

Abstract
Plasticity is a universal property of synapses. It is expressed in a variety of forms mediated by a multitude of mechanisms. Here we consider two broad kinds of plasticity that differ in their requirement for presynaptic activity during the induction. Homosynaptic plasticity occurs at synapses that were active during the induction. It is also called input specific or associative, and it is governed by Hebbian-type learning rules. Heterosynaptic plasticity can be induced by episodes of strong postsynaptic activity also at synapses that were not active during the induction, thus making any synapse at a cell a target to heterosynaptic changes. Both forms can be induced by typical protocols used for plasticity induction and operate on the same time scales but have differential computational properties and play different roles in learning systems. Homosynaptic plasticity mediates associative modifications of synaptic weights. Heterosynaptic plasticity counteracts runaway dynamics introduced by Hebbian-type rules and balances synaptic changes. It provides learning systems with stability and enhances synaptic competition. We conclude that homosynaptic and heterosynaptic plasticity represent complementary properties of modifiable synapses, and both are necessary for normal operation of neural systems with plastic synapses.
Keywords: synaptic plasticity, runaway dynamics, weight normalization, heterosynaptic plasticity, neocortex, neuron model
Introduction
Plasticity is a universal property of synapses. It is expressed in a variety of forms mediated by a multitude of mechanisms. In this review, we will consider two broad kinds of plasticity that differ in their requirement for presynaptic activity during the induction. Homosynaptic plasticity occurs at synapses that were directly involved in activation of a cell during the induction. Induction of homosynaptic plasticity at a synapse requires its presynaptic activation. This form of plasticity is also called input specific or associative. Heterosynaptic plasticity can be induced at synapses that were not active during the induction. Thus, any synapse at a cell can be subject to heterosynaptic plasticity after episodes of strong postsynaptic activity. Both forms of plasticity can be induced by typical protocols used for plasticity induction. The two forms of plasticity have differential computational properties and play different roles in learning systems. Homosynaptic and heterosynaptic changes represent complementary forms of plasticity, both necessary for normal operation of neural systems with plastic synapses.
Synaptic Plasticity: Homosynaptic and Heterosynaptic
Plasticity is a universal property of synapses, vital for fundamental operations of the nervous system. Synaptic plasticity mediates formation and refinement of connectivity patterns in the nervous system during development and effective adaptive behavior in changing environment throughout life. Hebb’s rule (Hebb 1949) formulates the basic principle according to which synaptic weights change: synaptic connections that repeatedly or persistently take part in firing of the postsynaptic neuron increase their strength. The original rule, suggested only for potentiation of synaptic connections, was later on extended to include depression: synapses that consistently do not take part in firing the postsynaptic neuron decrease their strength. After the discovery of the cellular basis of associative learning, the long-term potentiation (LTP) in the hippocampal formation (Bliss and Lomo 1973), the associativity rule has been complemented by the rules of cooperativity and input specificity (Bliss and Collingridge 1993). The rule of cooperativity states that activation should be strong, exceeding a certain threshold, to induce plastic changes. The rule of input specificity maintains that changes take place only at synapses that were activated during the induction but not at other synapses. Altogether, these Hebbian-type learning rules capture the associative nature of synaptic changes and explain a multitude of plastic phenomena in the nervous system, ranging from refinement of connectivity during development (“neurons that fire together wire together”) to extraction of casual relations between events in the environment in Pavlovian conditioning and other types of associative learning, motor learning, and acquisition of sequences of behavioral actions.
Spike-timing dependent plasticity (STDP) explicitly implements relative timing between firing of the neuron and activity at its inputs (presynaptic spikes) as a determinant of the direction and the magnitude of synaptic weight changes. In the STDP learning rule, synaptic inputs that were active shortly before a postsynaptic action potential (pre before post) are potentiated, whereas synaptic inputs active shortly after the postsynaptic spike (post before pre) are depressed (Abbott and Nelson 2000; Caporale and Dan 2008; Magee and Johnston 1997; Markram and others 1997). Generalization of this rule includes models that take into account effects of multiple presynaptic and one postsynaptic spikes (Gjorgjieva and others 2011; Pfister and Gerstner 2006) but the main principle remains—presynaptic activation is required to induce changes.
