

Abstract
This review focuses on research in epidemiology, neuropathology, molecular biology, and genetics regarding the hypothesis that pathogens interact with susceptibility genes and are causative in sporadic Alzheimer’s disease (AD). Sporadic AD is a complex multifactorial neurodegenerative disease with evidence indicating coexisting multi-pathogen and inflammatory etiologies. There are significant associations between AD and various pathogens, including Herpes simplex virus type 1 (HSV-1), Cytomegalovirus, and other Herpesviridae, Chlamydophila pneumoniae, spirochetes, Helicobacter pylori, and various periodontal pathogens. These pathogens are able to evade destruction by the host immune system, leading to persistent infection. Bacterial and viral DNA and RNA and bacterial ligands increase the expression of pro-inflammatory molecules and activate the innate and adaptive immune systems. Evidence demonstrates that pathogens directly and indirectly induce AD pathology, including amyloid-β (Aβ) accumulation, phosphorylation of tau protein, neuronal injury, and apoptosis. Chronic brain infection with HSV-1, Chlamydophila pneumoniae, and spirochetes results in complex processes that interact to cause a vicious cycle of uncontrolled neuroinflammation and neurodegeneration. Infections such as Cytomegalovirus, Helicobacter pylori, and periodontal pathogens induce production of systemic pro-inflammatory cytokines that may cross the blood-brain barrier to promote neurodegeneration. Pathogen-induced inflammation and central nervous system accumulation of Aβ damages the blood-brain barrier, which contributes to the pathophysiology of AD. Apolipoprotein E4 (ApoE4) enhances brain infiltration by pathogens including HSV-1 and Chlamydophila pneumoniae. ApoE4 is also associated with an increased pro-inflammatory response by the immune system. Potential antimicrobial treatments for AD are discussed, including the rationale for antiviral and antibiotic clinical trials.
Keywords: Alzheimer’s disease, ApoE4, amyloid, Cytomegalovirus, dementia, Herpes simplex, neurodegeneration, pathogen
THE ALZHEIMER’S DISEASE PATHOGEN HYPOTHESIS
Alzheimer’s disease (AD) is an inflammatory brain disease that affects 20 million people worldwide and the incidence is expected to rise. Current medical treatment is not optimal, and thus an effective treatment is very much needed. The disease is associated with a combination of environmental agents and genetic influences leading to inflammation of the brain, neuronal cell death, and progressive dementia [1].
AD is characterized by two main pathological features in the brain: senile plaques and neurofibrillary tangles (NFTs). Senile plaques are extracellular and are predominantly made up of amyloid-β (Aβ), a peptide cleaved from the much longer amyloid-β protein precursor (AβPP). Neurofibrillary tangles are intracellular and comprised of abnormally phosphorylated tau protein. Tau protein is normally associated with microtubules in neurons, and contributes to AD pathology in its phosphorylated state [2].
The AD pathogen hypothesis states that pathogens act as triggers, interacting with genetic factors to initiate the accumulation and/or formation of Aβ, hyperphosphorylated tau proteins, and inflammation in the AD brain. Herpes simplex virus type 1 (HSV-1) and other pathogens including Chlamydophila pneumoniae and Spirochetes are able to infect the brain, evade the host immune response, and are highly prevalent in the AD brain [3–9]. In vitro studies and animal models indicate that pathogens induce formation of Aβ, amyloid plaques, and hyperphosphorylated tau proteins [10–13]. Pathogens induce a glial inflammatory response and can directly and indirectly damage and destroy neurons [14–18]. Significant inflammatory cascades are activated in the brains of AD patients [19, 20]. Together, these processes result in neurodegeneration and disease progression.
This review examines evidence implicating HSV-1and Cytomegalovirus (CMV), both members of the Herpesviridae family, and the bacterial pathogens Chlamydia pneumoniae, spirochetes, periodontal pathogens, and Helicobacter pylori as causative in the pathogenesis of AD. Limited evidence is also presented regarding the Herpesviridae Epstein Barr Virus (EBV) and Human herpes virus 6 (HHV-6) as possible contributing factors in AD pathogenesis. The multi-pathogen AD hypothesis does not exclude toxins or other environmental co-factors that may be involved in the pathogenesis of AD and are reviewed elsewhere [21]. Pathogens were selected based on the degree of significant cumulative evidence identified in an extensive PubMed literaturesearch.
HERPES SIMPLEX VIRUS TYPE 1
HSV-1 is a neurotropic virus that infects most humans, attaining 90% prevalence by the sixth decade of life. Infection is life long, as the virus resides in the trigeminal ganglia of the peripheral nervous system in latent form with viral genome but no virions present. Reactivation leads to viral replication and acute infections known as herpes labialis, commonly referred to as cold sores [22].
In 1982, Melvin Ball hypothesized that HSV-1 was causative in AD. He proposed that latent HSV-1 located in the trigeminal ganglia could reactivate and ascend along known nerve pathways into the limbic system and areas of the brain most affected in AD [23].
Herpes simplex encephalitis and AD affect the same brain regions, including the frontal lobes, temporal lobes, and hippocampus. Herpes simplex encephalitis survivors show cognitive, memory, and behavioral decline. Other viruses implicated in neurological disease include measles in subacute sclerosing panencephalitis and human immunodeficiency virus in HIV-associated dementia [22]. As with AD, both subacute sclerosing panencephalitis [24] and HIV infection [25] are associated with the formation of phosphorylated tau protein and NFTs in the brain.
EPIDEMIOLOGICAL STUDIES: HSV-1 HUMORAL RESPONSE, COGNITIVE DECLINE, AND AD
Epidemiological studies show an association between viral infectious burden (IB) and cognitive decline. IB is defined as a composite serological measure of exposure to common pathogens [27]. Strandberg et al. measured seropositivity to HSV-1, HSV-2, CMV, Chlamydia pneumoniae, and Mycoplasma pneumoniae in 383 elderly patients with cardiovascular disease. Assessments including the Mini-Mental Status Examination (MMSE) and the Clinical Dementia Rating were used to define cognitive impairment. Having three positive viral titers was associated with a 2.5 times higher risk for cognitive impairment after 12 months [26]. Katan et al. [27] found an association between Herpesviridae and cognitive decline using a composite serologic measure of exposure to both bacterial (Chlamydia pneumoniae and Helicobacter pylori) and viral (CMV, HSV-1, and HSV-2) pathogens. As reviewed by Strandberg, the association was primarily driven by viral IB [28].
Letenneur et al. studied the risk of developing AD according to the presence or absence of serum anti-HSV IgG and IgM antibodies by following 512 elderly patients initially free of dementia for 14 years. The presence of anti-HSV IgM antibodies is associated with primary infection or recent reactivation of HSV. In contrast, the presence of anti-HSV IgG antibodies indicates lifelong HSV infection [29]. Subjects who were IgM-positive at baseline showed a significantly higher risk of developing AD (hazard ratio = 2.55). No significant increased risk for AD was found in IgG-positivesubjects. Among the 43 IgM-positive subjects, only 2 were IgG-negative, which supports recent HSV reactivation rather than primary infection in most of the IgM-positive subjects [29]. Similar results were obtained in a longitudinal study by Lövheim et al. involving 3,432 elderly patients with a mean follow-up time of 11.3 years. Baseline increased serum levels of anti-HSV IgM antibodies were associated with increased risk of developing AD by a factor of 2 [30]. Thus, HSV reactivation, as indicated by the presence of anti-HSV IgM antibodies, is highly correlated with incident AD [29, 30].
Kobayashi et al. [31] used the avidity index of anti-HSV-1 IgG antibodies as an indicator of HSV-1 reactivation. The study, involving patients with amnestic mild cognitive impairment (MCI), AD, and healthy controls evaluated the relationship between HSV-1 reactivation and the degree of cognitive impairment in AD. The avidity index is defined as the strength with which IgG attaches to antigen [32]. HSV reactivation is characterized by increased levels of high-avidity anti-HSV IgG antibodies compared to lower levels seen with initial HSV infections [31]. MMSE and frontal assessment battery were used to assess cognition. MCI patients had a higher anti-HSV-1 IgG antibody avidity index than AD patients or healthy controls implying that HSV-1 reactivation occurs more frequently in the MCI group than in the AD group or healthy control group. Differences in anti-HSV-1 IgG antibody titer and anti-HSV-1 avidity index readings between the MCI group and healthy controls also suggests that reactivation of HSV-1 contributes to progression from healthy state to MCI [31].
In a longitudinal nested case–control study, Lövheim et al. measured plasma HSV antibody samples taken on average 9.6 years before AD diagnosis. In the 360 patients who developed AD and who had a follow-up time of 6.6 years or more, past HSV infection (as indicated by the baseline presence of anti-HSV IgG antibodies) increased the risk of developing AD by a factor of 2.25 [33].
Schretlen et al. evaluated cognitive performance in a group of patients who had been diagnosed with schizophrenia with an average cohort age of 39 years [34]. Schizophrenia patients who were HSV-1 IgG antibody seropositive performed significantly worse on neuropsychological measures (including psychomotor speed, executive functioning, and explicit verbal memory) than the combined HSV-1 and HSV-2 IgG antibody seronegative control group. Patients who tested seropositive for HSV-1 had decreased grey matter volume in the anterior cingulate and cerebellum seen on morphometric magnetic resonance imaging (MRI) of the brain compared to the HSV-1 seronegative control group [34]. Poor cognitive test performance correlated with decreased grey matter volume in some of the same brain regions that distinguished the patient subgroups defined by HSV-1 status [34]. Several studies have confirmed significant cognitive impairment in HSV-1 IgG seropositive schizophrenia patients compared to HSV-1 IgG seronegative controls with average cohort ages reported as 38 to 42 years old [35–39]. A causal association between exposure to HSV-1 and increased risk for schizophrenia has not been proven [34]; however, HSV-1 exposure in this group of neuropsychiatric patients is associated with cognitive impairment and provides further supportive evidence for the role of HSV-1 in cognitive dysfunction.
Higher levels of HSV-1 humoral immune response appear to play a protective role in the early stages of AD. Analyses performed with voxel-based morphometric brain MRI in AD patients and healthy controls indicate the presence of significant correlations between the preservation of cortical bilateral temporal and orbitofrontal grey matter volumes with higher HSV-1 IgG serum antibody titers [40].
HSV-1 IS HIGHLY PREVALENT IN ELDERLY BRAINS
Polymerase chain reaction (PCR) methods used by Jamieson et al. to detect HSV-1 DNA in autopsy brain specimens confirmed that latent HSV-1 is present in a high proportion (70–100%) of sporadic AD and elderly normal brains [4]. HSV-1 was found in brain areas most affected by AD, namely the temporal cortices, frontal cortices, and hippocampus. The Jamieson et al. findings have been confirmed in several studies (Table 1) [41]. The virus was found in very low proportions in younger brains [42]. In addition, Mori et al. [43] and Rodriguez et al. [44] used PCR to identify HSV-1 DNA in AD brains. PCR improves sensitivity in HSV-1 detection when compared to previously applied techniques such as in situ hybridization [4]. Some PCR studies had lower detection rates than others, perhaps due to a lower prevalence of HSV-1 infection in Japan [45] or age not having been taken into account. For unknown reasons, Hemling et al. [46] and Marquis et al. [47] detected HSV-1 DNA in a very low proportion of brains.
Table 1.
Studies that have detected HSV-1 DNA using PCR in brain tissue from patients with AD and controls (non-neurological cases)
| Study | Primers used for PCR | Area of brain sample | HSV-1 DNA-positive individuals | |
| AD n (%) | Controls n (%) | |||
| Jamieson et al. [4] | TK | Temp, frontal cortex, hippocampus | 8 (100) | 6 (100) |
| Jamieson et al. [42] | TK | Temp, frontal cortex, hippocampus | 21 (67) | 15 (60) |
| Baringer and Pisani [271] | Various | Various | NR | 40 (35) |
| Gordon et al. [272] | Various | Hippocampus and frontal cortex | 30 (27) | |
| Itabashi et al. [45] | gD | Temporal and frontal cortex | 46 (30) | 23 (22) |
| Itzhaki et al. [49] | TK | Frontal and temporal cortex | 46 (67) | 44 (64) |
| Lin et al. [50] | TK | Frontal and temporal cortex | 61 (74) | 48 (63) |
| Bertrand et al. [273] | gD | Various | 98 (75) | 57 (72) |
| Cheon et al. [274] | gD | Frontal cortex | 10 (100) | 10 (100) |
HSV-1, herpes simples virus type 1; AD, Alzheimer’s disease; PCR, polymerase chain reaction; gD, glycoprotein D protein; TK, thymidine kinase; NR, not Reported. Table adapted from Itzhaki R (2004) Herpes simplex virus type 1, apolipoprotein E and Alzheimer’ disease. Herpes 11(Suppl 2), 77A-82A. [41] Reprinted with permission from Ruth Itzhaki.
Intrathecal HSV-1 IgG was found in 52% of an AD cohort and 69% of the age-matched normal group using enzyme-linked immunosorbent assay (ELISA) testing [48]. This data confirms the aforementioned PCR finding that HSV-1 DNA sequences are present in many elderly brains as a whole functional HSV-1 genome and provides evidence that the virus replicates in the brain [48].
HSV-1 IN THE BRAIN OF APOE-ɛ4 ALLELE CARRIERS INCREASES THE RISK FOR AD
Additional evidence for HSV-1 in AD involves the type-4 allele of the apolipoprotein E gene, known as APOE-ɛ4 or APOE4. A significantly increased risk for sporadic AD is associated with the presence of both HSV-1 in brain and carriage of the APOE-ɛ4 allele [49]. As shown in a study of AD postmortem brains by Itzhaki et al. [49] and confirmed by Lin et al. [50], neither HSV-1 nor the APOE-ɛ4 allele alone was found to be a risk factor for AD. However, the combination of HSV-1 with the APOE-ɛ4 allele increased the risk for AD by a factor of 12 [50]. HSV-1 in the brains of APOE-ɛ4 allele carriers accounted for over half of AD patients in the study (Table 2) [49]. The proportion of HSV-1 positive elderly controls was similar to that of HSV-1 positive AD patients, indicating that the AD brain is not predisposed to HSV-1 infection. Few HSV-1 positive elderly controls were positive for the APOE-ɛ4 allele, indicating that APOE-ɛ4 allele carriers are not predisposed to HSV-1 infection [49]. Itzhaki’s results were later confirmed by Itabashi and colleagues [45].
Table 2.
APOE genotypes of Alzheimer’s disease patients and aged non-Alzheimer’s disease patients positiveor negative for Herpes Simplex Virus Type 1 in all brain regions
| Overall data for all brain regions | ||||||
| Non-AD (n = 44) | AD (n = 46) | |||||
| HSV1+ | HSV1– | Total | HSV1+ | HSV1– | Total | |
| Genotype | ||||||
| ɛ2/ ɛ2 | 0 | 0 | 0 | 0 | 0 | 0 |
| ɛ2/ ɛ3 | 1 | 3 | 4 | 2 | 1 | 3 |
| ɛ2/ ɛ4 | 0 | 0 | 0 | 0 | 0 | 0 |
| ɛ3/ ɛ3 | 25 | 11 | 36 | 5 | 7 | 12 |
| ɛ3/ ɛ4 | 2 | 2 | 4 | 20 | 2 | 22 |
| ɛ4/ ɛ4 | 0 | 0 | 0 | 9 | 0 | 9 |
| Allele number | ||||||
| ɛ2 | 1 | 3 | 4 | 2 | 1 | 3 |
| ɛ3 | 53 | 27 | 80 | 32 | 17 | 49 |
| ɛ4 | 2 | 2 | 4 | 38 | 2 | 40 |
| APOE- ɛ4 | 3.6% | 6.3% | 4.5% | 52.8% | 10.0% | 43.4% |
HSV-1 in brains of APOE-ɛ4 allele carriers accounts for over 50% of AD brains with testing done postmortem. Table from Itzhaki RF, Lin WR, Shang D, Wilcock GK, Faragher B, Jamieson GA (1997) Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 349, 241-244 [49]. Copyright 1997. Reprinted with permission from Elsevier and Ruth Itzhaki.
APOLIPOPROTEIN E INFLUENCES HSV-1 VIRAL LOAD IN ANIMAL BRAIN STUDIES
Apolipoprotein E dosage and the presence of APOE-ɛ4 determine latent HSV-1 DNA concentrations in the mouse brain [51]. Burgos and colleagues inoculated mice with HSV-1 and measured brain viral DNA concentrations. Thirty-seven days after infection, the HSV-1 brain DNA concentrations for APOE+/+ wild-type mice were 13.7 times greater than those of APOE–/– knockout mice. HSV-1 brain DNA concentrations for human APOE4 transgenic mice were 13.6 times greater than those of APOE3 mice. Apolipoprotein E4 appeared to facilitate HSV-1 latency in the brain much more than apolipoprotein E3, and APOE dosage correlated directly with the concentration of HSV-1 in the brain [51]. Guzman-Sanchez and collaborators later confirmed that apolipoprotein interacts with HSV-1 in animal models to increase viral load in the brain. 2-month-old wild-type and APOE knock-out mice were infected with HSV-1 and followed for 16 months. Viral load was found to increase with age. Viral load in the brains of aged APOE+/+ wild-type female mice was 43 times that seen in knock-out APOE–/– male mice. Although no neuropathological or brain MRI morphological differences were detected between 18-month-old infected mice when compared to controls, the central nervous system (CNS) HSV-1 infected mice showed associated memory deficit and reduction in metabolic indicators of CNS health [52]. These animal studies which associate APOE4 with increased HSV-1 viral load in the brain may relate to Itzhaki’s human postmortem study, indicating that the combined presence of HSV-1 in brain and carriage of the APOE-ɛ4 allele are involved in the pathogenesis of AD [49].
GENOME-WIDE ASSOCIATION STUDIES
Two genome-wide association studies identified APOE, complement receptor 1 (CR1), clusterin (CLU), and phosphatidylinositol binding clathrin assembly protein (PICALM) as major susceptibility genes in AD [53]. These susceptibility genes are associated with the HSV life cycle, and relate either directly or indirectly to cellular entry, intracellular transport, nuclear egress, AβPP processing, and Aβ processing [53].
AD AMYLOID PLAQUES CONTAIN HSV-1 DNA
HSV-1 coexists with Aβ in AD amyloid plaques [3]. Using in situ PCR to detect HSV-1 DNA and immunohistochemistry or thioflavin S staining to detect amyloid plaques, Wozniak and coworkers discovered a striking co-localization of HSV-1 DNA and Aβ within senile plaques in postmortem brains (Fig. 1) [3]. In AD brains, 90% of the plaques contained HSV-1 DNA and 72% of the total brain HSV-1 DNA was associated with plaques. The HSV-1 DNA associated with plaques was much lower in aged normal brains than in AD brains (p < 0.001) [3]. The co-localization of HSV-1 DNA and Aβ within amyloid plaques in the AD brain places HSV-1 in direct juxtaposition with a highly significant AD biomarker and suggests a significant role for HSV-1 in AD pathogenesis.
Fig.1.

Co-localization of HSV-1 DNA and amyloid-β in AD plaques. A strong co-localization showing HSV-1 DNA (brown staining using PCR) and amyloid plaque (blue staining using immunohistochemistry) in a postmortem AD brain (G). Greater than 90% AD plaques contained viral DNA. Scale bar = 50μm. Figure from Wozniak, MA, Mee AP, Itzhaki RF (2009) Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J Pathol 217, 131-138 [3]. Copyright 2008. Reprinted with permission from John Wiley and Sons, Inc. and Ruth Itzhaki.
IN VITRO AND ANIMAL STUDIES: HSV-1 INFECTION INDUCES ELEVATED LEVELS OF Aβ AND P-TAU
Human cultured neuroblastoma cells infected with HSV-1 in vitro produce Aβ42 and Aβ40, and increased amounts of the enzymes β-site AβPP-cleaving enzyme (BACE-1) and nicastrin (a component of the γ-secretase enzyme) [10]. Both enzymes are involved in cleavage of the AβPP to produce Aβ. Rapid reduction of AβPP and a dramatic increase in Aβ42 and Aβ40 is seen in HSV-1-infected neuronal cell cultures [10]. Rat cortical neurons challenged with HSV-1 demonstrate hyperexcitability, membrane depolarization, and increased intracellular calcium levels with enhanced calcium dependent-AβPP phosphorylation and intracellular accumulation of Aβ42 [54]. Animal models also support HSV-1 as causative in the formation of Aβ. Mouse brain infected with HSV-1 produced marked increases in Aβ42 five days post-intranasal infection when compared to uninfected controls [10]. These studies indicate that HSV-1 exposure to neuronal cells results in cellular production of Aβ.
HSV-1 is able to induce tau phosphorylation, thus linking HSV-1 to the formation of another abnormal protein found in AD brains. Neuroblastoma cells infected with HSV-1 produce hyperphosphorylated tau protein and increased amounts of the enzymes that phosphorylate tau protein including glycogen synthase kinase-3β (GSK-3β) and protein kinase A [11]. Alvarez et al. demonstrated accumulation of hyperphosphylated tau protein within the nucleus of HSV-1 infected neuroblastoma cells [55]. Zambrano et al. showed that HSV-1 infection of murine neuronal cultures results in tau hyperphosphorylation and alterations in the microtubule dynamics of the neuronal cytoskeleton [56]. The ability of HSV-1 to induce phosphorylation of tau proteins in neuronal cells is significant because p-tau proteins contribute to the formation of NFTs in AD brains [2].
ADDITIONAL MOLECULAR EVIDENCE AND CELLULAR MECHANISMS RELATING HSV-1 TO AD
Additional studies show a structural link between HSV-1 and Aβ. Aβ34 - 42 is 67% identical to the HSV-1 envelope protein glycoprotein B (gB) peptide sequence, indicating peptide homology [57]. Synthetic peptides derived from the HSV-1 gB fragment self-assemble into thioflavin-positive fibrils and form β-pleated sheets that are ultrastucturally indistinguishable from Aβ [57].The gB fragment accelerates in vitro formation of Aβ fibrils that are toxic to primary cortical neurons at a dose comparable to Aβ [57].
HSV-1 travels inside the neuronal cytoplasm in association with AβPP [58]. In squid axons, HSV-1 travels with AβPP during fast anterograde transport from the nerve cell body down the axon [59]. The virus interferes with AβPP processing in HSV-1-infected neuronal cells, reducing the level of AβPP and increasing the level of a 55 kDa C-terminal AβPP fragment containing Aβ [60]. De Chiara et al. found that HSV-1 infection of neuroblastoma cells and rat cortical neurons induces multiple cleavages of AβPP with resultant neurotoxic intra and extracellular AβPP fragments that comprise portions of Aβ. Components of the amyloidogenic AβPP processing pathway, including host cell β-secretase, γ-secretase, and caspase-3 like enzymes, were shown to be involved in the AβPP cleavage process. These findings suggest that repeated HSV-1 reactivation in the presence of other risk factors may play a co-factorial role in the development of AD [61]. Cheng and colleagues evaluated HSV-1 interactions with AβPP using immune-fluorescence, immunogold electron microscopy, and live cell confocal imaging to visualize newly synthesized viral particles inside epithelial cells as they traveled to the cell surface. Cytoplasmic HSV-1 particles labeled with green fluorescent protein co-localized and traveled with AβPP inside living cells. Most intracellular HSV-1 particles interacted frequently with AβPP, which facilitated viral transport while interfering with normal AβPP transport and distribution. Intracellular HSV-1 interactions with AβPP provide a mechanistic basis for the association between HSV-1 seropositivity and AD [62].
