

Abstract
Growth related traits in fish are controlled by quantitative trait loci (QTL), but no QTL for growth have been detected in bighead carp (Hypophthalmichthys nobilis) due to the lack of high-density genetic map. In this study, an ultra-high density genetic map was constructed with 3,121 SNP markers by sequencing 117 individuals in a F1 family using 2b-RAD technology. The total length of the map was 2341.27 cM, with an average marker interval of 0.75 cM. A high level of genomic synteny between our map and zebrafish was detected. Based on this genetic map, one genome-wide significant and 37 suggestive QTL for five growth-related traits were identified in 6 linkage groups (i.e. LG3, LG11, LG15, LG18, LG19, LG22). The phenotypic variance explained (PVE) by these QTL varied from 15.4% to 38.2%. Marker within the significant QTL region was surrounded by CRP1 and CRP2, which played an important role in muscle cell division. These high-density map and QTL information provided a solid base for QTL fine mapping and comparative genomics in bighead carp.
High resolution linkage maps have become indispensable to many genetic studies, such as molecular marker-assisted selection (MAS)1, genome scaffolding and assembly2, comparative genomic analysis3 and fine-scale quantitative trait locus (QTL) mapping4. Large numbers of genetic markers available is a key prerequisite for constructing a high-resolution linkage map, especially single nucleotide polymorphisms (SNPs) markers, which also played an important role in ecological, genetics and evolutionary studies5,6,7,8. Next generation sequencing opened up new possibilities in SNP marker’s developing and has made it in a rapid and cost-effective manner6. Recent years, many genotyping-by-sequencing (GBS) methods which combined restriction enzymes and high-throughput next generation sequencing have been developed9,10,11,12 and used in aquaculture species12,13,14,15. In particular, restriction-sites associated DNA sequencing (RAD-seq) is convenient for non-model species16,17. Through the massively parallel sequencing of DNA sequences flanking restricted sites, RAD-seq identifies hundreds or thousands SNPs simultaneously for multiple samples. But there are a lot of pitfalls in RAD-seq, such as complicated manual operations and uneven genome coverage. Then, methods with simpler library preparation protocols were developed. For example, 2b-RAD method developed by Wang et al.10 has many benefits compared with other methods, such as easy to handle, less time needed for manual operation and even distribution on genome.
Bighead carp (Hypophthalmichthys nobilis) belongs to family of Cyprinidae. This species has 24 pairs of chromosomes and the genome size is about 0.9 Gb18. The annual production of bighead carp reached 3.25 million tons worldwide in 2014, and over 98% of the production was from China (FAO, 2014). Bighead carp is one of most important and valuable aquaculture species in China, with annual production rank the fourth among all freshwater species in 201219. Although considerable work had been done for bighead carp to increase production, some specific characters hindered achievement of the traditional selective breeding based on phenotypes, such as very long breeding cycle (i.e. 4 years for male and 4–5 years for female), lack of large-scale genomic resource and short of markers tightly associated with growth rate. These disadvantages made it infeasible to select phenotypes for strains or varieties with higher growth rate in a short time. MAS can overcome those disadvantages in traditional selection by using DNA markers tightly linked to some QTL to help phenotypic screening20,21. This method can increase the accuracy and efficiency of selection, thereafter is an ideal mean for traits that are difficult to be improved by traditional selection.
In bighead carp, some work had been done on development of co-dominate DNA markers and linkage map construction22,23,24. However, the resolution of these maps isn’t high enough to detect QTL. The reason for that is genotyping large numbers of simple sequence repeats (SSRs) needs a lot of time and human labor. Now, with the development of sequencing, rapid genotyping of tens of thousands of SNP markers is accessible, which makes construction of a high-density SNP genetic map easy to come6.
The main purpose of our present study is to construct a high-density linkage map with only SNP markers to facilitate QTL mapping for growth related traits, such as body weight (BW), body length (BL), total length (TL), head length (HL) and body height (BH). Here, we constructed a linkage map with 3,121 markers using 2b-RAD on Illumina HiSeq 2500 platform. A major QTL for body length were detected on linkage group (LG) 15. Besides, 37 suggestive QTL were detected for five growth-related traits, whose phenotypic variation explained (PVE) varied from 15.4% to 38.2%. The genetic map and QTL can be used in breeding populations for genetic improvement of bighead carp.
Results
2b-RAD Sequencing
All sequencing libraries for the full-sibling family were built with modified 2b-RAD methods10. After filtering low quality reads, the sequencing depth for the two parents was 4,852,376 and 5,162,352 reads, respectively. While the average depth for all progenies was 2.128 M reads per sample and total reads number for them was 249,204,191 reads (Supplementary Table S1). A total of 70,504 loci were detected after reads from parents clustering. The average depths for all the loci were 70.98-fold for parents and 30.17-fold for progenies. Finally, we mapped all the reads from parents and offspring to the loci and genotyped every locus with at least 80% of progenies presented. We found 7,786 loci were polymorphic and used them in the following map construction. Among these loci, 5,567 were co-dominant loci which were present in both parents. While the rest 2,219 were dominant loci which were present only in one of the parents.
