

Abstract
The N-terminal Src homology 3 (nSH3) domain of a signaling adaptor protein, CT-10 regulator of kinase II (CrkII), recognizes proline-rich motifs (PRMs) of binding partners, such as cAbl kinase. The interaction between CrkII and cAbl kinase is involved in the regulation of cell spreading, microbial pathogenesis, and cancer metastasis. Here, we report the detailed biophysical characterizations of the interactions between the nSH3 domain of CrkII and PRMs in cAbl. We identified that the nSH3 domain of CrkII binds to three PRMs in cAbl with virtually identical affinities. Structural studies, by using x-ray crystallography and NMR spectroscopy, revealed that the binding modes of all three nSH3:PRM complexes are highly similar to each other. Van ’t Hoff analysis revealed that nSH3:PRM interaction is associated with favorable enthalpy and unfavorable entropy change. The combination of experimentally determined thermodynamic parameters, structure-based calculations, and 15N NMR relaxation analysis highlights the energetic contribution of conformational entropy change upon the complex formation, and water molecules structured in the binding interface of the nSH3:PRM complex. Understanding the molecular basis of nSH3:PRM interaction will provide, to our knowledge, new insights for the rational design of small molecules targeting the interaction between CrkII and cAbl.
Introduction
The CT-10 regulator of kinase (Crk) oncogene was originally identified in avian retrovirus CT10 (1). Human Crk protein family consists of three isoforms, CrkI, CrkII, and CrkIII, and a homolog, CrkL (2, 3, 4). CrkII is a signaling adaptor protein that is involved in a variety of cellular processes, including proliferation, migration, invasion, and apoptosis, and its expression levels have been associated with aggressive lung, breast, and ovarian cancers (5, 6, 7, 8, 9, 10). CrkII consists of three Src homology (SH) domains: SH2, the N-terminal SH3 (nSH3), and the C-terminal SH3 (cSH3) domains. As a signaling adaptor, CrkII mediates a number of protein-protein interactions, through its SH2 and SH3 domains (10, 11). Among these three domains, the nSH3 plays a central role in the mediation of a number of protein-protein interactions, by recognizing proline-rich motifs (PRMs) (12, 13, 14, 15, 16, 17).
Cellular nonreceptor tyrosine kinase cAbl is one of the major binding partners of CrkII (18, 19, 20). cAbl is a proto-oncoprotein, and it is involved in various cellular processes (21). The N-terminal half of cAbl consists of SH3, SH2, and kinase domain, while the C-terminal half of cAbl mainly consists of structurally disordered regions, which contain a variety of posttranslational modifications and linear motifs, such as phosphorylation, PRMs, and nuclear localization/export signals (21). PRMs in cAbl are essential for the modulation of filopodium formation during the attachment to the fibronectin-coated surfaces, through the interactions with SH3 domain-containing proteins, such as CrkII, Nck, and Grb2 (19, 22, 23). The interaction between CrkII and cAbl through PRMs results in the phosphorylation of CrkII (19), which leads to the inhibition of Crk signaling during attachment and an increased rate of filopodium formation. In contrast, the overexpression of Crk family proteins decreases filopodium formation, while enhancing lamellipodium formation (20, 22).
The interactions between Crk family proteins and cAbl have bidirectional regulatory effects. For example, while the cAbl-mediated phosphorylation of CrkII regulates the function of CrkII, phosphorylated CrkII in turn transactivates cAbl kinase activity (24, 25). In addition, the interaction between CrkII and cAbl is important for the development of anticancer drugs that are targeting the oncogenic form of cAbl, Bcr-Abl, which causes the development of chronic myeloid leukemia (18, 26). Because of the importance of the interactions between CrkII and cAbl, they have been studied using various assays, such as yeast two-hybrid system, filter-binding assay, phage display screening, and pulldown assays (20, 22). These studies revealed the presence of multiple PRMs in cAbl (PRMcAbl), which mediate the interaction with the nSH3 domain of CrkII (nSH3CrkII). However, the quantitative and detailed biophysical studies of the interactions between the nSH3 domain and individual PRMs in cAbl have not been conducted.
The thermodynamic principles underlying the interaction between SH3 domains and PRMs (SH3:PRM interactions) are not well understood. Despite the largely hydrophobic binding interface, SH3:PRM interaction is associated with a large favorable enthalpy and unfavorable entropic penalty (i.e., ΔH°bind < 0 and −TΔS°bind > 0) (27, 28, 29, 30). Various mechanisms have been proposed to explain the anomalous binding thermodynamics of SH3:PRM interaction. For example, the change in conformational dynamics of SH3 domains and PRMs has been considered a source of unfavorable binding entropy change (29). Additionally, the importance of water molecules at the binding interface of SH3:PRM complex in the binding thermodynamics has been demonstrated (27, 31).
In this study, as a first step toward elucidating the interaction between CrkII and cAbl, we have carried out extensive studies of structures, thermodynamics, and conformational dynamics of nSH3CrkII:PRMcAbl interactions. We have investigated the binding of nSH3CrkII and all individual PRMs in the C-terminal disordered region of cAbl, and showed that the nSH3 domain binds to the three PRMs with similar affinity. To understand the binding mechanism of nSH3 and PRMs, we have determined the crystal structure of one of the three nSH3:PRM complexes. Moreover, the results of NMR chemical shift perturbation (CSP) study indicate that all three nSH3:PRM complexes have similar structures. The combination of the structure-based calculation of binding thermodynamics and NMR 15N relaxation analysis allows understanding the molecular origin of binding free energy between nSH3 and PRM. These results also provide insights into the energetic contribution of water molecules at the binding interface and the conformational entropy of protein in protein-ligand interactions.
Materials and Methods
Protein preparation
The protein samples used in this study were prepared using the Escherichia coli BL21 (DE3) expression system with a plasmid containing a gene for the nSH3 domain (residues 124–207) of human CrkII. The expression was induced with 0.5 mM IPTG (isopropyl β-D-1-thiogalactopyranoside) containing 15NH4Cl. For the NMR triple-resonance assignment experiments, 13C6 glucose was added to M9 medium, in addition to 15NH4Cl. The expressed protein was purified using Ni-sepharose (HisTrap HP column; GE Healthcare Life Sciences, Marlborough, MA) and digested with TEV-protease to remove N-terminal His-tag. The nSH3 domain was further purified on a second Ni-sepharose column and an anion exchange column (Mono-Q; GE Healthcare Life Sciences). Protein concentration was determined by measuring UV absorbance at 280 nm.
Peptide ligands
Synthetic PRMs used in this study were purchased from LifeTein (Somerset, NJ) in a crude form, and further purified using reverse-phase high performance liquid chromatography in our laboratory. The N- and C-termini of peptides were acetylated and amidated, respectively. The peptide concentration was determined by measuring the UV absorption of a single tyrosine at the N- or C-terminal ends of the peptide.
Binding assay
The dissociation constant, Kd, of nSH3:PRM complexes was measured by monitoring the change of tryptophan fluorescence signal. Excitation wavelength was 295 nm. All binding assays were performed in a stirred 1-cm path-length cuvette using a QM-400 fluorimeter (Photon Technology International/HORIBA Scientific, Edison, NJ). Protein concentration used for the fluorescence-based binding assays was 0.1 μM. All measurements were done in 20 mM sodium phosphate (pH 6.1) and 80 mM NaCl. All of the experiments were repeated two or three times. Kd was calculated by assuming a 1:1 complex, and by the global fitting of the repeatedly measured fluorescence intensities to Eq. 1:
| (1) |
where ΔF and ΔFmax are the signal change and the maximum amplitude of signal change, respectively. Pt is the total protein concentration and Lt is the total ligand concentration at each titration point.
