. This is consistent with previous studies showing that the active GTP-bound form of Rac1, compared with the inactive GDP-bound form of Rac1, has a higher binding affinity with PAK-PBD (38). Taken together, the binding study result suggests that, although both 6-TGDP and 6-TGTP are covalently attached to Rac1, their mechanistic roles in Rac1 activity are, respectively, identical to those of GDP and GTP, in which the 6-TGTP-Rac1 adduct is biologically active, but the 6-TGDP-Rac1 adduct is not.
FIGURE 4.

Although the 6-TGDP-Rac1 adduct is biologically inactive, the 6-TGTP-Rac1 adduct is biologically active. A, fluorescence rhodamine B-based binding assays of the rhodamine B-attached 6-TGNP-Rac1 adduct, the rhodamine B-attached 6-TGNP-bound Rac1, and IPed Rac1 with PAK-PBD in the presence and absence of RhoGAP were performed as described under “Experimental Procedures.” The addition of PAK-PBD followed by the addition of RhoGAP to the assay cuvette containing the rhodamine B-attached 6-TGTP-Rac1 adduct (▴), the rhodamine B-attached 6-TGDP-Rac1 adduct (▵), the rhodamine B-attached 6-TGTP-bound Rac1 (●), the rhodamine B-attached 6-TGDP-bound Rac1 (○), and the rhodamine B-attached IPed Rac1 from CD4+ cells treated with 6-TG and DETA/NO (□) was as indicated by the arrows. Note that the fluorescence spectrum of the rhodamine B-attached IPed Rac1 from CD4+ cells treated only with 6-TG was omitted because it overlapped with that of the rhodamine B-attached IPed Rac1 from CD4+ cells treated with 6-TG and DETA/NO. The rhodamine B-attached GTP-bound Rac1 that was not treated with PAK-PBD and/or RhoGAP (×) was used as a control. The rhodamine B-attached GDP-bound Rac1 that also was not treated with PAK-PBD and/or RhoGAP was used as a control. Nevertheless, its spectrum is almost identical to that of the rhodamine B-attached GTP-bound Rac1, and thus it is omitted for clarity. B, quantification of the Pi released from the 6-TGNP-bound Rac1, 6-TGNP-Rac1 adduct, and IPed Rac1 from CD4+ cells treated with 6-TG and DETA/NO with and without RhoGAP was determined by using malachite green as described under “Experimental Procedures.” The Pi quantities measured were then converted into the mole fractions (mole of Pi released/mol of 6-TGNP-bound Rac1 or 6-TGNP-Rac1 adduct). Note that the Pi value associated with its standard deviations determined for the IPed Rac1 from CD4+ cells treated only with 6-TG is identical to that for the IPed Rac1 from CD4+ cells treated with 6-TG and DETA/NO, and thus is not shown. The mean values data are represented with S.D. bars associated with three independent experiments. Statistical results from the ANOVA Tukey test were as follows: *, p < 0.01, versus the 6-TGTP-bound Rac1 treatment without RhoGAP; **, p < 0.01, versus the 6-TGTP-bound Rac1 treatment with RhoGAP. a, IPed Rac1, Rac1 IPed from CD4+ cells treated with 6-TG only.
Fig. 4A also shows that the IPed Rac1 proteins from CD4+ cells treated with 6-TG and/or DETA/NO show a minimal binding affinity for PAK-PBD (Fig. 4A). This is consistent with the finding of the inactive form of Rac1 in CD4+ cells treated with 6-TG and/or DETA/NO that was cellularly identified by the Rac1 G-LISA assays (see the above analyses of the effect of 6-TG and/or DETA/NO on CD4+ cells and Rac1 activities). These results in combination with the fact that the IPed Rac1 proteins are shown to be in the form of the 6-TGNP-Rac1 adduct (see the above MS analyses) permit a postulation that the 6-TGDP-Rac1 adduct is likely the inactive form of Rac1 in CD4+ cells treated with 6-TG and/or DETA/NO. The same reasoning excludes the likelihood of the 6-TGTP-Rac1 adduct in this role. This reasoning is further confirmed by the analyses below of RhoGAP activity on the 6-TGNP-Rac1 adduct and IPed Rac1.
Conservation of the Catalytic Function of RhoGAP on the 6-TGTP-Rac1 Adduct
Both GDP- and GTP-bound Rac1 exist in cells. By assuming that the metabolic process of 6-TG is similar to the guanine base, it is possible to speculate that the biologically active 6-TGTP-Rac1 adduct, in addition to the biologically inactive 6-TGDP-Rac1 adduct, may also exist in CD4+ cells treated with 6-TG and/or DETA/NO. If so, the mechanism of the conversion of the 6-TGTP-Rac1 adduct into the 6-TGDP-Rac1 adduct is of interest because only the 6-TGDP-Rac1 adduct is populated in CD4+ cells treated with 6-TG and/or DETA/NO. It has been shown that the catalytic action of RhoGAP on GTPase is the main factor that converts the active GTP-bound form of Rac1 into the inactive GDP-bound Rac1 (39, 40). Hence, the catalytic activity of RhoGAP on the 6-TGTP-Rac1 adduct was examined to recognize the mechanism for the dominant population of the 6-TGDP-Rac1 adduct in CD4+ cells treated with 6-TG and/or DETA/NO. As with the above fluorescence analysis, the 6-TGDP- and 6-TGTP-Rac1 adducts were prepared from Rac1 that was expressed in and purified from E. coli.
Similar to what happens in the catalytic action of RhoGAP on GTP bound to Rac1, RhoGAP is able to facilitate the hydrolysis of the 6-TGTP adduct linked to Rac1 to produce the 6-TGDP-Rac1 adduct (Fig. 5). This result indicates that, like the γ-phosphate of the Rac1-bound GTP, the γ-phosphate of the 6-TGTP-Rac1 adduct also is accessible to and labile for the RhoGAP action. Consequently, it is possible to suggest that, when the 6-TGTP-Rac1 adduct exists in cells, RhoGAP can convert it into the biologically inactive 6-TGDP-adduct.
FIGURE 5.