High-resolution linkage mapping
After discarding 4,463 SNPs with segregation distortion the rest 3,323 markers were used in sex-average linkage group assignment . Finally, 3,121 markers were placed onto 24 linkage groups (LGs) with a LOD (logarithm of odds threshold) of 11.0, designated as LG1-LG24, corresponding to the LG names of map constructed by micro-satellite markers23 (Fig. 1). The types of all 3,121 markers were illustrated in Supplementary Figure 2. The total length of the map was determined to be 2341.27 cM, with an average interval of 0.75 cM. About 97.4% of all markers’ interval was within 3 cM and 75.2% of them shorter than 1 cM. The sizes of linkage groups range from 68.53 cM to 145.42 cM (Table 1). For markers in each LG, map length calculations were performed based on recombination frequencies. The information of all markers on the LGs is presented in Supplementary Table S2, which includes marker name, sequence, and genetic position on each LG. The largest gap in the map was 14.55 cM in LG16, followed by 12.86 cM in LG13. Though we removed all skewed markers with P < 0.001, we found 184 skewed markers with P < 0.05 and 257 with P < 0.01 placed on the final map. An area containing four skewed markers was defined as a segregation distortion region (SDR). Four SDRs were detected in the map, and two of them were in LG13, which were separated by only one un-skewed marker. The other two were in LG21 and LG23. These SDRs may be related with preferential selection and would not affect the accuracy of the genetic map25,26. We mapped all 16 skewed markers to bighead carp genome (unpublished data) and got only three genes with define functions, abi1a, dtx4 and b4galt2.
Figure 1. Genetic lengths and marker distribution of 24 linkage groups in the sex-averaged linkage map of bighead carp.

A black bar indicated a 2b-RAD marker. The scaleplate on the left indicated genetic distance (centiMorgan as unit).
Table 1. Summary statistics of the sex-averaged linkage map of bighead carp.
| LG Name | Num of SNPs | LG length(cM) | Average Dist(cM) | Largest Gap(cM) |
|---|---|---|---|---|
| LG1 | 109 | 123.841 | 1.136155963 | 6.636 |
| LG2 | 76 | 77.742 | 1.022921053 | 9.164 |
| LG3 | 174 | 107.931 | 0.620293103 | 4.835 |
| LG4 | 80 | 68.534 | 0.856675 | 4.989 |
| LG5 | 118 | 102.407 | 0.867855932 | 4.838 |
| LG6 | 109 | 77.082 | 0.707174312 | 9.946 |
| LG7 | 109 | 76.585 | 0.702614679 | 8.759 |
| LG8 | 350 | 145.428 | 0.415508571 | 3.602 |
| LG9 | 192 | 102.288 | 0.53275 | 4.768 |
| LG10 | 77 | 92.693 | 1.203805195 | 5.343 |
| LG11 | 103 | 114.211 | 1.10884466 | 5.19 |
| LG12 | 170 | 90.881 | 0.534594118 | 4.955 |
| LG13 | 151 | 97.681 | 0.64689404 | 12.86 |
| LG14 | 95 | 89.481 | 0.941905263 | 6.064 |
| LG15 | 105 | 101.486 | 0.966533333 | 5.801 |
| LG16 | 121 | 109.233 | 0.902752066 | 14.552 |
| LG17 | 103 | 92.183 | 0.894980583 | 5.323 |
| LG18 | 103 | 91.019 | 0.883679612 | 4.093 |
| LG19 | 92 | 85.724 | 0.931782609 | 3.09 |
| LG20 | 93 | 82.143 | 0.883258065 | 8.631 |
| LG21 | 150 | 119.297 | 0.795313333 | 3.203 |
| LG22 | 240 | 109.188 | 0.45495 | 3.253 |
| LG23 | 82 | 86.766 | 1.058121951 | 7.271 |
| LG24 | 119 | 97.443 | 0.818848739 | 6.136 |
| TOTAL | 3121 | 2341.26 | 0.75 | 14.552 |
LG: linkage group; cM: centiMorgan.
To uncover the macro-collinearity between bighead carp and zebrafish, we mapped all 3,121 markers on final map to zebrafish genome and got 873 markers were located on 25 chromosomes of zebrafish (Supplementary Table S5). 23 pairs of 1 to 1 synteny on chromosome level were uncovered, except for LG 9 in bighead carp was mapped to Chr10 and Chr22 in zebrafish (Fig. 2). Within each pair of chromosomes, the map position of markers also showed good synteny with their corresponding loci in zebrafish genome.
Figure 2. Circos mapping of 24 linkage group of bighead carp (right semi-cirle) to the 25 linkage group of zebrafish (left semi-circle).

QTL mapping of growth-related traits
Pairwise comparisons among five growth-related traits (TL, BL, HL, BW, BH) using Pearson’s correlation revealed that all of them were highly correlated (P < 0.01) (Table 2). The highest correlation coefficients were 0.98 between TL and BL, and the lowest correlation coefficients were 0.82 between BH and HL.
Table 2. Pairwise correlation for all 5 growth-related traits in bighead carp.
| TL | BL | BH | HL | BW | |
|---|---|---|---|---|---|
| TL | 1 | ||||
| BL | 0.987885 | 1 | |||
| BH | 0.883167 | 0.871764 | 1 | ||
| HL | 0.911896 | 0.91138 | 0.82535 | 1 | |
| BW | 0.964491 | 0.968115 | 0.918774 | 0.872767 | 1 |
TL: total length. BL: body length. BH: body height. HL: head length. BW: body weight.
We used the phenotype data above to detect QTL affecting growth-related traits and found they were located on various linkage groups in linkage-group-wide scale (Table 3 and Fig. 3). TL had 9 QTL, and qTL15-a, the most prominent QTL on LG15, accounted for 30.2% of the PVE. Another two QTL on LG 11 within a region of 3 cM (position of 52.31 cM and 55.22 cM) accounted for 27.3% and 26.1% of PVE. The genetic effects of other QTL were comparatively smaller. Nine QTL for BL were identified, all of which were the same as that for TL. The PVE of these nine QTL varied form 18.2–29.5%. We also found 9 QTL for BH, the largest of which is qBH22-a on LG22 with 23.2% of the PVE. For HL, only 4 QTL were found, with the largest effect displayed by qHL15-a on LG15, explaining 38.3% of the PVE. Seven QTL were found for BW, and the largest effect was on qBW11-b, explaining 22.1% of the PVE.