Santana et al. demonstrated that HSV-1 infection of neuroblastoma cells induced significant intracellular accumulation of Aβ in autophagosomes and a marked decrease in Aβ secretion. Aβ failed to fuse with lysosomes in HSV-1-infected neuroblastoma cells, indicating the impaired degradation of Aβ localized in autophagic vesicles [63]. HSV-1 decreases autophagy using HSV-1 infected cell polypeptide 34.5 (ICP 34.5) which blocks the protein kinase R (PKR) and eukaryotic initiating factor 2α (eIF2α) signaling pathways [64]. This action inhibits HSV-1 degradation by interfering with autophagy of the virus [64, 65]. Itzhaki suggests that this may lead to a decrease in Aβ clearance and an accumulation of senile plaques in AD [66].
HSV-1 can both block and induce neuronal apoptosis. HSV-1 protein ICP34.5 dephosphorylates eIF2α to block both the shutdown of host cell protein synthesis and apoptosis [66, 67]. HSV-1 infection of murine neuronal cultures results in marked neurite damage and neuronal apoptosis [56].
HSV-1 INDUCES AD-LIKE INFLAMMATION AND OXIDATIVE STRESS
Elevated levels of pro-inflammatory cytokines are consistently found in the brains of AD patients [19, 20]. Infection by HSV-1 induces expression of cytokines and pro-inflammatory molecules, including interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), IL-6, IL-8, macrophage inflammatory protein 1-α (MIP-1 α), chemokine (C-C motif) ligand 5 (CCL5), and chemokine CXCL 10 in human microglial cells [17]. Persistent cytokine expression occurs in mouse trigeminal ganglion infected with HSV-1, including IL-2, IL-6, TNF-α, interferon- γ (IFN- γ), IL-10, and CCL5 [18]. The direct effects of HSV-1 on neurons and the host inflammatory response to infection can lead to oxidative damage due to increased formation of reactive oxygen and reactive nitrogen species [68].
Interactions between HSV-1 and oxidative stress promote neurodegenerative processes found in AD. In HSV-1 infected human neuroblastoma cells, experimentally induced oxidative stress was found to significantly enhance the accumulation of intracellular Aβ, inhibit Aβ secretion, and appeared to be mediated by HSV-1 infection [69]. Oxidative stress also potentiated the accumulation of autophagic compartments within the cell [69]. HSV-1 interactions with oxidative stress are significant because oxidative damage is thought to occur early in the pathogenesis of AD [70].
HSV-1 REACTIVATION IN THE BRAIN
Itzhaki points out the lack of methodology for detecting the hypothesized sub-clinical limited reactivation of HSV-1 in localized areas of the brain in AD patients [32]. This contrasts with clinically apparent acute HSV encephalitis, where detection of HSV-1 DNA in cerebrospinal fluid (CSF) is commonly used for diagnosis [32]. Mild forms of HSV-1 encephalitis in humans have been reported [71, 72]. These patients usually have less severe symptomatology and good prognoses when compared to patients with severe diffuse HSV-1 meningoencephalitis. Klapper et al. suggest that sub-acute HSV-1 encephalitis may be a more common and often missed sub-clinical presentation of the disease [71].
Peter and collaborators reviewed 3,200 randomly selected CSF specimens submitted for HSV testing and found a total of 62 HSV positive specimens. HSV-2 was detected more often than HSV-1 (36:26). However, the HSV-2: HSV-1 ratio reversed in the patients over age 60 with HSV-1 being more prominent (3:13). Female patients who were positive for HSV-1 predominated in the over-70 age group (10 female and 1 male with 90% of females positive for HSV-1). This study shows predominance in the reactivation of HSV-1 rather than HSV-2 in older females, a group known for having a higher incidence of AD [73].
Saldanha et al. found that HSV-1 reactivates in the brains of immunosuppressed patients. HSV-1 DNA was detected by in situ hybridization in postmortem frontal and temporal lobe human brain samples from immunosuppressed leukemia patients who were seropositive for HSV-1. HSV-1 DNA was not found in HSV-1 seronegative patients or in those who had not been immunosuppressed [74].
The reactivation rate of HSV-1 in the human brain is not known; however, animal and human studies involving the CNS and peripheral nervous system suggest that periodic sub-clinical reactivation may occur with subsequent immune response and neurodegeneration. Kaufman et al. measured the rate of asymptomatic HSV-1 reactivation and shedding in human tears from normal adults without signs of ocular herpetic disease. 74% of the 50 subjects were positive for HSV IgG by ELISA. 49/50 (98%) of subjects shed HSV-1 DNA at least one time during the course of the 30-day study [75].
Margolis and colleagues described spontaneous molecular reactivation of HSV-1 with viral protein expression, positive HSV-1 antigen staining, and infectious virus found in 6% of “latently” infected murine sensory ganglia 37 days post ocular infection [76]. Using in situ hybridization and immunohistochemistry, Feldman et al. found that HSV-1 spontaneous reactivation occurred in one neuron per 10 HSV-1 latently infected mouse trigeminal ganglia tested [77]. These neurons were surrounded by focal white cell infiltrate, indicating an inflammatory response. The authors estimate that this is equivalent to one neuron expressing high-level productive cycle viral genes in each ganglion every 10 days [77].
Asymptomatic reactivation of HSV-1 occurs in vivo in the CNS of mice and is associated with production of markers of neurodegeneration found in AD [78]. HSV-1 reactivation from the asymptomatic latent phase, was demonstrated by detection of viral ICP4 protein in the trigeminal ganglion and cerebral cortex of mice 60 days post-infection. Reactivation was accompanied by upregulation of both markersof neuroinflammation [(toll-like receptor (TLR)-4, interferon α/β, and phosphorylated interferon regulatory factor 3 (p-IRF3)] and early neurodegeneration (phospho-tau and caspase-3 cleaved tau proteins) [78]. These findings support the hypothesis that recurrent HSV-1 CNS reactivation could result in AD associated neurodegenerative processes.
PROPOSED MECHANISM OF HSV-1 PATHOGENESIS IN AD
The above data and studies support the hypothesis that HSV-1 in combination with APOE-ɛ4 allele carriage is a major cause of sporadic AD. As proposed by Itzhaki, this highly prevalent virus reactivates and enters the brain in older age by way of the peripheral nervous system or the olfactory route. HSV-1 becomes latent in the brain, but periodically reactivates in association with systemic infection, immunosuppression, or other stressors. The reactivated virus causes limited local damage via direct viral action and through inflammatory and oxidative effects. This leads to deposition of Aβ and abnormal phosphorylation of tau, which eventually forms amyloid plaques and NFTs. Defective autophagy due to aging and viral ICP34.5 action prevents degradation of HSV-1 and reduces the degradation of Aβ and phosphorylated tau protein. This results in decreased clearance of these proteins [66, 79, 80].
CYTOMEGALOVIRUS AND OTHER HERPESVIRDAE
Cytomegalovirus
CMV is a β-herpes virus prevalent in humans causing persistent lifelong asymptomatic infection in the immunocompetent host. Primary infection usually occurs early in life and is asymptomatic but occasionally causes a self-limiting mononucleosis-like syndrome [81]. CMV seropositivity in the human population ranges from 20%–100% depending on socioeconomic status and age [82]. The virus resides in the myeloid cell compartment, remaining latent in monocytes [83], but has tropism for numerous cell types such as endothelial cells, epithelial cells, fibroblasts, smooth muscle cells, neuronal cells, hepatocytes, trophoblasts, macrophages, and dendritic cells [81]. As with other members of the Herpesviridae family, CMV may reactivate under stress conditions or other stimuli. Other diseases associated with CMV infection in normal hosts include Guillain-Barre syndrome, meningoencephalitis, hemolytic anemia, and thrombocytopenia [81]. CNS infection by CMV in immunocompetent patients is rare. Most CMV brain infections occur in those who are immunocompromised, such as HIV-infected patients, transplant recipients, and infants with congenital CMV disease contracted in utero.
Epidemiological studies: CMV humoral response, cognitive decline, and AD
Several studies have shown an association between CMV infection and increased risk of both cognitive impairment and development of AD. Aiello et al. found that individuals with higher levels of IgG antibody to CMV at baseline experienced a more rapid rate of cognitive decline over a 4-year study period than those with lower levels [84]. Strandberg et al. [26] and Katan et al. [27] found that CMV was one of the viruses from the Herpesviridae family associated with cognitive decline as discussed in the HSV-1 section above. Carbone et al. studied a group of elderly patients and found baseline CMV IgG antibody levels to be significantly increased in patients who developed clinical AD over a 5-year follow-up period compared to patients who remained cognitively healthy [85]. Barnes et al. followed 849 participants and found that baseline CMV seropositivity doubled the risk of developing clinical AD over a 5-year follow-up period and noted a faster rate of decline in global cognition [86].
Tarter et al. studied cognitive impairment in various age groups in relation to CMV and HSV-1 seropositivity. Among children (ages 6–16 years), HSV-1 seropositivity was associated with lower reading and spatial reasoning test scores. Both HSV-1 and CMV seropositivity in middle–aged adults (ages 20–59 years) was associated with impaired coding speed. CMV seropositivity was also associated with impaired middle-aged learning and recall. Among older adults, HSV-1 seropositivity was associated with immediate memory impairment. The data indicated that HSV-1 may have a life course effect on cognition across all age groups, while CMV appeared to adversely affect cognition specifically in the middle aged. The authors suggest that individuals who acquire infection with these Herpesviridae earlier in life with more reactivations and subsequent immune activation may be at greater risk for developing social disparities in cognition, educational attainment, and social mobility across the life course [87].
Prevalence of CMV in the AD Brain
Data does not indicate a definitive direct infiltrative CNS role for CMV in AD. Using PCR, Lin et al. found CMV present in 16/45 (35.6%) of postmortem AD brains compared with 10/29 (34.5%) of non-AD controls, which was not statistically significant [88]. The authors point out that these values may be artificially high due to the possibility of CMV residing in lymphocytes within blood vessels rather than brain cells. In a more recent 2013 study, 93 AD brains were tested for CMV DNA using nested PCR and all samples were negative for CMV [85]. In contrast, Lin et al. found CMV in a very high proportion of postmortem vascular dementia brains [89]. CMV was found in 14/15 (93%) of brains from subjects who had been diagnosed with vascular dementia and was present in only 10/29 (34%) of age-matched normal controls. The results were statistically significant and suggest a possible role for CMV in vascular dementia [89].
CMV and immunosenescence: Impairment of the elderly immune system
CMV appears to be a strong causative factor in the development of immunosenescence by adversely affecting T cell immunity with resultant immune dysregulation and impairment in the elderly [90]. CMV has been implicated in T cell oligoclonal expansions, altered phenotypes and function of CMV specific CD8+ T cells, and decrease in the naïve and early memory T cell pool seen in the elderly [90]. Koch et al. reviews evidence for CMV involvement in immuno-senescence and suggests that the long-term attempt of the T-cell immune system to keep CMV from spreading results in reduction of the naïve T-cell pool, leading to deficits in the immunological response to new antigens in the aged [90]. Clonal expansions of CD8+ T cells directed against another Herpes virus, EBV, are also seen in the aged population [91].
Increased reactivation of CMV and other herpesviridae in the elderly
Using molecular and serological techniques, Stowe et al. found significant increases in reactivation of CMV and EBV in elderly subjects compared to younger subjects. Increases in CMV DNA in urine and Epstein Barr viral load in peripheral blood were demonstrated. In addition, elevated levels of CD8+ T cells directed against CMV and EBV were found in the elderly group. The authors concluded that the aged immune system is no longer able to control EBV and CMV reactivation, resulting in chronic infection and age-related clonal expansions of CD8+ T cells directed against EBV and CMV [91].
Evidence suggests that CMV infection may adversely influence the immune response, allowing for increased HSV-1 reactivation. Stowe et al. measured serum CMV and HSV-1 antibody levels in 1,454 multiethnic subjects. Higher HSV-1 IgG serum antibody levels, which presumably reflect higher rates of HSV reactivation, were more common in CMV seropositive subjects. Elevated antibody titers to latent HSV-l were significantly associated with both CMV seropositivity and high CMV antibody levels. Increases in HSV-1 antibodies by age occurred in CMV seropositive individuals but not CMV seronegative subjects. Among CMV seropositive subjects, increases in HSV-1 antibodies by age were only found in individuals with low CMV antibody levels, as those with high CMV antibodies already exhibited elevated HSV-1 antibodies. The results suggest chronic CMV infection is able to accelerate immunosenescence, leading to immune dysregulation with increased HSV-1 reactivation [92].
CMV is associated with elevated IFN-γ that associates with AD
CMV-specific CD8+ T cells have been shown to produce increased amounts of pro-inflammatory IFN-γ and very low levels of anti-inflammatory cytokines IL-2 and IL-4 with a potential shift to a pro-inflammatory cytokine profile in the elderly [93]. Westman et al. measured the cytokine response of peripheral blood mononuclear cells (PBMCs) from CMV seropositive and seronegative AD patients. PBMCs from CMV seropositive AD patients challenged by CMV antigens produced increased amounts of IFN-γ compared with CMV seronegative AD patients and CMV seropositive non-demented controls [94]. The authors suggest CMV acts as an inflammatory promoter in AD immunology.
In the Rush AD Center Religious Orders Study, Lurain and colleagues studied a clinical-pathological community cohort by evaluating CMV serum antibody levels, CSF IFN-γ levels, cryopreserved lymphocytes, and brain pathology from deceased and autopsied subjects [82]. CMV-specific serum IgG antibody levels were significantly associated with NFTs. CSF IFN-γ was detected in greater than 80% of CMV seropositive but not in CMV seronegative subjects. In the CMV seropositive subjects, CSF IFN-γ levels were associated with NFTs. Therefore, this study showed an association between CMV seropositivity and detection of IFN-γ in CSF, which in turn was associated with AD pathology in the form of NFTs. In addition, the percentage of senescent T cells (CD28- CD57+) from the peripheral circulation was significantly higher for CMV-seropositive as compared to CMV-seronegative subjects [82]. This study did not prove CMV presence in the brain of AD patients as CMV intrathecal antibody levels were not measured, and thus viral replication of CMV within the brain was not substantiated [95]. However, results from the Lurain et al. study support an association between CMV infection and the development of AD with CMV-induced inflammation as one potential mechanism for this association [96].
Human herpesvirus 6
HHV-6 is a neurotropic virus and exists in 2 forms: type A and type B. The HHV-6A variant is considered more neurotropic than type B [97]. HHV-6B primary infection is the cause of the common childhoodillness exanthem subitum, which is also known as roseola infantum or sixth disease. This illness affects infants and typically presents with self-limiting fever followed by a maculopapular rash. Febrile seizures occur in 10–15% of cases, and severe CNS complications have been reported in rare cases. The virus is highly seroprevalent, affecting nearly 100% of the population by age 3 [98]. HHV-6 can cause meningoencephalitis, and has been associated with multiple sclerosis, seizures, and temporal lobe epilepsy [99]. HHV-6 establishes latency in the brain and can reactivate under conditions of immunosuppression [97].
HHV-6 has been found in the brains of AD patients in various studies using PCR; however, increased incidence in AD patients versus controls has not been shown with consistent statistical significance. Lin and collaborators studied 50 postmortem AD brains and found HHV-6 in 72% of frontal and temporal cortex samples versus 40% of age-matched normal brain samples, which was statistically significant [88]. In the HHV-6 positive brains, 59% (17/29) had type B alone, 3% had type A alone, and 38% (11/29) had both types. No additional increased risk for AD was found in APOE-ɛ4 carriers who were HHV-6 positive. The authors reasoned that HHV-6 might enhance the damage caused by HSV-1 and APOE-ɛ4 in AD. However, it was not possible to exclude HHV-6 as an opportunistic secondary infection, or the possibility that HHV-6 DNA is present within leucocytes within the brain vasculature [88]. Hemling et al. examined autopsy brain samples from hippocampus, temporal cortex, frontal cortex, and anterior cingulate gyrus, and found HHV-6 in 88% of AD and 87.5% of normal controls, indicating no significant difference between the two groups. However, the number of specimens from the different brain regions tested was not specified [46]. Carbone and colleagues found HHV-6 in 17.3 % of frontal cortex samples from postmortem AD patients using qPCR with no APOE-ɛ4 carrier association found. The same group found that baseline HHV-6 DNA positivity in peripheral blood leukocytes (PBLs) was significantly associated with cognitive decline and development of AD at 5-year follow-up [85].
Epstein barr virus
EBV is a Herpes virus that infects 95% of humans early in life resulting in lifelong latent asymptomatic infection residing in B-lymphocytes [100]. Primary infection of the oropharynx often occurs during childhood and is generally asymptomatic, although the virus does cause acute infectious mononucleosis in a minority of immune competent subjects. Intermittent reactivation of the virus occurs throughout life within B cells, involving a lytic cycle at mucosal sites with low levels of asymptomatic viral shedding [101]. EBV is causatively linked to Hodgkin lymphoma, Burkitt lymphoma, and nasopharyngeal carcinoma [102, 103]. EBV is also associated with neurological diseases including encephalitis, myelitis, mononeuritis [104,105], and multiple sclerosis [106].
Although EBV-related AD data is limited, the virus may be a risk factor for development AD. Carbone et al. measured EBV DNA in PBLs and postmortem brain samples from a group of AD subjects and non-AD controls. 45% of PBLs were EBV DNA positive in AD patients compared to 31% of controls, which was statistically significant. Using qPCR, only 6% of AD brains were EBV DNA positive with all of these subjects found to be APOE-ɛ4 positive. The same researchers found that baseline EBV DNA positive PBLs and serum IgG levels for EBV antigens were significantly increased in a group of elderly individuals who developed AD during a subsequent 5-year follow-up period [85]. Thus, positive IgG levels for EBV and peripheral viral infection involving PBLs with either EBV or HHV-6 have been associated with increased risk for AD even though significant infiltrative CNS presence has not been demonstrated for EBV and has been equivocal for HHV-6.
Bacterial pathogens
Chlamydophila pneumoniae
C. pneumoniae is an obligate intracellular respiratory pathogen that can persist as a chronic infection in monocytes, macrophages, and other cell types for long periods of time. Serum antibody prevalence reaches 70% to 80% by 60 to 70 years of age [107]. Evidence indicates that C. pneumoniae crosses the blood-brain barrier (BBB) after infection of the respiratory mucosa, with subsequent hematogenous and lymphatic dissemination within infected monocytes [108, 109]. C. pneumoniae is also thought to enter the CNS via the olfactory route [80]. C. pneumoniae can evade the mechanisms of bactericidal and oxidative stress, activate endothelial cells with production of adhesion molecules, and induce cytokine overproduction [107].
C. pneumoniae has a biphasic life cycle. The elementary body is spore-like, infectious, metabolically inactive, and attaches to and enters the host cell. Elementary bodies then differentiate into reticulate bodies, which are the reproductive forms. The reticulate bodies undergo binary fission, differentiate back intoelementary bodies, and exit the cell either by cytolysis with apoptosis or by exocytosis, leaving the cell intact [107, 110].
C. pneumoniae interferes with the normal apoptotic signaling pathways and can both inhibit and induce cellular apoptosis. The bacterium can evade the host cell’s defense mechanisms, and exist as an acute infection or a chronic persistent infection [107, 111].
Under certain conditions, C. pneumoniae can enter into a chronic persistent phase characterized by aberrant reticulate bodies and other pleomorphic forms [112]. Metabolic activity is reduced and the organism is viable but non-cultivable, resulting in a chronic infection of the host cell [110, 112]. This persistent phase has been associated with several chronic diseases including asthma and chronic obstructive pulmonary disease [112].
Chlamydophila pneumoniae vascular infection and dissemination into the brain
Vascular infections with C. pneumoniae are associated with atheromasic plaques and may be an important factor in the development of brain infection with this pathogen. Using PCR and IHC techniques, Rassu et al. detected the presence of C. pneumoniae in atheromasic plaques sampled from five vascular sites in 18 autopsy cases (basilar artery, coronary artery, thoracic aorta, abdominal aorta, and renal arteries). The study showed 100% patient positivity with C. pneumoniae present at 2–5 sites for each case tested [113]. Di Pietro et al. investigated 19 postmortem cases with past chlamydial vascular infection using immunohistochemistry, PCR, in situ PCR and in situ reverse transcription PCR. C. pneumoniae was detected in the brain tissue of 16 out of 19 subjects (84.2%) who also had C. pneumoniae vascular infection. The organism was not detected in the brains of control subjects who were negative for C. pneumoniae vascular infection (p = 0.0002) [114]. These results provide evidence that a C. pneumoniae vascular infection can disseminate to the brain.
Prevalence of Chlamydophila pneumoniae in the AD brain
Balin et al. used PCR to identify C. pneumoniae in 17/19 (90%) of AD postmortem brain samples, and in only 1/19 (5%) of control brain samples, suggesting that infection with the organism is a risk factor for AD [6]. The results were confirmed using multiple methodologies. Electron microscopy and immunoelectron-microscopy studies identified chlamydial elementary and reticulate bodies in affected AD brain regions. C. pneumoniae was present in areas of the brain showing the typical AD neuropathology. Immunohistochemical tests on AD brains showed C. pneumoniae within pericytes, microglia, and astroglia. Reverse transcription (RT)-PCR assays using RNA from affected areas of AD brains confirmed the presence of transcripts from two important C. pneumoniae genes not seen in controls. Cultures were strongly positive for C. pneumoniae from a subset of affected AD brain tissues and negative in controls. C. pneumoniae was present, viable, and transcriptionally active in areas of neuropathology in the AD brain [6]. In addition to the standard morphological forms of the organism, pleomorphic forms of C. pneumoniae were later observed on ultrastructural analysis, suggesting an adaptive response and/or persistent state of infection for these organisms in AD [115]. As reviewed by Shima [116] and Balin [117], four studies [118–121] failed to detect significant C. pneumoniae in AD brains potentially due to sampling error, variable methodologies, and/or absence of standardized techniques.
Gerard and colleagues found C. pneumoniae in 20/25 (80%) of AD postmortem brain samples and 3/27 (11%) of controls (Fig. 2) [7]. Immunohistochemical analyses found that neurons, microglia, and astrocytes all served as host cells for C. pneumoniae [7]. Infected cells were seen in close proximity to senile neuritic plaques and NFTs [7]. In situ hybridization analysis in AD postmortem brains indicates an increase in the number of C. pneumoniae infected cells in APOE-ɛ4 carriers [123].
Fig.2.

Images demonstrating Chlamydophila pneumoniae in AD brain tissue by in situ hybridization. Figure (b) demonstrates C. pneumoniae from the hippocampus of an AD brain by in situ hybridization. Figures (d) and (e) show photographic enlargement of cells harboring C. pneumoniae inclusions identified in AD brain tissue. Arrows indicate the signal for C. pneumoniae. Image (b) was obtained using a x40 objective. Figure from Gérard HC, Dreses-Werringloer U, Wildt KS, Deka S, Oszust C, Balin BJ, Frey WH 2nd, Bordayo EZ, Whittum Hudson JA, Hudson AP (2006) Chlamydophila (Chlamydia) pneumoniae in the Alzheimer’s brain. FEMS Immunol Med Microbiol 48, 355-366 [7]. Copyright 2006. Reprinted with permission from John Wiley and Sons and permission from Brian Balin.