Van ’t Hoff analysis
The Kd values of the nSH3:PRM complexes were measured at seven different temperature points, from 10 to 40°C, using fluorescence-based method. The binding free energy, ΔG°, was calculated using ΔG° = −RT ln(Kd). Binding thermodynamic parameters, ΔH°, and ΔS°, were calculated by fitting the temperature-dependent ΔG° to Eq. 2 (32), with and without taking into account ΔCp:
| (2) |
An extensive Monte Carlo simulation study indicated that the errors of ΔH° and ΔS° obtained from van ’t Hoff analysis are proportional to the relative error of Kd, and the use of symmetric temperature range around the standard temperature (25°C) was suggested to reduce the effect of the error of Kd on the thermodynamic parameters (33).
Crystallization, data collection, processing, and structure determination
Crystallization conditions were found after screening several commercial (Hampton Research, Aliso Viejo, CA) and in-house screens. 4 mM nSH3 was mixed with 5 mM PRM758 in a molar ratio of 1:1 and incubated for 30 min for the crystallization trials. The best diffracting crystals were obtained by sitting drop vapor diffusion method in 0.1 M sodium acetate trihydrate (pH 4.5), 13% polyethylene glycol 3350, 3.0 M sodium chloride. Crystals grew approximately to dimensions of 0.3 × 0.2 × 0.2 mm. A 1.8 Å resolution data set was collected at 113 K using an R-AXIS IV2+ imaging-plate detector and Cu Kα radiation generated by a rotating-anode x-ray generator (Rigaku/MSC, The Woodlands, TX). The data were processed using HKL2000 (34) in space group P1211 with cell dimensions of a = 45.2 Å, b = 29.5 Å, and c = 45.8 Å. The solution was subjected to an initial cycle of model rebuilding using AutoBuild module of PHENIX (35) followed by several iterations of manual model building in COOT (36) and refinement in REFMAC (37). PRM758 was added to both copies of the model guided by the Fo-Fc electron density map.
NMR spectroscopy
All NMR experiments were conducted using protein samples in 20 mM sodium phosphate (pH 6.1), 80 mM NaCl, 0.02% sodium azide, 1 mM EDTA, 10 μM DSS (4,4-dimethyl-4-silapentane-sulfonate), and 10% D2O at 25°C. NMR spectra were acquired on an AVANCE 600 and 800 MHz spectrometers (Bruker, Billerica, MA), equipped with a cryogenic probe. NMR spectra were processed with NMRPipe (38) and analyzed with NMRViewJ (One Moon Scientific, Westfield, NJ) and CARA (39). The assignment of 1H, 13C, and 15N resonances was carried out using a set of experiments: (1H, 15N) heteronuclear single quantum coherence, (1H, 13C) heteronuclear single quantum coherence, HNCO (40), HNCACB (41), HN(CO)CA (42), HNCA (43), HN(CA)CO (44), CBCA(CO)NH (45), HBHA(CO)NH (46), and HC(C)H-TOCSY (47). 1H chemical shifts were referenced with respect to DSS, and 13C and 15N chemical shifts were referenced indirectly (48).
Measurement of relaxation parameters
All relaxation parameters were measured at 25°C on a 500 MHz NMR spectrometer (Bruker). For R1, five relaxation time points were taken between 100 ms and 1 s. For R2, five relaxation time points were taken between 4 and 120 ms. For R1 and R2 measurements, a recycle delay of 2 s was used between transitions. For the heteronuclear nuclear Overhauser effect measurements, a recycle delay of 10 s was used in reference experiment. The saturation of proton during steady state was performed by applying 180° pulses for 4 s (49). Errors of the relaxation parameters were estimated using duplicated measurements.
Model free analysis
The R1, R2, and heteronuclear nuclear Overhauser effect were analyzed according to the Lipari-Szabo model free formalism (50, 51) using the softwares Modelfree version 4.20 (52) and FAST-Modelfree (53). An initial estimation of the rotational diffusion tensor of PRM-bound state was performed using the programs Quadratic and Pdbinertia (http://cpmcnet.columbia.edu/dept/gsas/biochem/labs/palmer/software.html). The axially symmetric diffusion tensor was used for analysis of the complex. The isotropic diffusion model was used for analysis of the free nSH3 domain. Anisotropic model was also tested but the isotropic model yielded lower sum of squared errors.
Structure-based calculation of thermodynamic parameters
Thermodynamic parameters were calculated based on the change of the solvent-accessible surface area (SASA), upon the complex formation of the nSH3 domain and PRM. The structure of free nSH3 domain (PDB: 1M30) and the crystal structures of nSH3:PRM758 complex (PDB: 5IH2) were used for the calculations. The changes in polar and nonpolar solvent-accessible surface area (ΔSASApol and ΔSASAnp) were calculated using parameter-optimized surfaces web server (54, 55). All water molecules were removed from PDB files before calculation. The effect of structured water molecules on the SASA was calculated after adding the identified water molecules to the PDB file. The structured water molecules were considered absent from the free nSH3 domain.
Heat capacity change upon complexation, ΔCp, was calculated by the Eq. 3 using the estimated ΔSASApol and ΔSASAnp (56):
| (3) |
where a = 0.45 cal K−1 (Å2)−1 and b = −0.26 cal K−1 (Å2)−1. The enthalpy change according to ΔSASApol and ΔSASAnp was parameterized at 60°C, which is the midpoint of thermal unfolding of proteins. The enthalpy change, ΔH (60°C), was calculated using Eq. 4 (28):
| (4) |
where a(60) and b(60) are the empirical coefficients obtained from the regression analysis of protein thermal unfolding data set, and 31.4 cal (mol Å2)−1 and −8.44 cal (mol Å2)−1, respectively. The empirical parameterization of the binding enthalpy for protein-ligand interaction at 25°C resulted in Eq. 5 (57):
| (5) |
where a(25) = –7.35 cal (mol Å2)−1 and b(25) = 31.06 cal (mol Å2)−1, and ΔHconf represents the enthalpy change accompanied by the conformational change of protein and ligand upon binding. The entropy change of solvent, ΔS°solv, associated with the burial of hydrophobic surface after binding at a temperature T was calculated using the Eq. 6 (56):
| (6) |
where Tpol = 335.15 K and Tnp = 385.15 K are the polar and nonpolar reference temperatures at which the hydration entropy is equal to zero (28, 56).
Results and Discussion
Identification of Crk binding sites in Abl kinase
The C-terminal disordered region (from amino acid 512–1130 in Abl-1A) of cAbl contains seven PxxP motifs (Table S1 in the Supporting Material). In this study, we have investigated the binding of the nSH3 domain to all individual PRMs. We have tested seven PRMs and found that the nSH3 domain binds to three of them (Table 1 and Table S1). The amino-acid sequences of the PRMs that bind to the nSH3 domain are shown in Table 1. The number in a peptide name represents the amino-acid number of the first proline in PxxP motif in Abl-1A sequence. The binding between nSH3CrkII and PRMs was measured by monitoring the changes in tryptophan fluorescence intensity of the nSH3 domain (Fig. 1). The three nSH3:PRM complexes have similar Kd values, on the order of 1–2 μM. The affinities of the three peptides for nSH3CrkII are comparable to the affinity of PRM524 to CrkL (58). All three binding peptides have a common PxφPxK motif. Intriguingly, we did not observe the binding of nSH3 to PRM610, although it contains the PxxPxR motif. Similarly, the binding of nSH3 and PRM610 was identified by phage display screening, but it was not detected by GST-pulldown assay (22). The interaction between the nSH3 and PRM758 was confirmed unambiguously in our study. The functional significance of nSH3:PRM758 interaction was confirmed by the results showing that the deletion of two PRMs (PRM524 and PRM568) does not inhibit the phosphorylation of Crk by cAbl kinase (24).