Table 3. All QTL for five growth-related traits.
| Trait | QTL name | Marker | LG | Position(cM) | LOD | LOD Threshold |
PVE(%) | |
|---|---|---|---|---|---|---|---|---|
| Genome-wide | Group-wide | |||||||
| Total length | qTL15-a | ref-8593_27 | 15 | 23.97 | 8.28 | 8.4 | 3.9 | 30.2 |
| qTL11-b | ref-24929_7 | 11 | 52.318 | 7.33 | 8.4 | 4.6 | 27.3 | |
| qTL11-c | ref-65692 | 11 | 55.227 | 6.95 | 8.4 | 4.6 | 26.1 | |
| qTL11-d | ref-67070_2 | 11 | 96.315 | 6.86 | 8.4 | 4.6 | 25.8 | |
| qTL19-a | ref-20240_31 | 19 | 6.099 | 6.71 | 8.4 | 4.1 | 25.3 | |
| qTL18-a | ref-36051 | 18 | 51.107 | 5.87 | 8.4 | 4.5 | 22.5 | |
| qTL22-a | ref-34298 | 22 | 54.484 | 5.52 | 8.4 | 4.3 | 21.3 | |
| qTL3-b | ref-51859 | 3 | 27.12 | 5.09 | 8.4 | 4.3 | 19.8 | |
| qTL3-a | ref-66486_3 | 3 | 4.741 | 4.64 | 8.4 | 4.3 | 18.3 | |
| Body length | qBL15-a | ref-8593_27 | 15 | 23.97 | 8.05 | 8 | 4 | 29.5 |
| qBL11-c | ref-65692 | 11 | 55.227 | 7.55 | 8 | 4.8 | 27.9 | |
| qBL11-b | ref-24929_7 | 11 | 52.318 | 6.93 | 8 | 4.8 | 26 | |
| qBL11-d | ref-67070_2 | 11 | 96.315 | 6.52 | 8 | 4.8 | 24.7 | |
| qBL19-a | ref-20240_31 | 19 | 6.099 | 6.38 | 8 | 3.8 | 24.2 | |
| qBL18-b | ref-36051 | 18 | 51.107 | 5.76 | 8 | 4.3 | 22.1 | |
| qBL22-a | ref-34298 | 22 | 54.484 | 5.23 | 8 | 4.2 | 20.3 | |
| qBL3-b | ref-51859 | 3 | 27.12 | 5.07 | 8 | 4.2 | 19.8 | |
| qBL3-a | ref-66486_3 | 3 | 4.741 | 4.61 | 8 | 4.2 | 18.2 | |
| Body heigth | qBH22-a | ref-34298 | 22 | 54.484 | 6.07 | 7.4 | 4 | 23.2 |
| qBH11-b | ref-65692 | 11 | 55.227 | 5.96 | 7.4 | 4.2 | 22.8 | |
| qBH11-a | ref-24929_7 | 11 | 52.318 | 5.76 | 7.4 | 4.2 | 22.2 | |
| qBH15-a | ref-8593_27 | 15 | 23.97 | 5.23 | 7.4 | 3.9 | 20.3 | |
| qBH3-b | ref-15932_2 | 3 | 60.679 | 4.6 | 7.4 | 4.1 | 18.1 | |
| qBH3-a | ref-51859 | 3 | 27.12 | 4.48 | 7.4 | 4.1 | 17.7 | |
| qBH18-a | ref-36051 | 18 | 51.107 | 4.26 | 7.4 | 4 | 16.9 | |
| qBH11-c | ref-67070_2 | 11 | 96.315 | 4.22 | 7.4 | 4.2 | 16.7 | |
| qBH19-a | ref-20240_31 | 19 | 6.099 | 3.84 | 7.4 | 3.7 | 15.4 | |
| Head length | qHL15-a | ref-8593_27 | 15 | 23.97 | 11.08 | 12.3 | 4.1 | 38.2 |
| qHL22-a | ref-34298 | 22 | 54.484 | 5.96 | 12.3 | 4.7 | 22.8 | |
| qHL3-b | ref-51859 | 3 | 27.12 | 5.51 | 12.3 | 4.6 | 21.3 | |
| qHL3-a | ref-66486_3 | 3 | 4.741 | 4.96 | 12.3 | 4.6 | 19.4 | |
| Body | qBW11-b | ref-65692 | 11 | 55.227 | 5.75 | 5.8 | 4 | 22.1 |
| weight | qBW11-a | ref-24929_7 | 11 | 52.318 | 5.39 | 5.8 | 4 | 20.9 |
| qBW15-a | ref-8593_27 | 15 | 23.97 | 5.19 | 5.8 | 3.7 | 20.2 | |
| qBW11-c | ref-67070_2 | 11 | 96.315 | 4.88 | 5.8 | 4 | 19.1 | |
| qBW15-b | ref-4159 | 15 | 70.05 | 4.59 | 5.8 | 3.7 | 18.1 | |
| qBW18-a | ref-36051 | 18 | 51.107 | 4.51 | 5.8 | 3.8 | 17.8 | |
| qBW19-a | ref-20240_31 | 19 | 6.099 | 4.42 | 5.8 | 3.7 | 17.5 | |
PVE: phenotypic variance explained.
Figure 3. LOD scores along the 24 linkage groups for variation in 5 growth-related traits in bighead carp.

The red horizontal line on each trait indicates the LOD for genome-wide significances for that trait.