A statistically significant increase in CSF levels of C. pneumoniae DNA has been found in AD patients [122]. Miklossy found that combined data from studies attempting to isolate C. pneumoniae reached statistical significance in AD brains and AD brains plus CSF compared to controls (Table 3) [5].
Table 3.
Detection of Chlamydophila pneumoniae in Alzheimer’s disease
| Material | Number | Method | AD | Control | Ref |
| Brain | 38 | PCR, EM, IHC, RT-PCR, Cult | 17/19 | 1/19 | [6] |
| Brain | 25 | PCR, IHC | 0/25 | [118] | |
| Brain | 20 | PCR, IHC | 0/20 | [120] | |
| Brain | 20 | PCR, Cult | 2/15a | 1/5a | [119] |
| Brain | 21 | PCR, ISH | 21/21 | 0/1 | [123] |
| Brain | 52 | PCR, Cult, RT-PCR | 20/25 | 3/27 | [7] |
| CSF | 104 | PCR, Cult | 25/57 | 5/47 | [122] |
| Total Brain | 177 | p = 4.5×10–7, OR = 8.7 CI = 3.1–29.5 | 60/125 | 5/52 | |
| Brain and CSF | 281 | p = 9.8×10–11, OR = 7.8 CI = 3.7–17.8 | 85/182 | 10/99 |
AD, number of AD cases with positive detection/number of AD cases analyzed: Control, number of control cases with positive detection/number of control cases analyzed; PCR, polymerase chain reaction; CSF, cerebrospinal fluid; RT-PCR, reverse transcriptase-PCR; EM, electron microscopy; Cult, culture; P, exact value of significance following Fisher test; OR, odds ratio; CI, 95% confidence interval values; IHC, immunohistochemistry. aPositive in at least one of several samples. Table adapted from Miklossy J (2011) Emerging roles of pathogens in Alzheimer disease. Expert Rev Mol Med 13, e30 [5]. Copyright 2011. Reprinted with permission from Cambridge University Press and Judith Miklossy.
Chlamydophila pneumoniae induces inflammation, Aβ plaque formation, and neurodegeneration
Increased levels of cytokines IL1-β, IL-6 and TNF-α were found in supernatants of C. pneumoniae-infected murine microglial cells in vitro. Neurons exposed to the supernatants displayed a significant increase in apoptosis [124].
C. pneumoniae infection in the brains of BALB/c mice via the intranasal route induces a significant increase in Aβ plaques compared with non-infected mice [12]. Early treatment of infected mice with moxifloxacin decreased the number of Aβ plaques to levels similar to those seen in uninfected control mice. Infected untreated mice had 8-9 times more Aβ plaques than the antibiotic treatment group [117, 125].
Spirochetes
Spirochetes are Gram-negative, helical bacteria that possess endoflagella. Spirochetes cause a number of chronic diseases including syphilis (Treponema pallidum), Lyme disease (Borrelia burgdorferi), and periodontal disorders such as gingivitis (oral periodontal Treponema spirochetes such as T. sokranski and T. pectinovarum) [5]. Spirochetes can invade the brain and form chronic persistent infections. They are known to spread by hematogenous dissemination, through the lymphatic system, and along nerve fibers [126].
Prevalence of spirochetes in AD brain
Spirochetes have been detected using various methodologies with prevalence approaching 90% in AD brains (Fig. 3) [5, 126]. The association was statistically significant in postmortem AD brain studies of all types of spirochetes combined, oral spirochetes, and Borrelia burgdorferi. Combined, studies detecting all types of spirochetes and their specific species indicated a prevalence of 68% (90/131) in AD brains compared to 8.45% in controls [5]. The spirochete frequency detected in all studies reviewed by Miklossy was eight times higher in AD brains than in controls [5]. Miklossy’s extensive review of research data regarding spirochetes and AD indicate a probable causal relationship between neurospirochetosis and AD based on Koch’s and Hill’s criteria [126].
Fig.3.

Association of spirochetes with Alzheimer’s disease. The frequency of spirochetes is significantly higher in the brains of Alzheimer’s disease patients compared to controls. Graph from Miklossy J (2011) Alzheimer’s disease - a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J Neuroinflammation 8, 90 [126]. Copyright 2011. Reprinted with permission under the terms of the Creative Commons Attribution License, (http://creativecommons.org/licenses/by/2.0) and permission from Judith Miklossy.
Using dark field microscopy, Miklossy identified spirochetes in blood, CSF, and brain in 14/14 AD autopsy cases and in 0/13 non-AD controls [9]. In this study, spirochetes were cultured from the blood of four AD cases. Concurrent silver stained and anti-AβPP-immunostained frozen section AD brain specimens evaluated with electron microscopy revealed spirochetes located in areas of AD pathology. Immunohistochemistry using a specific antibody against B. burgdorferi identified spirochetes in senile plaques, neurons, neuropil threads, and in the leptomeningeal and cortical vessel walls in a patient with concurrent Lyme disease and AD [9]. Electron microscopy and atomic force microscopy techniques have been used to identify spirochetes isolated and cultured from postmortem AD brains [8]. PCR and immunohistochemistry identified oral spirochetes in 14/16 AD and 4/18 non-AD postmortem brains (Fig. 4) [127]. DNA identified within neuropil threads in AD brains using the florescent dye DAPI revealed a helically shaped morphology similar to the morphology and distribution in reference spirochete samples [128]. Spirochetes were detected in the brains of 8/8 postmortem AD cases and in the blood samples from five living AD patients [129]. Using immunohistochemical techniques, Borrelia antigens (including the outer surface protein A (OspA) of B. burgdorferi) and Borrelia genes were co-localized with Aβ deposits and NFTs in three AD brains from which B. burgdorferi was cultured [130]. Bacterial peptidoglycan has been immunolocalized to senile plaques and NFTs in autopsied brain specimens from 54 AD patients [131, 132]. In addition, peptidoglycan and was found co-localized with Aβ in AD brains but not in controls [131]. The synthetic peptide BH (9-10), which corresponds to a β-hairpin segment of the B. burgdorferi OspA protein, forms amyloid-like fibrils in vitro [133].
Fig.4.
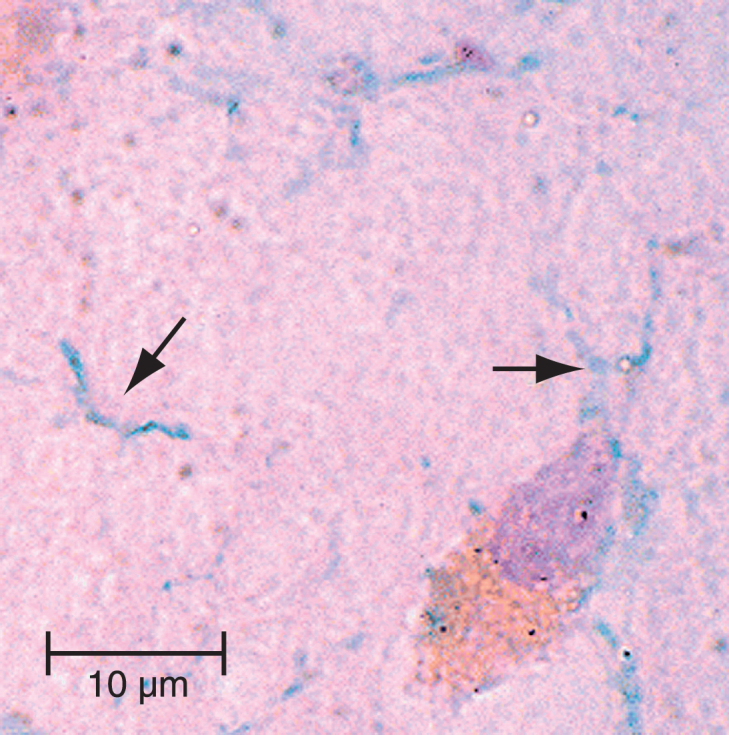
Image of oral spirochetes in Alzheimer’s disease brain. The oral spirochete T. pectinovorum stained dark blue (arrows) in a section from the hippocampus from an 84-year-old woman with Alzheimer’s disease. The section was incubated with monoclonal antibodies to T. pectinovorum, and binding was disclosed using biotinylated anti-mouse antibodies and avidin-peroxidase. The photomicrograph was taken at1000X. Scale bar = 10μm. Figure from Riviere GR, Riviere KH, Smith KS (2002) Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol Immunol 17, 113-118 [127]. Copyright 2002. Reprinted with permission from John Wiley and Sons, Inc. and George Riviere.
B. burgdorferi induces Aβ and p-tau formation, inflammation, and neurodegeneration
In vitro, B. burgdorferi invades mammalian neurons and glial cells to cause an AD-like host cell reaction. Aβ deposition is induced in vitro by exposure of mammalian neurons, astrocytes, microglial cells, and brain organotypic cell aggregates to Borrelia burgdorferi sensu strictu [13]. Histochemical and immunohistochemical analysis showed morphological changes including Aβ plaques with β-pleated sheet conformation and tangles. Intracytoplasmic granules found in astrocytes were similar to the granulovacuolar degeneration seen in AD neurons [13]. Increases in AβPP, Aβ, and hyperphosphorylated tau proteins were detected by western blot in these cells [13]. Nuclear fragmentation in rat astrocyte cells exposed to pleomorphic and cystic forms of B. burgdorferi suggests that the spirochete can cause functional damage and apoptosis [134].
Exposure of rat glial cells to B. burgdorferi recombinant lipidated outer surface protein A (L-OspA) induces astrocyte proliferation and apoptosis. Astrocytes produce IL-6 and TNF-α in response to L-OspA [14]. Ex vivo stimulation of monkey brain explants with B. burgdorferi induces the production of cytokines IL-6, IL-8, IL-1β, cox-2, and the chemokine B lymphocyte chemoattractant (CXCL13) by glial cells, with concomitant glial and neuronal apoptosis [16].
Additional periodontal pathogens
In addition to the oral spirochetes discussed above, Kamer et al. found that both the number of positive tests for IgG serum antibodies against periodontal bacteria commonly involved in periodontitis (A. actinomycetemcomitans, P. gingivalis, and T. forsythia) and plasma TNF-α level were elevated in AD patients compared to normal controls. Both endpoints were independently associated with AD [135]. Results from the NHANES III study involving a large community sample, showed that the extent of periodontal disease, as measured by gingival bleeding, loss of periodontal attachment, and loss of teeth, was associated with significantly decreased cognitive function in early-, mid-, and late-adult life [136]. Cognitive testing included the Symbol Digit substitution and the Serial digit Learning Tests among patients 20–59 years of age and a Story Recall test in participants aged 70 years of age or older. Worse scores on all three measures of oral health status were significantly associated with poorer performance on all three measures of cognitive function after adjustment for age. Level of education was found to be an important confounding factor. The authors concluded that poor oral health is associated with impaired cognitive function throughout adult life [136]. A separate study from NHANES III showed an association between high serum antibody levels against P. gingivalis and lower cognitive function with delayed verbal recall and impaired subtraction in subjects greater than 60 years of age [137]. Thus, exposure to oral pathogens is associated with systemic inflammation, cognitive decline, and AD.
Helicobacter pylori
H. pylori is a curved spiral Gram-negative bacterium which colonizes the gastric mucosa in more than 50% of humans worldwide. The bacterium causes gastric disorders including functional dyspepsia,gastritis, peptic ulcer disease, and gastric cancer [138, 139]. H. pylori infection is associated with extra-digestive disorders including idiopathic thrombocytopenic purpura, vitamin B12 deficiency, and iron deficiency anemia [140]. The bacterium is also associated with vascular disorders such as atherosclerosis, ischemic stroke, and coronary artery disease [141].
H. pylori gastric infections may be diagnosed with non-invasive procedures including urea breath test, serology, or whole blood antibody testing depending on clinical circumstances [142]. Diagnostic tests for H. pylori, which require upper endoscopy with biopsy sampling of the gastric mucosa, include rapid urease test, histology, bacterial culture, and polymerase chain reaction technique [143].
Epidemiological studies: H. pylori infection, cognitive decline, and AD
Epidemiological studies support an association between H. pylori infection and both MCI and AD. Kountouras et al. studied sixty-three patients with amnestic MCI who underwent upper gastrointestinal endoscopy with histological and serological testing for H. pylori infection. Significantly increased H. pylori gastric infection, higher mean serum anti-H pylori IgG concentrations, and higher plasma total homocysteine titers were found in MCI patients compared to non-MCI anemic controls [144]. In another study, Kountouras and colleagues found a significantly higher rate of histologically proven H. pylori gastric infection among 50 AD patients compared to thirty non-AD anemic controls [145]. A longitudinal study by Roubaud-Baudron et al. followed 603 subjects who were initially free of dementia and 65 years or older. Baseline seropositivity for H. pylori IgG antibody was associated with a 1.46 times increased risk for the development of dementia over the 20 year follow-up period compared to non-infected controls [146]. In a prospective non-randomized study, Kountouras et al. found that AD patients had significantly higher levels of anti-H. pylori-specific IgG antibodies in CSF and serum than age-matched cognitively normal controls. The severity of AD, as indicated by lower MMSE scores, correlated with higher levels of anti-H. pylori IgG antibodies in the CSF of these patients. The authors concluded that the data appears to link H. pylori infection to the pathophysiology of AD. They could not exclude the passage of H. pylori IgG and antibodies through an AD-related dysfunctional blood-CSF barrier to explain their findings [147].
In vitro and animal models: H. pylori induces formation of Aβ42 and P-tau
Mouse neuroblastoma N2a cells transfected with human AβPP are found to overexpress AβPP. Incubation of H. pylori filtrate with these cells resulted in increased production of presenilin-2 (a component of the gamma secretase enzyme complex) and Aβ42. In the same study, intraperitoneal injection of H. pylori filtrate resulted in spatial learning and memory deficits in rats, abnormal hippocampal dendritic spine maturation, and increased presenilin-2 and Aβ42 in rat brain hippocampus and cortex [148].
H. pylori filtrate induced significant tau hyperphosphorylation at several AD-related tau phosphorylation sites in mouse neuroblastoma N2a cells through activation of glycogen synthase kinase-3β. In the same study, intraperitoneal injection of H. pylori filtrate in rats resulted in significant tau hyperphosphorylation in hippocampal areas of rat brain. Microglial activation and elevated brain/plasma cytokine levels were not found. The authors concluded that soluble exotoxins of H. pylori may induce tau hyperphosphorylation and that H. pylori eradication may be beneficial in the prevention of tauopathy [149]. These studies provide evidence which links H. pylori infection with AD-like Aβ and p-tau pathology.
Potential H. pylori pathogenic mechanisms in AD
Evidence for direct brain infiltration by H. pylori is lacking, and exactly how a gastrointestinal infection like H. pylori might influence neurodegeneration in AD is unknown. However, gastric inflammation has been found in patients infected by H. pylori, with increased production of IL-1, IL-6, IL-12, IL-18, TNF-α, and IFN-γ [150]. Lagunes-Servin et al. found that H. pylori gastric mucosa infection in children was associated with upregulation of toll-like receptors TLR2, TLR4, TLR5, and TLR9, and overproduction of cytokines, including TNF-α, IL-10, and IL-8 [151]. These findings are potentially significant because increased levels of pro-inflammatory cytokines and TLR-induced cell signaling cascades are implicated in AD pathogenesis [19, 20].
As reviewed by Kountouras and collaborators, additional proposed mechanisms for H. pylori related AD pathogenesis include: i) influences on neuronal apoptosis through molecular mimicry, in which homologous H. pylori epitopes induce humoral and cellular immune responses, which then cross-react with components of nerves; ii) molecular mimicry between H. pylori and endothelial antigens; iii) mononuclear cell production of a tissue factor-like pro-coagulant that converts fibrinogen into fibrin; iv) production of reactive oxygen species and circulating lipid peroxidases; v) platelet activation and aggregation; and vi) reduced levels of vitamin B12 and folate secondary to chronic atrophic gastritis, resulting in elevated serum homocysteine levels and subsequent endothelial damage and neurodegeneration [144, 152].
NEUROINFLAMMATION, PATHOGENS, AND NEURODEGENERATION
The innate immune system: Glial cells and the vicious cycle of inflammation
Gao and Hong [1] and Griffin [153] have advanced the hypothesis that uncontrolled inflammation drives neurodegenerative disease (Fig. 5) [1]. They propose that CNS pathological processes are initiated by environmental insults interacting with genetic susceptibility. Interactions between damaged neurons and deregulated, over activated microglia create a vicious self-propagating cycle causing uncontrolled long-term inflammation and progression of chronic neurodegenerative disease [1, 153].
Fig.5.

The vicious cycle of neurodegeneration. Neuroinflammation when controlled is reparative and self-limiting but when uncontrolled forms a vicious cycle and leads to chronic neurodegeneration. Figure from Gao HM, Hong JS (2008) Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol 29, 357-365 [1]. Copyright 2008. Reprinted with permission from Elsevier and John Hong.
Chronic overexpression of IL-1β is found in AD brains [20] and has been induced by pathogens in vitro [17,124] and ex vivo [16]. IL-1β has been shown to increase neuronal AβPP production [153, 154], apolipoprotein E levels, and astrocyte-mediated S100β protein levels [153]. BACE-1 levels in neurons are increased by cytokines [155], oxidative stress molecules [155], and pathogens such as HSV-1 [10]. Along with γ-secretase, BACE-1 catalyzes the conversion of AβPP to Aβ, resulting in elevated levels of toxic forms of Aβ [155]. Aβ activates microglial RAGE receptors, which appear to mediate the proinflammatory response to Aβ. Fibrillar Aβ activates microglia cell surface protein CD36 and scavenger receptors A and B (SR-A and SR-B). Activation of these receptors induces production of reactive oxygen species by microglia [156].
The cycle is further accelerated by neuronal injury and neuron cell membrane breakdown products, cytosolic compounds, and glutamate excess, which further activates microglia [1]. There is loss of homeostasis from a tightly controlled and regulated process where anti-inflammatory cytokines are utilized for tissue repair and recovery of function. The resultant uncontrolled inflammation and amplified cytokine cycle induces neuronal injury, apoptosis, and chronic disease progression [1, 153, 157].
Microglia function as innate immune cells in the brain [156]. Pattern recognition receptors located on the microglia cell surface identify pathogen associated molecular patterns on viruses and bacteria, leading to microglial production of pro-inflammatory molecules [5]. Pathogen associated molecular patterns include lipopolysaccharide (LPS), peptidoglycan, lipoteichoic acid, flagellin, bacterial lipoprotein, and nucleic acid structures such as bacterial DNA or viral RNA [5]. Examples of pattern recognition receptors located on the microglia cell surface include TLRs1-9, scavenger receptors (SRA, SRB, Macrophage receptor with collagenous domain (MARCO), CD36) and receptors for advanced glycogen end products (RAGE) [156].
Additional receptors include Major histocompatibility complex II (MHCII), cytokine receptors (CD40) and chemokine receptors (CCR3, CCR5) [20]. The NADPH oxidase receptor is a membrane-bound enzyme that catalyzes the production of superoxide from oxygen. This receptor is activated in the AD brain, and is associated with neurodegeneration [156]. Many of these receptors are upregulated in regions of typical AD brain pathology [19]. Increases inthe levels of pattern recognition receptors in animal models and cell cultures are associated with neurodegeneration [19].
Microglia produce pro-inflammatory molecules via intracellular signaling pathways in cell cultures and animal models. For example, pathogens activate microglia TLRs leading to activation of nuclear factor κB, the mitogen activated protein kinases, and jun kinase. Pathogens can also induce activation of a second microglial pathway involving interferon regulatory factor-3 [5]. These pathways lead to the induction of inflammatory genes that produce cytokines and other pro-inflammatory compounds [5].
Microglia and astroglia are consistently found surrounding amyloid plaques in AD brains [157]. Amyloid deposition causes a microglial-mediated inflammatory response [19]. Pro-inflammatory molecules have been shown to be involved in pathways of neuronal apoptosis [20]. Aβ stimulated microglia secrete TNF-α and glutamate in vitro, resulting in simultaneous activation of neuronal TNF-α and N-methyl-D-aspartate (NMDA) receptors and subsequent neuronal apoptosis [158].
Pro-inflammatory compounds produced by glial cells and upregulated in AD brains include cytokines (IL-1α, IL-1β, IL-6, TNF-α), chemokines including macrophage inflammatory protein-1β and monocyte chemotactic protein-1, prostaglandins (cox 2), growth factors such as macrophage colony stimulating factor, and complement components (C1q, and C1 to C9) [19, 20]. Additional neurotoxic compounds produced by activated microglia include superoxide, hydrogen peroxide, and nitrous oxide [156]. Oxidative stress (lipid oxidation, protein oxidation, DNA oxidation, and glycol-oxidation) contributes to neurodegeneration in AD [159]. Associated pathological processes include endoplasmic reticulum stress associated with change in endoplasmic reticulum calcium homeostasis [160, 161], release of free electrons from dysfunctional mitochondria [162], and formation of reactive oxygen species [163].
Infection with either HSV-1 [10] or C. pneumoniae [12] induces Aβ42 deposits and plaques, and H. pylori filtrate [148] results in elevated levels of Aβ42 in animal brain models. In vitro infection by HSV-1 [10], B. burgdorferi [13], and H. pylori filtrate [148] induces Aβ deposition in mammalian neuronal or neuronal/glial cell models.
Aβ has been shown in vitro to be an anti-microbial peptide against eight specific microorganisms, including Escherichia coli, Streptococcus pneumonia, and Candida albicans. AD whole brain homogenates have significantly higher antimicrobial activity compared to age matched non-AD control samples [164]. Aβ42 has shown anti-microbial peptide properties by attenuating HSV-1–induced miRNA-146a levels in human neuronal-glial cell cultures and significantly reducing pathological HSV-1 effects on cultured brain cells [165]. Aβ production may be part of the CNS immune response to infection with eventual harmful effects to neurons due to overproduction of Aβ [166].
Evidence supporting the role of the adaptive immune system in AD
Lynch has proposed that BBB permeability, which is increased in AD, together with the creation of a chemotactic gradient, leads to infiltration of IFN-γ-producing T cells into the AD brain [167]. T cell production of IFN-γ induces classical microglia activation, which leads to inflammatory cytokine and chemokine production. This in turn results in increased AβPP processing, Aβ accumulation, further BBB permeability, and infiltration of more T cells, leading to a continuous cycle of neurodegeneration (Fig. 6) [167].
Fig.6.

The Lynch Hypothesis: T-cell lymphocytes infiltrate the brain and secrete IFN-γ which induces microglia activation and contributes to neurodegeneration in AD. Proposed sequence of events leading to amyloid pathology and microglial activation in AD. T lymphocytes cells activated peripherally cross the BBB and secrete IFN-γ and other cytokines, interact with microglia, and influence the neurodegenerative processes involved in AD. Figure from Lynch MA (2014) The impact of neuroimmune changes on development of amyloid pathology; relevance to Alzheimer’s disease. Immunology 141, 292-301 [167]. Copyright 2014. Reprinted with permission from John Wiley and Sons, Inc. andMarina Lynch.