Table 1.
The Thermodynamic Parameters for the Interactions between the nSH3 Domain and PRMs: T = 298 K
| Peptides | Kd (μM) | ΔG° (kcal mol−1) | ΔH° (kcal mol−1) | –TΔS° (kcal mol−1) |
|---|---|---|---|---|
| PRM524 QAPELPTKTRTSY | 1.72 ± 0.47 | −7.86 ± 0.45 | −15.37 ± 0.31 | 7.51 ± 0.32 |
| PRM568 VSPLLPRKERGY | 2.90 ± 0.19 | −7.55 ± 0.99 | −16.78 ± 0.70 | 9.23 ± 0.70 |
| PRM758 YEKPALPRKR | 1.72 ± 0.48 | −7.86 ± 0.44 | −11.50 ± 0.31 | 3.64 ± 0.31 |
Figure 1.

Binding isotherms of the nSH3 domain to PRM524 (solid circles), PRM568 (triangles), and PRM758 (open circles) of cAbl.
The structure determination of nSH3:PRM758 complex
We have determined the crystal structure of the nSH3:PRM758 complex (Fig. 2) at 1.8 Å resolution (Table S2). The complex crystallizes in the space group P1211 with cell dimensions of a = 45.2 Å, b = 29.5 Å, and c = 45.8 Å. Molecular replacement in Phaser (59) using nSH3 domain (PDB: 1CKA) (12) as a search model could output a unique solution. The solution contained two copies of the protein in the asymmetric unit. A Matthews coefficient (VM) of 2.18 for two copies of the protein also indicated the same. Water molecules were added after one more round of real and reciprocal space refinement. The final structure has R/Rfree = 17%/24%.
Figure 2.

The structure of the nSH3:PRM758 complex. (A) Representation of the 2Fo-Fc electron density map of PRM758 contoured at 1σ. The amino-acid sequence of PRM758 is shown at the bottom. Salt-bridges between K-3 of PRM758 and three acidic residues (D147, E149, and D150) of the nSH3 domain are shown in red. Salt-bridges between the acidic residues in nSH3 domain and K-3 of PRM758 are shown as dashed lines. Overlaid structures of (B) two nSH3 domains (gray and orange for molecules A and B in the PDB file, respectively) and (C) bound PRMs in an asymmetric unit of the nSH3:PRM758 complex crystal. PRMs bound to molecules A and B are shown in cyan and orange color, respectively. To see this figure in color, go online.
We adopted the notation where φ in PxφPxK motif is designated as P0 position (φ represents hydrophobic residues) (60). The residues located at the C-terminal end of P0 were designated as P−1, P−2, etc. The residues located at the N-terminal of P0 were designated as P1, P2, etc. In that complex, the side chains of Y5, E4, A1, and R−2 of PRM758 do not interact directly with the nSH3 domain. The hydrophobic binding interface of the complex is formed by P2, L0, and P−1 in the PRM, and F141, F143, W169, P183, P185, and Y186 in the nSH3 domain. A salt-bridge network was observed between K−3 in PRM and a cluster of acidic residues (D147, E149, and D150) in the RT-loop of the nSH3 domain (Fig. 2). This salt-bridge network is critical for the binding affinity and the recognition of specific PRMs (12).
Two molecules per asymmetric unit were observed, and although these two complex molecules are highly similar (average Cα RMSD = 0.4 Å), the conformations of RT-loop and nSrc-loop show considerable difference from each other (Fig. 2). These loops are crucial for the recognition of a specific PRM sequence (12, 60). This structural heterogeneity is due to the low packing density in these parts of the molecule: the contacts between the nSH3 domain and PRM represent salt-bridge interactions between the terminal charged groups of long side chains. The similar dynamic behavior of RT-loop was suggested by a thorough analysis using molecular dynamics simulation of the complex between the nSH3 domain and PRM derived from SOS protein (61).
To gain structural insights into two other nSH3:PRMcAbl complexes, we conducted NMR CSP analysis (Fig. 3). Three peaks corresponding to the E149, D150, and W169 show significantly large CSPs, compared to others. Because large CSPs of these three residues considerably affect the overall mean and standard deviation, we did not include the CSPs of these residues for the calculation of CSP statistics. Then, the residues showing the CSP larger than the sum of the mean and standard deviation are identified, and the excluded residues were added afterwards. Fig. 3 shows the identified residues for each complex.
Figure 3.

NMR CSP plots of backbone amide resonances of the nSH3 domain upon binding to (A) PRM524, (B) PRM568, and (C) PRM758. (D) Standard deviation of NMR CSPs of the three complexes. Key binding interfacial residues (D147, E149, D150, D169, and Y184) are marked by the ∗ symbol.
Despite overall similarity, the magnitudes of CSPs of the complexes differ considerably among each other. Fig. 3 D shows the standard deviation of the CSPs from these three complexes. The residues showing standard deviations larger than the average value are mapped on the binding interface (Fig. 4). This reflects the variability of amino-acid sequences in the bound PRMs. The CSPs of E149 and D150 show the largest standard deviations. These residues form a salt-bridge network with the K-3 of bound PRM (Fig. 2 A). This may indicate considerable structural heterogeneity of the salt-bridge interactions in different complexes, and this is consistent with the structural variation in the same region between the two complex structures in the asymmetric unit, as well.
Figure 4.

Mapping of binding interfaces between the nSH3 domain and PRMs. Significant NMR CSPs upon binding of (A) PRM524, (B) PRM568, and (C) PRM758 are shown in red. The structure of PRM758 is shown in (C). To see this figure in color, go online.
Thermodynamic characterization of ligand binding to nSH3 domain
To measure the thermodynamic parameters (ΔH° and ΔS°) associated with the binding of nSH3 and PRMs, we conducted van ’t Hoff analysis. When properly executed, there is no systematic difference between the thermodynamic parameters obtained by van ’t Hoff analysis and calorimetric measurement (62). The Kd values of the nSH3:PRMcAbl complexes were measured using fluorescence spectroscopy as a function of temperature ranging from 10 to 40°C (Fig. 5). Thermodynamic parameters are reported in Table 1. As described in Materials and Methods, our experimental temperature range is symmetrical, with respect to the standard state temperature (25°C). This also helps reduce the error of ΔH° and ΔS° estimated by van ’t Hoff analysis.
Figure 5.

Van ’t Hoff plots of the interactions between the nSH3 domain and (A) PRM524, (B) PRM568, and (C) PRM758. The lines are linear regression fits using Eq. 2 with ΔCp = 0.
The results of the van ’t Hoff analysis indicate that the binding of nSH3 and all three PRMs is driven by favorable enthalpy, accompanied by unfavorable entropy changes at 298 K (Table 1). The obtained ΔH° and ΔS° for all three complexes are within the typical range for SH3:PRM interactions (30). Although the binding free energies of all three complexes are similar to each other, ΔH° and –TΔS° vary considerably. ΔH° and –TΔS° of nSH3:PRM758 binding is notably smaller than for the other two complexes. This is especially interesting because all three PRMs contain the identical core sequences (PxLPxK), and the other residues in the PRM do not interact with nSH3 domain.
Because PRMs form type-II polyproline (PPII) helix in the complex, we further investigated whether the difference in the PPII helix propensities of PRMs can explain the differences in binding thermodynamic parameters of the three nSH3:PRM complexes. To calculate the PPII propensity of each PRM, we simply summed up the propensities of individual constituent amino acids, determined by various methods (63, 64, 65). We have not found a correlation between the calculated PPII propensity of PRMscAbl and any of the thermodynamic parameters. The absence of correlation between PPII propensity and thermodynamic parameters has been observed for other SH3:PRM interactions (66, 67, 68). However, the effect of PPII propensity to binding thermodynamics was shown in the binding of PRMSos to Sem-5 cSH3 domain (69, 70). Because we tested only three PRMs in this study, the effect of PPII helix propensity on binding affinity should be further tested with a larger data set.