However, in these 38 QTL, only one QTL was of the genome-wide level of significance. After counting the frequency of the QTL in the five traits, we found 11 different loci on the genome (Fig. 4). Within which, one co-localization QTL (qBW15-a) on LG15 was common to all five traits, while 7 other co-localization QTL were common to four traits, and the rest 3 QTL were associated with 3 and 1 traits, respectively (Supplementary Table S3). After mapping 11 markers in these QTL to bighead carp genome (unpublished data), we found 12 genes closing to the mapping position. Then these genes were blasted to the protein database of zebrafish and got the corresponding protein. Co-localization QTL for five traits (Marker: ref-8593_27) were surrounded by CRP1 and CRP2, which had good synteny compared with zebrafish genome. The closest gene next to co-localization QTL for four traits (Marker: ref-67070_2) was SASH1, which regulates cell proliferation and apoptosis in human27,28.
Figure 4. Genomic QTL distribution on 6 different chromosomes.

Discussion
In this work, we constructed a high-density genetic map for bighead carp via 2b-RAD and used it to detect growth-related QTL. Linkage map is an important pre-requisite for mapping QTL of interested traits for a given species. In bighead carp, two genetic maps have been published previously: one was constructed by SSR and AFLP markers22, the other was constructed solely by SSR markers23. Table 4 summarized the main differences in the parameters derived from them. For the first one, 120 co-dominate markers were used and resulted in 39 LG, which is much more than the number of haploid chromosomes in bighead carp (N = 24)29. The second generation genetic map made great improvement, which involved 659 co-dominate markers and formed exactly 24 LG. And the map resolution also narrowed from 3.9 cM to 2.9 cM and the map length doubled from 852.0 cM to 1,917.3 cM23. Although the density and length of linkage map had been improved in the last few years, it’s still far from satisfaction for genetic studies and MAS in bighead carp. The emergency and development of next generation sequencing (NGS) technology has mad RAD-seq a feasible way to genotyping the whole genome in a short time. However, library construction for RAD-seq is a labor-intensive and time-consuming work. Therefore, a lot of modifications had been proposed, such as ddRAD30, SLAF12,31 and 2b-RAD10. 2b-RAD provided a streamlined alternative to existing RAD-seq library construction method; for it can be done in 4 hours with all reactions happened consecutively in a single well. At the same time, 2b-RAD can detected almost every restriction site in the genome in parallel whereas other RAD-seq methods can only get a subset of total sites due to the multiple times’ size selection9,10. The third advantage that 2b-RAD provided is its flexible choice of selective adaptor, which can adjust the marker density in the genome. This choice can balance level of genotyping detail against sequencing throughput depending on various types of study13.
Table 4. Comparison of genome feature for three bighead carp genetic linkage maps.
| This study | Liao et al.22 | Zhu et al.32 | |
|---|---|---|---|
| Number of linkage groups recovered | 24 | 30 | 24 |
| Genome size estimate | 2379.5 | 1209.0 | 2067.1 |
| Genome coverage estimate | 98.3% | 82.8% | 92.8% |
| Number of markers used for map | 3121 SNPs | 153(27 SSRs & 126 AFLPs) | 659 SSRs |
| Map length [cM] | 2341.3 | 852.0 | 1917.3 |
| Average length of marker interval | 0.75 | 7.0 | 2.9 |
| Linkage group size range [cM] | 68.5–145.4 | 2.2–74.9 | 52.2–133.5 |
In this study, we used a crossing strategy of a male and a female bighead carp and their 117 progenies as mapping population. Then genotyping SNPs for the population with 2b-RAD and got a high-density linkage map of 3,121 markers. This number is almost five times of that in previous linkage map constructed by SSR markers, and the average distance between markers dropped drastically from 2.9 cM to 0.75 cM. With more markers used in our map, the observed genome length was 2341.27 cM for the consensus map, which is 22% longer than previous map constructed by SSRs23. The synteny analysis between this genetic map and zebrafish genome showed 23 pairs of ortholog chromosomes, except for the LG9 in bighead carp corresponding to Chr10 and Chr22 in zebrafish genome. This result of a good macro-collinearity is consistent with previous study on comparative analysis of bighead carp genome and zebrafish genome32 Meanwhile, the macro-collinearity above justified the LOD used in this study is adequate in high-density SNP genetic map construction33, although they are much higher compared with those LOD value used in SSR genetic map23,34.
To increase the genomic coverage on the final genetic map, skewed markers were also used in the map construction. In total, 16 skewed markers in four segregation distortion regions were found. The annotation of these markers found three genes with known function, abi1a, dtx4 and b4galt2. Gene abi1 play a role in the progression of several malignancies including colon cancer35, ovarian cancer36 and breast cancer37. Gene b4galt2 act as a target gene of p53-mediated HeLa cell apoptosis38. Marker distortion might be caused by special selection on some type of genotype, such as double recessive of some lethal genes, or caused by zygotic selection39. It has been reported that skewed markers can be beneficial to map construction if used properly, for these areas containing skewed markers may have higher recombination ratio for specific traits40.
In addition to the high recovery of type IIB restriction fragments of BcgI, RAD typing41 was used to genotype all dominant (presented in only one parent) and co-dominant (presented in both parents) markers. As more than half of 2b-RAD markers were made up of dominant markers13, using software only genotyping co-dominate markers like Stacks42 will greatly waste genetic information offered by sequencing reads. In this study, we constructed 2,219 (28.49%) dominant out of 7,786 reference markers. In the final 3,121 markers, 1,474 (47.22%) were dominant markers, and this proportion is similar to that in bivalve mollusc13. At the same time, high macro-collinearity among 2b-RAD markers of our map and the zebrafish genome indicated that the genotyping of dominant and co-dominant markers is free from error32.