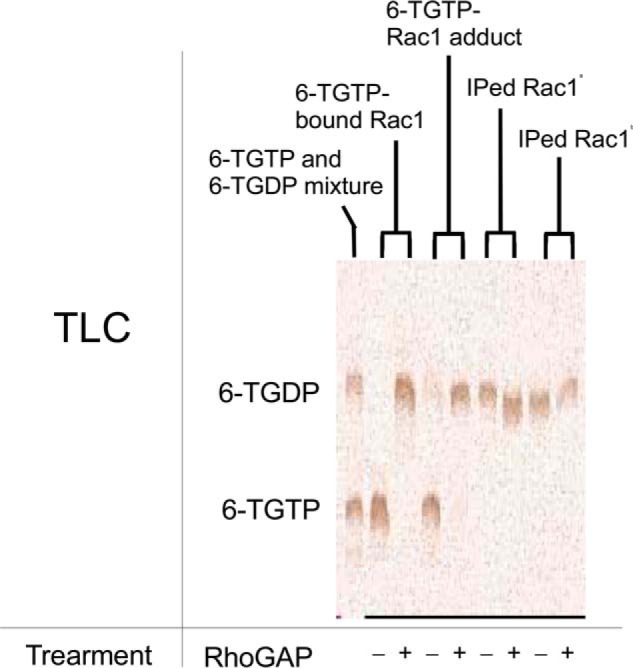
Catalytic function of RhoGAP on the 6-TGTP-Rac1 adduct to produce the 6-TGDP-Rac1 adduct and free phosphate is preserved. TLC analysis for the quantification of 6-TGNP adducted to and bound to Rac1 as well as to the IPed Rac1 after the treatment with and without RhoGAP were conducted as described under “Experimental Procedures.” The far left lane represents the standard TLC indicator of free 6-TGTP and 6-TGDP. The fractions of the 6-TGDP and 6-TGTP were expressed, respectively, as normalized values against the sample value of 6-TGTP-bound Rac1 treatment with and without RhoGAP, in which their band intensities were set at 100%. Hence, the band intensities of fractions of the 6-TGTP-bound Rac1 without RhoGAP were estimated to be 6-TGTP (100%) and 6-TGDP (0%); with RhoGAP, they were estimated to be 6-TGTP (0%) and 6-TGDP (100%). The band intensities of fractions of the 6-TGTP-Rac1 adduct without RhoGAP were estimated to be 6-TGTP (98%) and 6-TGDP (3%); and with RhoGAP they were estimated to be 6-TGTP (1%) and 6-TGDP (89%). The band intensities of fractions of the IPed Rac1 from CD4+ cells treated with only 6-TG without RhoGAP were estimated to be 6-TGTP (0%) and 6-TGDP (78%). With RhoGAP, they were estimated to be 6-TGTP (0%) and 6-TGDP (85%). The band intensities of fractions of the IPed Rac1 from CD4+ cells treated with 6-TG and DETA/NO without RhoGAP were estimated to be 6-TGTP (0%) and 6-TGDP (77%); and with RhoGAP, they were estimated to be 6-TGTP (0%) and 6-TGDP (52%). Standard deviations of the densitometry analyses of the three independent measurements were less than 20% of the values shown. Note that the single band intensity and the sum of the band intensities of 6-TGTP and 6-TGDP are often less than 100%. This is likely because of imperfect spotting concentrations of each sample on the TLC plate. However, the key significance of this analysis is the concentration comparison of 6-TGTP versus 6-TGDP within the lanes but not the concentration comparison of 6-TGTP versus 6-TGDP between the lanes. Therefore, the analysis figures and their corresponding values are certainly valid. a, IPed Rac1, Rac1 IPed from CD4+ cells treated only with 6-TG; and b, IPed Rac1, Rac1 IPed from CD4+ cells treated with 6-TG and DETA/NO.
Notably, Fig. 5 shows that the 6-TGNP adducted to the IPed Rac1 is 6-TGDP, which is insensitive to the catalytic action of RhoGAP. This IPed Rac1 was isolated from CD4+ cells treated with 6-TG and/or DETA/NO. It is unlikely that the IPed Rac1 was originally in the form of the 6-TGTP-Rac1 adduct and subsequently converted into the 6-TGDP-Rac1 adduct during the preparation of the sample. This unlikelihood is because the IPed Rac1 samples used in the analyses in Fig. 5 were prepared at 0 °C, which minimizes the hydrolysis of the temperature-sensitive labile γ-phosphate of 6-TGTP adducted to Rac1 (if it exists). Therefore, detection of 6-TGDP adducted to Rac1 in CD4+ cells (Fig. 5) further supports a conclusion that the biologically inactive form of Rac1 in CD4+ cells treated with 6-TG and/or DETA/NO is yet again the 6-TGDP-Rac1 adduct.
Intriguingly, RhoGAP is also able to perturb the 6-TGTP-Rac1 adduct-binding interaction with PAK-PBD (Fig. 4A). The most plausible reason for such a perturbation is that the RhoGAP-mediated conversion of the 6-TGTP-Rac1 adduct into the 6-TGDP-Rac1 adduct results in a decrease in the population of the 6-TGTP-Rac1 adduct complexed with PAK-PBD. This plausibility takes into account the catalytic function of RhoGAP on the 6-TGTP-Rac1 adduct and the preference of PAK-PBD to bind to the 6-TGTP-Rac1 adduct but not to the 6-TGDP-Rac1 adduct. However, one concern is that the presence of PAK-PBD may block the catalytic function of RhoGAP on the 6-TGTP-Rac1 adduct to produce the 6-TGDP-Rac1 adduct. Nevertheless, detection of the release of Pi from the mixture of the 6-TGTP-Rac1 adduct and PAK-PBD in the presence of RhoGAP, but not from the mixture of the 6-TGTP-Rac1 adduct and PAK-PBD without RhoGAP (Fig. 4B), suggests that despite the presence of PAK-PBD, RhoGAP is able to promote the hydrolysis of the γ-phosphate of the 6-TGTP-Rac1 adduct to produce the 6-TGDP-Rac1 adduct. This then leads to a proposed potential mechanism for the RhoGAP-mediated perturbation of the 6-TGTP-Rac1 adduct binding interaction with PAK-PBD; the catalytic action of RhoGAP on the 6-TGTP-Rac1 adduct produces the 6-TGDP-Rac1 adduct that in turn decreases the population of the 6-TGTP-Rac1 adduct that binds to PAK-PBD.
An alternative possibility that the binding competition between RhoGAP and PAK-PBD for the 6-TGTP-Rac1 adduct results in perturbation of the 6-TGTP-Rac1 binding interaction with PAK-PBD should also be considered. The molecular mass of the RhoGAP used within this study (the catalytic core domain, 29 kDa) is more than that of PAK-PBD (6 kDa). Therefore, the fluorescence intensity of the complex of the 6-TGTP-Rac1 adduct with RhoGAP exceeds that of the complex of the 6-TGTP-Rac1 adduct with PAK-PBD. Accordingly, if RhoGAP competitively displaces the 6-TGTP-Rac1 adduct-bound PAK-PBD to produce the complex of the 6-TGTP-Rac1 adduct with PAK-PBD, this should result, because of the addition of RhoGAP, in an increase in the fluorescence intensity of the spectrum of the 6-TGTP-Rac1 adduct with PAK-PBD. However, after the addition of RhoGAP, the actual fluorescence intensity of the 6-TGTP-Rac1 adduct with PAK-PBD declined (Fig. 4B). This decline makes it highly unlikely that RhoGAP is a factor in the potential competitive-based perturbation of the 6-TGTP-Rac1 adduct-binding interaction with PAK-PBD. Taken together, although further studies are necessary to clarify the mechanism of its occurrence, the RhoGAP-mediated conversion of the 6-TGTP-Rac1 adduct into the 6-TGDP-Rac1 adduct is likely responsible for perturbation of the 6-TGTP-Rac1 adduct binding interaction with PAK-PBD. This concept implies that RhoGAP may play a key role in generation of the biologically inactive 6-TGDP-Rac1 adduct in CD4+ cells treated with 6-TG and/or DETA/NO.
Moreover, Fig. 4B also shows that only minimal Pi was detected from the RhoGAP-treated IPed Rac1 that lacks a binding interaction with PAK-PBD. The results again support the notion that Rac1 in CD4+ cells treated with 6-TG and/or DETA/NO is in the form of the biologically inactive 6-TGDP-Rac1 adduct that does not produce Pi in the presence of RhoGAP.
Minimal Effect of a Redox Agent on the Formation of 6-TGNP-Rac1 Adduct in CD4+ Cells
An endogenously released cellular redox agent could play a role in the formation of the 6-TGNP-Rac1 adduct in cells. This possibility arises because a thiol (or thiolate) does not react with another thiol to produce a disulfide bond unless a radical redox agent (e.g. •NO2 or O2˙̄) is present (13). Cell-based and in vitro biochemical analyses of the effects of an NO donor DETA/NO on the formation of the 6-TGNP-Rac1 adduct were performed to observe any enhancement effect of an exogenously added redox agent on the formation of the 6-TGNP-Rac1 adduct in CD4+ cells.