Resident cells in the brain produce only limited IFN-γ [167]. Under normal conditions, T cell entry into the CNS is limited and thought to be related to T cell immuno-surveillance [168, 169]. Significant infiltration of immune cells does occur in neuroinflammatory conditions [170] and T cells have been localized in the brains of AD patients [171–178]. Breakdown of the BBB (see BBB section below), increased expression of T cell attractant chemokines such as interferon-γ-inducible protein 10 (IP-10), and corresponding chemokine receptors such as CXCR3 on neuronal cells have been found in AD brains [179] and may contribute to infiltration of T cells into the AD brain [167].
T cells can interact with microglia to modulate their function, as demonstrated by in vitro co-culture experiments [167]. Resting microglia cells from BALB/c mice developed features of antigen presenting cells, including strongly upregulated surface expression of MHCII, CD40, and CD54 when co-cultured with type 1 T helper cells (Th1) [180]. Conversely, mouse microglia induce Th1 cells to release IFN-γ [180]. Supernatants produced by allo-antigen and myelin basic protein-specific human pro-inflammatory Th1 T-cell lines augmented expression of cell surface molecules MHC class II, CD80, CD86, CD40, and CD54, enhanced the functional antigen-presenting cell capacity in a mixed lymphocyte reaction, and increased cytokine/chemokine secretion (TNF-α, IL-6, and CXCL10/IP-10) by CNS-derived human microglia [181]. Co-culture of Aβ-specific Th1 or Th17 cells and microglia induced pro-inflammatory cytokine production (IL1-β, TNF-α, and IL-6) and antigen presenting cell capacity of microglia in a murine model [182]. IFN-γ activates murine microglial cells and results in microglial production of TNF-α and inducible nitric oxide synthase (i-NOS) in vitro [183].
Browne and colleagues investigated the role of Aβ-specific T cells on Aβ accumulation in transgenic mice that overexpress AβPP and presenilin 1 (AβPP/PS1 mouse model), and found significant infiltration of T cells in these brains. Aβ-specific CD4+ T cells were generated by immunization with Aβ and a TLR agonist and polarized in vitro to Th1-, Th2-, or IL-17-producing CD4+ T cells. A proportion of these T cells secreted IFN-γ or IL-17. These Aβ-specific T cells were then adoptively transferred to AβPP/PS1 mice at 6 to 7 months of age. At 5 weeks, Th1 cells, but not Th2 or IL-17-producing CD4+ T cells had infiltrated into these brains. Additionally, there was increased microglial activation and CNS Aβ deposition. All of these findings were associated with impaired cognitive function. Treatment of the AβPP/PS1 mice with an anti-IFN-γ antibody attenuated the Th1 cell effects. The authors suggest that the release of IFN-γ from infiltrating Th1 cells significantly accelerates markers of diseases in an animal model of AD [184]. Murphy et al. demonstrated that murine Th17 and CD4+ lymphocytes, which produce IL-17 and IFN-γ, induce microglial production of IL-1β, IL-6, and TNF-α in experimental autoimmune encephalomyelitis, the animal model of multiple sclerosis [185]. The combination of IFN-γ and TNF-α has been shown to induce the production of Aβ peptides and inhibit the secretion of soluble AβPP by human neuronal and extraneuronal cells in vitro [186].
These findings lend support to the hypothesis that T lymphocytes activated peripherally may cross the BBB and secrete IFN-γ and other cytokines, interact with microglia, and influence the neurodegenerative processes involved in AD. Thus, T cells may be an important link between the systemic adaptive immune system and the innate immune system in the AD brain.
Infection-induced acute or chronic inflammation exacerbates tau pathology in vivo
Sy et al. demonstrated that acute inflammation induced by viral infection or chronic inflammation induced by bacterial LPS resulted in AD-like pathology in animal brains using the triple transgenic AD mouse model (3xTg-AD). Aged 3xTg-AD and non-Tg mice infected with a single dose of mouse hepatitis virus (MHV) by injection into the hippocampus developed similar acute neuroinflammatory responses with increased infiltration into the brain by macrophages, CD4+ T cells, CD8+ T cells, and activation of microglia. After viral clearance at 2 and 4 weeks post-infection, MHV-infected 3xTg-AD mice showed a marked increase in phosphorylated tau protein levels in the brain. In addition, increased activation of GSK-3β, one of the enzymes that phosphorylates tau protein, was found. This effect was not seen in MHV infected non-Tg mice. Sustained brain inflammation was induced in 3xTg-AD aged mice by intraperitoneal injection of lipopolysaccharide, which is an outer membrane Gram-negative bacterial endotoxin that simulates bacterial infection. Injection of LPS twice weekly for 6 weeks resulted in significant elevation of phosphorylated tau protein levels, increased GSK-3β activity, and cognitive decline compared with saline injected 3xTg-AD aged control mice. Based on these findings, the authors suggest that certain microbial infections may act as comorbid factors in the pathogenesis of AD by inducing inflammation, increasing levels of phosphorylated tau proteins, and exacerbating cognitive decline [187].
Infectious burden is associated with systemic inflammation and serum Aβ levels in AD
Bu and co-workers studied IB, serum cytokine, and Aβ levels, and cognition in 128 AD patients and 135 healthy controls. IB consisted of serum antibody levels to CMV, HSV-1, B. burgdorferi, C. pneumoniae, and H. pylori. Total IB, bacterial burden, and viral burden were independently associated with AD after adjusting for age, gender, education, APOE genotype, and other comorbidities. They found a significant association between AD and an increasing number of pathogens to which an individual had been exposed, with an OR of 3.988 in patients seropositive for 4-5 pathogens compared with those seropositive for 0–2 pathogens. Furthermore, higher IB was associated with higher serum Aβ levels in both AD and healthy controls with seropositivity to 3 or more pathogens. There were significantly higher serum levels of IFN- γ, TNF-a, IL-1β, and IL-6 in participants seropositive for 4-5 pathogens than those seropositive for 0–2 pathogens when including all cases. AD patients seropositive to 4-5 pathogens exhibited significantly higher levels of IFN-γ, TNF-α, and IL-6 when compared with AD patients seropositive to 0–2 pathogens. The authors suggest that higher levels of IB may promote the development of AD by infection-induced inflammation and elevated Aβ levels [188].
APOLIPOPROTEIN E OVERVIEW
Apolipoprotein E (ApoE) is a 299 amino acid protein component of lipoproteins. The primary metabolic role for ApoE is to shuttle and distribute lipids from one tissue or cell type to another and to regulate lipid metabolism [189]. Various ApoE isoforms appear to play a role in disease susceptibility and outcome of certain infections, with evidence also supporting involvement in immune regulation [189]. The liver synthesizes the majority of ApoE; however, 20–40% of ApoE is produced by extra-hepatic cells including glial cells [190] and neurons [191].
ApoE associates with lipoproteins including VLDL, LDL, and HDL during systemic transport of triglycerides and cholesterol and is a primary carrier of lipids across the BBB into the brain [190]. When carrying lipids, ApoE binds to members of the low density lipoprotein receptor (LDLR) family [191]. A secondary proposed ApoE binding site involves the heparan sulphate proteoglycan (HSPG)/LDL-C receptor-related protein pathway [192].
The human APOE gene is located on chromosome 19 as a single gene locus with three major allele isoforms designated ɛ4, ɛ3, and ɛ2 [189]. APOE allele frequencies vary between ethnicities, with the APOE-ɛ4 allele variant occurring at a frequency of 5–30% , the APOE-ɛ3 allele variant at 50–90% , and the APOE-ɛ2 allele variant at 0–15% [190]. The corresponding products of these alleles are the ApoE isoforms named ApoE4, ApoE3, and ApoE2. The 6 resultant ApoE phenotypes include 3 homozygous phenotypes (E4/4, E3/3, and E2/2) and 3 heterozygous phenotypes (E4/3, E4/2, and E3/2) [190].
The APOE-ɛ4 allele impacts outcomes in certain infections
As discussed above, Itzhaki demonstrated increased risk of AD by a factor or 12 in APOE-ɛ4 carriers who have HSV-1 in the brain [49]. Patients with the APOE-ɛ4 allele had a higher rate of oral herpetic lesions compared to non-APOE-ɛ4 allele carriers with a relative risk of 4.64 [193]. Transgenic APOE4 mice infected with HSV-1 demonstrate higher CNS viral loads compared with APOE3 [51]. HSV-1 binds to HSPG located on the target cell membrane to facilitate entry into the cell [194]. Itzhaki et al. suggest that ApoE4 may compete with HSV-1 for binding to cell surface HSPG receptors, and that ApoE4 is less competitive than ApoE3 or ApoE2, allowing more virus particles to gain entry into the cell [191].
C. pneumoniae elementary bodies bind to human astrocytoma cells possessing the APOE-ɛ4 allele at levels 3-fold higher than non-APOE-ɛ4 allele bearing cells. A separate line of astrocytoma cells transfected with plasmids expressing the ɛ4 coding sequence had 3-fold more C. pneumoniae elementary body attachment than astrocytoma cells encoding ApoE3. These findings demonstrate that expression of ApoE4 enhances attachment of C. pneumoniae elementary bodies to target host cells, which may enhance infectivity [195].
APOE-ɛ4 allele carriage is associated with a reduced risk of acquiring chronic hepatitis C virus (HCV) [190, 196]. Carriage is also protective against severe liver disease caused by HCV [197]. The exact mechanism for this protective effect has yet to be elucidated; however, HCV associates with serum lipoproteins including apoE to enter cells via the LDLR [198]. The expression of LDLR on the cell surface of hepatocytes is inversely related to the concentration of LDL in serum [199]. One hypothesis suggests that a higher serum LDL concentration in APOE-ɛ4 carriers leads to a lower LDLR expression, which potentially decreases virus entry and spread between hepatocytes [197].
Corder and colleagues phenotyped sera from HIV patients for ApoE and found that patients whopossessed a single copy of the ApoE4 isoform had significantly higher rates of dementia and peripheral neuropathy compared to those who were ApoE4 negative [200]. Burt et al. demonstrated that HIV positive patients with the APOE-ɛ4/ɛ4 genotype have both accelerated disease progression and progression to death compared to APOE-ɛ3/APOE-ɛ3 carriers. An association between APOE-ɛ4/ɛ4 genotype and HIV-associated dementia was not confirmed in this study. However, using an in vitro cell model with specialized HeLa cells, the authors found that the HIV infection rate was significantly higher in the presence of ApoE4 compared with ApoE3 [201].
The target cells of HIV include CD4+ T-cells, macrophages, and microglia cells. HIV initiates entry into the cell by attaching to the HSPG receptor on the target cell membrane [190]. HIV envelope glycoproteins then bind to the CD4 receptor. Fusion to the cell membrane requires cholesterol in HIV particles and lipid rafts, which are cell membrane micro domains enriched in certain lipids, cholesterol and proteins [190]. The mechanism by which ApoE isoforms impact HIV disease outcomes is not known. Research has focused on the differential effects of ApoE isoforms on HIV particle binding activity and uptake involving the LDL-R and the HSPG receptors [190]. One proposed mechanism is that ApoE4 may be less competitive compared with HIV at the target cell HSPG receptor than ApoE3 or ApoE2 resulting in increased HIV cell entry [190]. An additional hypothesis is that ApoE4 on the HIV viral envelope promotes HIV binding activity at the low-density lipoprotein receptor of the target cell [190].
APOE-ɛ4 allele is associated with enhanced human innate immune responses
Gale et al. demonstrated that carriage of the APOE-ɛ4 allele is associated with enhanced in vivo innate immune responses in human subjects [202]. Whole blood from healthy genotyped APOE-ɛ3/APOE-ɛ4 volunteers exposed ex vivo to TLR2, TLR4, or TLR5 ligands induced significantly higher levels of TNF-α than blood from APOE-ɛ3/APOE-ɛ3 carriers. Blood from APOE-ɛ3/APOE-ɛ4 carriers also induced significantly higher levels of IL1-β, IL-6, IFN-γ, and additional cytokines and chemokines after exposure to TLR2 or TLR4 when compared with blood from APOEɛ3/APOEɛ3 carriers. No difference was seen between the two ApoE phenotypes regarding production of anti-inflammatory compounds IL-4 and IL-1 receptor antagonist. Thus, ApoE4 is associated with a broad pro-inflammatory response to TLR cascades. Human APOE-ɛ3/APOE-ɛ4 subjects exposed to an intravenous LPS challenge demonstrated enhanced immune responses with significantly higher plasma levels of TNF-α and greater sustained body temperatures compared with APOE-ɛ3/APOE-ɛ3 subjects [202].
Differences in monocyte lipid rafts have been found in APOE-ɛ3/APOE-ɛ4 human peripheral blood monocytes compared with APOE-ɛ3/APOE-ɛ3 monocytes [202]. Lipid rafts within the cell membrane organize cellular signaling events [203]. Several TLR cascades are initiated in lipid rafts, which and are enhanced by cholesterol loading [202]. ApoE4 is less efficient at inducing cholesterol efflux and removing cholesterol in mouse macrophages [204]. Gale et al. used a fluoro-tagged cholera toxin B subunit lipid raft probe to study in vitro CD14 monocytes. Augmented lipid raft assembly in APOE-ɛ3/APOE-ɛ4 monocytes compared to APOE-ɛ3/APOE-ɛ3 monocytes was demonstrated. The authors suggest that ApoE4 may enhance cholesterol loading in monocyte lipid raft structures due to decrease in cholesterol efflux, which in turn enhances TLR responses with pro-inflammatory consequences [202].
The APOE-ɛ4 genotype with resultant ApoE4 phenotype appears to influence disease outcome for several infections and elevate the pro-inflammatory innate immune response. Evidence supporting ApoE4 associated enhanced infectivity and brain infiltration would explain the increased risk of AD in patients who are co-factor positive for HSV-1 in the brain and the APOE-ɛ4 allele [50]. Increased binding of C. pneumoniae elementary bodies to APOE-ɛ4 target host cells suggests increased infectivity as a mechanism to explain elevated numbers of C. pneumoniae infected cells in APOE-ɛ4 allele carrier AD brains [123]. An increased ApoE4 associated pro-inflammatory response may contribute to AD pathogenesis by both increasing BBB permeability and elevating levels of pro-inflammatory cytokines.
THE BLOOD-BRAIN BARRIER AND THE AD PATHOGEN HYPOTHESIS: BBB IMPAIRMENT IN AD
Breakdown of the BBB with increased permeability is involved in the pathophysiology of AD [205, 206]. Postmortem studies have shown BBB damage in AD with accumulation of blood-derived proteins including albumin, fibrinogen, thrombin, and immunoglobulins in the hippocampus and cortex [207]. BBB breakdown appears to be an early event in the aging human brain that begins in the hippocampus and may contribute to cognitive impairment [207]. Montagne and collaborators used an advanced dynamic contrast enhanced MRI protocol with high spatial and temporal resolutions to quantify regional BBB permeability in the living human brain. In doing so, they demonstrated an age-dependent BBB breakdown in the hippocampus, an area of the brain affected early in AD. The BBB breakdown in the hippocampus and its CA1 and dentate gyrus subdivisions worsened with MCI that correlated with injury to BBB-associated pericytes, as shown by the CSF analysis [207]. Evidence suggests that pro-inflammatory cytokines, Aβ, and APOE4 genetics are contributing factors in the breakdown of the BBB [212–214], with data indicating pathogen association and interactions with each of these factors as previously discussed.
Composition of the blood-brain barrier
The BBB is located at the level of the cerebral microvasculature and serves as the largest interface for blood-brain exchange [208]. The BBB forms a physical, enzymatic, and transport barrier between the vasculature and the brain parenchyma [209]. Brain microvascular endothelial cells (BMECs) line cerebral blood vessels to form a barrier on the endothelial side of the vessel. Pericytes and astrocyte end feet form a continuous barrier in association with the basement membrane on the adluminal vessel surface [210]. BMECs form intercellular contacts via two types of junctions known as adherens junctions (AJs) and tight junctions (TJs). TJs in BMECs are composed of the membrane proteins occludin, claudins, junctional adhesion molecules, and cell-selective adhesion molecules, which are linked to the actin cytoskeleton by cytoplasmic proteins ZO-1, ZO-2, ZO-3, and cingulin [210]. AJs contain cadherins bound to actin microfilaments by a submembranous zone of catenins [211]. These junctions promote high transendothelial electrical resistance that restricts paracellular permeability [210]. This in turn blocks the transport of a wide range of molecules and regulates the passage of ions, macromolecules, and polar molecules from the systemic circulation [210]. Endothelial cells of the BBB lack fenestrations and have a reduced number of pinocytotic vesicles, which restricts trans-cellular flux [208]. However, some molecules and solutes are transported across the BBB by various mechanisms including transporters, receptor and/or adsorption-mediated transcytosis, and passive diffusion [208].
The blood-brain barrier: Pathogens and neuroinflammation
Certain pathogens may cross the BBB either transcellularly, paracellularly, or by means of infected phagocytes by a Trojan horse mechanism [215]. N. meningitidis exemplifies a bacterium which crosses the BBB transcellularly [215]. Evidence suggests that B. burgdorferi may cross the BBB paracellularly [215], whereas C. pneumoniae may cross by way of the “Trojgan horse” mechanism, intracellulary within monocytes or macrophages [108, 109]. Other pathogens including HSV-1 cross the BBB via the olfactory nerve and/or trigeminal nerve [5, 208]. In addition to pathogens that cross the BBB directly, blood-born cytokines including IL-1α, IL1-β, IL-6, TNF-α, and others can cross BBB via transport systems and act directly on brain tissue as demonstrated in animal models [209, 216].
CNS infections and other diseases associated with elevated pro-inflammatory cytokine levels increase the permeability of the BBB [214]. Cytokines such as IL-1 act on brain endothelial cells to increase the expression of endothelial adhesion molecules and chemokines, including CC chemokine ligand-2 (CCL2), selectins, and intracellular adhesion molecule-1 (ICAM-1), which contribute to BBB permeability [217]. Pro-inflammatory cytokines also increase expression of matrix metalloproteinases (MMPs), which degrade extracellular matrix components in the endothelial basement membrane and tight junctions, resulting in increased BBB permeability [218, 219]. Labus et al. found that IL-1β induced an inflammatory response and breakdown of the endothelial layer in an in vitro BBB model. IL-1β induced endothelial cells expression of adhesion molecule ICAM-1, IL-6, IL-8, and TNF- α. IL-1β also reduced the expression of tight junction protein ZO-1. Increases in both paracellular permeability and leukocytes crossing the cell layer of the BBB model were demonstrated [214].
Aβ and the blood-brain barrier
Evidence indicates that Aβ and BBB disruption interact to mutually promote their effects on AD pathogenesis [213]. Accumulation of Aβ in the brain may contribute to the breakdown of the BBB. Conversely, dysfunction of the BBB may result in Aβ accumulation due to BBB leakage or decreased clearance [213]. P-glycoprotein, an efflux pump highly expressed on the luminal surface of brain capillary endothelial cells of the BBB, has been shown to transport Aβ from the brain to the blood [220, 221]. Decreased function of this transporter has been reported in AD brains [221]. Aβ42 is able to modify the expression of TJs and alter the functionality of an epithelial BBB in vitro model [222]. In addition, Aβ accumulation has been shown to cause BBB dysfunction by inducing endothelial cell toxicity both in vitro and in vivo in animal studies, human studies, and human postmortem studies [223]. Soluble Aβ also stimulates BBB endothelial cells to increase monocyte adhesion, which has been hypothesized to increase monocyte permeability across the BBB leading to monocyte transmigration into the brain parenchyma [223].
APOE-ɛ4 and the blood-brain barrier
Animal models support a role for genetic susceptibility in the breakdown of the BBB. Using APOE transgenic mice, Bell et al. found that expression of ApoE4 and lack of murine ApoE lead to BBB breakdown by activating a pro-inflammatory cyclophilin A (CypA)-nuclear factor-κB-MMP-9 pathway in pericytes through a lipoprotein receptor. This was followed by neuronal uptake of neurotoxic proteins and reductions in microvascular and cerebral blood flow. Breakdown in the BBB preceded neuronal dysfunction and the initiation of neurodegenerative changes [224].
In a postmortem study, Halliday and colleagues demonstrated accelerated pericyte degeneration in AD APOE-ɛ4 carriers > AD APOE-ɛ3 carriers > non-ADcontrols, which correlated with the magnitude of BBB breakdown as measured by permeability to immunoglobulin G and fibrin. Accumulation of CypA and MMP-9 in pericytes and endothelial cells was greater in AD subjects who were APOE-ɛ4 carriers than those who were APOE-ɛ3 carriers. Levels of the ApoE lipoprotein receptor, low-density lipoprotein receptor-related protein-1 (LRP1), were reduced in AD APOE-ɛ4 and APOE-ɛ3 carriers. This data suggests that possession of APOE-ɛ4 leads to accelerated pericyte loss and enhanced activation of LRP1-dependent CypA-MMP-9 BBB-degrading pathway in pericytes and endothelial cells. These processes mediate BBB damage, which is more severe in AD APOE-ɛ4 carriers than AD APOE-ɛ3 carriers [212].
POTENTIAL ANTIMICROBIAL TREATMENTS FOR AD CLINICAL TRIALS
Treatment of HSV-1: Acyclovir and valacyclovir
Acyclovir decreases HSV-1-induced Aβ accumulation in cultured neuroblastoma cells [166]. Aβ in cell cultures was reduced by 70% at a 200μM concentration of acyclovir. In addition, acyclovir inhibits HSV-1-induced abnormal tau phosphorylation in vitro. Abnormal tau phosphorylation was reduced nearly 100% at a 200μM concentration of acyclovir (Fig. 7) [166]. Acyclovir decreased Aβ by reducing cellular viral spreading and decreased tau phosphorylation by interfering with viral replication [166]. Penciclovir and foscarnet, which also inhibit viral DNA replication, were shown to reduce phosphorylated tau and Aβ, with foscarnet being less effective than acyclovir and penciclovir [166].
Fig.7.

Quantification of HSV-1 proteins (A), amyloid-β (B), and abnormal tau phosphorylation (C) in HSV-1-infected cells after acyclovir treatment. HSV-1 infected vero cell cultures treated with 0μM, 50μM, 100μM, or 200μM acyclovir (ACV). ACV significantly inhibited replication of HSV-1 as shown by a decrease in HSV-1 proteins (A). Aβ in cell cultures was reduced by 70% at a 200μM concentration of acyclovir (B). Abnormal tau phosphorylation was reduced nearly 100% at a 200μM concentration of acyclovir (C). p < 0.0001 compared to controls at all ACV concentrations for A and C and at 100μM and 200μM ACV concentrations for B. Graph from Wozniak MA et al. (2011) Antivirals reduce the formation of key Alzheimer’s disease molecules in cell cultures acutely infected with Herpes simplex virus type 1. PLoS One 6, e25152 [166]. Copyright 2011. Reprinted under the terms of the Creative Commons Attribution License, (http://creativecommons.org/licenses/by/2.0) and permission from Ruth Itzhaki.
Acyclovir is a nucleoside analogue that interferes with HSV-1 replication and reactivation. Viral thymidine kinase is required to convert acyclovir into acyclo-guanosine monophosphate. Subsequently, the monophosphate form is converted to the active tri-phosphate form by cellular kinases. As a substrate, acyclo-guanosine triphosphate is incorporated into viral DNA, resulting in premature chain termination. Acyclovir is able to cross the BBB [225]. The drug would directly target a potential cause of AD, act on infected cells only, and would not affect the normal metabolism of infected neurons [166].