Growth-related traits are economically and ecologically important traits and their genetic basis is thought to be highly polygenic in most organisms. Mapping QTL for growth-related traits is critically important in molecular breeding. Many QTL studies of growth-related traits have been conducted in common carp43,44, Asian seabass45, nine-spined stickleback46 and Atlantic salmon47. Only one study was carried out about the QTL of growth-related traits in a hybrid progenies between bighead carp and silver carp48. In this study, 5 morphometric body traits were measured and QTL responded for them were detected.
Here, we have found one significant QTL and 37 suggestive QTL for growth-related traits in bighead carp. For these QTL, we found most of their locations were overlapped on linkage groups (Supplementary Table S3). For example, marker ref-8593_27 locating on LG15 corresponded to five QTL, including qTL15-a, qBL15-b, qBH15-a, qHL15-a and qBW15-c. Meanwhile, each of seven locations on LG19, LG11, LG22, LG18, LG3 were corresponding to four QTL. For the rest of five QTL, three were overlapped in one genomic location and other two QTL were on two different genomic locations. One reason for this result might be the high correlation coefficients among the 5 traits in bighead carp. As can be seen from Table 2, the lowest correlation coefficients were 0.82 between BH and HL and the highest correlation coefficients were 0.98 between TL and BL. Based on this, we could conclude that there was a strong almost linear relationship among the 5 traits. And even further, QTL in one trait might be response for all other four traits. In bighead carp breeding, selection for one of the five traits may also result in improvement of others without use all measurement traits. Another reason for this result might be growth-related traits of bighead carp were controlled by only a few major areas in the genome. This pattern was similar to the pattern of QTL for growth-related traits in nine-spined stickleback46, common carp44 and chicken49. There are two patterns of growth-related QTL in fish, one is single major QTL accounted for significant portion of a trait and the other is many QTL with small or intermediate effects on the trait. Previous study compared growth rate in two pufferfish found that juvenile body size was mapped to only one QTL, which indicated single major gene mode of inheritance50. However, more studies uncovered that several QTL influenced growth in the Mexican cavefish51, the lake whitefish52 and nine-spined stickleback46. In this study, our results demonstrated that there are 4–9 QTL for each of the five traits, which indicated that bighead carp may be putative example of multiple QTL influencing on phenotypic variations of growth rate.
It has been reported that early growth in most tissues was caused by cell division, whereas in later stage growth was caused by an increase in cell size53. As the sex maturation for bighead carp can take 4–5 years, the sample time in this study (270 dpf) should be considered as early stage of the bighead carp lifespan. To study the functional background of these candidates QTL, we mapped all markers in QTL regions to bighead carp genome and searched their ortholog in zebrafish genome. For the QTL common to all five traits, we found it was surrounded by CRP1 and CRP2, which are potent smooth muscle differentiation cofactors that may play roles in muscle cell division54. Besides, we also found a QTL common to four traits was involved in SASH1, which regulates cell proliferation and apoptosis in human27,28.
This study represents the first ultra-high density genetic map for bighead carp, which has 3,121 SNP markers in it. This map was constituted by 24 linkage groups and they showed good synteny information with zebrafish genome. The average marker spacing was 0.75 cM. Using this linkage group, we mapped 38 QTL for five growth related traits. Five out of these 38 QTL were located on the same location on LG15, and QTL on this place was significant on genome wide level for body length. According to the mapping result of the markers in LG15, we found it was surrounded by CRP1 and CRP2, which played roles in muscle cell division. Markers flanking the QTL can be used to select fishes with better growth related traits.
Methods
Ethics statement
All experimental procedures involving the fish in this study were approved by the Committee for Animal Experiments of the Institute of Hydrobiology of the Chinese Academy of Sciences, China. The methods used in this study were carried out in accordance with the Laboratory Animal Management Principles of China. The rearing activities of bighead carp in Wuhan, Hubei were approved by the owner of the pond.
Mapping families and phenotypic data
The fish formed the parental generation (F0) were collected from Zhangdu Lake (Wuhan; 30° 40′ 03″N, 114° 43′ 51″E) in 2009. The female fish was crossed in vitro with the male one in May, 2011 and the resulting F1 –offspring was group-reared in one mud pond. At the same time, the mud pond cultured grass carp (Ctenopharyngodon idella) and common carp (Cyprinus carpio). Although bighead carp mainly feed on filtering algae in the water, feed was put into the pond for the growth of common carp. A total of 117 full-sib F1 progeny were sampled to construct a linkage map. Growth related traits including body length (BL), body height (BH), body weight (BW), head length (HL) and total length (TL) were measured only once for all 117 offspring at 240 dph (days post hatch). This family exhibited high within family variation of growth related traits and therefore was used for linkage construction and QTL analysis.(Supplementary Table S4).
2b-RAD sequencing
Libraries for 2b-RAD were prepared for two parents and 117 progenies according to the protocol developed by Wang et al.10 but did some modifications (Supplementary Figure 1). Fin clips cut from all samples were preserved in 95% ethanol at room temperature for DNA samples. Genomic DNA was extracted following the standard phenol-chloroform protocol55. The concentration of extracted DNA was determined using a spectrophotometer. We used BcgI enzyme (NEB, UK) to digest genomic DNA of parents and offspring. The sequences cut by BcgI enzyme were ligated to adaptors with 5′- NN - 3′ overhangs. A unique six base pair barcode was used in each library during library preparation, and then all libraries were mixed together for Illumina single-end sequencing (1*50 bp). The sequencing was carried out by Shanghai Sangon Inc, Shanghai. To maximize the chances of discovering segregating SNPs, the parents were sequenced at a substantially higher depth than the progenies (Supplementary Table 2). All the 2b-RAD sequencing reads were deposited in the NCBI sequence read archive (SRA) database (accession no. SRP070967).