Additional treatment of cells with DETA/NO, in addition to 6-TG, apparently only slightly increases the fraction of the 6-TGNP-Rac1 adduct (Fig. 3B). However, the increment of the fraction of the 6-TGNP-Rac1 adduct is within the range of 20% of the errors of the values given (from 29 to 38%) (Fig. 3B). These results are essentially similar to the results of the direct measurements of Rac1 activity in CD4+ (Fig. 1B) in which the treatment of CD4+ cells with DETA/NO in addition to 6-TG further diminishes Rac1 activity, yet remains within the range of the errors. Therefore, even if the additional exogenous redox agent NO increases the formation of the 6-TGNP-Rac1 adduct in CD4+ cells, the increment is likely to be marginal. Furthermore, the very few increases in ERM dephosphorylation and IFN-γ secretion of CD4+ cells (Fig. 1B) also were coupled with a negligible increase in the quantity of the 6-TGNP-Rac1 adduct in CD4+ cells by treatment of the CD4+ cells with DETA/NO in addition to 6-TG (Fig. 3B). One possible reason for this insignificant effect of the exogenous redox agent on the production of the cellular 6-TGNP-Rac1 adduct may be that the endogenous redox agents are sufficient to almost maximize the cellular production of the 6-TGNP-Rac1 disulfide adduct; the result of such a maximizing effect may be minimization of the effect of the exogenous redox agent on the 6-TGNP-Rac1 disulfide adduct production in CD4+ cells. Further studies are necessary to clarify this observed negligible effect of the exogenous •NO2 on the formation of the 6-TGNP-Rac1 disulfide adduct in CD4+ cells.
Redox-dependent Formation and Disruption of the 6-TGNP-Rac1 Adduct
Although formation of the 6-TGNP-Rac1 adduct has been detected in CD4+ cells treated with 6-TG, the key chemical feature of the disulfide bond between Rac1 and the bound 6-TGNP necessary to form the 6-TGNP-Rac1 adduct has not been characterized. Redox biochemical analysis for the IPed Rac1 from CD4+ cells has been performed to assess the chemical feature of the key disulfide bond within the 6-TGNP-Rac1 adduct.
The fraction of the 6-TGNP-free IPed Rac1 (Fig. 3B) can be converted into a form of the 6-TGDP-Rac1 adduct by a sequential treatment with 6-TGDP and NO in the presence of Vav in vitro (Fig. 3C). The conversion of the 6-TGNP-free IPed Rac1 into a form of the 6-TGTP-Rac1 adduct can also be achieved by a sequential treatment with 6-TGTP and NO in the presence of Vav (data not shown). Note that neither 6-TG nor a redox agent alters the Vav activity (see below). Therefore, we think that the conversion of the 6-TGNP-free IPed Rac1 into the 6-TGNP-Rac1 adduct could be because the IPed Rac1 was in a native form that allows Vav to enhance the guanine nucleotide exchange (GNE) of the bound GNP with 6-TGNP in the presence of NO to produce the 6-TGNP-bound Rac1. Significantly, treatment with DTT can strip from the IPed Rac1 all 6-TGDPs covalently attached to it (Fig. 3D). It is also true that treatment with DTT can strip from the IPed Rac1 all of the 6-TGTPs covalently attached to it (data not shown). Our hypothesis that 6-TGNP is covalently attached to Rac1 via a disulfide bond certainly explains this result as follows: (i) DTT disrupts the disulfide bond between 6-TGNP and Rac1; (ii) the process of SDS-PAGE, followed by Western blot, perturbed the remaining binding interaction between 6-TGNP and Rac1, and consequently 6-TGNP was released from Rac1 and the liberated 6-TGNP was washed out; (iii) thus, no 6-TGNP remained on the nitrocellulose membrane associated with Rac1 for the anti-6-TGNP antibody to detect. Note that the SDS-PAGE-mediated perturbation of the binding interaction between 6-TGNP and Rac1 is because the SDS-PAGE analysis includes a heat denaturation step. Therefore, as noted above, the result of DTT treatment of the IPed Rac1 after the SDS-PAGE analysis, followed by Western blot (Fig. 3D), should be interpreted as meaning that only DTT is able to disrupt the disulfide bond between 6-TGNP and Rac1, and the SDS-PAGE analysis is then responsible for the release of 6-TGNP from Rac1.
Lack of Inhibitory Effects of 6-TP Prodrugs on the Vav Activity of CD4+ Cells
In addition to the formation of the 6-TGDP-Rac1 adduct, other modes of inactivating 6-TG-mediated Rac1 are possible. These include potential 6-TG-mediated down-regulations of Vav at the gene level and/or by impairment of the catalytic function of the 6-TGNP-bound Rac1. To examine any of these additional possibilities, studies have been performed of the cellular and kinetic properties of Vav from CD4+ cells treated with 6-TG.
Although Rac1 was inactivated by CD4+ cells treated with 6-TG for 3 days (Fig. 1A), the cellular level of Vav expression was conversely increased (Fig. 6A). The fraction of the active form of Vav, pVav (33, 41), was also proportionally increased with the increase in the Vav expression (Fig. 6A). This 6-TP-mediated increase in Vav expression as well as its phosphorylation were also observed in the previous study (11). The unit catalytic activity of the Vav (per nanogram of protein) purified from the 6-TG-treated CD4+ cells on the substrate GDP-bound Rac1 was essentially the same as that of the Vav purified from untreated CD4+ cells (Fig. 6B). This result indicates that treatment with 6-TG has not altered the specific activity of Vav in CD4+ cells. This result in combination with the detection of the biologically inactive 6-TGNP-Rac1 adduct (see above) suggests that 6-TG down-regulates Rac1 activity by directly targeting Rac1 but not Vav.
FIGURE 6.
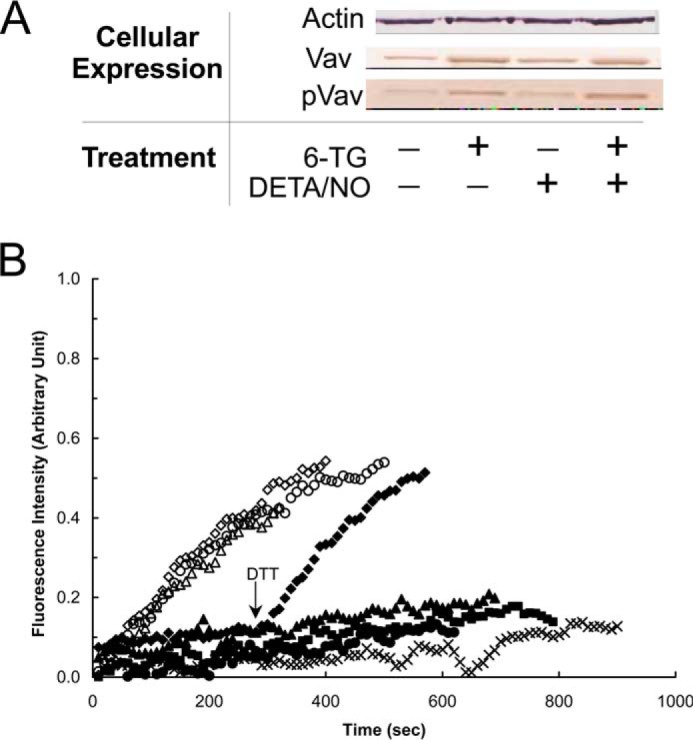
6-TG enhances the expression and phosphorylation of Vav but does not inhibit the specific activity of Vav in CD4+ cells. A, Western analyses were performed for Vav and pVav, respectively, by using anti-Vav and anti-pVav antibodies as described under “Experimental Procedures.” Actin expression was shown as a control. B, fluorescence mant-based kinetic properties of the 6-TGNP-Rac1 adduct, in comparison with the GDP-Rac1, in the presence and absence of various Vav proteins, were examined as described under “Experimental Procedures.” Both the GDP-bound Rac1 and the 6-TGDP-Rac1 adducts were used as substrates of Vav. Three different forms of Vav proteins also were used as follows: (i) Vav from nontreated; (ii) treated only with 6-TG; and (iii) 6-TG and DETA/NO treated CD4+ cells. Accordingly, six different kinetic assays were conducted as follows: the GDP-bound Rac1 with Vav from nontreated CD4+ cells (▵); the GDP-bound Rac1 with Vav from CD4+ cells treated with 6-TG (♢); the GDP-bound Rac1 with Vav from CD4+ cells treated with both 6-TG and DETA/NO (○); the 6-TGDP-Rac1 adduct with Vav from nontreated CD4+ cells (▴); the 6-TGDP-Rac1 adduct with Vav from CD4+ cells treated with 6-TG (♦); and the 6-TGDP-Rac1 adduct with Vav from CD4+ cells treated with both 6-TG and DETA/NO (●). The kinetic assay controls, the GDP-bound Rac1 without Vav (■) and the 6-TGDP-Rac1 adduct without Vav (×), were also conducted. Note that DTT (10 mm) was added to the assay cuvette containing the 6-TGDP-Rac1 adduct with Vav from CD4+ cells treated with 6-TG at time 300 s as indicated by an arrow. All fluorescence spectra were normalized against the spectrum of the GDP-bound Rac1 with Vav from 6-TG and DETA/NO treated CD4+ cells because it shows the highest fluorescence intensity at time 900 s.