Structure-based calculations of thermodynamic parameters
Because of the hydrophobic binding interface, the binding of the SH3 domain and PRM is associated with a change in the heat capacity (ΔCp), which leads to the appearance of nonlinear curve in the van ’t Hoff plot. Therefore, the change in ΔCp can be obtained by measuring the nonlinearity of van ’t Hoff plot. Although the fitting with nonlinear model (Eq. 2) provides a smaller sum of squared errors compared to a linear model, the improvement of the fit was not statistically significant, based on the F-test with α = 0.05. Table 1 shows the results of the application of the simple linear model (i.e., ΔCp = 0).
We further investigated why the van ’t Hoff plot of the nSH3:PRMcAbl interaction does not show the curvature, despite the hydrophobic binding interface shown in the analysis of crystal structure. The changes in solvent-accessible surface (ΔSASA) are associated with protein-ligand interaction, which is a major source of the heat capacity change (28, 57). Therefore, we first tested whether the results obtained by van ’t Hoff analysis are consistent with the change in ΔSASA upon the nSH3:PRM complexation. We measured the change in polar and nonpolar solvent-accessible surface area, ΔSASApol and ΔSASAnp, between NMR-derived structure of free nSH3 and the crystal structure of nSH3:PRM758 complex. SASA was calculated by taking the average of all structures in each PDB file. Applying ΔSASA to Eq. 3, we determined that ΔCp = −204.6 cal mol−1 K−1. We fit van ’t Hoff plot with this calculated ΔCp as a restraint. While the regression curve displayed the curvature, the fitted values of ΔH° and ΔS° were virtually identical to the values obtained by linear van ’t Hoff equation. A similar result was obtained when we applied a different ΔCp = −255.4 cal mol−1 K−1, which is calculated based on the ΔSASA estimated by the crystal structure in the presence and absence of PRM. The calculated ΔCp values are also similar to the experimentally determined values for different SH3:PRM interactions (27, 28). For example, a negative ΔCp = −166 cal mol−1 K−1, was obtained for the interaction of Sem-5 SH3 and PRM (28). Taken together, these results indicate that ΔSASA, upon the binding of nSH3 domain and PRM, is too small for nonlinear van ’t Hoff plot to be observed in the investigated temperature range (from 283 to 310 K). However, due to the concern of protein stability at higher temperatures, we did not extend our temperature range. This test also implies that the measured enthalpy and entropy values for the nSH3:PRMcAbl are reliable.
Molecular origin of the anomalous thermodynamic parameters
Understanding the molecular origin of thermodynamic parameters is one of the major goals in molecular biophysics and it is important for the rational ligand design as well. However, the calculations of the thermodynamic parameters of SH3:PRM interactions have generally resulted in serious inconsistency with experimental outcomes. Therefore, we sought to identify the molecular basis of the discrepancy between the experimental and the calculated results.
We first calculated the binding enthalpy, ΔH°bind, based on the structural change using Eq. 4. However, we found that the use of this equation yields ΔH°bind values that significantly deviate from the experimental result, regardless of our choice of coefficients. For example, when a = –8.43 and b = 31.06 are applied, this calculation yields ΔH°bind = 4.7 kcal mol−1, which is drastically different from the experimental value (ΔH°bind = −11.5 kcal mol−1). The choice of free nSH3 domain structure can affect the calculated ΔH°bind because of the structural rearrangements upon the complexation. Here, however, the calculation of ΔSASApol and ΔSASAnp using crystal structure, with and without bound ligand, yielded similar results as when NMR and crystal structures were used as a model for free and PRM-bound states, respectively. It should be noted that this result does not indicate that the nSH3 domain undergoes minor structural changes upon binding to ligand. Instead, it indicates that the structural change does not result in considerable SASA changes. The selection of a different free nSH3 structure from 20 lowest-energy NMR structures results in a larger change in SASA. To avoid any bias stemming from the selection of a specific free nSH3 structure, we simply used the average value of all available free nSH3 structures.
The change in protonation state of protein and ligand can affect ΔH°bind. However, the detailed studies of several other SH3:PRM interactions showed that this effect is typically minor (27, 28). To further test the effects of ionization on ΔH°bind, we measured the Kd of nSH3:PRM758, at pH 7.5 in HEPES buffer (Fig. S1 in the Supporting Material). The obtained Kd value was shown to be very similar to the results of our choice of experimental conditions in this study: phosphate buffer at pH 6.1. Although further tests are necessary, this result supports our assumption that ΔH°ion does not contribute greatly to the difference between the experimental and theoretical ΔH°bind.
Next, we tested whether the observed difference is due to fact that these coefficients are derived from protein (un)folding data, and not from protein-ligand binding data. Luque and Freire (57) reparameterized the coefficients based on the binding data of HIV-1 protease and inhibitors at 25°C. When we applied Eq. 5, the calculation yielded ΔH°bind = –3.42 kcal mol−1, which is much closer to the experimental value. This improvement might be due to the relatively smaller binding interface of SH3:PRM complex compared to ΔSASA associated with protein (un)folding process.
Water molecules at the binding interface of nSH3 and PRM
There is a growing interest in the role of water molecules at the binding interface of protein and ligand in the binding thermodynamics (31, 71, 72, 73). It has been suggested that an incomplete dehydration of binding interface can result in considerable favorable enthalpy and unfavorable entropy changes (57, 74). Therefore, the atypical ΔH° and –TΔS° of nSH3:PRM interaction can be attributed to the water molecules located at the binding interface. Recent studies indicated that the binding interfaces of many SH3:PRM complexes include structured interfacial water molecules (31, 72, 75). The localization of water molecules was identified in three regions, including the 310-helix, nSrc-loop, and RT-loop regions (75). The enthalpy contribution of water molecules in the 310-helix region of SH3cAbl:PRMp41 complex was estimated to be –4.78 kcal mol−1 (27).
We investigated whether interfacial water molecules are responsible for the discrepancy between the experimental and calculated thermodynamic parameters of nSH3:PRMcAbl interactions. We compared nSH3CrkII:PRM758 structure with SH3cAbl:PRMp41 complex, where five specific interfacial water molecules were identified. However, nSH3:PRM758 complex does not contain water molecules at the sites identified in SH3cAbl:PRMp41 complex. Only one interfacial water molecule in nSH3:PRM758 is located at a similar site in the nSrc loop region, but this water site is characterized as a transient binding site, and it underwent fast exchange with bulk water in the molecular dynamic simulation of SH3cAbl:PRMp41 complex (75).
Furthermore, we searched for a set of interfacial water molecules, conserved specifically in the nSH3:PRM complexes. Because of the diversity of amino-acid sequences and the structures of SH3:PRM interfaces, it is not unreasonable to expect that there are interfacial water sites unique to each SH3 domain. To identify the conserved interfacial water molecules, we superimposed all available crystal structures of nSH3:PRM complexes (PDB: 1CKA, 1CKB, and 5IH2). We found eight water molecules that are located at very similar sites in all three complexes. Additionally, we identified several more water molecules per complex that mediate the interactions between the bound ligand and the nSH3 domain. However, only a small number of interfacial water molecules in each complex showed the characteristics of buried water molecules that might affect binding energetics: i.e., low normalized B-factors (<0.4 Å2) and SASA (<10 Å2) (76). Consequently, we identified two water molecules (w34 and w53) that are conserved in all nSH3:PRM complexes and have low normalized B-factor and SASA values (Fig. 6).
Figure 6.