STDP, and Hebbian-type learning rules in general, extract casual relations between events by using temporal order of their occurrence. This dictates the necessity of activity of both presynaptic and postsynaptic neurons for plasticity induction. Furthermore, the rule of input specificity explicitly restricts plasticity induction to synaptic inputs that were active during the induction. Thus, plasticity governed by Hebbian-type learning is input-specific, or homosynaptic (Fig. 1).
Figure 1.

Homosynaptic and heterosynaptic plasticity. In a typical plasticity experiment, a set of inputs to a neuron is stimulated to induce plasticity, for example, at high frequency to induce potentiation, or a low frequency to induce depression, or paired with bursts of postsynaptic spikes. Homosynaptic plasticity refers to changes of transmission at synapses that were activated during the induction (red inputs). Heterosynaptic plasticity—changes at synapses that were not active during the induction (black inputs, green question marks).
Functional Roles for Homosynaptic Plasticity
Synaptic plasticity governed by Hebbian-type learning rules mediates formation and refinement of neuronal connectivity during development and thus plays a pivotal role in establishing the hardware for future neuronal computations. Similar mechanisms may be involved in trauma-induced compensatory and repair processes. During the whole life, Hebbian-type plasticity mediates a multitude of processes underlying the abilities of organisms to adapt their behavior to the changing environment. Such processes include learning of specific and repeatedly occurring in the environment relations between sensory stimuli and behaviorally relevant events, associations between sensory stimuli, learning of motor programs, and sequences of behavioral actions. Ultimately, homosynaptic plasticity is believed to provide a cellular mechanism underlying processes of new memory formation and learning.
Why Heterosynaptic Plasticity Is Required in Addition to Hebbian-Type Learning Rules
However, learning systems in which all plastic changes are governed by Hebbian-type learning rules have two major drawbacks. First, Hebbian-type learning rules have intrinsic positive feedback. Potentiation, by making synapses stronger, increases their chances to take part in firing a neuron, and thus increases probability of these synapses to be potentiated further. Similarly, synapses weakened by depression have a lower chance to lead to spikes, and thus increased probability for being further depressed. Because of this positive feedback loop the synaptic weights tend to be either potentiated to the maximal value or depressed to zero. The runaway of synaptic weights leads to overexcitability or silencing of neurons, which compromises computational abilities of neuronal networks by disturbing input–output relations of neurons and inducing runaway activity. Second, Hebbian-type learning rules introduce only a weak degree of competition between synapses. Although synaptic weights can change in both directions, these changes require repetition of specific patterns of the input activity. For two groups of inputs to change in opposite directions, both groups should be repeatedly activated either with different patterns, low frequency for induction of long-term depression (LTD) but high frequency for induction of LTP, or be systematically active at different timing relative to postsynaptic activity to change by STDP mechanism. Although possible in theory, such a scenario imposes strict requirements on the relations between specifics of plasticity rules and patterns of input activity.