The 6-TGDP-Rac1 adduct was minimally affected by the catalytic activity of the Vav directly purified from CD4+ cells treated with 6-TG; however, adding DTT enables this purified Vav to dissociate 6-TGDP from Rac1 (Fig. 6B). Similarly, the Vav protein was not able to dissociate 6-TGTP from Rac1 in the absence of DTT but was able to dissociate 6-TGTP from Rac1 in the presence of DTT (data not shown). We contend that the disulfide bond between 6-TGNP and Rac1 hinders the catalytic action of the Vav purified from CD4+ cells and that whenever the disulfide bond is disrupted (i.e. by DTT), the Vav can enhance Rac1 6-TGNP dissociation.
Lack of Effects of a Redox Agent on the Vav Expression and Activity in CD4+ Cells
Given that a redox agent also is involved in the formation of the 6-TGDP-Rac1 adduct, further investigation by using cell-based biochemical analyses has been performed to examine any potential effect of a redox agent with and without 6-TG on the Vav activity. This is necessary to comprehensively understand the effect of 6-TG in combination with NO on Rac1 inactivation in CD4+ cells.
DETA/NO, regardless of the presence and absence of 6-TG, had a minor effect on the expression and phosphorylation of the Vav in CD4+ cells (Fig. 6). Similarly, DETA/NO had no effect on the specific activity of the Vav in CD4+ cells (Fig. 6). This is consistent with the previous result that showed that the catalytic function of Vav is redox-insensitive (42). These results together suggest that the redox signaling associated with NO and its derivatives (e.g. •NO2) has no role in either the expression of Vav or in the regulation of its activity.
Minimal Effects of 6-TG and/or a Redox Agent on the RhoA and Cdc42 Activity of CD4+ Cells
To a limited extent, the essential structural feature of Rac1 for the 6-TG-mediated Rac1 inactivation in CD4+ cells is the redox-sensitive GXXXXGK(S/T)C motif. Intriguingly, however, RhoA and CDc42 that are likewise involved in the regulation of CD4+ cell activity (43, 44) also possess the GXXXXGK(S/T)C motif (23). Our IP-based ESI-MS and MS/MS also identified the 6-TGNP-RhoA and 6-TGNP-Cdc42 disulfide adduct from the CD4+ cells treated with 6-TG (data not shown). Hence, it is reasonable to postulate that 6-TGNP targets and reacts with RhoA and CDc42 to produce the 6-TGNP-RhoA and -Cdc42 disulfide adducts, respectively. These adduct formations could result in inactivation of RhoA or Cdc42. If so, this would then induce inactivation of CD4+ cells. To assess whether the formation of the 6-TGNP-RhoA and -Cdc42 adducts causes CD4+ cell inactivation, the effects of 6-TG on Rac1 activity in CD4+ cells and on RhoA and Cdc42 activities as well as the production of cytokine IFN-γ were examined after the CD4+ cells were treated with 6-TG and a redox agent.
The IFN-γ secretion of CD4+ cells was decreased by inhibition of the activity of RhoA and Cdc42 by using their specific inhibitor (Fig. 7). This result suggests that, in addition to Rac1 (11), the inactivation of RhoA and Cdc42 results in reduction of the secretion of IFN-γ. Intriguingly, 6-TG with DETA/NO significantly reduced the IFN-γ secretion of CD4+ cells (Fig. 7). It is, however, notable that 6-TG with DETA/NO affected the activity of RhoA and Cdc42 in CD4+ cells (Fig. 7), but this effect was comparatively less significant than the effect on the activity of Rac1 (Fig. 1). Accordingly, the declination of the IFN-γ secretion was because of the inactivation of RhoA and Cdc42. Accounting for this (see above) and in the previous study (11), the reduction of the IFN-γ secretion was likely because of the Rac1 inactivation.
FIGURE 7.

6-TG with or without DETA/NO does not inhibit the activity of RhoA and Cdc42 of CD4+ cells. The RhoA and Cdc42 activities as well as the secretion of IFN-γ from CD4+ cells in each treatment were determined as described under “Experimental Procedures.” As positive controls, CD4+ cells treated with 6-TG and/or DETA/NO were also treated with C3 exotoxin (purified C3 transferase; the RhoA inhibitor; 30 μg/ml), and/or secramine B (the Cdc42 inhibitor; 20 μm) for 3 days. Total expression of RhoA and Cdc42 was determined by Western analyses using RhoA and Cdc42. The treatment of these inhibitors did not alter the expression level of total RhoA and Cdc42 (data not shown). The value of the activity of RhoA and Cdc42 as well as the quantity of IFN-γ determined from the untreated sample was set as 100%, and other results were then expressed as normalized values against the value associated with the untreated sample. Data are shown with the mean values and S.D. bars associated with the three independent experiments. Statistical results obtained by using the ANOVA Tukey test were as follows: *, p < 0.01, versus the RhoA activity of the untreated sample; **, p < 0.01, versus the Cdc42 activity of the untreated sample; and ***, p < 0.05, versus the quantity of IFN-γ secreted from the untreated sample.
Nevertheless, these analyses suggest that, unlike in the case of Rac1, although both RhoA and Cdc42 have the GXXXXGK(S/T)C motif, they are not the main target of 6-TG with a redox agent. This enigmatic result can be explained by a previous three-part suggestion (11). This suggestion is as follows: (i) unlike with the Rac1 GEF Vav, the GEF(s) of RhoA and Cdc42 was (were) minimally activated in CD4+ cells; (ii) 6-TGNP was minimally loaded onto RhoA and Cdc42, and this minimal loading diminishes formation of the 6-TGNP-RhoA and -Cdc42 disulfide adduct in CD4+ cells; and (iii) the diminished formation of this adduct lessens the effect of 6-TGNP on the activity of RhoA and Cdc42.
Inhibitory Effects of 6-TG and a Redox Agent on Development of CD4+ Cells
The previous sections investigated how 6-TG with a redox agent inactivates Rac1 and its consequent inactivation of CD4+ cells. Questions could also be posed about the effects of 6-TG with a redox agent on other CD4+ cell characteristics, such as T-cell development. T-cell proliferation and survival are feature indicators of T-cell development. Accordingly, the effects of 6-TG with a redox agent on the development, proliferation, and survival of CD4+ cells after treatment with 6-TG and/or a redox agent have been examined.