Acyclovir is FDA approved and widely used for the treatment of HSV infections. The side effect profile is mild; however it would be necessary to monitor renal function [225]. Rarely, treatment is complicated by reversible neuropsychiatric symptoms occurring more frequently in patients with pre-existing renal impairment [225].
Valacylovir is the biodrug of acyclovir. Following oral administration, valacyclovir is rapidly hydrolyzed to acyclovir via first-pass intestinal and hepatic metabolism [225]. Valacyclovir has better oral absorption than acyclovir and is able to cross the BBB as acyclovir following hydrolysis [226]. Oral valacyclovirhas been used to successfully treat herpes simplex encephalitis [227]. In a multiple sclerosis trial evaluating HHV-6, valacyclovir at a dose of 3 grams per day for 2 years was shown to be safe with no patient discontinuation due to side effects or toxicity [228]. Both acyclovir and valacyclovir have been found safe during long-term use in patients being treated for HSV suppression [229]. Furthermore, resistance rates are low (<0.5%) among immunocompetent patients[229].
Prasad et al. conducted a clinical trial involving 24 HSV-1 IgG seropositive schizophrenia patients who were treated with valacyclovir 1.5 grams twice daily for 18 weeks. The treatment group demonstrated significantly improved cognition, specifically in verbal memory, working memory, and visual object learning, compared with the HSV-1 IgG seronegative schizophrenia control group. Both treatment and control groups were treated with anti-psychotic medication. While psychotic symptoms did not improve with valacyclovir, the study did show that valacyclovir improved cognition in a select group of HSV-1 exposed neuropsychiatric patients [230].
Intravenous immunoglobulin (IVIG)
Natural antibodies to Aβ in human IVIG promote microglial recognition and removal of natively formed human Aβ deposits ex vivo in an AβPP/PS1 mouse model of AD [231]. IVIG administered peripherally in vivo crossed the mouse BBB, reaching highest concentrations in the hippocampus. The antibodies bound selectively to Aβ deposits and co-localized with microglia [231]. Preclinical and small clinical studies treating early stage AD patients with IVIG showed decreases in CSF Aβ levels and increases in serum Aβ levels compared to baseline, with reductions in cognitive decline compared to controls [232, 233]. However, the Gamma Globulin Alzheimer’s Partnership (GAP) study, a large double blind IVIG clinical trial treating mild to moderate AD patients, did not meet primary outcome objectives regarding cognitive function and activities of daily living. Biomarker studies did indicate dose dependent increases in plasma and CSF immunoglobulins and decreases in plasma Aβ42 levels [234].
IVIG has antiviral activity against HSV-1 and can neutralize extracellular virus [235]. Additionally, IVIG in conjunction with lymphocytes is able to destroy cells infected with HSV-1 [235–237]. IVIG (Privigen) treated HSV-1 infected Vero cells demonstrated statistically significant reductions of Aβ, phosphorylated tau, and HSV-1 proteins [238]. Privagen appeared to prevent viral entry into cells and act synergistically with acyclovir, leading the authors to suggest that the combination of IVIG and acyclovir may be beneficial in the treatment of AD [32, 238].
HSV-1 vaccine
Lin and coworkers showed that a vaccine of mixed HSV-1 glycoproteins had a protective effect against HSV-1 infection of mouse brain. The vaccine significantly reduced HSV-1 latency in the CNS of mice that had been infected peripherally with HSV-1 [239]. Although not yet developed, a human vaccine for HSV-1 administered to prevent HSV-1 infiltration into the brain might prevent some cases of AD [239].
Treatment of CMV, EBV, and HHV-6
Anti-viral therapy for asymptomatic CMV infection in immunocompetent patients is not feasible because of toxicity, limited number of approved drugs, and the potential for drug resistance [82]. Thus, there is no available suppressive therapy for CMV comparableto the usage of valacyclovir or acyclovir for HSV suppression. Ganciclovir (GCV) and valganciclovir, the oral prodrug of GCV, are nucleoside analogues. GCV is phosphorylated to its active form in CMV-infected cells. These medications are currently the most commonly prescribed drugs for the prevention and treatment of CMV in immunocompromised patients [240]. Although limited, current evidence suggests that targeted antiviral therapy with GCV or valganciclovir is appropriate for severe CMV disease in immunocompetent adults [241]. There are no FDA approved medications for the treatment of EBV, and anti-viral therapy is generally ineffective for this virus [242]. No therapies are approved for the treatment of HHV-6 currently; however, small studies and individual case reports have reported intermittent success with drugs such as cidofovir, GCV, and foscarnet [243]. To date there are no FDA approved vaccines for CMV, EBV, or HHV-6 [240].
Treatment of Chlamydophila pneumoniae
Acute C. pneumoniae infections are susceptible to antibiotics that interfere with DNA and protein synthesis, including tetracyclines, macrolides, quinolones, and rifamycins [110]. All of these antibiotics cross the BBB except for the macrolides [244]. C. pneumoniae becomes spontaneously persistent following infection of monocytes [112]. C. pneumoniae within infected lymphocytes in vitro demonstrated resistance to single antibiotics including minocycline and tosufloxacin and did not show uniform susceptibility within infected monocytes [245]. Nine months of combination doxycycline and rifampin were used in a successful clinical trial as treatment for chronic Chlamydia-induced reactive arthritis. This study suggests that persistent chlamydia infection responds poorly to single antibiotic therapy but appears to be susceptible to combination antibiotic regimens [246].
Tetracyclines possess both immunomodulatory and antiapoptotic properties [247] and have been shown to be anti-inflammatory and neuroprotective in various models of neurodegenerative disease [248]. Tetracycline and doxycycline exhibit anti-amyloidogenic activity in vitro by inhibiting the self-aggregation capacity of Aβ42 and disassembling pre-formed Aβ42 fibrils [249]. The semi-synthetic, second generation tetracycline analog minocycline inhibited increases in p-eIF2α and reduced neuronal cell death in Aβ42 peptide treated nerve growth factor-differentiated rat pheochromocytoma (PC12) cells in vitro [161]. Minocycline also reduced neuronal cell death and attenuated deficits in learning and memory after Aβ42 infusion in a rat model of AD [161].
Rifampin crosses the BBB [244, 250] and attenuates all chlamydial gene transcription [246]. Rifampin-induced upregulation of LRP1 and p-glycoprotein at the BBB enhances Aβ clearance from the mouse brain [251].
In a clinical trial, Loeb et al. treated probable AD patients diagnosed with mild to moderate dementia with doxycycline and rifampin versus placebo for 3 months. There was significantly less decline in cognition as measured by the Standardized Alzheimer’s Disease Assessment Scale cognitive subscale at 6 months in the antibiotic treatment group compared to the placebo group [252]. In a clinical trial by Molloy and colleagues, mild to moderate AD patients were treated for 12 months with doxycycline and rifampin. These drugs, given both alone or in combination, had no effect on cognition or function [253]. Pre-clinical AD patients and biomarker endpoints were not assessed in this study. In addition, these medications have not been tested in combination with valacyclovir.
Treatment of spirochetes
The CNS infection Lyme neuroborreliosis caused by the spirochete B. burgdorferi has been successfully treated in clinical trials with 10–14 days of intravenous ceftriaxone or oral doxycycline [254, 255]. In one trial, 79% of the ceftriaxone-treated patients and 72% of the doxycycline-treated patients had completely recovered by 6 month follow-up. Ceftriaxone and oral doxycycline used as single agents were found to be effective, safe, and convenient for treatment of Lyme neuroborreliosis [254]. Abramson et al. studied the bactericidal activity of antimicrobial agents in vitro for 17 strains of treponemes. Most treponemes, including human oral spirochetes such as T. dentolytica, were sensitive to tetracycline, doxycycline, and cephalothin [256].
Treatment of periodontal pathogens
Improved oral hygiene and meticulous dental care may be beneficial for patients with dementia. Kikutani et al. studied the effects of oral care in a group of nursing home vascular dementia and AD patients. The oral care group had a high level of dental care, as nurses and caregivers cleaned the patients’ teeth and mouth with a toothbrush for approximately 5 minutes after each meal for one year. The oral care group had significantly less decline in MMSE scores compared to the non-oral care group at six months and one year [257].
Systemic antibiotics are widely used in the treatment of periodontal infections; however, clear guidelines for the use of systemic antibiotics to treat periodontitis in general clinical practice are not yet available [258, 259]. Adjunctive antibiotic treatment in moderate periodontal disease for 14 days with either amoxicillin or metronidazole in addition to traditional scaling and root planing has been shown to markedly reduce bacterial counts for pathogenic species including B. forsythus, P. gingivalis, and T. denticola, which remained lower than baseline at one year post-treatment [259]. Several clinical trials treating patients with mild to moderate periodontal disease have shown microbiological and/or clinical benefits by using azithromycin or metronidazole combined with scaling and root planing or pocket reduction surgery when compared to scaling and root planing alone [260–262], or surgery alone [263]. Subantimicrobial dose doxycycline (20 mg doxycycline twice daily) has also been used successfully to treat periodontitis [247]. Feres et al. extensively reviewed randomized clinical trials performed over the last decade involving antibiotic treatment of periodontitis where advanced microbial diagnostic testing had been performed. Patients treated with adjunctive antibiotic therapy had improved microbiological and clinical outcomes [258]. The authors recommended that patients with advanced or very advanced periodontitis should be treated with the adjunctive use of metronidazole or combination metronidazole plus amoxicillin for 14 days in addition to traditional scaling and root planing [258]. Antibiotic treatment does not appear to create lasting changes in the percentage of resistant isolates or sites harboring resistant species [260, 264].
Treatment of Helicobacter pylori
Treatment of H. pylori involves triple or quadruple regimens using a proton pump inhibitor such as omeprazole plus various combinations of amoxicillin, Clarithromycin, metronidazole, and tetracycline taken orally for 7 to 14 days. Sequential, concomitant, or hybrid regimens are chosen depending on resistance rates and other clinical factors [265, 266]. Clarithromycin and metronidazole resistance has been increasing in certain populations. Levaquin triple based and bismuth quadruple based regimens have been used successfully to treat resistant H. pylori. Patient-specific therapy is based on factors such as cost, allergy history, and known or suspected patterns of resistance [266]. Treatment success rates with current regimens are variable, ranging from 50% to over 95% in clinical trials depending on factors such as length of treatment, rates of antibiotic resistance and patient specific cytochrome P450 2C19 genotype [265].
Kountouras et al. evaluated AD patients with gastric biopsy-proven H. pylori infection. Patients were treated with combination omeprazole, clarithromycin, and amoxicillin triple based therapy to eradicate H. pylori from the gastric mucosa. At the 2-year clinical endpoint, cognitive (MMSE and Cambridge Cognitive Test) and functional (Functional Rating Scale for Symptoms of Dementia) parameters significantly improved in the subgroup of AD patients with successful H. pylori eradication, but not in the subgroup of AD patients in which H. pylori eradication was not achieved [152]. In another clinical trial, successful treatment and eradication of H. pylori in a group of probable AD patients was associated with a significantly lower 5-year mortality risk than a control group of probable AD patients who did not achieve eradication of the bacterium [267].
CONCLUSION
Evidence supports the hypothesis that pathogens interact with susceptibility genes and are causative in AD. Pathogen induced neurodegeneration occurs by both direct effects on brain cells and indirect inflammatory and oxidative effects. HSV-1, C. pneumoniae, and spirochetes are able to infect the brain and induce formation of Aβ and hyperphosphylated tau proteins. Chronic CNS infections lead to glial cell activation, resulting in neuroinflammation and neuronal apoptosis. Peripheral infections including CMV, periodontal pathogens, and H. pylori induce systemic inflammation with elevated levels of pro-inflammatory molecules. Pathogen induced pro-inflammatory cytokines are transported across the BBB into the brain where they induce CNS inflammation and contribute to AD pathology in genetically susceptible individuals. In addition, pathogen induced pro-inflammatory cytokines and Aβ interact with genetic susceptibility factors to damage the BBB, resulting in increased BBB permeability. Chronic CMV infection with reactivation in peripheral blood or organ systems induces systemic activation of IFN-γ-producing T cells, which may enter the brain and contribute to the pathogenesis of AD. CMV also plays a role in immunosenescence, which damages the immune system and is associated with the reactivation of other Herpesviridae such as HSV-1. Thus, in addition to direct pathogen effects, AD pathogenesis results from interactions involving both the innate and adaptive immune responses to both CNS and peripheral systemic pathogens.
As proposed, the AD pathogen hypothesis would explain the multiple epidemiological studies showing increased risk for development of cognitive impairment and AD in patients with various CNS infiltrative and systemic chronic infections. Peripheral viral reactivation with resultant systemic inflammation is a potential mechanism involved in the association between HSV-1 seropositivity and cognitive impairment in younger patients ages 6 to 16 as found by Tarter et al. [87]. Systemic HSV-1 induced inflammation may also explain the association between HSV-1 positivity, cognitive impairment, and neurodegeneration on MRI in relatively younger schizophrenic patients as demonstrated by Schretlen et al. [34]. Younger subjects have been shown not to have HSV-1 in the brain [107] so presumably, the patients in these studies did not have direct HSV-1 CNS infiltration by the virus. Limited studies to date have failed to detect HSV in postmortem brain tissue from patients with schizophrenia as well [268], including one study using nested PCR [269].
The APOE-ɛ4 allele with resultant ApoE4 phenotype impacts the pathophysiology of AD by increasing the pathogen load in the brain, specifically for HSV-1 and C. pneumoniae. ApoE4 also interacts with pathogens to enhance the human innate pro-inflammatory response and contributes to the breakdown of the BBB.
HSV-1 may be a primary CNS infiltrative pathogen in the pathophysiology of AD. There is substantial evidence for HSV-1 causation in AD involving studies in epidemiology, neuropathology, and molecular biology as cited in this review. There is the high prevalence of HSV-1 in both elderly normal brains and AD brains [4, 41] (Table 1) and the presence of both HSV-1 in brain and carriage of the APOE-ɛ4 allele significantly increases the risk for sporadic AD [49]. Data for both C. pneumoniae and spirochetes (Table 3 and Fig. 3) shows a high prevalence of these pathogens in AD brains only [5, 126], suggesting that secondary C. pneumoniae and/or spirochete infection of brain may occur after HSV-1 and other co-factors have already initiated AD pathogenesis. Additional studies need to be done to confirm thishypothesis.
A comprehensive antimicrobial treatment strategy for AD must be developed and tested in clinical trials focusing on pre-clinical or early onset AD. Itzhaki [270] and Strandberg [28] have proposed a randomized controlled antiviral clinical trial using oral valacyclovir. The initial study would test the concept that HSV-1 in APOE-ɛ4 carriers is causative in AD [80]. Additional AD clinical trials could then evaluate the AD multi-pathogen hypothesis by testing the effectiveness of valacyclovir combined with the appropriate antibiotics used to treat spirochetes, chronic persistent C. pneumoniae, and systemic AD associated pathogens such as H. pylori and periodontalinfections.
Safe, effective, and less toxic treatments for CMV and other Herpesviridae must be developed. Antimicrobial medication in combination with anti-inflammatory treatments may also be beneficial in the treatment of AD. Vaccines against CMV, HSV-1, and other Herpesviridae must be developed, as reducing primary infection or reactivation may be useful in AD prevention.
Treatment of HSV-1 and other pathogens present in the AD brain and peripherally may be the most efficacious way to reduce CNS inflammation. The goal of such treatment would be to lower production of pro-inflammatory molecules, reduce accumulation of Aβ, and lower the levels of hyperphosphorylated tau proteins. Antimicrobial therapy could decrease neuronal damage and apoptosis and ultimately aid in the prevention and treatment of AD.
ACKNOWLEDGMENTS
There has been no financial or material support involved in the writing of this article. The authors did not receive support for this work from any for-profit entity, including licensing agreements. Special thanks to Dr. Ramesh Shah for administrative assistance.
The author’s disclosure is available online (http://j-alz.com/manuscript-disclosures/14-2853r2).
REFERENCES
- 1.Gao HM, Hong JS. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008;29:357–365. doi: 10.1016/j.it.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De-Paula VJ, Radanovic M, Diniz BS, Forlenza OV. Alzheimer’s disease. Subcell Biochem. 2012;65:329–352. doi: 10.1007/978-94-007-5416-4_14. [DOI] [PubMed] [Google Scholar]
- 3.Wozniak MA, Mee AP, Itzhaki RF. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J Pathol. 2009;217:131–138. doi: 10.1002/path.2449. [DOI] [PubMed] [Google Scholar]
- 4.Jamieson GA, Maitland NJ, Wilcock GK, Craske J, Itzhaki RF. Latent herpes simplex virus type 1 in normal and Alzheimer’s disease brains. J Med Virol. 1991;33:224–227. doi: 10.1002/jmv.1890330403. [DOI] [PubMed] [Google Scholar]
- 5.Miklossy J. Emerging roles of pathogens in Alzheimer disease. Expert Rev Mol Med. 2011;13:e30. doi: 10.1017/S1462399411002006. [DOI] [PubMed] [Google Scholar]
- 6.Balin BJ, Gérard HC, Arking EJ, Appelt DM, Branigan PJ, Abrams JT, Whittum-Hudson JA, Hudson AP. Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med Microbiol Immunol. 1998;187:23–42. doi: 10.1007/s004300050071. [DOI] [PubMed] [Google Scholar]
- 7.Gérard HC, Dreses-Werringloer U, Wildt KS, Deka S, Oszust C, Balin BJ, Frey WH, 2nd, Bordayo EZ, Whittum Hudson JA, Hudson AP. Chlamydophila(Chlamydia) pneumoniae in the Alzheimer’s brain. FEMS Immunol Med Microbiol. 2006;48:355–366. doi: 10.1111/j.1574-695X.2006.00154.x. [DOI] [PubMed] [Google Scholar]
- 8.Miklossy J, Kasas S, Janzer RC, Ardizzoni F, Van der Loos H. Further ultrastructural evidence that spirochaetes may play a role in the etiology of Alzheimer’s disease. Neuroreport. 1994;5:1201–1204. doi: 10.1097/00001756-199406020-00010. [DOI] [PubMed] [Google Scholar]
- 9.Miklossy J. Alzheimer’s disease–a spirochetosis? Neuroreport. 1993;4:841–848. [PubMed] [Google Scholar]
- 10.Wozniak MA, Itzhaki RF, Shipley SJ, Dobson CB. Herpes simplex virus infection causes cellular β amyloid accumulation and secretase up-regulation. Neurosci Lett. 2007;429:95–100. doi: 10.1016/j.neulet.2007.09.077. [DOI] [PubMed] [Google Scholar]
- 11.Wozniak MA, Frost AL, Itzhaki RF. Alzheimer’s disease-specific tau phosphorylation is induced by herpes simplex virus type 1. J Alzheimers Dis. 2009;16:341–350. doi: 10.3233/JAD-2009-0963. [DOI] [PubMed] [Google Scholar]
- 12.Little CS, Hammond CJ, MacIntyre A, Balin BJ, Appelt DM. Chlamydia pneumoniae induces Alzheimer-like amyloid plaques in brains of BALB/c mice. Neurobiol Aging. 2004;25:419–429. doi: 10.1016/S0197-4580(03)00127-1. [DOI] [PubMed] [Google Scholar]
- 13.Miklossy J, Kis A, Radenovic A, Miller L, Forro L, Martins R, Reiss K, Darbinian N, Darekar P, Mihaly L, Khalili K. Beta-amyloid deposition and Alzheimer’s type changes induced by Borrelia spirochetes. Neurobiol Aging. 2006;27:228–236. doi: 10.1016/j.neurobiolaging.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 14.Ramesh G, Alvarez AL, Roberts ED, Dennis VA, Lasater BL, Alvarez X, Philipp MT. Pathogenesis of Lyme neuroborreliosis: Borrelia burgdorferi lipoproteins induce both proliferation and apoptosis in rhesus monkey astrocytes. Eur J Immunol. 2003;33:2539–2550. doi: 10.1002/eji.200323872. [DOI] [PubMed] [Google Scholar]
- 15.Boelen E, Steinbusch HW, Bruggeman CA, Stassen FR. The inflammatory aspects of Chlamydia pneumoniae- induced brain infection. Drugs Today (Barc) 2009;45(Suppl B):159–164. [PubMed] [Google Scholar]