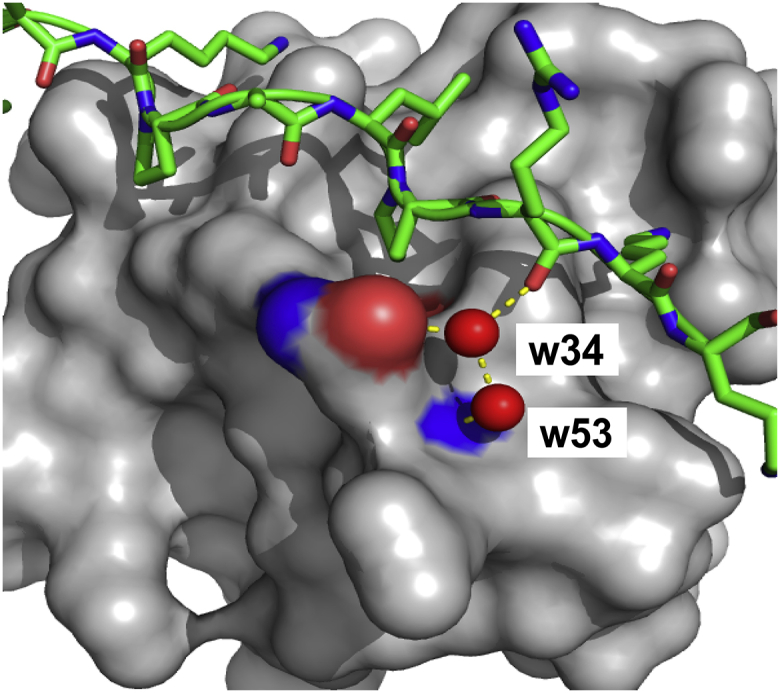
Water molecules at the nSH3:PRM758 binding interface. The nSH3 domain is shown as a surface representation. Two water molecules (w34 and w53) are hydrogen-bonded with the side-chain and backbone amides of Q168, respectively, in the nSH3 domain and mediate the interactions with the backbone carbonyl oxygen of R−2 in PRM758. To see this figure in color, go online.
To estimate the effects of the two interfacial water molecules on the binding thermodynamics, we recalculated ΔSASA taking into consideration the interfacial water molecules as a part of bound ligand. This resulted in the increase of ΔSASApol by ∼200 Å2 upon the complexation. The calculation based on this ΔSASA resulted in ΔH°bind = –9.4 kcal mol−1, which is a significant improvement compared to the result obtained without considering the water molecules. The increase in ΔSASApol improved the agreement between the calculations and the experimental results, regardless of the choice of the coefficients and equations. This result indicates that the interfacial water can contribute considerably to the binding thermodynamics. However, it should be noted that these water molecules have higher B-factors than well-buried water. Therefore, the validity of these water molecules as buried (or structured) remains to be further investigated. We speculate that our results represent the upper bound of the contribution of interfacial water in nSH3:PRM complex.
Role of conformational entropy in nSH3:PRM interaction
In the calculation of ΔH°bind we ignored ΔH°conf, which represents the structural rearrangement of protein and ligand upon complexation. This suggests that the difference between experimental and calculated ΔH°bind can be due to conformational rearrangement of protein and ligand. It was shown that the ΔH°conf, obtained as a fitting parameter of Eq. 5 to several protein-ligand binding data, ranges from 0.1 to 5.9 kcal mol−1 (57). However, most of the ligands used in the calculations are small molecules. For ∼10 amino-acid-long peptides used in this study, ΔH°conf could be considerably larger than the reported values. In addition, the effect of conformational flexibility on the estimation of binding thermodynamic parameters was well highlighted in the study of the Sem-5 cSH3 domain (77, 78).
We have estimated the contribution of the conformational entropy, −TΔS°conf, accompanied by the rearrangement of protein and ligand, to the binding thermodynamic parameters. The two major components of binding entropy change are the conformational entropy of protein and ligand, and solvent (or solvation) entropy (ΔSbind = ΔSconf + ΔSsolv). Therefore, if we compare the –TΔSbind and –TΔSsolv, we can indirectly estimate –TΔSconf. We first calculated the solvent entropy change upon the nSH3:PRM758 interaction, using Eq. 6. The calculated –TΔSsolv was –15.8 kcal mol−1 at 298 K, and differed by >18.0 kcal mol−1 compared to the experimentally obtained binding entropy change. Furthermore, the sign of the calculated –TΔSsolv is opposite to –TΔSbind. A considerable difference between –TΔSbind and calculated –TΔSsolv was also observed for other SH3:PRM interactions (27, 28). These results indicate that the conformational entropy change contributes significantly to –TΔSbind, and consequently to the overall binding thermodynamics. It is also important to point out that our estimation of –TΔSsolv does not take into account the interfacial water molecules. –TΔSconf associated with the ordering of a water molecule was estimated to be on the order of 2 kcal mol−1 at 298 K (74). Therefore, the interfacial water can considerably affect the estimation of –TΔSsolv, but this does not affect our conclusion that conformational entropy contributes significantly to the binding thermodynamics of nSH3 and PRM.
NMR dynamic studies provided a quantitative estimation of conformational entropy contribution (79, 80, 81). To further determine the contribution of conformational entropy to the binding thermodynamics, we have measured backbone N-H bond order parameters (O2) of the nSH3 domain in free and PRM758-bound states, using the Lipari-Szabo model-free approach (50, 51). Although structural change upon PRM-binding is mainly localized in nSrc- and RT-loops, PRM-induced changes in O2 are observed beyond the ligand binding interface (Fig. 7 A). These results may indicate that favorable binding energy is redistributed to optimize overall backbone hydrogen-bond geometry of SH3 domain as observed for other SH3:PRM interactions (29, 82).
Figure 7.

(A) Difference of backbone N-H order parameters (O2) between the free and PRM-bound nSH3 domains. (B) Conformational entropy change of N-H bonds in the nSH3 domain upon binding of PRM758 (T = 298 K).
The change in O2 values upon ligand-binding can be converted to the change in TΔSconf of individual N-H bonds (Fig. 7 B) (79, 80). The sum of all individual NMR-derived TΔSconf yields −4.79 kcal mol−1 at 298 K. Similarly, the binding of PRM to the Src SH3 domain resulted in conformational entropy change by –1.5 ± 0.6 kcal mol−1 (29). Here, we excluded the flexible N- (residues 126–133) and C-terminal (residues 190–207) tails. In addition, we did not take into account the change in motions in timescales slower than the overall tumbling time of protein molecule. Although this calculation has some limitations, the result clearly indicates that the conformational entropy contributes significantly to overall binding entropy. A large portion of –TΔSsolv is compromised by ordering backbone N-H bonds in the nSH3 domain. The sum of ΔSsolv and ΔSconf is still considerably different from the ΔSbind. However, it should be emphasized that here we only considered backbone N-H bonds of the nSH3 domain. The folding of the disordered PRM to PPII conformation upon binding contributes substantially to the conformational entropy change. A remarkable example of this is the energetic contribution of conformational change of PRM upon binding to the Sem-5 SH3 domain (28). The authors found that the thermodynamic signature corresponding to the formation of PPII helix by PRMSos is similar to the difference between the experimental and calculated thermodynamics based on static structures. Furthermore, the change in side-chain motion upon complexation contributes considerably to this as well (83).
In this study, we observed that the binding thermodynamics of the nSH3:PRMscAbl complexes differ considerably, despite the fact that they have similar binding interfaces. It is noteworthy that Ferreon et al. (77) and Manson et al. (78) demonstrated that the conformational fluctuation of RT-loop can affect the binding thermodynamics between the cSH3Sem5 domain and PRMSos considerably. Hence, it raises the possibility that the difference in the binding thermodynamics among the nSH3:PRMscAbl complexes is due to the differences in the conformational heterogeneity between the complexes. Furthermore, it is equally possible that the differences in the conformational dynamics among the PRMscAbl are responsible for the difference in binding thermodynamics.