Proliferation of CD4+ cells was suppressed by treatment with 6-TG (Fig. 8). Viability of CD4+ cells also was diminished by treatment with 6-TG (Fig. 9A). Nevertheless, CD4+ cells treated with 6-TG increased the caspase activity of CD4+ cells (Fig. 9B). The decrease in T-cell viability and the increase in the caspase activity signify the diminution of T-cell survival. These results suggest that 6-TG inhibits T-cell development. Studies show that the Rac1 activity is involved in T-cell proliferation and viability (45–47). Within this study, we provide evidence that the 6-TG-mediated Rac1 inactivation is because of the formation of the inactive 6-TGDP-Rac1 adduct in CD4+ cells. Taken together, although other possibilities are open to investigation, the results lead to a suggestion that, at a minimum, the 6-TG-mediated formation of the inactive 6-TGDP-Rac1 adduct deters or inhibits T-cell development by suppressing the proliferation and diminution of the viability of T-cells.
FIGURE 8.

6-TG blocks the DETA/NO-mediated T-cell stimulation. Fluorescence-based determination of CD4+ cell proliferations was performed as indicated under “Experimental Procedures.” The fluorescence value of the untreated sample was set as 1, and all other results were then expressed as normalized values against the untreated sample value initially established. Data are shown with the mean values and S.D. bars associated with the three independent experiments. The statistical ANOVA Tukey test results were as follows: *, p < 0.05, versus the result of the untreated sample; and **, p < 0.01, versus the result of the sample treated only with DETA/NO.
FIGURE 9.

6-TG in combination with DETA/NO decreases viability and increases apoptosis of CD4+ cells. A, MTT-based CD4+ cell viability assays in each treatment were conducted as described under “Experimental Procedures.” The viability value of CD4+ cells of the untreated sample was set as 100%, and other results were then expressed as normalized values against the untreated sample value. Data are shown with the mean and S.D. bars associated with three independent experiments. Statistics were performed with a Tukey ANOVA. *, p < 0.05, versus the result of the untreated sample. B, colorimetric-based apoptotic assays of CD4+ cells in each treatment were performed as described under “Experimental Procedures.” The caspase activity of a sample treated with the apoptotic inducer was set as 100%, and other results were then expressed as normalized values against this value. Data are shown with the mean values and S.D. bars associated with the three independent experiments. Statistics were performed with a Tukey ANOVA: *, p < 0.01, versus the result of the untreated sample; and **, p < 0.05, versus the result of the apoptotic inducer-treated sample.
Proliferation of CD4+ cells was significantly enhanced by treatment with DETA/NO alone (Fig. 8). The effect of NO can be blocked by treatment with the soluble guanylyl cyclase inhibitor, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (Fig. 8). Hence, T-cell proliferation mediated by DETA/NO is likely because NO, released from DETA/NO, targets soluble guanylyl cyclase, resulting in stimulation of CD4+ cell proliferation (48). Unlike with CD4+ cell proliferation, NO had almost no effect on the viability and caspase activity of CD4+ cells (Fig. 9, A and B). These results together suggest that NO alone enhances CD4+ cell proliferation but does not alter CD4+ cell survival.
Interestingly, 6-TG-mediated suppression of CD4+ cell proliferation was not enhanced by additional treatment with DETA/NO (Fig. 8). 6-TG-mediated diminution of CD4+ cell survival also was not enhanced by additional treatment with DETA/NO (Fig. 9, A and B). The notion that the inactive 6-TGDP-Rac1 adduct is at least to some extent responsible for the diminution of CD4+ cell survival can be explained by evidence that the endogenous redox agents may be sufficient to produce the maximal content of the 6-TGNP-Rac1 disulfide adduct in CD4+ cells (see under “Minimal Effect of a Redox Agent on the Formation of 6-TGNP-Rac1 Adduct in CD4+ Cells”). However, abolition of the stimulatory NO effect on CD4+ cell proliferation by 6-TG treatment cannot be explained by the above notion. It is possible that the stimulatory effect of NO on CD4+ cell proliferation is somehow overwhelmed by the 6-TG-mediated suppressive effect of Rac1 inactivation. Nevertheless, further studies are necessary to better understand the combined effects of 6-TG and a redox agent on T-cell development.
Discussion
This study has shown that 6-TGNP converted from 6-TP prodrugs in CD4+ cells targets and reacts with Rac1 to form the 6-TGNP-Rac1 adduct, resulting in a blockade of the Rac1 activation.
Proposed Molecular Mechanism of 6-TP-mediated Blockage of Rac1 Activation
We have shown that 6-TGNP that is cellularly converted from the 6-TP prodrug targets Rac1 to produce the 6-TGNP-Rac1 adduct. The feature form of this adduct is a covalent link between the redox-sensitive sulfur atom of the side chain of Cys18 of the Rac1 GXXXXGK(S/T)C motif and the sulfur atom of the bound 6-TGNP. The crystal structure of the GDP-bound Rac1 shows that the sulfur atom of the side chain of Cys18 of Rac1 is located at the Rac1 nucleotide-binding site. This site is ∼6.9 Å apart from the bound GDP position C-6 oxygen atom (49). Because the C-6 oxygen atom of GDP is equivalent to the sulfur atom of the bound 6-TGDP, its equivalency supports the structural possibility that a disulfide bond will be formed between the sulfur atom of the side chain of Cys18 of Rac1 and the sulfur atom of the bound 6-TGNP. Given that formation of a protein disulfide bond is an oxidation process that is facilitated by a redox agent (13), the formation of the 6-TGNP-Rac1 adduct is likely redox-dependent. We also have shown that the formation of the 6-TGNP-Rac1 adduct dovetails with the inactivation of CD4+ cells. We have further shown that, unless the disulfide bond between 6-TGNP and Rac1 is otherwise disrupted by a reduction agent, a RhoGEF, such as Vav, is unable to displace the covalently linked 6-TGNP to Rac1 with the free cellularly available GNP and/or 6-TGNP.
Based on these results and analyses, we propose a detailed chemistry-based molecular mechanism for the 6-TP-mediated blockage of the Rac1 GNE in CD4+ cells (Fig. 10). When 6-TGNP binds to Rac1 instead of GNP, the sulfur atom of the bound 6-TGNP is near the sulfur atom of the side chain of Cys18 of Rac1. When a redox agent is available in these conditions, the 6-TGNP sulfur atom reacts with the sulfur atom of the Cys18 side chain of Rac1 to produce a 6-TGNP-Rac1 disulfide adduct. This disulfide then blocks the GEF-mediated GNE of Rac1. This proposed chemistry-based molecular mechanism explains the previous enigmatic observation in which the observed blockage of the Vav-mediated GNE of the 6-TGNP-bound Rac1 (5, 11) is due to the reactivity of 6-TGNP with the Rac1 Cys18 to produce a 6-TGNP-Rac1 adduct that, in turn, blocks the action of Vav for Rac1 GNE.
FIGURE 10.

Chemistry-based molecular mechanism of the formation of the Rac1–6-TGNP disulfide adduct is proposed. 6-TGNP and a disulfide linkage are represented in blue and red, respectively. The dotted lines in black represent putative hydrogen bond interactions between protein residues and 6-TGNP. Red, reduction; and Ox, oxidation.