- 16.Ramesh G, Borda JT, Dufour J, Kaushal D, Ramamoorthy R, Lackner AA, Philipp MT. Interaction of the Lyme disease spirochete Borrelia burgdorferi with brain parenchyma elicits inflammatory mediators from glial cells as well as glial and neuronal apoptosis. Am J Pathol. 2008;173:1415–1427. doi: 10.2353/ajpath.2008.080483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lokensgard JR, Hu S, Sheng W, vanOijen M, Cox D, Cheeran MC, Peterson PK. Robust expression of TNF alpha, IL-1beta, RANTES, and IP-10 by human microglial cells during nonproductive infection with herpes simplex virus. J Neurovirol. 2001;7:208–219. doi: 10.1080/13550280152403254. [DOI] [PubMed] [Google Scholar]
- 18.Halford WP, Gebhardt BM, Carr DJ. Persistent cytokine expression in trigeminal ganglion latently infected with herpes simplex virus type 1. J Immunol. 1996;157:3542–3549. [PubMed] [Google Scholar]
- 19.Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012;2:a006346. doi: 10.1101/cshperspect.a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho GJ, Drego R, Hakimian E, Masliah E. Mechanisms of cell signaling and inflammation in Alzheimer’s disease. Curr Drug Targets Inflamm Allergy. 2005;4:247–256. doi: 10.2174/1568010053586237. [DOI] [PubMed] [Google Scholar]
- 21.Dosunmu R, Wu J, Basha MR, Zawia NH. Environmental and dietary risk factors in Alzheimer’s disease. Expert Rev Neurother. 2007;7:887–900. doi: 10.1586/14737175.7.7.887. [DOI] [PubMed] [Google Scholar]
- 22.Itzhaki RF (2011) Herpes simplex and Alzheimer’s -Time to Think Again? Alzforum, http://www.alzforum.org/webinars/herpes-simplex-and-alzheimers-time-thinkagain?liveID=188, Posted 24 Feb 2011, Accessed on March 3, 2013
- 23.Ball MJ. Limbic predilection in Alzheimer dementia: Is reactivated herpes virus involved? Can J Neurol Sci. 1982;9:303–306. doi: 10.1017/s0317167100044115. [DOI] [PubMed] [Google Scholar]
- 24.McQuaid S, Allen IV, McMahon J, Kirk J. Association of measles virus with neurofibrillary tangles in subacute sclerosing panencephalitis: A combinedhybridization and immunocytochemical investigation. Neuropathol Appl Neurobiol. 1994;20:103–110. doi: 10.1111/j.1365-2990.1994.tb01168.x. [DOI] [PubMed] [Google Scholar]
- 25.Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE. Accelerated tau deposition in the brains of individuals infected with human immunodeficiency virus-1 before and after the advent of highly active anti-retroviral therapy. Acta Neuropathol. 2006;111:529–538. doi: 10.1007/s00401-006-0037-0. [DOI] [PubMed] [Google Scholar]
- 26.Strandberg T, Pitkala K, Linnavuori K, Tilvis R. Impact of viral and bacterial burden on cognitive impairment in elderly persons with cardiovascular disease. Stroke. 2003;34:2126–2131. doi: 10.1161/01.STR.0000086754.32238.DA. [DOI] [PubMed] [Google Scholar]
- 27.Katan M, Moon YP, Paik MC, Sacco RL, Wright CB, Elkind MS. Infectious burden and cognitive function: The Northern Manhattan Study. Neurology. 2013;80:1209–1215. doi: 10.1212/WNL.0b013e3182896e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strandberg TE, Aiello AE. Is the microbe-dementia hypothesis finally ready for a treatment trial? Neurology. 2013;80:1182–1183. doi: 10.1212/WNL.0b013e3182897126. [DOI] [PubMed] [Google Scholar]
- 29.Letenneur L, Pérès K, Fleury H, Garrigue I, Barberger-Gateau P. Seropositivity to herpes simplex virus antibodies and risk of Alzheimer’s disease: A population-based cohort study. PLoS One. 2008;3:e3637. doi: 10.1371/journal.pone.0003637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lövheim H, Gilthorpe J, Adolfsson R, Nilsson LG, Elgh F. Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimers Dement. 2015;11:593–599. doi: 10.1016/j.jalz.2014.04.522. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi N, Nagata T, Shinagawa S, Oka N, Shimada K, Shimizu A, Tatebayashi Y, Yamada H, Nakayama K, Kondo K. Increase in the IgG avidity index due to herpes simplex virus type 1 reactivation and its relationship with cognitive function in amnestic mild cognitive impairment and Alzheimer’s disease. Biochem Biophys Res Commun. 2013;430:907–911. doi: 10.1016/j.bbrc.2012.12.054. [DOI] [PubMed] [Google Scholar]
- 32.Itzhaki RF. Herpes simplex virus type 1 and Alzheimer’s disease: Increasing evidence for a major role of the virus. Front Aging Neurosci. 2014;6:202. doi: 10.3389/fnagi.2014.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lövheim H, Gilthorpe J, Johansson A, Eriksson S, Hallmans G, Elgh F. Herpes simplex infection and the risk of Alzheimer’s disease-A nested case-control study. Alzheimers Dement. 2015;11:587–592. doi: 10.1016/j.jalz.2014.07.157. [DOI] [PubMed] [Google Scholar]
- 34.Schretlen DJ, Vannorsdall TD, Winicki JM, Mushtaq Y, Hikida T, Sawa A, Yolken RH, Dickerson FB, Cascella NG. Neuroanatomic and cognitive abnormalities related to herpes simplex virus type 1 in schizophrenia. Schizophr Res. 2010;118:224–231. doi: 10.1016/j.schres.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 35.Prasad KM, Watson AM, Dickerson FB, Yolken RH, Nimgaonkar VL. Exposure to herpes simplex virus type 1 and cognitive impairments in individuals with schizophrenia. Schizophr Bull. 2012;38:1137–1148. doi: 10.1093/schbul/sbs046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dickerson FB, Boronow JJ, Stallings C, Origoni AE, Ruslanova I, Yolken RH. Association of serum antibodies to herpes simplex virus 1 with cognitive deficits in individuals with schizophrenia. Arch Gen Psychiatry. 2003;60:466–472. doi: 10.1001/archpsyc.60.5.466. [DOI] [PubMed] [Google Scholar]
- 37.Shirts BH, Prasad KM, Pogue-Geile MF, Dickerson F, Yolken RH, Nimgaonkar VL. Antibodies to cytomegalovirus and herpes simplex virus 1 associated with cognitive function in schizophrenia. Schizophr Res. 2008;106:268–274. doi: 10.1016/j.schres.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yolken RH, Torrey EF, Lieberman JA, Yang S, Dickerson FB. Serological evidence of exposure to herpes simplex virus type 1 is associated with cognitive deficits in the CATIE schizophrenia sample. Schizophr Res. 2011;128:61–65. doi: 10.1016/j.schres.2011.01.020. [DOI] [PubMed] [Google Scholar]
- 39.Dickerson F, Stallings C, Origoni A, Vaughan C, Khushalani S, Yolken R. Additive effects of elevated C-reactive protein and exposure to herpes simplex virus type 1 on cognitive impairment in individuals with schizophrenia. Schizophr Res. 2012;134:83–88. doi: 10.1016/j.schres.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 40.Mancuso R, Baglio F, Cabinio M, Calabrese E, Hernis A, Nemni R, Clerici M. Titers of herpes simplex virus type 1 antibodies positively correlate with grey matter volumes in Alzheimer’s disease. J Alzheimers Dis. 2014;38:741–745. doi: 10.3233/JAD-130977. [DOI] [PubMed] [Google Scholar]
- 41.Itzhaki R. Herpes simplex virus type 1, apolipoprotein E and Alzheimer’s disease. Herpes. 2004;11(Suppl 2):77A–82A. [PubMed] [Google Scholar]
- 42.Jamieson GA, Maitland NJ, Wilcock GK, Yates CM, Itzhaki RF. Herpes simplex virus type 1 DNA is present in specific regions of brain from aged people with and without senile dementia of the Alzheimer type. J Pathol. 1992;167:365–368. doi: 10.1002/path.1711670403. [DOI] [PubMed] [Google Scholar]
- 43.Mori I, Kimura Y, Naiki H, Matsubara R, Takeuchi T, Yokochi T, Nishiyama Y. Reactivation of HSV-1 in the brain of patients with familial Alzheimer’s disease. J Med Virol. 2004;73:605–611. doi: 10.1002/jmv.20133. [DOI] [PubMed] [Google Scholar]
- 44.Rodriguez JD, Royall D, Daum LT, Kagan-Hallet K, Chambers JP. Amplification of herpes simplex type 1 and human herpes type 5 viral DNA from formalin-fixed Alzheimer brain tissue. Neurosci Lett. 2005;390:37–41. doi: 10.1016/j.neulet.2005.07.052. [DOI] [PubMed] [Google Scholar]
- 45.Itabashi S, Arai H, Matsui T, Higuchi S, Sasaki H. Herpes simplex virus and risk of Alzheimer’s disease. Lancet. 1997;349:1102. doi: 10.1016/S0140-6736(05)62325-2. [DOI] [PubMed] [Google Scholar]
- 46.Hemling N, Röyttä M, Rinne J, Pöllänen P, Broberg E, Tapio V, Vahlberg T, Hukkanen V. Herpesviruses in brains in Alzheimer’s and Parkinson’s diseases. Ann Neurol. 2003;54:267–271. doi: 10.1002/ana.10662. [DOI] [PubMed] [Google Scholar]
- 47.Marques AR, Straus SE, Fahle G, Weir S, Csako G, Fischer SH. Lack of association between HSV-1 DNA in the brain, Alzheimer’s disease and apolipoprotein E4. J Neurovirol. 2001;7:82–83. doi: 10.1080/135502801300069773. [DOI] [PubMed] [Google Scholar]
- 48.Wozniak MA, Shipley SJ, Combrinck M, Wilcock GK, Itzhaki RF. Productive herpes virus in brain of elderly normal subjects and Alzheimer’s disease patients. J Med Virol. 2005;75:300–306. doi: 10.1002/jmv.20271. [DOI] [PubMed] [Google Scholar]
- 49.Itzhaki RF, Lin WR, Shang D, Wilcock GK, Faragher B, Jamieson GA. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet. 1997;349:241–244. doi: 10.1016/S0140-6736(96)10149-5. [DOI] [PubMed] [Google Scholar]
- 50.Lin WR, Graham J, MacGowan SM, Wilcock GK, Itzhaki RF. Alzheimer’s disease, herpes virus in brain, apolipoprotein E4 and herpes labialis. Alzheimers Rep. 1998;1:173–178. [Google Scholar]
- 51.Burgos J, Ramirez C, Sastre I, Valdivieso F. Effect of Apolipoprotein E on the cerebral load of latent herpes simplex virus type 1 DNA. J Virol. 2006;80:5383–5387. doi: 10.1128/JVI.00006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guzman-Sanchez F, Valdivieso F, Burgos JS. Aging-related neurostructural, neuropathological, and behavioral changes associated with herpes simplex virus type 1 brain infection in mice. J Alzheimers Dis. 2012;30:779–790. doi: 10.3233/JAD-2012-120070. [DOI] [PubMed] [Google Scholar]
- 53.Carter CJ. APP, APOE, complement receptor 1, clusterin and PICALM and their involvement in the herpes simplex life cycle. Neurosci Lett. 2010;483:96–100. doi: 10.1016/j.neulet.2010.07.066. [DOI] [PubMed] [Google Scholar]
- 54.Piacentini R, Civitelli L, Ripoli C, Marcocci ME, De Chiara G, Garaci E, Azzena GB, Palamara AT, Grassi C. HSV-1 promotes Ca2+-mediated APP phosphorylation and Aβ accumulation in rat cortical neurons. Neurobiol Aging. 2011;32:2323.e13–2323.e26. doi: 10.1016/j.neurobiolaging.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 55.Alvarez G, Aldudo J, Alonso M, Santana S, Valdivieso F. Herpes simplex virus type 1 induces nuclear accumulation of hyperphosphorylated tau in neuronal cells. J Neurosci Res. 2012;90:1020–1029. doi: 10.1002/jnr.23003. [DOI] [PubMed] [Google Scholar]
- 56.Zambrano A, Solis L, Salvadores N, Cortés M, Lerchundi R, Otth C. Neuronal cytoskeletal dynamic modification and neurodegeneration induced by infection with herpes simplex virus type 1. J Alzheimers Dis. 2008;14:259–269. doi: 10.3233/jad-2008-14301. [DOI] [PubMed] [Google Scholar]
- 57.Cribbs DH, Azizeh BY, Cotman CW, LaFerla FM. Fibril formation and neurotoxicity by a herpes simplex virus glycoprotein B fragment with homology to the Alzheimer’s A beta peptide. Biochemistry. 2000;39:5988–5994. doi: 10.1021/bi000029f. [DOI] [PubMed] [Google Scholar]
- 58.Bearer EL. HSV, axonal transport and Alzheimer’s disease: In vitro and in vivo evidence for causal relationships. Future Virol. 2012;7:885–899. doi: 10.2217/fvl.12.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Satpute-Krishnan P, DeGiorgis JA, Bearer EL. Fast anterograde transport of herpes simplex virus: Role for the amyloid precursor protein of Alzheimer’s disease. Aging Cell. 2003;2:305–318. doi: 10.1046/j.1474-9728.2003.00069.x. Erratum in: Aging Cell9 (2010), 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shipley SJ, Parkin ET, Itzhaki RF, Dobson CB. Herpes simplex virus interferes with amyloid precursor protein processing. BMC Microbiol. 2005;5:48. doi: 10.1186/1471-2180-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Chiara G, Marcocci ME, Civitelli L, Argnani R, Piacentini R, Ripoli C, Manservigi R, Grassi C, Garaci E, Palamara AT. APP processing induced by herpes simplex virus type 1 (HSV-1) yields several APP fragments in human and rat neuronal cells. PLoS One. 2010;5:e13989. doi: 10.1371/journal.pone.0013989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheng SB, Ferland P, Webster P, Bearer EL. Herpes simplex virus dances with amyloid precursor protein while exiting the cell. PLoS One. 2011;6:e17966. doi: 10.1371/journal.pone.0017966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Santana S, Recuero M, Bullido MJ, Valdivieso F, Aldudo J. Herpes simplex virus type I induces the accumulation of intracellular β-amyloid in autophagic compartments and the inhibition of the non-amyloidogenic pathway in human neuroblastoma cells. Neurobiol Aging. 2012;33:e19–e33. doi: 10.1016/j.neurobiolaging.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 64.Tallóczy Z, Virgin HW, 4th, Levine B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy. 2006;2:24–29. doi: 10.4161/auto.2176. [DOI] [PubMed] [Google Scholar]
- 65.Tallóczy Z, Jiang W, Virgin HW, 4th, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A. 2002;99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Itzhaki RF, Cosby SL, Wozniak MA. Herpes simplex virus type 1 and Alzheimer’s disease: The autophagy connection. J Neurovirol. 2008;14:1–4. doi: 10.1080/13550280701802543. [DOI] [PubMed] [Google Scholar]
- 67.Chou J, Chen JJ, Gross M, Roizman B. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with gamma 134.5- mutants of herpes simplex virus 1. Proc Natl Acad Sci U S A. 1995;92:10516–10520. doi: 10.1073/pnas.92.23.10516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Valyi-Nagy T, Dermody TS. Role of oxidative damage in the pathogenesis of viral infections of the nervous system. Histol Histopathol. 2005;20:957–967. doi: 10.14670/HH-20.957. [DOI] [PubMed] [Google Scholar]
- 69.Santana S, Sastre I, Recuero M, Bullido MJ, Aldudo J. Oxidative stress enhances neurodegeneration markers induced by herpes simplex virus type 1 infection in human neuroblastoma cells. PLoS One. 2013;8:e75842. doi: 10.1371/journal.pone.0075842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bonda DJ, Wang X, Perry G, Nunomura A, Tabaton M, Zhu X, Smith MA. Oxidative stress in Alzheimer disease: A possibility for prevention. Neuropharmacology. 2010;59:290–294. doi: 10.1016/j.neuropharm.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 71.Klapper PE, Cleator GM, Longson M. Mild forms of herpes encephalitis. J Neurol Neurosurg Psychiatry. 1984;47:1247–1250. doi: 10.1136/jnnp.47.11.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marton R, Gotlieb-Stematsky T, Klein C, Lahat E, Arlazoroff A. Mild form of acute herpes simplex encephalitis in childhood. Brain Dev. 1995;17:360–361. doi: 10.1016/0387-7604(95)00075-m. [DOI] [PubMed] [Google Scholar]
- 73.Peter JB, Sevall JS. Review of 3200 serially received CSF samples submitted for type-specific HSV detection by PCR in the reference laboratory setting. Mol Cell Probes. 2001;15:177–182. doi: 10.1006/mcpr.2001.0356. [DOI] [PubMed] [Google Scholar]
- 74.Saldanha J, Sutton RN, Gannicliffe A, Farragher B, Itzhaki RF. Detection of HSV1 DNA byhybridisation in human brain after immunosuppression. J Neurol Neurosurg Psychiatry. 1986;49:613–619. doi: 10.1136/jnnp.49.6.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kaufman HE, Azcuy AM, Varnell ED, Sloop GD, Thompson HW, Hill JM. HSV-1 DNA in tears and saliva of normal adults. Invest Ophthalmol Vis Sci. 2005;46:241–247. doi: 10.1167/iovs.04-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Margolis TP, Elfman FL, Leib D, Pakpour N, Apakupakul K, Imai Y, Voytek C. Spontaneous reactivation of herpes simplex virus type 1 in latently infected murine sensory ganglia. J Virol. 2007;81:11069–11074. doi: 10.1128/JVI.00243-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Feldman LT, Ellison AR, Voytek CC, Yang L, Krause P, Margolis TP. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proc Natl Acad Sci U S A. 2002;99:978–983. doi: 10.1073/pnas.022301899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Martin C, Aguila B, Araya P, Vio K, Valdivia S, Zambrano A, Concha MI, Otth C. Inflammatory and neurodegeneration markers during asymptomatic HSV-1 reactivation. J Alzheimers Dis. 2014;39:849–859. doi: 10.3233/JAD-131706. [DOI] [PubMed] [Google Scholar]
- 79.Itzhaki RF, Wozniak MA. Herpes simplex virus type 1 in Alzheimer’s disease: The enemy within. J Alzheimers Dis. 2008;13:393–405. doi: 10.3233/jad-2008-13405. [DOI] [PubMed] [Google Scholar]
- 80.Itzhaki RF, Wozniak MA, Appelt DM, Balin BJ. Infiltration of the brain by pathogens causes Alzheimer’s disease. Neurobiol Aging. 2004;25:619–627. doi: 10.1016/j.neurobiolaging.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 81.Varani S, Landini MP. Cytomegalovirus-induced immunopathology and its clinical consequences. Herpesviridae. 2011;2:6. doi: 10.1186/2042-4280-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lurain NS, Hanson BA, Martinson J, Leurgans SE, Landay AL, Bennett DA, Schneider JA. Virological and immunological characteristics of human cytomegalovirus infection associated with Alzheimer disease. J Infect Dis. 2013;208:564–572. doi: 10.1093/infdis/jit210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Taylor-Wiedeman J, Sissons JG, Borysiewicz LK, Sinclair JH. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J Gen Virol. 1991;72:2059–2064. doi: 10.1099/0022-1317-72-9-2059. [DOI] [PubMed] [Google Scholar]
- 84.Aiello AE, Haan M, Blythe L, Moore K, Gonzalez JM, Jagust W. The influence of latent viral infection on rate of cognitive decline over 4 years. J Am Geriatr Soc. 2006;54:1046–1054. doi: 10.1111/j.1532-5415.2006.00796.x. [DOI] [PubMed] [Google Scholar]
- 85.Carbone I, Lazzarotto T, Ianni M, Porcellini E, Forti P, Masliah E, Gabrielli L, Licastro F. Herpes virus in Alzheimer’s disease: Relation to progression of the disease. Neurobiol Aging. 2014;35:122–129. doi: 10.1016/j.neurobiolaging.2013.06.024. [DOI] [PubMed] [Google Scholar]
- 86.Barnes LL, Capuano AW, Aiello AE, Turner AD, Yolken RH, Torrey EF, Bennett DA. Cytomegalovirus infection and risk of Alzheimer disease in older black and white individuals. J Infect Dis. 2015;211:230–237. doi: 10.1093/infdis/jiu437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tarter KD, Simanek AM, Dowd JB, Aiello AE. Persistent viral pathogens and cognitive impairment across the life course in the third national health and nutrition examination survey. J Infect Dis. 2014;209:837–844. doi: 10.1093/infdis/jit616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lin WR, Wozniak MA, Cooper RJ, Wilcock GK, Itzhaki RF. Herpesviruses in brain and Alzheimer’s disease. J Pathol. 2002;197:395–402. doi: 10.1002/path.1127. [DOI] [PubMed] [Google Scholar]
- 89.Lin WR, Wozniak MA, Wilcock GK, Itzhaki RF. Cytomegalovirus is present in a very high proportion of brains from vascular dementia patients. Neurobiol Dis. 2002;9:82–87. doi: 10.1006/nbdi.2001.0465. [DOI] [PubMed] [Google Scholar]
- 90.Koch S, Solana R, Dela Rosa O, Pawelec G. Human cytomegalovirus infection and T cell immunosenescence: A mini review. Mech Ageing Dev. 2006;127:538–543. doi: 10.1016/j.mad.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 91.Stowe RP, Kozlova EV, Yetman DL, Walling DM, Goodwin JS, Glaser R. Chronic herpesvirus reactivation occurs in aging. Exp Gerontol. 2007;42:563–570. doi: 10.1016/j.exger.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stowe RP, Peek MK, Cutchin MP, Goodwin JS. Reactivation of herpes simplex virus type 1 is associated with cytomegalovirus and age. J Med Virol. 2012;84:1797–1802. doi: 10.1002/jmv.23397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Almanzar G, Schwaiger S, Jenewein B, Keller M, Herndler-Brandstetter D, Würzner R, Schönitzer D, Grubeck-Loebenstein B. Long-term cytomegalovirus infection leads to significant changes in the composition of the CD8+ T-cell repertoire, which may be the basis for an imbalance in the cytokine production profile in elderly persons. J Virol. 2005;79:3675–3683. doi: 10.1128/JVI.79.6.3675-3683.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Westman G, Berglund D, Widén J, Ingelsson M, Korsgren O, Lannfelt L, Sehlin D, Lidehall AK, Eriksson BM. Increased inflammatory response in cytomegalovirus seropositive patients with Alzheimer’s disease. PLoS One. 2014;9:e96779. doi: 10.1371/journal.pone.0096779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Itzhaki RF, Klapper P. Cytomegalovirus: An improbable cause of Alzheimer disease. J Infect Dis. 2014;209:972–973. doi: 10.1093/infdis/jit665. [DOI] [PubMed] [Google Scholar]
- 96.Lurain NS, Hanson BA, Martinson J, Leurgans SE, Landay AL, Bennett DA, Schneider JA. Reply to Itzhaki and Klapper. J Infect Dis. 2014;209:974. doi: 10.1093/infdis/jit666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yao K, Gagnon S, Akhyani N, Williams E, Fotheringham J, Frohman E, Stuve O, Monson N, Racke MK, Jacobson S. Reactivation of human herpesvirus-6 in natalizumab treated multiple sclerosis patients. PLoS One. 2008;3:e2028. doi: 10.1371/journal.pone.0002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stone RC, Micali GA, Schwartz RA. Roseola infantum and its causal human herpesviruses. Int J Dermatol. 2014;53:397–403. doi: 10.1111/ijd.12310. [DOI] [PubMed] [Google Scholar]