Bioinformatics pipeline and SNP calling
The process for generating SNP genotype in this study was as follows. Firstly, raw reads were assigned to each sample according to their unique six base pair barcode. Secondly, raw reads were trimmed to remove adaptors and the terminal 2-bp positions were discarded to eliminate artifacts that might be arisen by ligation. Finally, reads contained no restriction site, long homo-polymer regions (>10 bp), ambiguous bases (N) or reads of low quality (>2 bp with quality less than Q20) were removed. The remaining high quality reads were used for further genotyping. Denovo genotyping was performed by RADtyping v1.3.041 with default parameters. This software used stringent criteria in filtering candidate markers. It removed markers with too much coverage compared with the average depth of all markers. Only these loci with at least 6 reads and the minority allele had at least 2 reads supporting were kept in the following analysis.
Linkage map construction and synteny analysis
Markers genotyped in at least 80% of all the samples were considered as high-quality markers and used for further analysis. Markers with significant segregation distortion (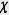 2 test, P < 0.001, df = 2) were excluded from the following map construction. Linkage map was created using JoinMap 4.0 software56. Linkage groups were identified with an independence logarithm of odds (LOD) threshold of 11.0. Unlinked markers and small linkage groups with less than 3 markers were not included from further analysis. The regression mapping algorithm was used for map construction. Map distances were estimated for each linkage group using Kosambi function. The map was visualized using MapChart ver.2.21.
2 test, P < 0.001, df = 2) were excluded from the following map construction. Linkage map was created using JoinMap 4.0 software56. Linkage groups were identified with an independence logarithm of odds (LOD) threshold of 11.0. Unlinked markers and small linkage groups with less than 3 markers were not included from further analysis. The regression mapping algorithm was used for map construction. Map distances were estimated for each linkage group using Kosambi function. The map was visualized using MapChart ver.2.21.
To uncover the synteny information between bighead carp and zebrafish, we mapped all tags within 24 LGs to zebrafish genome to get their corresponding locus. As the type IIB restriction fragment was only 32 bp, it’s difficult to be used as query in searching zebrafish genome. Therefore, we firstly mapped the short sequences to bighead carp genome (Shunping He et al., unpublished data) and then we extracted 100 bp in each end flanking the mapping site of every uniquely mapped marker and used all 232 bp sequences as query to blast the zebrafish genome.
Identification of segregation distortion region
After filtering these skewed markers with P < 0.001, we carefully check the position of markers with P < 0.01. If there are four consecutive skewed markers in one region, we defined this place as segregation distortion region. To uncover the genes within these regions, we mapped all skewed markers to bighead carp genome (Shunping He et al. unpublished data). Based on the position of all annotated genes and the skewed markers, we found genes located nearby these skewed markers. The position information about these skewed markers can be obtained upon request.
QTL analysis
QTL mapping for growth traits was carried out with MapQTL version 6.057. Multiple QTL Mapping (MQM) method was used to detect any significant associations between marker loci and growth-related traits. LOD statistics were calculated at an interval of 1 cM. The genome-wide (significance level) and linkage group-wide (suggestive level) LOD threshold were estimated using the permutation test (10,000 replicates) with a confidence interval of 95%. Graphic illustrations of the QTL profiles were generated using the program MapChart 2.258.
Additional Information
How to cite this article: Fu, B. et al. A high-density genetic map and growth related QTL mapping in bighead carp (Hypophthalmichthys nobilis). Sci. Rep. 6, 28679; doi: 10.1038/srep28679 (2016).
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grants 31502153) and the Knowledge Innovation Program of the Chinese Academy of Science. We thank Prof Shunping He for providing the unpublished bighead carp genome sequences.
Footnotes
The authors declare no competing financial interests.
Author Contributions B.F. and J.T. designed the project. B.F. and X.Y. performed the experiments. B.F. and H.L. analyzed the data. B.F. and J.T. wrote the manuscript. B.F. and H.L. prepared figures and tables. All authors reviewed and approved the manuscript.
References
- Fuji K. et al. Identification of a single major genetic locus controlling the resistance to lymphocystis disease in Japanese flounder (Paralichthys olivaceus). Aquaculture 254, 203–210, 10.1016/j.aquaculture.2005.11.024 (2006). [DOI] [Google Scholar]
- Jia J. et al. Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496, 91–95, 10.1038/nature12028 (2013). [DOI] [PubMed] [Google Scholar]
- Kakioka R., Kokita T., Kumada H., Watanabe K. & Okuda N. A RAD-based linkage map and comparative genomics in the gudgeons (genus Gnathopogon, Cyprinidae). BMC Genomics 14, 32, 10.1186/1471-2164-14-32 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K. et al. High-density genetic map construction and QTLs analysis of grain yield-related traits in Sesame (Sesamum indicum L.) based on RAD-Seq techonology. BMC Plant Biol 14, 274, s12870-014-0274-7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henning F., Lee H. J., Franchini P. & Meyer A. Genetic mapping of horizontal stripes in Lake Victoria cichlid fishes: benefits and pitfalls of using RAD markers for dense linkage mapping. Mol Ecol 23, 5224–5240, 10.1111/mec.12860 (2014). [DOI] [PubMed] [Google Scholar]