The need for additional mechanisms that maintain stability of synaptic weights and neuronal activity, and support strong synaptic competition in learning systems with Hebbian-type rules, has been appreciated since early theoretical studies (Miller 1996; Miller and MacKay 1993; Von der Malsburg 1973). The requirements for both stabilization and competition can be solved by introducing heterosynaptic plasticity—changes at synapses that were not active during plasticity induction (Fig. 1). This can be achieved by normalization: after a weight change at any synapse, all synaptic weights are normalized so that their total remains constant (Von der Malsburg 1973). This mechanism, although not eliminating the possibility of saturating potentiation or depression of an individual synapse, effectively prevents runaway of activity, and introduces synaptic competition that does not require repetition of a specialized LTD-inducing activity pattern at a synapse to decrease its weight. Later studies elaborated the use of normalization as mechanism of stability and synaptic competition (e.g., Elliott and Shadbolt 2002; Finelli and others 2008; Kempter and others 2001; Wu and Yamaguchi 2006) and further corroborated the requirement for stabilizing and balancing mechanisms in STDP-based models of learning and decision making in the framework of the complex tasks (Skorheim and others 2014). A recent theoretical study by Zenke and others (2013) established one further requirement for heterosynaptic plasticity: to be able to prevent runaway dynamics in neuronal networks and achieve robust stability of their operation, it should operate on the time scale of seconds to minutes, similar to the time scale on which homosynaptic plasticity is induced.
Thus, whereas the necessity of heterosynaptic plasticity to solve the issues associated with homosynaptic plasticity has been recognized since early theoretical studies, those normalization rules remained a theoretical concept until discovery of the heterosynaptic LTD—a form of plasticity involving synapses that were not activated presynaptically—in the hippocampus (Lynch and others 1977).
Experimental Evidence for Heterosynaptic Plasticity
Heterosynaptic LTD accompanying induction of LTP has been first described in the hippocampus shortly after the phenomenon of LTP was discovered (Fig. 2A; Lynch and others 1977). Induction of LTP at inputs to apical dendrites of CA1 pyramidal neurons, formed by Schaffer collateral-commissural fibers, was accompanied by an LTD at inputs to basal dendrites made by commissural fibers that were not stimulated during the induction. And vice versa, induction of LTP at the inputs to the basal dendrites was accompanied by LTD of inputs to the apical dendrites. Because of the clear separation of the presynaptic fibers and synapses mediating the two groups of inputs, this work presented a strong case for heterosynaptic plasticity. Heterosynaptic LTD accompanying the induction of homosynaptic LTP clearly has potential for both balancing plastic changes and supporting synaptic competition.
Figure 2. Experimental evidence for heterosynaptic plasticity.

(A) Heterosynaptic LTD in the hippocampus (Lynch and others 1977, field potential recording). Induction of LTP at Schaffer collateral-commissural inputs to apical dendrites of CA1 pyramidal neurons was accompanied by an LTD at commissural inputs to basal dendrites that were not stimulated during the induction. And vice versa, induction of LTP at inputs to basal dendrites was accompanied by LTD of inputs to the apical dendrites. (B) Distributed plasticity and breakage of input specificity at short distances (Bonhoeffer and others 1989; Engert and Bonhoeffer 1997; Kossel and others 1990; Schuman and Madison 1994). After pairing of one input to a CA1 neuron, synapses formed by nearby fibers on that cell and even on nearby neurons were potentiated too (pink circle). (C) Mexican hat profile of plasticity (Royer and Paré 2003; White and others 1990). In structures with regular organization of the inputs, such as the hippocampus or amygdala, induction of LTP was accompanied by a weaker LTP at nearby inputs (pink circle) and heterosynaptic LTD at more distant inputs (green annulus), with the amplitude of LTD decreasing with distance. Symmetrical profile was observed around the site of LTD induction. This resulted in a Mexican hat profile of plasticity, with balanced potentiation and depression (Royer and Paré 2003). (D) Long-term plastic changes can be induced by rises of intracellular calcium concentration produced by photolysis of caged calcium (Neveu and Zucker 1996; Yang and others 1999). LTP could be induced by large increases of intracellular calcium, and LTD by smaller amplitude prolonged increases.
Heterosynaptic LTP was first discovered at synapses adjacent to potentiated inputs (Fig. 2B). After pairing of one input to a CA1 neuron, synapses formed by nearby fibers on that cell and even on nearby neurons were potentiated too (Bonhoeffer and others 1989; Engert and Bonhoeffer 1997; Kossel and others 1990; Schuman and Madison 1994). These results demonstrated that input specificity breaks down at short distances: LTP protocols induce plasticity not only at the activated synapses but also at those not active during the induction.