- 99.Yao K, Crawford JR, Komaroff AL, Ablashi DV, Jacobson S. Review part 2: Human herpesvirus-6 in central nervous system diseases. J Med Virol. 2010;82:1669–1678. doi: 10.1002/jmv.21861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Licastro F, Carbone I, Raschi E, Porcellini E. The 21st century epidemic: Infections as inductors of neuro-degeneration associated with Alzheimer’s disease. Immun Ageing. 2014;11:22. doi: 10.1186/s12979-014-0022-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Landais E, Saulquin X, Houssaint E. The human T cell immune response to Epstein-Barr virus. Int J Dev Biol. 2005;49:285–292. doi: 10.1387/ijdb.041947el. [DOI] [PubMed] [Google Scholar]
- 102.Kutok JL, Wang F. Spectrum of Epstein-Barr virus-associated diseases. Annu Rev Pathol. 2006;1:375–404. doi: 10.1146/annurev.pathol.1.110304.100209. [DOI] [PubMed] [Google Scholar]
- 103.Schmidt CW, Misko IS. The ecology and pathology of Epstein-Barr virus. Immunol Cell Biol. 1995;73:489–504. doi: 10.1038/icb.1995.79. [DOI] [PubMed] [Google Scholar]
- 104.Kleines M, Schiefer J, Stienen A, Blaum M, Ritter K, Häusler M. Expanding the spectrum of neurological disease associated with Epstein-Barr virus activity. Eur J Clin Microbiol Infect Dis. 2011;30:1561–1569. doi: 10.1007/s10096-011-1261-7. [DOI] [PubMed] [Google Scholar]
- 105.Connelly KP, DeWitt LD. Neurologic complications of infectious mononucleosis. Pediatr Neurol. 1994;10:181–184. doi: 10.1016/0887-8994(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 106.Pender MP, Burrows SR. Epstein-Barr virus and multiple sclerosis: Potential opportunities for immunotherapy. Clin Transl Immunology. 2014;3:e27. doi: 10.1038/cti.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Contini C, Seraceni S, Cultrera R, Castellazzi M, Granieri E, Fainardi E. Chlamydophila pneumoniae infection and its role in neurological disorders. Interdiscip Perspect Infect Dis. 2010;2010:273573. doi: 10.1155/2010/273573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Moazed TC, Kuo CC, Grayston JT, Campbell LA. Evidence of systemic dissemination of Chlamydiapneumoniae via macrophages in the mouse. J Infect Dis. 1998;177:1322–1325. doi: 10.1086/515280. [DOI] [PubMed] [Google Scholar]
- 109.MacIntyre A, Abramov R, Hammond CJ, Hudson AP, Arking EJ, Little CS, Appelt DM, Balin BJ. Chlamydia pneumoniae infection promotes the transmigration of monocytes through human brain endothelial cells. J Neurosci Res. 2003;1:740–750. doi: 10.1002/jnr.10519. [DOI] [PubMed] [Google Scholar]
- 110.Hammerschlag MR, Kohlhoff SA. Treatment of chlamydial infections. Expert Opin Pharmacother. 2012;13:545–552. doi: 10.1517/14656566.2012.658776. [DOI] [PubMed] [Google Scholar]
- 111.Appelt DM, Roupas MR, Way DS, Bell MG, Albert EV, Hammond CJ, Balin BJ. Inhibition of apoptosis in neuronal cells infected with Chlamydophila (Chlamydia) pneumoniae. BMC Neurosci. 2008;9:13. doi: 10.1186/1471-2202-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hogan RJ, Mathews SA, Mukhopadhyay S, Summersgill JT, Timms P. Chlamydial persistence: Beyond the biphasic paradigm. Infect Immun. 2004;72:1843–1855. doi: 10.1128/IAI.72.4.1843-1855.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rassu M, Cazzavillan S, Scagnelli M, Peron A, Bevilacqua PA, Facco M, Bertoloni G, Lauro FM, Zambello R, Bonoldi E. Demonstration of Chlamydia pneumoniae in atherosclerotic arteries from various vascular regions. Atherosclerosis. 2001;158:73–79. doi: 10.1016/s0021-9150(01)00411-7. [DOI] [PubMed] [Google Scholar]
- 114.Di Pietro M, Filardo S, Cazzavillan S, Segala C, Bevilacqua P, Bonoldi E, D’Amore ES, Rassu M, Sessa R. Could past Chlamydial vascular infection promote the dissemination of Chlamydia pneumoniae to the brain? J Biol Regul Homeost Agents. 2013;27:155–164. [PubMed] [Google Scholar]
- 115.Arking EJ, Appelt DM, Abrams JT, Kolbe S, Hudson AP, Balin BJ. Ultrastructural analysis of Chlamydia pneumoniae in the Alzheimer’s brain. Pathogenesis (Amst) 1999;1:201–211. [PMC free article] [PubMed] [Google Scholar]
- 116.Shima K, Kuhlenbäumer G, Rupp J. Chlamydia pneumoniae infection and Alzheimer’s disease: A connection to remember? Med Microbiol Immunol. 2010;199:283–289. doi: 10.1007/s00430-010-0162-1. [DOI] [PubMed] [Google Scholar]
- 117.Balin BJ, Little CS, Hammond CJ, Appelt DM, Whittum-Hudson JA, Gérard HC, Hudson AP. Chlamydophila pneumoniae and the etiology of late-onset Alzheimer’s disease. J Alzheimers Dis. 2008;13:371–380. doi: 10.3233/jad-2008-13403. [DOI] [PubMed] [Google Scholar]
- 118.Nochlin D, Shaw CM, Campbell LA, Kuo CC. Failure to detect Chlamydia pneumoniae in brain tissues of Alzheimer’s disease. Neurology. 1999;53:1888. doi: 10.1212/wnl.53.8.1888-a. [DOI] [PubMed] [Google Scholar]
- 119.Ring RH, Lyons JM. Failure to detect Chlamydia pneumoniae in the late-onset Alzheimer’s brain. J Clin Microbiol. 2000;38:2591–2594. doi: 10.1128/jcm.38.7.2591-2594.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gieffers J, Reusche E, Solbach W, Maass M. Failure to detect Chlamydia pneumoniae in brain sections of Alzheimer’s disease patients. J Clin Microbiol. 2000;38:881–882. doi: 10.1128/jcm.38.2.881-882.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Taylor GS, Vipond IB, Paul ID, Matthews S, Wilcock GK, Caul EO. Failure to correlate C. pneumoniae with late onset Alzheimer’s disease. Neurology. 2002;59:142–143. doi: 10.1212/wnl.59.1.142. [DOI] [PubMed] [Google Scholar]
- 122.Paradowski B, Jaremko M, Dobosz T, Leszek J, Noga L. Evaluation of CSF-Chlamydia pneumoniae, CSF-tau and CSF-Abeta42 in Alzheimer’s disease and vascular dementia. J Neurol. 2007;254:154–159. doi: 10.1007/s00415-006-0298-5. [DOI] [PubMed] [Google Scholar]
- 123.Gérard HC, Wildt KL, Whittum-Hudson JA, Lai Z, Ager J, Hudson AP. The load of Chlamydia pneumoniae in the Alzheimer’s brain varies with APOE genotype. Microb Pathog. 2005;39:19–26. doi: 10.1016/j.micpath.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 124.Boelen E, Steinbusch HW, Pronk I, Stassen FR. Inflammatory responses following Chlamydia pneumoniae infection of glial cells. Eur J Neurosci. 2007;25:753–760. doi: 10.1111/j.1460-9568.2007.05339.x. [DOI] [PubMed] [Google Scholar]
- 125.Hammond C, Little CS, Longo N, Procacci C, Appelt DM, Balin BJ. Antibiotic alters inflammation in the mouse brain during persistentinfection. In: Iqbal K, Winblad B, Avila J, editors. Alzheimer’s Disease: New Advances. Medimond; 2006. pp. 295–299. [Google Scholar]
- 126.Miklossy J. Alzheimer’s disease - a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J Neuroinflammation. 2011;8:90. doi: 10.1186/1742-2094-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Riviere GR, Riviere KH, Smith KS. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol Immunol. 2002;17:113–118. doi: 10.1046/j.0902-0055.2001.00100.x. [DOI] [PubMed] [Google Scholar]
- 128.Miklossy J, Gern L, Darekar P, Janzer RC, Van der, Loos H. Senile plaques, neurofibrillary tangles and neuropil threads contain DNA? J Spirochetal Tick Borne Dis. 1995;2:1–5. [Google Scholar]
- 129.Miklossy J. The spirochetal etiology of Alzheimer’s disease: A putative therapeutic approach. In: Giacobini E, Becker R, editors. In Alzheimer Disease: Therapeutic Strategies. Proceedings of the Third International Springfield Alzheimer Symposium. Birkhauser; Boston: 1994. pp. 41–48. [Google Scholar]
- 130.Miklossy J, Khalili K, Gern L, Ericson RL, Darekar P, Bolle L, Hurlimann J, Paster BJ. Borrelia burgdorferi persists in the brain in chronic lyme neuroborreliosis and may be associated with Alzheimer disease. J Alzheimers Dis. 2004;6:639–649. doi: 10.3233/jad-2004-6608. discussion 673–681. [DOI] [PubMed] [Google Scholar]
- 131.Miklossy J. Chronic inflammation and amyloidogenesis in Alzheimer’s disease: Putative role of bacterial peptidoglycan, a potent inflammatory and amyloidogenic factor. Alzheimers Rev. 1998;3:45–51. [Google Scholar]
- 132.Miklossy J, Darekar P, Gern L, Janzer RC, Bosman FT. Bacterial peptidoglycan in neuritic plaque in Alzheimer’s disease. Alzheimers Res. 1996;2:95–100. [Google Scholar]
- 133.Ohnishi S, Koide A, Koide S. Solution conformation and amyloid-like fibril formation of a polar peptide derived from a beta-hairpin in the OspA single-layer beta-sheet. J Mol Biol. 2000;11:477–489. doi: 10.1006/jmbi.2000.3980. [DOI] [PubMed] [Google Scholar]
- 134.Miklossy J, Kasas S, Zurn AD, McCall S, Yu S, McGeer PL. Persisting atypical and cystic forms of Borrelia burgdorferi and local inflammation in Lyme neuroborreliosis. J Neuroinflammation. 2008;5:40. doi: 10.1186/1742-2094-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kamer AR, Craig RG, Pirraglia E, Dasanayake AP, Norman RG, Boylan RJ, Nehorayoff A, Glodzik L, Brys M, de Leon MJ. TNF-alpha and antibodies to periodontal bacteria discriminate between Alzheimer’s disease patients and normal subjects. J Neuroimmunol. 2009;216:92–97. doi: 10.1016/j.jneuroim.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Stewart R, Sabbah W, Tsakos G, D’Aiuto F, Watt RG. Oral health and cognitive function in the Third National Health and Nutrition Examination Survey (NHANES III) Psychosom Med. 2008;70:936–941. doi: 10.1097/PSY.0b013e3181870aec. [DOI] [PubMed] [Google Scholar]
- 137.Noble JM, Borrell LN, Papapanou PN, Elkind MS, Scarmeas N, Wright CB. Periodontitis is associated with cognitive impairment among older adults: Analysis of NHANES-III. J Neurol Neurosurg Psychiatry. 2009;80:1206–1211. doi: 10.1136/jnnp.2009.174029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kuipers EJ. Helicobacter pylori and the risk and management of associated diseases: Gastritis, ulcer disease, atrophic gastritis and gastric cancer. Aliment Pharmacol Ther. 1997;11(Suppl 1):71–88. doi: 10.1046/j.1365-2036.11.s1.5.x. [DOI] [PubMed] [Google Scholar]
- 139.NIH Consensus Conference. Helicobacter pylori in peptic ulcer disease. NIH Consensus Development Panel on Helicobacter pylori in Peptic Ulcer Disease. JAMA. 1994;272:65–69. [PubMed] [Google Scholar]
- 140.Papastergiou V, Georgopoulos SD, Karatapanis S. Treatment of Helicobacter pylori infection: Past, present and future. World J Gastrointest Pathophysiol. 2014;5:392–399. doi: 10.4291/wjgp.v5.i4.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.He C, Yang Z, Lu NH. Helicobacter pylori-an infectious risk factor for atherosclerosis? J Atheroscler Thromb. 2014;21:1229–1242. doi: 10.5551/jat.25775. [DOI] [PubMed] [Google Scholar]
- 142.Hahn M, Fennerty MB, Corless CL, Magaret N, Lieberman DA, Faigel DO. Noninvasive tests as a substitute for histology in the diagnosis of Helicobacter pylori infection. Gastrointest Endosc. 2000;52:20–26. doi: 10.1067/mge.2000.106686. [DOI] [PubMed] [Google Scholar]
- 143.Burette A. How (who?) and when to test or retest for H. pylori. Acta Gastroenterol Belg. 1998;61:336–343. [PubMed] [Google Scholar]
- 144.Kountouras J, Tsolaki M, Boziki M, Gavalas E, Zavos C, Stergiopoulos C, Kapetanakis N, Chatzopoulos D, Venizelos I. Association between Helicobacter pylori infection and mild cognitive impairment. Eur J Neurol. 2007;14:976–982. doi: 10.1111/j.1468-1331.2007.01827.x. [DOI] [PubMed] [Google Scholar]
- 145.Kountouras J, Tsolaki M, Gavalas E, Boziki M, Zavos C, Karatzoglou P, Chatzopoulos D, Venizelos I. Relationship between Helicobacter pylori infection and Alzheimer disease. Neurology. 2006;66:938–940. doi: 10.1212/01.wnl.0000203644.68059.5f. [DOI] [PubMed] [Google Scholar]
- 146.Roubaud Baudron C, Letenneur L, Langlais A, Buissonnière A, Mégraud F, Dartigues JF, Salles N. Personnes Agées QUID Study. Does Helicobacter pylori infection increase incidence of dementia? The Personnes Agées QUID Study. J Am Geriatr Soc. 2013;61:74–78. doi: 10.1111/jgs.12065. [DOI] [PubMed] [Google Scholar]
- 147.Kountouras J, Boziki M, Gavalas E, Zavos C, Deretzi G, Grigoriadis N, Tsolaki M, Chatzopoulos D, Katsinelos P, Tzilves D, Zabouri A, Michailidou I. Increased cerebrospinal fluid Helicobacter pylori antibody in Alzheimer’s disease. Int J Neurosci. 2009;119:765–777. doi: 10.1080/00207450902782083. [DOI] [PubMed] [Google Scholar]
- 148.Wang XL, Zeng J, Feng J, Tian YT, Liu YJ, Qiu M, Yan X, Yang Y, Xiong Y, Zhang ZH, Wang Q, Wang JZ, Liu R. Helicobacter pylori filtrate impairs spatial learning and memory in rats and increases β-amyloid by enhancing expression of presenilin-2. Front Aging Neurosci. 2014;6:66. doi: 10.3389/fnagi.2014.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wang XL, Zeng J, Yang Y, Xiong Y, Zhang ZH, Qiu M, Yan X, Sun XY, Tuo QZ, Liu R, Wang JZ. Helicobacter pylori filtrate induces Alzheimer-like tau hyperphosphorylation by activating glycogen synthase kinase-3β. J Alzheimers Dis. 2015;43:153–165. doi: 10.3233/JAD-140198. [DOI] [PubMed] [Google Scholar]
- 150.Romero-Adrián TB, Leal-Montiel J, Monsalve-Castillo F, Mengual-Moreno E, McGregor EG, Perini L, Antúnez A. Helicobacter pylori: Bacterial factors and the role of cytokines in the immune response. Curr Microbiol. 2010;60:143–155. doi: 10.1007/s00284-009-9518-4. [DOI] [PubMed] [Google Scholar]
- 151.Lagunes-Servin H, Torres J, Maldonado-Bernal C, Pérez-Rodríguez M, Huerta-Yépez S, Madrazo de la Garza A, Muñoz-Pérez L, Flores-Luna L, Ramón-García G, Camorlinga-Ponce M. Toll-like receptors and cytokines are upregulated during Helicobacter pylori infection in children. Helicobacter. 2013;18:423–432. doi: 10.1111/hel.12067. [DOI] [PubMed] [Google Scholar]
- 152.Kountouras J, Boziki M, Gavalas E, Zavos C, Grigoriadis N, Deretzi G, Tzilves D, Katsinelos P, Tsolaki M, Chatzopoulos D, Venizelos I. Eradication of Helicobacter pylori may be beneficial in the management of Alzheimer’s disease. J Neurol. 2009;256:758–767. doi: 10.1007/s00415-009-5011-z. [DOI] [PubMed] [Google Scholar]
- 153.Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer’s disease: The potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Griffin WS. Neuroinflammatory cytokine signaling and Alzheimer’s disease. N Engl J Med. 2013;368:770–771. doi: 10.1056/NEJMcibr1214546. [DOI] [PubMed] [Google Scholar]
- 155.Chami L, Checler F. BACE1 is at the crossroad of a toxic vicious cycle involving cellular stress and β-amyloid production in Alzheimer’s disease. Mol Neurodegener. 2012;7:52. doi: 10.1186/1750-1326-7-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 157.Cai Z, Hussain MD, Yan LJ. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int J Neurosci. 2014;124:307–321. doi: 10.3109/00207454.2013.833510. [DOI] [PubMed] [Google Scholar]
- 158.Floden AM, Li S, Combs CK. Beta-amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor alpha and NMDA receptors. J Neurosci. 2005;25:2566–2575. doi: 10.1523/JNEUROSCI.4998-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhao Y, Zhao B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid Med Cell Longe. 2013;2013:316523. doi: 10.1155/2013/316523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Katayama T, Imaizumi K, Manabe T, Hitomi J, Kudo T, Tohyama M. Induction of neuronal death by ER stress in Alzheimer’s disease. J Chem Neuroanat. 2004;28:67–78. doi: 10.1016/j.jchemneu.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 161.Choi Y, Kim HS, Shin KY, Kim EM, Kim M, Kim HS, Park CH, Jeong YH, Yoo J, Lee JP, Chang KA, Kim S, Suh YH. Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer’s disease models. Neuropsychopharmacology. 2007;32:2393–2404. doi: 10.1038/sj.npp.1301377. [DOI] [PubMed] [Google Scholar]
- 162.Parker WD., Jr Cytochrome oxidase deficiency in Alzheimer’s disease. Ann N Y Acad Sci. 1991;640:59–64. doi: 10.1111/j.1749-6632.1991.tb00191.x. [DOI] [PubMed] [Google Scholar]
- 163.Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta. 2014;1842:1240–1247. doi: 10.1016/j.bbadis.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, Moir RD. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5:e9505. doi: 10.1371/journal.pone.0009505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lukiw WJ, Cui JG, Yuan LY, Bhattacharjee PS, Corkern M, Clement C, Kammerman EM, Ball MJ, Zhao Y, Sullivan PM, Hill JM. Acyclovir or Aβ42 peptides attenuate HSV-1-induced miRNA-146a levels in human primary brain cells. Neuroreport. 2010;21:922–927. doi: 10.1097/WNR.0b013e32833da51a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wozniak MA, Frost AL, Preston CM, Itzhaki RF. Antivirals reduce the formation of key Alzheimer’s disease molecules in cell cultures acutely infected with Herpes simplex virus type 1. PLoS One. 2011;6:e25152. doi: 10.1371/journal.pone.0025152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lynch MA. The impact of neuroimmune changes on development of amyloid pathology; relevance to Alzheimer’s disease. Immunology. 2014;141:292–301. doi: 10.1111/imm.12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hickey WF. Migration of hematogenous cells through the blood-brain barrier and the initiation of CNS inflammation. Brain Pathol. 1991;1:97–105. doi: 10.1111/j.1750-3639.1991.tb00646.x. [DOI] [PubMed] [Google Scholar]
- 169.Hickey WF. Basic principles of immunological surveillance of the normal central nervous system. Glia. 2001;36:118–124. doi: 10.1002/glia.1101. [DOI] [PubMed] [Google Scholar]
- 170.Engelhardt B, Ransohoff RM. Capture, crawl, cross: The T cell code to breach the blood-brain barriers. Trends Immunol. 2012;33:579–589. doi: 10.1016/j.it.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 171.Rogers J, Luber-Narod J, Styren SD, Civin WH. Expression of immune system-associated antigens by cells of the human central nervous system: Relationship to the pathology of Alzheimer’s disease. Neurobiol Aging. 1988;9:339–349. doi: 10.1016/s0197-4580(88)80079-4. [DOI] [PubMed] [Google Scholar]
- 172.Itagaki S, McGeer PL, Akiyama H. Presence of T-cytotoxic suppressor and leucocyte common antigen positive cells in Alzheimer’s disease brain tissue. Neurosci Lett. 1988;91:259–264. doi: 10.1016/0304-3940(88)90690-8. [DOI] [PubMed] [Google Scholar]
- 173.Parachikova A, Agadjanyan MG, Cribbs DH, Blurton-Jones M, Perreau V, Rogers J, Beach TG, Cotman CW. Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol Aging. 2007;28:1821–1833. doi: 10.1016/j.neurobiolaging.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.McGeer PL, Akiyama H, Itagaki S, McGeer EG. Immune system response in Alzheimer’s disease. Can J Neurol Sci. 1989;16(4 Suppl):516–527. doi: 10.1017/s0317167100029863. [DOI] [PubMed] [Google Scholar]
- 175.Hartwig M. Immune ageing and Alzheimer’s disease. Neuroreport. 1995;6:1274–1276. doi: 10.1097/00001756-199506090-00011. [DOI] [PubMed] [Google Scholar]
- 176.Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, Kato M, Oda T, Tsuchiya K, Kosaka K. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J Neuroimmunol. 2002;124:83–92. doi: 10.1016/s0165-5728(01)00496-9. [DOI] [PubMed] [Google Scholar]
- 177.Town T, Tan J, Flavell RA, Mullan M. T-cells in Alzheimer’s disease. Neuromolecular Med. 2005;7:255–264. doi: 10.1385/NMM:7:3:255. [DOI] [PubMed] [Google Scholar]
- 178.Pirttilä T, Mattinen S, Frey H. The decrease of CD8-positive lymphocytes in Alzheimer’s disease. J Neurol Sci. 1992;107:160–165. doi: 10.1016/0022-510x(92)90284-r. [DOI] [PubMed] [Google Scholar]
- 179.Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT. Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: In vitro ERK1/2 activation and role in Alzheimer’s disease. J Neuroimmunol. 2000;108:227–235. doi: 10.1016/s0165-5728(00)00285-x. [DOI] [PubMed] [Google Scholar]
- 180.Aloisi F, De Simone R, Columba-Cabezas S, Penna G, Adorini L. Functional maturation of adult mouse resting microglia into an APC is promoted by granulocyte-macrophage colony-stimulating factor and interaction with Th1 cells. J Immunol. 2000;164:1705–1712. doi: 10.4049/jimmunol.164.4.1705. [DOI] [PubMed] [Google Scholar]
- 181.Séguin R, Biernacki K, Prat A, Wosik K, Kim HJ, Blain M, McCrea E, Bar-Or A, Antel JP. Differential effects of Th1 and Th2 lymphocyte supernatants on human microglia. Glia. 2003;42:36–45. doi: 10.1002/glia.10201. [DOI] [PubMed] [Google Scholar]
- 182.McQuillan K, Lynch MA, Mills KH. Activation of mixed glia by Abeta-specific Th1 and Th17 cells and its regulation by Th2 cells. Brain Behav Immun. 2010;24:598–607. doi: 10.1016/j.bbi.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 183.Kim HS, Whang SY, Woo MS, Park JS, Kim WK, Han IO. Sodium butyrate suppresses interferon-gamma-, but not lipopolysaccharide-mediated induction of nitric oxide and tumor necrosis factor-alpha in microglia. J Neuroimmunol. 2004;151:85–93. doi: 10.1016/j.jneuroim.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 184.Browne TC, McQuillan K, McManus RM, O’Reilly JA, Mills KH, Lynch MA. IFN-γ production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer’s disease. J Immunol. 2013;190:2241–2251. doi: 10.4049/jimmunol.1200947. [DOI] [PubMed] [Google Scholar]
- 185.Murphy AC, Lalor SJ, Lynch MA, Mills KH. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun. 2010;24:641–651. doi: 10.1016/j.bbi.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 186.Blasko I, Marx F, Steiner E, Hartmann T, Grubeck-Loebenstein B. TNFalpha plus IFNgamma induce the production of Alzheimer beta-amyloid peptides and decrease the secretion of APPs. FASEB J. 1999;13:63–68. doi: 10.1096/fasebj.13.1.63. [DOI] [PubMed] [Google Scholar]
- 187.Sy M, Kitazawa M, Medeiros R, Whitman L, Cheng D, Lane TE, Laferla FM. Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol. 2011;178:2811–2822. doi: 10.1016/j.ajpath.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bu XL, Yao XQ, Jiao SS, Zeng F, Liu YH, Xiang Y, Liang CR, Wang QH, Wang X, Cao HY, Yi X, Deng B, Liu CH, Xu J, Zhang LL, Gao CY, Xu ZQ, Zhang M, Wang L, Tan XL, Xu X, Zhou HD, Wang YJ. A study on the association between infectious burden and Alzheimer’s disease. Eur J Neurol. 2014. 10.1111/ene.12477 [Epub ahead of print] PubMed PMID: 24910016. [DOI] [PubMed]