Biological implication of nSH3CrkII:PRMcAbl interactions
We have examined the binding of the nSH3 domain of CrkII to all putative binding PRMs in cAbl. Our study shows that three PRMs in cAbl mediate the interactions with CrkII. It remains unknown why cAbl contains multiple PRMs for CrkII interactions. While the interaction of CrkII and cAbl results in the phosphorylation of CrkII, the deletion of two PRMs (PRM524 and PRM568) does not inhibit the phosphorylation of Crk by cAbl kinase (24). This indicates that a single PRM may be enough to induce the phosphorylation of CrkII. It may possible that the binding of CrkII to each of the PRMs in cAbl enables different biological roles.
Signaling adapters containing SH3 domain, such as CrkII, Grb, and Nck, bind to these PRMs in cAbl (20, 22). For example, Grb2 can bind to PRM524 and PRM568 (20), and Nck binds to PRM610 (20, 22). This raises the possibility that these adaptor proteins bind simultaneously to cAbl. In light of these results, Antoku et al. (22) proposed an interesting model in which the binding of CrkII to multiple PRMs regulates the binding of other proteins to cAbl. For example, it was suggested that the binding competition of CrkII and Nck for the PRMs in cAbl regulates cell spreading (22). Although the C-terminal region of cAbl contains three nuclear localization signals, it is not exclusively localized to the nucleus (84), and the PRMs are located near nuclear localization signal sites. This suggests that CrkII binding may block the translocation of cAbl to the nucleus (20). In fact, PRM758 overlaps with nuclear localization signal (LPRKRAGE). Some PRMs in cAbl overlap with phosphorylation sites. The investigations of the functional relationship between PRM-mediated interactions and posttranslational modifications are of great importance.
Conclusions
In this study, we have investigated structures and thermodynamics of the interactions mediated by nSH3 domain of CrkII and PRMs of cAbl kinase. Understanding the molecular basis of the nSH3:PRM interaction is pivotal to successful design of small molecules that inhibit the interaction between CrkII and cAbl kinase. Despite its importance, binding free energy alone does not provide an insight into the mechanism by which protein-ligand interactions are achieved. Mechanistic insight into protein-ligand interaction requires the understanding of principles underlying the thermodynamic signatures of protein-ligand interactions. Our results, obtained by combination of structural, thermodynamic, and conformational dynamics analysis, highlight the role of interfacial water molecules and the conformational entropy in the SH3:PRM interactions. PRMs are one of the most abundant motifs of the eukaryotic proteome (85). Therefore, it is important to elucidate the mechanism underlying the selectivity of SH3:PRM-mediated protein-protein interactions. We expect that the results from this study will be useful to identify the mechanism underlying the ligand-selectivity of SH3 domains.
Author Contributions
V.S.B. and J.-H.C. conceived the research; V.S.B., D.Z., I.K., and J.-H.C. conducted experiments; V.S.B., D.Z., I.K., J.C.S., and J.-H.C. analyzed data; and J.-H.C. wrote the article.
Acknowledgments
Work in the J.-H.C. lab was supported by start-up funds from Texas A&M University.
Editor: David Eliezer.
Footnotes
Veer S. Bhatt and Danyun Zeng contributed equally to this work.
One figure and two tables are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(16)30285-5.
Supporting Citations
Reference (86) appears in the Supporting Material.
Supporting Material
References
- 1.Mayer B.J., Hamaguchi M., Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature. 1988;332:272–275. doi: 10.1038/332272a0. [DOI] [PubMed] [Google Scholar]
- 2.Matsuda M., Tanaka S., Shibuya M. Two species of human CRK cDNA encode proteins with distinct biological activities. Mol. Cell. Biol. 1992;12:3482–3489. doi: 10.1128/mcb.12.8.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.ten Hoeve J., Morris C., Groffen J. Isolation and chromosomal localization of CRKL, a human crk-like gene. Oncogene. 1993;8:2469–2474. [PubMed] [Google Scholar]
- 4.Prosser S., Sorokina E., Sorokin A. CrkIII: a novel and biologically distinct member of the Crk family of adaptor proteins. Oncogene. 2003;22:4799–4806. doi: 10.1038/sj.onc.1206714. [DOI] [PubMed] [Google Scholar]
- 5.Miller C.T., Chen G., Beer D.G. Increased C-CRK proto-oncogene expression is associated with an aggressive phenotype in lung adenocarcinomas. Oncogene. 2003;22:7950–7957. doi: 10.1038/sj.onc.1206529. [DOI] [PubMed] [Google Scholar]
- 6.Rodrigues S.P., Fathers K.E., Park M. CrkI and CrkII function as key signaling integrators for migration and invasion of cancer cells. Mol. Cancer Res. 2005;3:183–194. doi: 10.1158/1541-7786.MCR-04-0211. [DOI] [PubMed] [Google Scholar]
- 7.Linghu H., Tsuda M., Tanaka S. Involvement of adaptor protein Crk in malignant feature of human ovarian cancer cell line MCAS. Oncogene. 2006;25:3547–3556. doi: 10.1038/sj.onc.1209398. [DOI] [PubMed] [Google Scholar]
- 8.Fathers K.E., Bell E.S., Park M. Crk adaptor proteins act as key signaling integrators for breast tumorigenesis. Breast Cancer Res. 2012;14:R74. doi: 10.1186/bcr3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sriram G., Birge R.B. Emerging roles for crk in human cancer. Genes Cancer. 2010;1:1132–1139. doi: 10.1177/1947601910397188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feller S.M. Crk family adaptors-signalling complex formation and biological roles. Oncogene. 2001;20:6348–6371. doi: 10.1038/sj.onc.1204779. [DOI] [PubMed] [Google Scholar]
- 11.Birge R.B., Kalodimos C., Tanaka S. Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun. Signal. 2009;7:13. doi: 10.1186/1478-811X-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu X., Knudsen B., Kuriyan J. Structural basis for the specific interaction of lysine-containing proline-rich peptides with the N-terminal SH3 domain of c-Crk. Structure. 1995;3:215–226. doi: 10.1016/s0969-2126(01)00151-4. [DOI] [PubMed] [Google Scholar]
- 13.Birge R.B., Fajardo J.E., Hanafusa H. Identification and characterization of a high-affinity interaction between v-Crk and tyrosine-phosphorylated paxillin in CT10-transformed fibroblasts. Mol. Cell. Biol. 1993;13:4648–4656. doi: 10.1128/mcb.13.8.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oda T., Heaney C., Druker B.J. Crkl is the major tyrosine-phosphorylated protein in neutrophils from patients with chronic myelogenous leukemia. J. Biol. Chem. 1994;269:22925–22928. [PubMed] [Google Scholar]