- Davey J. W. et al. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet 12, 499–510, 10.1038/nrg3012 (2011). [DOI] [PubMed] [Google Scholar]
- Hohenlohe P. A. et al. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLos Genet 6, e1000862, 10.1371/journal.pgen.1000862 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L. et al. Identification of domestication-related loci associated with flowering time and seed size in soybean with the RAD-seq genotyping method. Sci Rep 5, 9350, 10.1038/srep09350 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird N. A. et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLos One 3, e3376, 10.1371/journal.pone.0003376 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Meyer E., McKay J. K. & Matz M. V. 2b-RAD: a simple and flexible method for genome-wide genotyping. Nat Methods 9, 808–810, 10.1038/nmeth.2023 (2012). [DOI] [PubMed] [Google Scholar]
- Poland J. A., Brown P. J., Sorrells M. E. & Jannink J. L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLos One 7, e32253, 10.1371/journal.pone.0032253 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X. et al. SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLos One 8, e58700, 10.1371/journal.pone.0058700 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao W. et al. High-resolution linkage and quantitative trait locus mapping aided by genome survey sequencing: building up an integrative genomic framework for a bivalve mollusc. DNA Res 21, 85–101, 10.1093/dnares/dst043 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonen S. et al. Linkage maps of the Atlantic salmon (Salmo salar) genome derived from RAD sequencing. BMC Genomics 15, 166, 10.1186/1471-2164-15-166 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaiokostas C. et al. Mapping and validation of the major sex-determining region in Nile tilapia (Oreochromis niloticus L.) Using RAD sequencing. PLos One 8, e68389, 10.1371/journal.pone.0068389 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegarty M. et al. Genotyping by RAD sequencing enables mapping of fatty acid composition traits in perennial ryegrass (Lolium perenne (L.)). Plant Biotechnol J 11, 572–581, 10.1111/pbi.12045 (2013). [DOI] [PubMed] [Google Scholar]
- Baxter S. W. et al. Linkage mapping and comparative genomics using next-generation RAD sequencing of a non-model organism. PLos One 6, e19315, 10.1371/journal.pone.0019315 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyeno T., Hakubutsukan T. K., Hakubutsukan K. K. & Gakkai N. G. Indo-Pacific fish biology: proceedings of the Second International Conference on Indo-Pacific Fishes: conducted at the Tokyo National Museum Ueno Park, Tokyo, July 29-August 3, 1985. (Ichthyological Society of Japan, 1986). [Google Scholar]
- Bureau of Fishery, M. o. A. China Fishery Statistical Yearbook 2013. 1 edn, 138 (2013). [Google Scholar]
- Tong J. & Sun X. Genetic and genomic analyses for economically important traits and their applications in molecular breeding of cultured fish. Sci China Life Sci 58, 178–186, 10.1007/s11427-015-4804-9 (2015). [DOI] [PubMed] [Google Scholar]
- Collard B. C. & Mackill D. J. Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 363, 557–572, 10.1098/rstb.2007.2170 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M. et al. Development of silver carp (Hypophthalmichthys molitrix) and bighead carp (Aristichthys nobilis) genetic maps using microsatellite and AFLP markers and a pseudo-testcross strategy. Anim Genet 38, 364–370, 10.1111/j.1365-2052.2007.01617.x (2007). [DOI] [PubMed] [Google Scholar]
- Zhu C. et al. A second-generation genetic linkage map for bighead carp (Aristichthys nobilis) based on microsatellite markers. Anim Genet 45, 699–708, 10.1111/age.12194 (2014). [DOI] [PubMed] [Google Scholar]
- Lamer J. T. et al. Restriction site-associated DNA sequencing generates high-quality single nucleotide polymorphisms for assessing hybridization between bighead and silver carp in the United States and China. Mol Ecol Resour 14, 79–86, 10.1111/1755-0998.12152 (2014). [DOI] [PubMed] [Google Scholar]
- Zhang J. et al. High-density genetic map construction and identification of a locus controlling weeping trait in an ornamental woody plant (Prunus mume Sieb. et Zucc). DNA Res 22, 183–191, 10.1093/dnares/dsv003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J. L. Informativeness of human (dC-dA)n.(dG-dT)n polymorphisms. Genomics 7, 524–530 (1990). [DOI] [PubMed] [Google Scholar]
- Meng Q. et al. SASH1 regulates proliferation, apoptosis, and invasion of osteosarcoma cell. Molecular and cellular biochemistry 373, 201–210, 10.1007/s11010-012-1491-8 (2013). [DOI] [PubMed] [Google Scholar]
- Lin S. et al. Effects of SASH1 on melanoma cell proliferation and apoptosis in vitro. Molecular medicine reports 6, 1243–1248, 10.3892/mmr.2012.1099 (2012). [DOI] [PubMed] [Google Scholar]
- Cui J., Ren X. & Yu Q. Nuclear DNA Content Variation in Fishes. CYTOLOGIA 56, 425–429, 10.1508/cytologia.56.425 (1991). [DOI] [Google Scholar]
- Kai W. et al. A ddRAD-based genetic map and its integration with the genome assembly of Japanese eel (Anguilla japonica) provides insights into genome evolution after the teleost-specific genome duplication. BMC Genomics 15, 233, 10.1186/1471-2164-15-233 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q. et al. An SNP-based saturated genetic map and QTL analysis of fruit-related traits in cucumber using specific-length amplified fragment (SLAF) sequencing. BMC Genomics 15, 1158, 10.1186/1471-2164-15-1158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C., Tong J., Yu X. & Guo W. Comparative mapping for bighead carp (Aristichthys nobilis) against model and non-model fishes provides insights into the genomic evolution of cyprinids. Mol Genet Genomics 290, 1313–1326, 10.1007/s00438-015-0992-z (2015). [DOI] [PubMed] [Google Scholar]