Proposed Cellular Mechanism of 6-TP-mediated CD4+ Cell Inactivation
We have shown that, among the 6-TGNP-Rac1 adduct species, the 6-TGDP- and the 6-TGTP-Rac1 adducts, the 6-TGDP-Rac1 adduct is biologically inactive, but the 6-TGTP-Rac1 adduct is biologically active. An inclusive five-step cellular mechanism (Fig. 11) of the action of 6-TP for the inactivation of CD4+ cells can be proposed based on previous studies (5, 11) and on the proposed chemistry-based molecular mechanism of the formation of the 6-TGNP-Rac1 adduct (Fig. 10) in combination with the catalytic action of RhoGAP on the 6-TGTP-Rac1 adduct. In this scenario, (i) the treated 6-TP prodrugs are converted into 6-TGNP in cells by (ii) the action of RhoGEF, and Rac1 is loaded with 6-TGNP to produce a 6-TGNP-bound Rac1; (iii) next a cellular redox agent facilitates the reaction of the bound 6-TGNP with the side chain of Cys18 of Rac1 to produce a 6-TGNP-Rac1 disulfide adduct; (iv) whenever the covalently linked 6-TGNP is in a form of 6-TGTP, by the action of RhoGAP, the linked 6-TGTP is converted into 6-TGDP to produce a biologically inactive 6-TGDP-bound Rac1; and (v) because the 6-TGDP is covalently linked to Rac1, RhoGEF is unable to displace the linked 6-TGDP with the cellular free GNP and/or 6-TGNP. The continuation of the cycle of i–v accumulates the biologically inactive 6-TGDP-Rac1 adduct, resulting in inactivation of CD4+ cells. The covalently linked 6-TGDP can be liberated by the action of cellular reducing agents. However, the cellular regulatory mechanism of the action of reducing agents on the 6-TGDP-Rac1 disulfide adduct remains to be clarified.
FIGURE 11.

Cellular mechanism of 6-TP-mediated inactivation CD4+ cells.
Potential Mechanism-based Improvement of 6-TP Prodrugs and Scheme of New Drugs That Target Rac1
The proposed mechanism (Fig. 10) revealed that a redox agent is also necessary to cross-link 6-TGNP with Rac1. Despite a cellularly present redox agent, such as the •NO2 derived from the NO released from nitric-oxide synthase, additional exogenous redox agents may enhance the disulfide adduction formation between 6-TGNP and Rac1 by administering an NO-releasing agent in combination with 6-TP prodrugs. Such a potential optimization of 6-TP prodrugs will also need to be investigated.
The mechanistic features of the inhibitory action of 6-TGNP on Rac1 (Fig. 10) also open possibilities for the design of new drugs. This possibility includes GNP analogs that can bind to Rho GTPases, yet they have a functional group capable of reacting with the redox-sensitive cysteine residue in the GTPase GXXXXGK(S/T)C motif to form a GTPase adduct. Further investigation is expected of the mechanism-based development of new drugs that target Rho GTPases.
Potential Cytotoxicities of 6-TP Prodrugs That Target RhoA and Cdc42
This study shows that, in addition to Rac1, other Rho GTPases, such as RhoA and Cdc42 in CD4+ cells, are also targets of 6-TGNP. We think this is because both RhoA and Cdc42 also possess the GXXXXGK(S/T)C motif. We have also previously observed that 6-TGNP forms a disulfide bond with the side chain of Cys20 (equivalent to Rac1 Cys18) in the GXXXXGK(S/T)C motif of RhoC in tumor cells (derived from inflammatory breast cancer). This disulfide bond terminates the RhoC-mediated cell metastasis (28). These results together suggest that 6-TGNP does not just target a specific Rho GTPase (e.g. Rac1) but also any GTPases that have the GXXXXGK(S/T)C motif. Our postulation for the apparent 6-TGNP preference for Rac1 over RhoA and Cdc42 of 6-TGNP in T-cells is simply because T-cells have more Rac1 than they do RhoA and Cdc42. Consequently, by chance, 6-TGNP targets Rac1 more often than it does RhoA and Cdc42.
Because both RhoA and Cdc42 are also targets of 6-TGNP, cytotoxicities associated with the 6-TGNP-mediated inactivation of RhoA and Cdc42 are certainly possible. These cytotoxicities could be potentially significant if RhoA and/or Cdc42 is involved in a critical cell signaling event(s) that controls the function of key organs. For example, it is expected that 6-TGNP derived from 6-TP prodrugs is present not only in T-cells but also in the cells of other organs in patients, such as in their hearts and blood vessels. Notably, RhoA has diverse functions in the vasculature. Consequently, its misregulation results in various vascular portal hypertensions (50). Accordingly, it can be speculated that 6-TGNP derived from 6-TP induces vascular diseases through its targeting of RhoA in the cells of the heart and blood vessels. In fact, cardiovascular disease and portal hypertension in patients on 6-TP prodrugs are regarded as two of their key adverse cytotoxicities (51, 52). However, no one has yet investigated the association between 6-TP prodrugs and the 6-TGNP targeting of RhoA in terms of the mechanism by which these vascular diseases develop. Further study is necessary to determine whether the action of 6-TGNP is truly the culprit in these diseases so as to avoid or at least control these presumed cytotoxicities associated with 6-TP prodrugs.
Unlike the sulfur atom of the redox-sensitive cysteine residue (i.e. Cys18 of Rac1) in the GXXXXGK(S/T)C motif of GTPases, the sulfur atom of the redox-sensitive cysteine residue (i.e. of Cys118 of Ras) in the NKCD motif of GTPases is remote from the GTPase nucleotide-binding site (53). Hence, formation of a disulfide bond between the bound 6-TGNP and the redox-sensitive cysteine residue in the NKCD motif of GTPases is implausible. Also, our recent separate study suggests that 6-TGNP in combination with a redox agent rather enhances the Ras GNE to produce 5-guanidino-4-nitroimidazole diphosphate but does not produce a 6-TGNP-Ras adduct (54). Hence, formation of the TGNP-GTPase disulfide (e.g. a 6-TGNP-Rac1 adduct) at the nucleotide-binding site is likely to be specific for GTPases that possess the GXXXXGK(S/T)C motif but not the NKCD motif. Accordingly, given that many Rho and Rab GTPases possess the GXXXXGK(S/T)C motif, the findings within this study are significant because generation of the 6-TGNP-GTPase adduct by a combination of 6-TP and a redox agent could be a useful strategy for treating diseases specifically related to misregulation of Rho and Rab GTPases.
Other Potential Targets and Cytotoxicities of 6-TP Prodrugs
This study first tried to link the direct Rac1-targeting action of 6-TG to production of the inactive 6-TGDP-Rac1 adduct with the diminution of the proliferation and survival of T-cells by the 6-TG treatment. However, many other possibilities, such as an indirect 6-TG-mediated Rac1 down-regulation to diminish the proliferation and survival of T-cells, also must be considered. For example, as noted above, 6-TG inactivates Ras via targeting its NKCD motif, and Ras is an upstream of Rac1 (13). Therefore, it is possible that the 6-TG-mediated Ras inactivation cascades down to further inhibit Rac1 activity. It is also possible that the 6-TG-mediated decrease in T-cell proliferation and survival may be because of the DNA-targeting therapeutic action of 6-TG in which incorporation of 6-TG into DNA interferes with the action of the mismatch repair system (7, 8, 10). Further studies to configure the mechanism of the 6-TG-mediated diminution of the proliferation and survival of T-cells will be necessary.