- 15.Matsuda M., Ota S., Kurata T. Interaction between the amino-terminal SH3 domain of CRK and its natural target proteins. J. Biol. Chem. 1996;271:14468–14472. doi: 10.1074/jbc.271.24.14468. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka S., Morishita T., Nagashima K. C3G, a guanine nucleotide-releasing protein expressed ubiquitously, binds to the Src homology 3 domains of CRK and GRB2/ASH proteins. Proc. Natl. Acad. Sci. USA. 1994;91:3443–3447. doi: 10.1073/pnas.91.8.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasegawa H., Kiyokawa E., Matsuda M. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol. Cell. Biol. 1996;16:1770–1776. doi: 10.1128/mcb.16.4.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brehme M., Hantschel O., Superti-Furga G. Charting the molecular network of the drug target Bcr-Abl. Proc. Natl. Acad. Sci. USA. 2009;106:7414–7419. doi: 10.1073/pnas.0900653106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feller S.M., Knudsen B., Hanafusa H. c-Abl kinase regulates the protein binding activity of c-Crk. EMBO J. 1994;13:2341–2351. doi: 10.1002/j.1460-2075.1994.tb06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren R., Ye Z.S., Baltimore D. Abl protein-tyrosine kinase selects the Crk adapter as a substrate using SH3-binding sites. Genes Dev. 1994;8:783–795. doi: 10.1101/gad.8.7.783. [DOI] [PubMed] [Google Scholar]
- 21.Hantschel O., Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat. Rev. Mol. Cell Biol. 2004;5:33–44. doi: 10.1038/nrm1280. [DOI] [PubMed] [Google Scholar]
- 22.Antoku S., Saksela K., Mayer B.J. A crucial role in cell spreading for the interaction of Abl PxxP motifs with Crk and Nck adaptors. J. Cell Sci. 2008;121:3071–3082. doi: 10.1242/jcs.031575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaul Y. c-Abl: activation and nuclear targets. Cell Death Differ. 2000;7:10–16. doi: 10.1038/sj.cdd.4400626. [DOI] [PubMed] [Google Scholar]
- 24.Shishido T., Akagi T., Hanafusa H. Crk family adaptor proteins trans-activate c-Abl kinase. Genes Cells. 2001;6:431–440. doi: 10.1046/j.1365-2443.2001.00431.x. [DOI] [PubMed] [Google Scholar]
- 25.Sriram G., Reichman C., Birge R.B. Phosphorylation of Crk on tyrosine 251 in the RT loop of the SH3C domain promotes Abl kinase transactivation. Oncogene. 2011;30:4645–4655. doi: 10.1038/onc.2011.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Savitski M.M., Reinhard F.B., Drewes G. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014;346:1255784. doi: 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- 27.Palencia A., Cobos E.S., Luque I. Thermodynamic dissection of the binding energetics of proline-rich peptides to the Abl-SH3 domain: implications for rational ligand design. J. Mol. Biol. 2004;336:527–537. doi: 10.1016/j.jmb.2003.12.030. [DOI] [PubMed] [Google Scholar]
- 28.Ferreon J.C., Hilser V.J. Thermodynamics of binding to SH3 domains: the energetic impact of polyproline II (PII) helix formation. Biochemistry. 2004;43:7787–7797. doi: 10.1021/bi049752m. [DOI] [PubMed] [Google Scholar]
- 29.Wang C., Pawley N.H., Nicholson L.K. The role of backbone motions in ligand binding to the c-Src SH3 domain. J. Mol. Biol. 2001;313:873–887. doi: 10.1006/jmbi.2001.5083. [DOI] [PubMed] [Google Scholar]
- 30.Ladbury J.E., Arold S.T. Energetics of Src homology domain interactions in receptor tyrosine kinase-mediated signaling. Methods Enzymol. 2011;488:147–183. doi: 10.1016/B978-0-12-381268-1.00007-0. [DOI] [PubMed] [Google Scholar]
- 31.Zafra-Ruano A., Luque I. Interfacial water molecules in SH3 interactions: getting the full picture on polyproline recognition by protein-protein interaction domains. FEBS Lett. 2012;586:2619–2630. doi: 10.1016/j.febslet.2012.04.057. [DOI] [PubMed] [Google Scholar]
- 32.Fersht A.R. W.H. Freeman; New York: 1999. Structure and Mechanism in Protein Science. [Google Scholar]
- 33.Zhukov A., Karlsson R. Statistical aspects of van ’t Hoff analysis: a simulation study. J. Mol. Recognit. 2007;20:379–385. doi: 10.1002/jmr.845. [DOI] [PubMed] [Google Scholar]
- 34.Otwinowski Z., Minor W. Processing of x-ray crystallographic data in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 35.Adams P.D., Afonine P.V., Zwart P.H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Murshudov G.N., Skubák P., Vagin A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delaglio F., Grzesiek S., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 39.Keller R. CANTINA; Reiskirchen, Germany: 2004. The Computer-Aided Resonance Assignment Tutorial. [Google Scholar]
- 40.Ikura M., Kay L.E., Bax A. A novel approach for sequential assignment of 1H, 13C, and 15N spectra of proteins: heteronuclear triple-resonance three-dimensional NMR spectroscopy. Application to calmodulin. Biochemistry. 1990;29:4659–4667. doi: 10.1021/bi00471a022. [DOI] [PubMed] [Google Scholar]
- 41.Grzesiek S., Bax A. An efficient experiment for sequential backbone assignment of medium-sized isotopically enriched proteins. J. Magn. Reson. 1992;99:201–207. [Google Scholar]
- 42.Bax A., Ikura M. An efficient 3D NMR technique for correlating the proton and 15N backbone amide resonances with the α-carbon of the preceding residue in uniformly 15N/13C enriched proteins. J. Biomol. NMR. 1991;1:99–104. doi: 10.1007/BF01874573. [DOI] [PubMed] [Google Scholar]
- 43.Kay L.E., Ikura M., Bax A. Three-dimensional triple-resonance NMR spectroscopy of isotopically enriched proteins. 1990. J. Magn. Reson. 2011;213:423–441. doi: 10.1016/j.jmr.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 44.Clubb R.T., Thanabai V., Wagner G. A constant-time three-dimensional triple-resonance pulse scheme to correlate intraresidue 1HN, 15N, and 13C′ chemical shifts in 15N–13C labeled proteins. J. Magn. Reson. 1992;97:213–217. [Google Scholar]
- 45.Wittekind M., Mueller L. HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the α- and β-carbon resonances in proteins. J. Magn. Reson. B. 1993;B101:201–205. [Google Scholar]
- 46.Grzesiek S., Bax A. Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N-enriched proteins. J. Biomol. NMR. 1993;3:185–204. doi: 10.1007/BF00178261. [DOI] [PubMed] [Google Scholar]
- 47.Bax A., Clore G.M., Gronenborn A.M. 1H-1H correlation via isotropic mixing of 13C magnetization, a new three-dimensional approach for assigning 1H and 13C spectra of 13C-enriched proteins. J. Magn. Reson. 1990;88:425–431. [Google Scholar]
- 48.Wishart D.S., Bigam C.G., Sykes B.D. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 49.Ferrage F., Reichel A., Ghose R. On the measurement of 15N-1H nuclear Overhauser effects. 2. Effects of the saturation scheme and water signal suppression. J. Magn. Reson. 2010;207:294–303. doi: 10.1016/j.jmr.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lipari G., Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 1982;104:4546–4559. [Google Scholar]
- 51.Lipari G., Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 1982;104:4559–4570. [Google Scholar]
- 52.Mandel A.M., Akke M., Palmer A.G., 3rd Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J. Mol. Biol. 1995;246:144–163. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- 53.Cole R., Loria J.P. FAST-Modelfree: a program for rapid automated analysis of solution NMR spin-relaxation data. J. Biomol. NMR. 2003;26:203–213. doi: 10.1023/a:1023808801134. [DOI] [PubMed] [Google Scholar]
- 54.Fraternali F., Cavallo L. Parameter optimized surfaces (POPS): analysis of key interactions and conformational changes in the ribosome. Nucleic Acids Res. 2002;30:2950–2960. doi: 10.1093/nar/gkf373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cavallo L., Kleinjung J., Fraternali F. POPS: a fast algorithm for solvent accessible surface areas at atomic and residue level. Nucleic Acids Res. 2003;31:3364–3366. doi: 10.1093/nar/gkg601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murphy K.P., Freire E. Thermodynamics of structural stability and cooperative folding behavior in proteins. Adv. Protein Chem. 1992;43:313–361. doi: 10.1016/s0065-3233(08)60556-2. [DOI] [PubMed] [Google Scholar]