Further studies of the spatial distribution of plastic changes induced in structures with regular organization of the inputs, such as the hippocampus or amygdala, revealed a Mexican hat type profile of plasticity (Fig. 2C; Royer and Paré 2003; White and others 1990). Induction of LTP at a set of synapses was accompanied by a biphasic profile of heterosynaptic changes: a weaker LTP at nearby inputs, LTD at more distant inputs, and finally no changes at yet more distantly located synapses. A symmetrical profile was observed around the site of LTD induction: weaker LTD at close distances and LTP at more distant inputs (Royer and Paré 2003). Importantly, the spatial profiles of plastic changes were balanced, so that neither LTP nor LTD induction at activated synapses resulted in net changes of the total synaptic input. Induction of balanced profiles of plastic changes can provide a powerful local mechanism of both normalization of synaptic weights and synaptic competition.
Heterosynaptic plasticity can be induced by purely postsynaptic protocols that do not involve any presynaptic stimulation during the induction. Long-term plasticity can be induced by rises of intracellular calcium concentration produced by photolysis of caged calcium (Fig. 2D; Neveu and Zucker 1996; Yang and others 1999). The direction of plastic changes in these experiments depended on the dynamics of intracellular calcium signal: large increases of [Ca2+]in could induce LTP, and smaller amplitude prolonged [Ca2+]in increases could lead to LTD.
Long-term plastic changes can be also induced by intracellular tetanization—bursts of spikes evoked by short depolarizing pulses applied through the recording electrode (Fig. 3A; Chistiakova and Volgushev 2009; Kuhnt and others 1994; Lee and others 2012; Volgushev and others 1994; Volgushev and others 1997; Volgushev and others 1999; Volgushev and others 2000; see also references to work from other labs in the section on mechanisms of heterosynaptic plasticity). The rationale behind the intracellular tetanization protocol is the following. Each neuron in the neocortex receives thousands of synaptic inputs. Repetitive activation of a fraction of these inputs, few dozens to hundreds, can lead to repetitive firing of a cell and under certain conditions induce synaptic plasticity. During the induction, all synapses but for those of the activated fraction will experience postsynaptic activity without activation of their presynaptic fibers. This situation, postsynaptic activity without presynaptic activation, is mimicked by the intracellular tetanization. Because no synaptic inputs were stimulated during the intracellular tetanization, any synaptic changes induced can be considered heterosynaptic.
Figure 3.

Long-term synaptic plasticity induced by intracellular tetanization. (A) A scheme of intracellular tetanization experiment. Bursts of short depolarizing pulses (5 pulses at 100 Hz; 10 bursts at 1 Hz, 3 trains of 10 bursts) were applied through the recording electrode without presynaptic stimulation to induce bursts of action potentials. Synaptic responses were recorded before and after the intracellular tetanization. Because no inputs were stimulated during the induction, plasticity at all synapses can be considered heterosynaptic. (B) Examples of inputs that underwent potentiation (top), depression (middle), or did not change after intracellular tetanization. Time courses of amplitudes of EPSPs evoked by the first pulse in a paired-pulse paradigm. The timing of intracellular tetanization is indicated by the arrows above each plot. Insets show averaged responses to paired pulse stimuli before and after intracellular tetanization, from color-coded time intervals. In this example, LTP and LTD were induced simultaneously at two inputs to the same neuron. Note that input resistance of neurons measured by responses to small hyperpolarizing pulses applied before synaptic stimuli remained unchanged. (C) Correlation between changes of EPSP amplitude after intracellular tetanization and initial paired-pulse ratio. Data for N = 136 inputs to pyramidal neurons in slices of visual cortex (N = 60 inputs) and auditory cortex (N = 76 inputs). Green symbols (star, square, and triangle) refer to the example inputs from B (Modified, with permission, from Chen and others 2013b).