Author Contributions
J. Y. S., M. W., and H. G. U. designed, performed, and analyzed the experiments shown in all figures. X. S. and J. S. provided technical assistance and contributed to the preparation of the figures. J. H. planned and coordinated the study and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by National Institutes of Health Grant 1R15AI096146-01A1 (to J. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- 6-TP
- 6-thiopurine
- 6-MP
- 6-mercaptopurine
- 6-TG
- 6-thioguanine
- 6-TGDP
- 6-thioguanosine diphosphate
- 6-TGNP
- 6-thioguanine nucleotide
- 6-TGNP
- 6-thioguanosine phosphate
- 6-TGTP
- 6-thioguanosine triphosphate
- 6-TGTP
- 6-thioguanosine triphosphate
- ANOVA
- analysis of variance
- DETA/NO
- diethylenetriamine/nitric oxide adduct
- ERM
- Ezrin-Radixin-Moesin
- ESI-MS
- electrospray mass spectrometry
- GAP
- GTPase-activating protein
- GEF
- guanine nucleotide exchange factor
- GNE
- guanine nucleotide exchange
- IPed
- immunoprecipitated
- mant
- 2′,3′-O-(N′-methylanthraniloyl)
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- N2O3
- dinitrogen trioxide
- •NO2
- nitrogen dioxide
- O2˙̄
- superoxide anion radical
- pERM
- ERM phosphorylation
- pVav
- phosphorylated Vav
- PAK-PBD
- p21 activated kinase 1 protein-binding domain.
References
- 1. Langmuir P. B., Aplenc R., and Lange B. J. (2001) Acute myeloid leukaemia in children. Best Pract. Res. Clin. Haematol. 14, 77–93 [DOI] [PubMed] [Google Scholar]
- 2. Elion G. B. (1989) The purine path to chemotherapy. Science 244, 41–47 [DOI] [PubMed] [Google Scholar]
- 3. Gearry R. B., and Barclay M. L. (2005) Azathioprine and 6-mercaptopurine pharmacogenetics and metabolite monitoring in inflammatory bowel disease. J. Gastroenterol. Hepatol. 20, 1149–1157 [DOI] [PubMed] [Google Scholar]
- 4. McDonald E. R. 3rd., Wu G. S., Waldman T., and El-Deiry W. S. (1996) Repair defect in p21 WAF1/CIP1−/− human cancer cells. Cancer Res. 56, 2250–2255 [PubMed] [Google Scholar]
- 5. Tiede I., Fritz G., Strand S., Poppe D., Dvorsky R., Strand D., Lehr H. A., Wirtz S., Becker C., Atreya R., Mudter J., Hildner K., Bartsch B., Holtmann M., Blumberg R., et al. (2003) CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J. Clin. Invest. 111, 1133–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Boer N. K., van Bodegraven A. A., Jharap B., de Graaf P., and Mulder C. J. (2007) Drug insight: pharmacology and toxicity of thiopurine therapy in patients with IBD. Nat. Clin. Pract. Gastroenterol. Hepatol. 4, 686–694 [DOI] [PubMed] [Google Scholar]
- 7. Lage H., and Dietel M. (1999) Involvement of the DNA mismatch repair system in antineoplastic drug resistance. J. Cancer Res. Clin. Oncol. 125, 156–165 [DOI] [PubMed] [Google Scholar]
- 8. Yan T., Berry S. E., Desai A. B., and Kinsella T. J. (2003) DNA mismatch repair (MMR) mediates 6-thioguanine genotoxicity by introducing single-strand breaks to signal a G2-M arrest in MMR-proficient RKO cells. Clin. Cancer Res. 9, 2327–2334 [PubMed] [Google Scholar]
- 9. Karran P. (2006) Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br. Med. Bull. 79–80, 153–170 [DOI] [PubMed] [Google Scholar]
- 10. Karran P., and Attard N. (2008) Thiopurines in current medical practice: molecular mechanisms and contributions to therapy-related cancer. Nat. Rev. Cancer 8, 24–36 [DOI] [PubMed] [Google Scholar]
- 11. Poppe D., Tiede I., Fritz G., Becker C., Bartsch B., Wirtz S., Strand D., Tanaka S., Galle P. R., Bustelo X. R., and Neurath M. F. (2006) Azathioprine suppresses ezrin-radixin-moesin-dependent T cell-APC conjugation through inhibition of Vav guanosine exchange activity on Rac proteins. J. Immunol. 176, 640–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wennerberg K., Rossman K. L., and Der C. J. (2005) The Ras superfamily at a glance. J. Cell Sci. 118, 843–846 [DOI] [PubMed] [Google Scholar]
- 13. Heo J. (2011) Redox control of GTPases: from molecular mechanisms to functional significance in health and disease. Antioxid. Redox Signal. 14, 689–724 [DOI] [PubMed] [Google Scholar]
- 14. Sato M., Kawamata H., Harada K., Nakashiro K., Ikeda Y., Gohda H., Yoshida H., Nishida T., Ono K., Kinoshita M., and Adachi M. (1997) Induction of cyclin-dependent kinase inhibitor, p21WAF1, by treatment with 3,4-dihydro-6-[4-(3,4)-dimethoxybenzoyl)-1-piperazinyl]-2(1H)-quinoline (vesnarinone) in a human salivary cancer cell line with mutant p53 gene. Cancer Lett. 112, 181–189 [DOI] [PubMed] [Google Scholar]
- 15. Geyer M., and Wittinghofer A. (1997) GEFs, GAPs, GDIs and effectors: taking a closer (3D) look at the regulation of Ras-related GTP-binding proteins. Curr. Opin. Struct. Biol. 7, 786–792 [DOI] [PubMed] [Google Scholar]
- 16. Bos J. L., Rehmann H., and Wittinghofer A. (2007) GEFs and GAPs: critical elements in the control of small G proteins. Cell 129, 865–877 [DOI] [PubMed] [Google Scholar]
- 17. Budzyn K., Marley P. D., and Sobey C. G. (2006) Targeting Rho and Rho-kinase in the treatment of cardiovascular disease. Trends Pharmacol. Sci. 27, 97–104 [DOI] [PubMed] [Google Scholar]
- 18. Shimokawa H., and Rashid M. (2007) Development of Rho-kinase inhibitors for cardiovascular medicine. Trends Pharmacol. Sci. 28, 296–302 [DOI] [PubMed] [Google Scholar]
- 19. van Golen K. L., Davies S., Wu Z. F., Wang Y., Bucana C. D., Root H., Chandrasekharappa S., Strawderman M., Ethier S. P., and Merajver S. D. (1999) A novel putative low-affinity insulin-like growth factor-binding protein, LIBC (lost in inflammatory breast cancer), and RhoC GTPase correlate with the inflammatory breast cancer phenotype. Clin. Cancer Res. 5, 2511–2519 [PubMed] [Google Scholar]
- 20. Nakamoto M., Teramoto H., Matsumoto S., Igishi T., and Shimizu E. (2001) K-ras and rho A mutations in malignant pleural effusion. Int. J. Oncol. 19, 971–976 [DOI] [PubMed] [Google Scholar]
- 21. Suwa H., Ohshio G., Imamura T., Watanabe G., Arii S., Imamura M., Narumiya S., Hiai H., and Fukumoto M. (1998) Overexpression of the rhoC gene correlates with progression of ductal adenocarcinoma of the pancreas. Br. J. Cancer 77, 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abe K., Rossman K. L., Liu B., Ritola K. D., Chiang D., Campbell S. L., Burridge K., and Der C. J. (2000) Vav2 is an activator of Cdc42, Rac1, and RhoA. J. Biol. Chem. 275, 10141–10149 [DOI] [PubMed] [Google Scholar]
- 23. Heo J., and Campbell S. L. (2005) Mechanism of redox-mediated guanine nucleotide exchange on redox-active Rho GTPases. J. Biol. Chem. 280, 31003–31010 [DOI] [PubMed] [Google Scholar]
- 24. Heo J., Raines K. W., Mocanu V., and Campbell S. L. (2006) Redox regulation of RhoA. Biochemistry 45, 14481–14489 [DOI] [PubMed] [Google Scholar]
- 25. Stubbe J., and van Der Donk W. A. (1998) Protein radicals in enzyme catalysis. Chem. Rev. 98, 705–762 [DOI] [PubMed] [Google Scholar]