- 57.Luque I., Freire E. Structural parameterization of the binding enthalpy of small ligands. Proteins. 2002;49:181–190. doi: 10.1002/prot.10208. [DOI] [PubMed] [Google Scholar]
- 58.Jankowski W., Saleh T., Kalodimos C.G. Domain organization differences explain Bcr-Abl’s preference for CrkL over CrkII. Nat. Chem. Biol. 2012;8:590–596. doi: 10.1038/nchembio.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McCoy A.J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lim W.A., Richards F.M., Fox R.O. Structural determinants of peptide-binding orientation and of sequence specificity in SH3 domains. Nature. 1994;372:375–379. doi: 10.1038/372375a0. [DOI] [PubMed] [Google Scholar]
- 61.Xue Y., Yuwen T., Skrynnikov N.R. Role of electrostatic interactions in binding of peptides and intrinsically disordered proteins to their folded targets. 1. NMR and MD characterization of the complex between the c-Crk N-SH3 domain and the peptide Sos. Biochemistry. 2014;53:6473–6495. doi: 10.1021/bi500904f. [DOI] [PubMed] [Google Scholar]
- 62.Horn J.R., Russell D., Murphy K.P. Van ’t Hoff and calorimetric enthalpies from isothermal titration calorimetry: are there significant discrepancies? Biochemistry. 2001;40:1774–1778. doi: 10.1021/bi002408e. [DOI] [PubMed] [Google Scholar]
- 63.Brown A.M., Zondlo N.J. A propensity scale for type II polyproline helices (PPII): aromatic amino acids in proline-rich sequences strongly disfavor PPII due to proline-aromatic interactions. Biochemistry. 2012;51:5041–5051. doi: 10.1021/bi3002924. [DOI] [PubMed] [Google Scholar]
- 64.Elam W.A., Schrank T.P., Hilser V.J. Evolutionary conservation of the polyproline II conformation surrounding intrinsically disordered phosphorylation sites. Protein Sci. 2013;22:405–417. doi: 10.1002/pro.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tran H.T., Wang X., Pappu R.V. Reconciling observations of sequence-specific conformational propensities with the generic polymeric behavior of denatured proteins. Biochemistry. 2005;44:11369–11380. doi: 10.1021/bi050196l. [DOI] [PubMed] [Google Scholar]
- 66.Ruzza P., Siligardi G., Borin G. 4-Fluoroproline derivative peptides: effect on PPII conformation and SH3 affinity. J. Pept. Sci. 2006;12:462–471. doi: 10.1002/psc.750. [DOI] [PubMed] [Google Scholar]
- 67.McDonald C.B., Seldeen K.L., Farooq A. SH3 domains of Grb2 adaptor bind to PXpsiPXR motifs within the Sos1 nucleotide exchange factor in a discriminate manner. Biochemistry. 2009;48:4074–4085. doi: 10.1021/bi802291y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rubini C., Ruzza P., Donella-Deana A. Recognition of lysine-rich peptide ligands by murine contacting SH3 domain: CD, ITC, and NMR studies. Biopolymers. 2010;94:298–306. doi: 10.1002/bip.21350. [DOI] [PubMed] [Google Scholar]
- 69.Ferreon J.C., Hilser V.J. The effect of the polyproline II (PPII) conformation on the denatured state entropy. Protein Sci. 2003;12:447–457. doi: 10.1110/ps.0237803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hamburger J.B., Ferreon J.C., Hilser V.J. Thermodynamic mechanism and consequences of the polyproline II (PII) structural bias in the denatured states of proteins. Biochemistry. 2004;43:9790–9799. doi: 10.1021/bi049352z. [DOI] [PubMed] [Google Scholar]
- 71.Breiten B., Lockett M.R., Whitesides G.M. Water networks contribute to enthalpy/entropy compensation in protein-ligand binding. J. Am. Chem. Soc. 2013;135:15579–15584. doi: 10.1021/ja4075776. [DOI] [PubMed] [Google Scholar]
- 72.Martin-Garcia J.M., Ruiz-Sanz J., Luque I. Interfacial water molecules in SH3 interactions: a revised paradigm for polyproline recognition. Biochem. J. 2012;442:443–451. doi: 10.1042/BJ20111089. [DOI] [PubMed] [Google Scholar]
- 73.Young T., Abel R., Friesner R.A. Motifs for molecular recognition exploiting hydrophobic enclosure in protein-ligand binding. Proc. Natl. Acad. Sci. USA. 2007;104:808–813. doi: 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li Z., Lazaridis T. Thermodynamics of buried water clusters at a protein-ligand binding interface. J. Phys. Chem. B. 2006;110:1464–1475. doi: 10.1021/jp056020a. [DOI] [PubMed] [Google Scholar]
- 75.Palencia A., Camara-Artigas A., Luque I. Role of interfacial water molecules in proline-rich ligand recognition by the Src homology 3 domain of Abl. J. Biol. Chem. 2010;285:2823–2833. doi: 10.1074/jbc.M109.048033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lu Y., Wang R., Wang S. Analysis of ligand-bound water molecules in high-resolution crystal structures of protein-ligand complexes. J. Chem. Inf. Model. 2007;47:668–675. doi: 10.1021/ci6003527. [DOI] [PubMed] [Google Scholar]
- 77.Ferreon J.C., Volk D.E., Hilser V.J. Solution structure, dynamics, and thermodynamics of the native state ensemble of the Sem-5 C-terminal SH3 domain. Biochemistry. 2003;42:5582–5591. doi: 10.1021/bi030005j. [DOI] [PubMed] [Google Scholar]
- 78.Manson A., Whitten S.T., Hilser V.J. Characterizing the role of ensemble modulation in mutation-induced changes in binding affinity. J. Am. Chem. Soc. 2009;131:6785–6793. doi: 10.1021/ja809133u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Akke M., Bruschweiler R., Palmer A.G. NMR order parameters and free energy: An analytical approach and its application to cooperative Ca2+ binding by calbinding D9k. J. Am. Chem. Soc. 1993;115:9832–9833. [Google Scholar]
- 80.Yang D., Kay L.E. Contributions to conformational entropy arising from bond vector fluctuations measured from NMR-derived order parameters: application to protein folding. J. Mol. Biol. 1996;263:369–382. doi: 10.1006/jmbi.1996.0581. [DOI] [PubMed] [Google Scholar]
- 81.Igumenova T.I., Lee A.L., Wand A.J. Backbone and side chain dynamics of mutant calmodulin-peptide complexes. Biochemistry. 2005;44:12627–12639. doi: 10.1021/bi050832f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ferreon J.C., Hilser V.J. Ligand-induced changes in dynamics in the RT loop of the C-terminal SH3 domain of Sem-5 indicate cooperative conformational coupling. Protein Sci. 2003;12:982–996. doi: 10.1110/ps.0238003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee A.L., Wand A.J. Microscopic origins of entropy, heat capacity and the glass transition in proteins. Nature. 2001;411:501–504. doi: 10.1038/35078119. [DOI] [PubMed] [Google Scholar]
- 84.van Etten R.A., Jackson P., Baltimore D. The mouse type IV c-abl gene product is a nuclear protein, and activation of transforming ability is associated with cytoplasmic localization. Cell. 1989;58:669–678. doi: 10.1016/0092-8674(89)90102-5. [DOI] [PubMed] [Google Scholar]
- 85.Rubin G.M., Yandell M.D., Lewis S. Comparative genomics of the eukaryotes. Science. 2000;287:2204–2215. doi: 10.1126/science.287.5461.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Engh R.A., Huber R. Accurate bond and angle parameters for x-ray protein structure refinement. Acta Crystallogr. A. 1991;47:392–400. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.