- 189.Mahley RW, Rall SC., Jr Apolipoprotein E: Far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 190.Kuhlmann I, Minihane AM, Huebbe P, Nebel A, Rimbach G. Apolipoprotein E genotype and hepatitis C, HIV and herpes simplex disease risk: A literature review. Lipids Health Dis. 2010;9:8. doi: 10.1186/1476-511X-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Itzhaki RF, Wozniak MA. Herpes simplex virus type 1, apolipoprotein E, and cholesterol: A dangerous liaison in Alzheimer’s disease and other disorders. Prog Lipid Res. 2006;45:73–90. doi: 10.1016/j.plipres.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 192.Mahley RW, Ji ZS. Remnant lipoprotein metabolism: Key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res. 1999;40:1–16. [PubMed] [Google Scholar]
- 193.Koelle DM, Margaret A, Warren T, Schellenberg GD, Wald A. APOE genotype is associated with oral herpetic lesions but not genital or oral herpes simplex virus shedding. Sex Transm Infect. 2010;86:202–206. doi: 10.1136/sti.2009.039735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.WuDunn D, Spear PG. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J Virol. 1989;63:52–58. doi: 10.1128/jvi.63.1.52-58.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Gérard HC, Fomicheva E, Whittum-Hudson JA, Hudson AP. Apolipoprotein E4 enhances attachment of Chlamydophila (Chlamydia) pneumoniae elementary bodies to host cells. Microb Pathog. 2008;44:279–285. doi: 10.1016/j.micpath.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 196.Price DA, Bassendine MF, Norris SM, Golding C, Toms GL, Schmid ML, Morris CM, Burt AD, Donaldson PT. Apolipoprotein epsilon3 allele is associated with persistent hepatitis C virus infection. Gut. 2006;55:715–718. doi: 10.1136/gut.2005.079905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wozniak MA, Itzhaki RF, Faragher EB, James MW, Ryder SD, Irving WL. HCV Study Group. Apolipoprotein E-epsilon 4 protects against severe liver disease caused by hepatitis C virus. Hepatology. 2002;36:456–463. doi: 10.1053/jhep.2002.34745. [DOI] [PubMed] [Google Scholar]
- 198.Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc Natl Acad Sci U S A. 1999;96:12766–12771. doi: 10.1073/pnas.96.22.12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Davignon J, Gregg RE, Sing CF. Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis. 1988;8:1–21. doi: 10.1161/01.atv.8.1.1. [DOI] [PubMed] [Google Scholar]
- 200.Corder EH, Robertson K, Lannfelt L, Bogdanovic N, Eggertsen G, Wilkins J, Hall C. HIV-infected subjects with the E4 allele for APOE have excess dementia and peripheral neuropathy. Nat Med. 1998;4:1182–1184. doi: 10.1038/2677. [DOI] [PubMed] [Google Scholar]
- 201.Burt TD, Agan BK, Marconi VC, He W, Kulkarni H, Mold JE, Cavrois M, Huang Y, Mahley RW, Dolan MJ, McCune JM, Ahuja SK. Apolipoprotein (apo) E4 enhances HIV-1 cell entry in vitro, and the APOE epsilon4/epsilon4 genotype accelerates HIV disease progression. Proc Natl Acad Sci U S A. 2008;105:8718–8723. doi: 10.1073/pnas.0803526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gale SC, Gao L, Mikacenic C, Coyle SM, Rafaels N, Murray Dudenkov T, Madenspacher JH, Draper DW, Ge W, Aloor JJ, Azzam KM, Lai L, Blackshear PJ, Calvano SE, Barnes KC, Lowry SF, Corbett S, Wurfel MM, Fessler MB. APOɛ4 is associated with enhanced in vivo innate immune responses in human subjects. J Allergy Clin Immunol. 2014;134:127–134. doi: 10.1016/j.jaci.2014.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Fessler MB, Parks JS. Intracellular lipid flux and membrane microdomains as organizing principles in inflammatory cell signaling. J Immunol. 2011;187:1529–1535. doi: 10.4049/jimmunol.1100253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Okoro EU, Zhao Y, Guo Z, Zhou L, Lin X, Yang H. Apolipoprotein E4 is deficient in inducing macrophage ABCA1 expression and stimulating the Sp1 signaling pathway. PLoS One. 2012;7:e44430. doi: 10.1371/journal.pone.0044430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Miyakawa T, Kimura T, Hirata S, Fujise N, Ono T, Ishizuka K, Nakabayashi J. Role of blood vessels in producing pathological changes in the brain with Alzheimer’s disease. Ann N Y Acad Sci. 2000;903:46–54. doi: 10.1111/j.1749-6632.2000.tb06349.x. [DOI] [PubMed] [Google Scholar]
- 206.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, Harrington MG, Chui HC, Law M, Zlokovic BV. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296–302. doi: 10.1016/j.neuron.2014.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Dando SJ, Mackay-Sim A, Norton R, Currie BJ, St John JA, Ekberg JA, Batzloff M, Ulett GC, Beacham IR. Pathogens penetrating the central nervous system: Infection pathways and the cellular and molecular mechanisms of invasion. Clin Microbiol Rev. 2014;27:691–726. doi: 10.1128/CMR.00118-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Pan W, Stone KP, Hsuchou H, Manda VK, Zhang Y, Kastin AJ. Cytokine signaling modulates blood-brain barrier function. Curr Pharm Des. 2011;17:3729–3740. doi: 10.2174/138161211798220918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.van Sorge NM, Doran KS. Defense at the border: The blood-brain barrier versus bacterial foreigners. Future Microbiol. 2012;7:383–394. doi: 10.2217/fmb.12.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Minagar A, Alexander JS. Blood-brain barrier disruption in multiple sclerosis. Mult Scler. 2003;9:540–549. doi: 10.1191/1352458503ms965oa. [DOI] [PubMed] [Google Scholar]
- 212.Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA, Zlokovic BV. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab. 2015. 10.1038/jcbfm.2015.44 [Epub ahead of print] PubMed PMID: 25757756. [DOI] [PMC free article] [PubMed]
- 213.Zhang F, Jiang L. Neuroinflammation in Alzheimer’s disease. Neuropsychiatr Dis Treat. 2015;11:243–256. doi: 10.2147/NDT.S75546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Labus J, Häckel S, Lucka L, Danker K. Interleukin-1β induces an inflammatory response and the breakdown of the endothelial cell layer in an improved human THBMEC-based in vitro blood-brain barrier model. J Neurosci Methods. 2014;228:35–45. doi: 10.1016/j.jneumeth.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 215.Kim KS. Mechanisms of microbial traversal of the blood-brain barrier. Nat Rev Microbiol. 2008;6:625–634. doi: 10.1038/nrmicro1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Banks WA. Blood-brain barrier transport of cytokines: A mechanism for neuropathology. Curr Pharm Des. 2005;11:973–984. doi: 10.2174/1381612053381684. [DOI] [PubMed] [Google Scholar]
- 217.Simi A, Tsakiri N, Wang P, Rothwell NJ. Interleukin-1 and inflammatory neurodegeneration. Biochem Soc Trans. 2007;35:1122–1126. doi: 10.1042/BST0351122. [DOI] [PubMed] [Google Scholar]
- 218.Rosenberg GA. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009;8:205–216. doi: 10.1016/S1474-4422(09)70016-X. [DOI] [PubMed] [Google Scholar]
- 219.Kim YS, Joh TH. Matrix metalloproteinases, new insights into the understanding of neurodegenerative disorders. Biomol Ther (Seoul) 2012;20:133–143. doi: 10.4062/biomolther.2012.20.2.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Syvänen S, Eriksson J. Advances in PET imaging of P-glycoprotein function at the blood-brain barrier. ACS Chem Neurosci. 2013;4:225–237. doi: 10.1021/cn3001729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.van Assema DM, Lubberink M, Bauer M, van der Flier WM, Schuit RC, Windhorst AD, Comans EF, Hoetjes NJ, Tolboom N, Langer O, Müller M, Scheltens P, Lammertsma AA, van Berckel BN. Blood-brain barrier P-glycoprotein function in Alzheimer’s disease. Brain. 2012;135:181–189. doi: 10.1093/brain/awr298. [DOI] [PubMed] [Google Scholar]
- 222.Gheorghiu M, Enciu AM, Popescu BO, Gheorghiu E. Functional and molecular characterization of the effect of amyloid-β42 on an in vitro epithelial barrier model. J Alzheimers Dis. 2014;38:787–798. doi: 10.3233/JAD-122374. [DOI] [PubMed] [Google Scholar]
- 223.Burgmans S, van de Haar HJ, Verhey FR, Backes WH. Amyloid-β interacts with blood-brain barrier function in dementia: A systematic review. J Alzheimers Dis. 2013;35:859–873. doi: 10.3233/JAD-122155. [DOI] [PubMed] [Google Scholar]
- 224.Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Smith JP, Weller S, Johnson B, Nicotera J, Luther JM, Haas DW. Pharmacokinetics of acyclovir and its metabolites in cerebrospinal fluid and systemic circulation after administration of high-dose valacyclovir in subjects with normal and impaired renal function. Antimicrob Agents Chemother. 2010;54:1146–1151. doi: 10.1128/AAC.00729-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lycke J, Malmeström C, Ståhle L. Acyclovir levels in serum and cerebrospinal fluid after oral administration of valacyclovir. Antimicrob Agents Chemother. 2003;47:2438–2441. doi: 10.1128/AAC.47.8.2438-2441.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Pouplin T, Pouplin JN, Van Toi P, Lindegardh N, Rogier van Doorn H, Hien TT, Farrar J, Török ME, Chau TT. Valacyclovir for herpes simplex encephalitis. Antimicrob Agents Chemother. 2011;55:3624–3626. doi: 10.1128/AAC.01023-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Friedman JE, Zabriskie JB, Plank C, Ablashi D, Whitman J, Shahan B, Edgell R, Shieh M, Rapalino O, Zimmerman R, Sheng D. A randomized clinical trial of valacyclovir in multiple Sclerosis. Mult Scler. 2005;11:286–295. doi: 10.1191/1352458505ms1185oa. [DOI] [PubMed] [Google Scholar]
- 229.Tyring SK, Baker D, Snowden W. Valacyclovir for herpes simplex virus infection: Long-term safety and sustained efficacy after 20 years’ experience with acyclovir. J Infect Dis. 2002;186(Suppl 1):S40–S46. doi: 10.1086/342966. [DOI] [PubMed] [Google Scholar]
- 230.Prasad KM, Eack SM, Keshavan MS, Yolken RH, Iyengar S, Nimgaonkar VL. Antiherpes virus-specific treatment and cognition in schizophrenia: A test-of-concept randomized double-blind placebo-controlled trial. Schizophr Bull. 2013;39:857–866. doi: 10.1093/schbul/sbs040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Magga J, Puli L, Pihlaja R, Kanninen K, Neulamaa S, Malm T, Härtig W, Grosche J, Goldsteins G, Tanila H, Koistinaho J, Koistinaho M. Human intravenous immunoglobulin provides protection against Aβ toxicity by multiple mechanisms in a mouse model of Alzheimer’s disease. J Neuroinflammation. 2010;7:90. doi: 10.1186/1742-2094-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Dodel RC, Du Y, Depboylu C, Hampel H, Frölich L, Haag A, Hemmeter U, Paulsen S, Teipel SJ, Brettschneider S, Spottke A, Nölker C, Möller HJ, Wei X, Farlow M, Sommer N, Oertel WH. Intravenous immunoglobulins containing antibodies against beta-amyloid for the treatment of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2004;75:1472–1474. doi: 10.1136/jnnp.2003.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Relkin NR, Szabo P, Adamiak B, Burgut T, Monthe C, Lent RW, Younkin S, Younkin L, Schiff R, Weksler ME. 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging. 2009;30:1728–1736. doi: 10.1016/j.neurobiolaging.2007.12.021. [DOI] [PubMed] [Google Scholar]
- 234.Relkin N. Intravenous immunoglobulin for Alzheimer’s disease. Clin Exp Immunol. 2014;178(Suppl 1):27–29. doi: 10.1111/cei.12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Kohl S, Loo LS. In vitro and in vivo antibody-dependent cellular cytotoxicity of intravenous immunoglobulin G preparations against herpes simplex virus. Rev Infect Dis. 1986;8(Suppl 4):S446–S448. doi: 10.1093/clinids/8.supplement_4.s446. [DOI] [PubMed] [Google Scholar]
- 236.Erlich KS, Mills J. Passive immunotherapy for encephalitis caused by herpes simplex virus. Rev Infect Dis. 1986;8(Suppl 4):S439–S445. doi: 10.1093/clinids/8.supplement_4.s439. [DOI] [PubMed] [Google Scholar]
- 237.Masci S, De Simone C, Famularo G, Gravante M, Ciancarelli M, Andreassi M, Amerio P, Santini G. Intravenous immunoglobulins suppress the recurrences of genital herpes simplex virus: A clinical and immunological study. Immunopharmacol Immunotoxicol. 1995;17:33–47. doi: 10.3109/08923979509052718. [DOI] [PubMed] [Google Scholar]
- 238.Wozniak MA, Itzhaki RF. Intravenous immunoglobulin reduces β amyloid and abnormal tau formation caused by herpes simplex virus type 1. J Neuroimmunol. 2013;257:7–12. doi: 10.1016/j.jneuroim.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 239.Lin WR, Jennings R, Smith TL, Wozniak MA, Itzhaki RF. Vaccination prevents latent HSV1 infection of mouse brain. Neurobiol Aging. 2001;22:699–703. doi: 10.1016/s0197-4580(01)00239-1. [DOI] [PubMed] [Google Scholar]
- 240.Field HJ, Vere Hodge RA. Recent developments in anti-herpesvirus drugs. Br Med Bull. 2013;106:213–249. doi: 10.1093/bmb/ldt011. [DOI] [PubMed] [Google Scholar]
- 241.Lancini D, Faddy HM, Flower R, Hogan C. Cytomegalovirus disease in immunocompetent adults. Med J Aust. 2014;201:578–580. doi: 10.5694/mja14.00183. [DOI] [PubMed] [Google Scholar]
- 242.Cohen JI. Optimal treatment for chronic active Epstein-Barr virus disease. Pediatr Transplant. 2009;13:393–396. doi: 10.1111/j.1399-3046.2008.01095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Prichard MN, Whitley RJ. The development of new therapies for human herpesvirus 6. Curr Opin Virol. 2014;9:148–153. doi: 10.1016/j.coviro.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Nau R, Sörgel F, Eiffert H. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin Microbiol Rev. 2010;23:858–883. doi: 10.1128/CMR.00007-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Yamaguchi H, Friedman H, Yamamoto M, Yasuda K, Yamamoto Y. Chlamydia pneumoniae resists antibiotics in lymphocytes. Antimicrob Agents Chemother. 2003;47:1972–1975. doi: 10.1128/AAC.47.6.1972-1975.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Carter JD, Espinoza LR, Inman RD, Sneed KB, Ricca LR, Vasey FB, Valeriano J, Stanich JA, Oszust C, Gerard HC, Hudson AP. Combination antibiotics as a treatment for chronic Chlamydia-induced reactive arthritis: A double-blind, placebo-controlled, prospective trial. Arthritis Rheum. 2010;62:1298–1307. doi: 10.1002/art.27394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Sadarangani SP, Estes LL, Steckelberg JM. Non-anti-infective effects of antimicrobials and their clinical applications: A review. Mayo Clin Proc. 2015;90:109–127. doi: 10.1016/j.mayocp.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 248.Kim HS, Suh YH. Minocycline and neurodegenerative diseases. Behav Brain Res. 2009;196:168–179. doi: 10.1016/j.bbr.2008.09.040. [DOI] [PubMed] [Google Scholar]
- 249.Forloni G, Colombo L, Girola L, Tagliavini F, Salmona M. Anti-amyloidogenic activity of tetracyclines: Studies in vitro. FEBS Lett. 2001;487:404–407. doi: 10.1016/s0014-5793(00)02380-2. [DOI] [PubMed] [Google Scholar]
- 250.Nau R, Prange HW, Menck S, Kolenda H, Visser K, Seydel JK. Penetration of rifampicin into the cerebrospinal fluid of adults with uninflamed meninges. J Antimicrob Chemother. 1992;29:719–724. doi: 10.1093/jac/29.6.719. [DOI] [PubMed] [Google Scholar]
- 251.Qosa H, Abuznait AH, Hill RA, Kaddoumi A. Enhanced brain amyloid-β clearance by rifampicin and caffeine as a possible protective mechanism against Alzheimer’s disease. J Alzheimers Dis. 2012;31:151–165. doi: 10.3233/JAD-2012-120319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Loeb MB, Molloy DW, Smieja M, Standish T, Goldsmith CH, Mahony J, Smith S, Borrie M, Decoteau E, Davidson W, McDougall A, Gnarpe J, O’DONNell M, Chernesky M. A randomized, controlled trial of doxycycline and rifampin for patients with Alzheimer’s disease. J Am Geriatr Soc. 2004;52:381–387. doi: 10.1111/j.1532-5415.2004.52109.x. [DOI] [PubMed] [Google Scholar]
- 253.Molloy DW, Standish TI, Zhou Q, Guyatt G. DARAD Study Group. A multicenter, blinded, randomized, factorial controlled trial of doxycycline and rifampin for treatment of Alzheimer’s disease: The DARAD trial. Int J Geriatr Psychiatry. 2013;28:463–470. doi: 10.1002/gps.3846. [DOI] [PubMed] [Google Scholar]
- 254.Borg R, Dotevall L, Hagberg L, Maraspin V, Lotric-Furlan S, Cimperman J, Strle F. Intravenous ceftriaxone compared with oral doxycycline for the treatment of Lyme neuroborreliosis. Scand J Infect Dis. 2005;37:449–454. doi: 10.1080/00365540510027228. [DOI] [PubMed] [Google Scholar]
- 255.Ljøstad U, Skogvoll E, Eikeland R, Midgard R, Skarpaas T, Berg A, Mygland A. Oral doxycycline versus intravenous ceftriaxone for European Lyme neuroborreliosis: A multicentre, non-inferiority, double- blind, randomised trial. Lancet Neurol. 2008;7:690–695. doi: 10.1016/S1474-4422(08)70119-4. [DOI] [PubMed] [Google Scholar]
- 256.Abramson IJ, Smibert RM. Bactericidal activity of antimicrobial agents for treponemes. Br J Vener Dis. 1971;47:413–418. doi: 10.1136/sti.47.6.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Kikutani T, Yoneyama T, Nishiwaki K, Tamura F, Yoshida M, Sasaki H. Effect of oral care on cognitive function in patients with dementia. Geriatr Gerontol Int. 2010;10:327–328. doi: 10.1111/j.1447-0594.2010.00637.x. [DOI] [PubMed] [Google Scholar]
- 258.Feres M, Figueiredo LC, Soares GM. Faveri M. Systemic antibiotics in the treatment of periodontitis. Periodontol 2000. 2015;67:131–186. doi: 10.1111/prd.12075. [DOI] [PubMed] [Google Scholar]
- 259.Feres M, Haffajee AD, Allard K, Som S, Socransky SS. Change in subgingival microbial profiles in adult periodontitis subjects receiving either systemically-administered amoxicillin or metronidazole. J Clin Periodontol. 2001;28:597–609. doi: 10.1034/j.1600-051x.2001.028007597.x. [DOI] [PubMed] [Google Scholar]
- 260.Haffajee AD, Patel M, Socransky SS. Microbiological changes associated with four different periodontal therapies for the treatment of chronic periodontitis. Oral Microbiol Immunol. 2008;23:148–157. doi: 10.1111/j.1399-302X.2007.00403.x. [DOI] [PubMed] [Google Scholar]
- 261.Haffajee AD, Torresyap G, Socransky SS. Clinical changes following four different periodontal therapies for the treatment of chronic periodontitis: 1-year results. J Clin Periodontol. 2007;34:243–253. doi: 10.1111/j.1600-051X.2006.01040.x. [DOI] [PubMed] [Google Scholar]
- 262.Oteo A, Herrera D, Figuero E, O’Connor A, González I, Sanz M. Azithromycin as an adjunct to scaling and root planing in the treatment of Porphyromonas gingivalis-associated periodontitis: A pilot study. J Clin Periodontol. 2010;37:1005–1015. doi: 10.1111/j.1600-051X.2010.01607.x. [DOI] [PubMed] [Google Scholar]
- 263.Dastoor SF, Travan S, Neiva RF, Rayburn LA, Giannobile WV, Wang HL. Effect of adjunctive systemic azithromycin with periodontal surgery in the treatment of chronic periodontitis in smokers: A pilot study. J Periodontol. 2007;78:1887–1896. doi: 10.1902/jop.2007.070072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Feres M, Haffajee AD, Allard K, Som S, Goodson JM, Socransky SS. Antibiotic resistance of subgingival species during and after antibiotic therapy. J Clin Periodontol. 2002;29:724–735. doi: 10.1034/j.1600-051x.2002.290809.x. [DOI] [PubMed] [Google Scholar]
- 265.O’Connor A, Gisbert JP, McNamara D, O’Morain C. Treatment of Helicobacter pylori infection 2011. Helicobacter. 2011;16(Suppl 1):53–58. doi: 10.1111/j.1523-5378.2011.00881.x. [DOI] [PubMed] [Google Scholar]
- 266.Graham DY. Helicobacter pylori Update: Gastric Cancer, Reliable Therapy, and Possible Benefits. Gastroenterology. 2015;148:719–731. doi: 10.1053/j.gastro.2015.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Kountouras J, Boziki M, Gavalas E, Zavos C, Deretzi G, Chatzigeorgiou S, Katsinelos P, Grigoriadis N, Giartza-Taxidou E, Venizelos I. Five-year survival after Helicobacter pylori eradication in Alzheimer disease patients. Cogn Behav Neurol. 2010;23:199–204. doi: 10.1097/WNN.0b013e3181df3034. [DOI] [PubMed] [Google Scholar]
- 268.Yolken R. Viruses and schizophrenia: A focus on herpes simplex virus. Herpes. 2004;11(Suppl 2):83A–88A. [PubMed] [Google Scholar]
- 269.Taller AM, Asher DM, Pomeroy KL, Eldadah BA, Godec MS, Falkai PG, Bogert B, Kleinman JE, Stevens JR, Torrey EF. Search for viral nucleic acid sequences in brain tissues of patients with schizophrenia using nested polymerase chain reaction. Arch Gen Psychiatry. 1996;53:32–40. doi: 10.1001/archpsyc.1996.01830010034006. [DOI] [PubMed] [Google Scholar]
- 270.Itzhaki RF, Wozniak MA. Could antivirals be used to treat Alzheimer’s disease? Future Microbiol. 2012;7:307–309. doi: 10.2217/fmb.12.10. [DOI] [PubMed] [Google Scholar]
- 271.Baringer JR, Pisani P. Herpes simplex virus genomes in human nervous system tissue analyzed by polymerase chain reaction. Ann Neurol. 1994;36:823–829. doi: 10.1002/ana.410360605. [DOI] [PubMed] [Google Scholar]
- 272.Gordon L, McQuaid S, Cosby SL. Detection of herpes simplex virus (types 1 and 2) and human herpesvirus 6 DNA in human brain tissue by polymerase chain reaction. Clin Diagn Virol. 1996;6:33–40. doi: 10.1016/0928-0197(95)00203-0. [DOI] [PubMed] [Google Scholar]
- 273.Bertrand P, Guillaume D, Hellauer L, Dea D, Lindsay J, Kogan S, gauthier s, Poirier J. Distribution of herpes simplex virus type 1 DNA in selected areas of normal and Alzheimer’s disease brains: A PCR study. Neurodegeneration. 1993;2:201–208. [Google Scholar]
- 274.Cheon MS, Bajo M, Gulesserian T, Cairns N, Lubec G. Evidence for the relation of herpes simplex virus type1 to Down syndrome and Alzheimer’s disease. Electrophoresis. 2001;22:445–448. doi: 10.1002/1522-2683(200102)22:3<445::AID-ELPS445>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]