- Wang L. et al. Construction of a high-density linkage map and fine mapping of QTL for growth in Asian seabass. Sci Rep 5, 16358, 10.1038/srep16358 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. et al. A consensus linkage map of oil palm and a major QTL for stem height. Sci Rep 5, 8232, 10.1038/srep08232 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinestel K. et al. Expression and Y435-phosphorylation of Abelson interactor 1 (Abi1) promotes tumour cell adhesion, extracellular matrix degradation and invasion by colorectal carcinoma cells. Molecular cancer 13, 145, 10.1186/1476-4598-13-145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. et al. Upregulation of Abelson interactor protein 1 predicts tumor progression and poor outcome in epithelial ovarian cancer. Human pathology 46, 1331–1340, 10.1016/j.humpath.2015.05.015 (2015). [DOI] [PubMed] [Google Scholar]
- Wang C. et al. Expression of Abl interactor 1 and its prognostic significance in breast cancer: a tissue-array-based investigation. Breast cancer research and treatment 129, 373–386, 10.1007/s10549-010-1241-0 (2011). [DOI] [PubMed] [Google Scholar]
- Zhou J. et al. Identification of beta1,4GalT II as a target gene of p53-mediated HeLa cell apoptosis. Journal of biochemistry 143, 547–554, 10.1093/jb/mvn003 (2008). [DOI] [PubMed] [Google Scholar]
- Wang C., Zhu C., Zhai H. & Wan J. Mapping segregation distortion loci and quantitative trait loci for spikelet sterility in rice (Oryza sativa L.). Genetical research 86, 97–106, 10.1017/S0016672305007779 (2005). [DOI] [PubMed] [Google Scholar]
- Xu S. Quantitative trait locus mapping can benefit from segregation distortion. Genetics 180, 2201–2208, 10.1534/genetics.108.090688 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X. et al. RADtyping: an integrated package for accurate de novo codominant and dominant RAD genotyping in mapping populations. PLos One 8, e79960, 10.1371/journal.pone.0079960 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen J., Hohenlohe P. A., Bassham S., Amores A. & Cresko W. A. Stacks: an analysis tool set for population genomics. Mol Ecol 22, 3124–3140, 10.1111/mec.12354 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laghari M. et al. Quantitative trait loci (QTL) associated with growth rate trait in common carp (Cyprinus carpio). Aquacult Int 21, 1373–1379, 10.1007/s10499-013-9639-4 (2013). [DOI] [Google Scholar]
- Zheng X. et al. A consensus linkage map of common carp (Cyprinus carpio L.) to compare the distribution and variation of QTLs associated with growth traits. Sci China Life Sci 56, 351–359, 10.1007/s11427-012-4427-3 (2013). [DOI] [PubMed] [Google Scholar]
- Wang C. M. et al. A high-resolution linkage map for comparative genome analysis and QTL fine mapping in Asian seabass, Lates calcarifer. BMC Genomics 12, 174, 10.1186/1471-2164-12-174 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine V. N., Shikano T., Herczeg G., Vilkki J. & Merila J. Quantitative trait loci for growth and body size in the nine-spined stickleback Pungitius pungitius L. Mol Ecol 22, 5861–5876, 10.1111/mec.12526 (2013). [DOI] [PubMed] [Google Scholar]
- Reid D. P., Szanto A., Glebe B., Danzmann R. G. & Ferguson M. M. QTL for body weight and condition factor in Atlantic salmon (Salmo salar): comparative analysis with rainbow trout (Oncorhynchus mykiss) and Arctic charr (Salvelinus alpinus). Heredity (Edinb) 94, 166–172, 10.1038/sj.hdy.6800590 (2005). [DOI] [PubMed] [Google Scholar]
- Wang J., Yang G. & Zhou G. Quantitative trait loci for morphometric body measurements of the hybrids of silver carp (Hypophthalmichthys molitrix) and bighead carp (H. nobilis). Acta Biol Hung 64, 169–183, 10.1556/ABiol.64.2013.2.4 (2013). [DOI] [PubMed] [Google Scholar]
- Podisi B. K., Knott S. A., Burt D. W. & Hocking P. M. Comparative analysis of quantitative trait loci for body weight, growth rate and growth curve parameters from 3 to 72 weeks of age in female chickens of a broiler-layer cross. BMC Genet 14, 22, 10.1186/1471-2156-14-22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoya S. et al. The genetic architecture of growth rate in juvenile Takifugu species. Evolution; international journal of organic evolution 67, 590–598, 10.1111/j.1558-5646.2012.01781.x (2013). [DOI] [PubMed] [Google Scholar]
- Protas M. et al. Multi-trait evolution in a cave fish, Astyanax mexicanus. Evolution & development 10, 196–209, 10.1111/j.1525-142X.2008.00227.x (2008). [DOI] [PubMed] [Google Scholar]
- Rogers S. M. & Bernatchez L. The genetic architecture of ecological speciation and the association with signatures of selection in natural lake whitefish (Coregonus sp. Salmonidae) species pairs. Molecular biology and evolution 24, 1423–1438, 10.1093/molbev/msm066 (2007). [DOI] [PubMed] [Google Scholar]
- Atchley W. R. & Zhu J. Developmental quantitative genetics, conditional epigenetic variability and growth in mice. Genetics 147, 765–776 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D. F. et al. Cysteine-rich LIM-only proteins CRP1 and CRP2 are potent smooth muscle differentiation cofactors. Developmental cell 4, 107–118 (2003). [DOI] [PubMed] [Google Scholar]
- Sambrook J. & Russell D. W. Molecular Cloning: A Laboratory Manual. (Cold Spring Harbor Laboratory Press, 2001). [Google Scholar]
- JOINMAP 4.0, Software for the calculation of genetic linkage maps in experimental populations (Kyamza BV, Wageningen, Netherland, 2006).
- Van Ooijen J. & Kyazma B. MapQTL 6. Software for the mapping of quantitative trait loci in experimental populations of diploid species. Kyazma BV: Wageningen, Netherlands (2009).
- Voorrips R. E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J Hered 93, 77–78, DOI 10.1093/jhered/93.1.77 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.