- 26. Augusto O., Bonini M. G., Amanso A. M., Linares E., Santos C. C., and De Menezes S. L. (2002) Nitrogen dioxide and carbonate radical anion: two emerging radicals in biology. Free Radic. Biol. Med. 32, 841–859 [DOI] [PubMed] [Google Scholar]
- 27. Heo J., Prutzman K. C., Mocanu V., and Campbell S. L. (2005) Mechanism of free radical nitric oxide-mediated Ras guanine nucleotide dissociation. J. Mol. Biol. 346, 1423–1440 [DOI] [PubMed] [Google Scholar]
- 28. Heo J., Wey M., and Hong I. (2011) Insight into the 6-thiopurine-mediated termination of the invasive motility of tumor cells derived from inflammatory breast cancer. Biochemistry 50, 5731–5742 [DOI] [PubMed] [Google Scholar]
- 29. Heo J., Thapar R., and Campbell S. L. (2005) Recognition and activation of Rho GTPases by vav1 and vav2 guanine nucleotide exchange factors. Biochemistry 44, 6573–6585 [DOI] [PubMed] [Google Scholar]
- 30. Heo J., and Campbell S. L. (2006) Ras regulation by reactive oxygen and nitrogen species. Biochemistry 45, 2200–2210 [DOI] [PubMed] [Google Scholar]
- 31. Downward J., Graves J. D., Warne P. H., Rayter S., and Cantrell D. A. (1990) Stimulation of p21ras upon T-cell activation. Nature 346, 719–723 [DOI] [PubMed] [Google Scholar]
- 32. Geladopoulos T. P., Sotiroudis T. G., and Evangelopoulos A. E. (1991) A malachite green colorimetric assay for protein phosphatase activity. Anal. Biochem. 192, 112–116 [DOI] [PubMed] [Google Scholar]
- 33. Crespo P., Schuebel K. E., Ostrom A. A., Gutkind J. S., and Bustelo X. R. (1997) Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vav proto-oncogene product. Nature 385, 169–172 [DOI] [PubMed] [Google Scholar]
- 34. Lenzen C., Cool R. H., Prinz H., Kuhlmann J., and Wittinghofer A. (1998) Kinetic analysis by fluorescence of the interaction between Ras and the catalytic domain of the guanine nucleotide exchange factor Cdc25Mm. Biochemistry 37, 7420–7430 [DOI] [PubMed] [Google Scholar]
- 35. Vistica D. T., Skehan P., Scudiero D., Monks A., Pittman A., and Boyd M. R. (1991) Tetrazolium-based assays for cellular viability: a critical examination of selected parameters affecting formazan production. Cancer Res. 51, 2515–2520 [PubMed] [Google Scholar]
- 36. Cernuda-Morollón E., Millán J., Shipman M., Marelli-Berg F. M., and Ridley A. J. (2010) Rac activation by the T-cell receptor inhibits T cell migration. PLoS One 5, e12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li B., Yu H., Zheng W., Voll R., Na S., Roberts A. W., Williams D. A., Davis R. J., Ghosh S., and Flavell R. A. (2000) Role of the guanosine triphosphatase Rac2 in T helper 1 cell differentiation. Science 288, 2219–2222 [DOI] [PubMed] [Google Scholar]
- 38. Azim A. C., Barkalow K. L., and Hartwig J. H. (2000) Determination of GTP loading on Rac and Cdc42 in platelets and fibroblasts. Methods Enzymol. 325, 257–263 [DOI] [PubMed] [Google Scholar]
- 39. Lamarche N., and Hall A. (1994) GAPs for rho-related GTPases. Trends Genet. 10, 436–440 [DOI] [PubMed] [Google Scholar]
- 40. Zalcman G., Dorseuil O., Garcia-Ranea J. A., Gacon G., and Camonis J. (1999) RhoGAPs and RhoGDIs, (His)stories of two families. Prog. Mol. Subcell. Biol. 22, 85–113 [DOI] [PubMed] [Google Scholar]
- 41. Michel F., Grimaud L., Tuosto L., and Acuto O. (1998) Fyn and ZAP-70 are required for Vav phosphorylation in T cells stimulated by antigen-presenting cells. J. Biol. Chem. 273, 31932–31938 [DOI] [PubMed] [Google Scholar]
- 42. Wey M., Phan V., Yepez G., and Heo J. (2014) Superoxide inhibits guanine nucleotide exchange factor (GEF) action on Ras, but not on Rho, through desensitization of Ras to GEF. Biochemistry 53, 518–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Smits K., Iannucci V., Stove V., Van Hauwe P., Naessens E., Meuwissen P. J., Ariën K. K., Bentahir M., Plum J., and Verhasselt B. (2010) Rho GTPase Cdc42 is essential for human T-cell development. Haematologica 95, 367–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Skelton T. S., Tejpal N., Gong Y., Kloc M., and Ghobrial R. M. (2010) Downregulation of RhoA and changes in T cell cytoskeleton correlate with the abrogation of allograft rejection. Transpl. Immunol. 23, 185–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guo F., Cancelas J. A., Hildeman D., Williams D. A., and Zheng Y. (2008) Rac GTPase isoforms Rac1 and Rac2 play a redundant and crucial role in T-cell development. Blood 112, 1767–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dumont C., Corsoni-Tadrzak A., Ruf S., de Boer J., Williams A., Turner M., Kioussis D., and Tybulewicz V. L. (2009) Rac GTPases play critical roles in early T-cell development. Blood 113, 3990–3998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hofbauer S. W., Krenn P. W., Ganghammer S., Asslaber D., Pichler U., Oberascher K., Henschler R., Wallner M., Kerschbaum H., Greil R., and Hartmann T. N. (2014) Tiam1/Rac1 signals contribute to the proliferation and chemoresistance, but not motility, of chronic lymphocytic leukemia cells. Blood 123, 2181–2188 [DOI] [PubMed] [Google Scholar]
- 48. Niedbala W., Wei X. Q., Campbell C., Thomson D., Komai-Koma M., and Liew F. Y. (2002) Nitric oxide preferentially induces type 1 T cell differentiation by selectively up-regulating IL-12 receptor β2 expression via cGMP. Proc. Natl. Acad. Sci. U.S.A. 99, 16186–16191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hirshberg M., Stockley R. W., Dodson G., and Webb M. R. (1997) The crystal structure of human rac1, a member of the rho-family complexed with a GTP analogue. Nat. Struct. Biol. 4, 147–152 [DOI] [PubMed] [Google Scholar]
- 50. Loirand G., and Pacaud P. (2010) The role of Rho protein signaling in hypertension. Nat. Rev. Cardiol. 7, 637–647 [DOI] [PubMed] [Google Scholar]
- 51. López-Martín C., Chaparro M., Espinosa L., Bejerano A., Maté J., and Gisbert J. P. (2011) Adverse events of thiopurine immunomodulators in patients with inflammatory bowel disease. Gastroenterol. Hepatol. 34, 385–392 [DOI] [PubMed] [Google Scholar]
- 52. Thapa S. D., Hadid H., Schairer J., Imam W., and Jafri S. M. (2015) Effect of inflammatory bowel disease-related characteristics and treatment interventions on cardiovascular disease incidence. Am. J. Med. Sci. 350, 175–180 [DOI] [PubMed] [Google Scholar]
- 53. Pai E. F., Kabsch W., Krengel U., Holmes K. C., John J., and Wittinghofer A. (1989) Structure of the guanine-nucleotide-binding domain of the Ha-ras oncogene product p21 in the triphosphate conformation. Nature 341, 209–214 [DOI] [PubMed] [Google Scholar]
- 54. Heo J., and Hong I. (2010) Ras-targeting action of thiopurines in the presence of reactive nitrogen species. Biochemistry 49, 3965–3976 [DOI] [PubMed] [Google Scholar]