Figure 3B shows examples of long-term plastic changes induced by intracellular tetanization in pyramidal neurons from slices of rat visual cortex. Amplitudes of synaptic responses could increase, decrease, or do not change after tetanization. In the illustrated example, intracellular tetanization simultaneously induced LTP in one input and LTD in the other (top and middle plots in Fig. 3B). The direction of plastic changes was correlated with initial paired-pulse ratio, which is inversely related to the release probability (Fig. 3C; Chen and others 2013b; Lee and others 2012; Volgushev and others 1997; Volgushev and others 2000). Inputs that expressed high initial paired-pulse ratio, and thus had low release probability, were typically potentiated. Inputs that expressed low initial paired-pulse ratio, and thus had high release probability, were typically depressed or did not change. We have suggested that the weight-dependence of plasticity reflected history-dependent predispositions of synaptic inputs to undergo potentiation or depression (Chistiakova and Volgushev 2009; Volgushev and others 1997; Volgushev and others 2000). Weak synaptic inputs with low release probability, for example, because they underwent depression in the past, have a stronger predisposition for potentiation. Strong synapses with high release probability, such as those recently potentiated, have higher predisposition for depression. The notion of predisposition of synapses for plastic changes is closely related to the ideas of sliding threshold between depression and potentiation in the BCM rule (Bienenstock and others 1982; Yeung and others 2004) and metaplasticity—history dependent changes of the abilities of synapses to undergo potentiation or depression (Abraham and Bear 1996). One further parallel to properties of homosynaptic plasticity is the dependence of the magnitude of plastic changes induced by intracellular tetanization on initial paired-pulse ratio, which is indicative of initial synaptic weight. Weight dependence of synaptic plasticity has been suggested and analyzed in a theoretical study by Oja (1982). In this learning rule, the magnitude of potentiation is larger when synaptic weight is small, but decreases, eventually reaching zero, for stronger synapses. Weight-dependence of potentiation has been also reported for tetanization or pairing induced LTP in the hippocampus and neocortex (Hardingham and others 2007; Sjöström and others 2001; Van Rossum and others 2000; also our data—see Fig. 7). Further evidence for heterosynaptic plasticity is discussed below, in the sections on induction and mechanisms of heterosynaptic plasticity.
Figure 7.
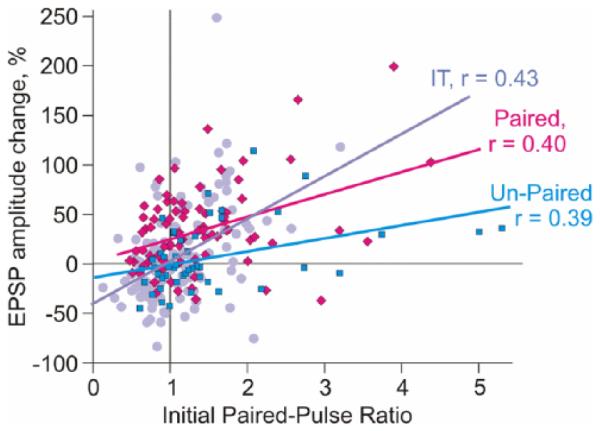
Homosynaptic and heterosynaptic plasticity induced by pairing and by intracellular tetanization. Correlation between long-term changes of EPSP amplitude and initial paired-pulse ratio for three groups of inputs. First, paired inputs (N = 81, magenta, diamond symbols): plasticity was induced by 50 pairings (1/s) of EPSPs with bursts of 5 action potentials produced by injection of short depolarizing current steps (5 ms duration at 100 Hz). During pairing, first spike in the burst was generated ~10 ms after the onset of the EPSP. Second, unpaired inputs (N = 50, cyan, square symbols): simultaneously recorded inputs to the same cells, but no synaptic responses were evoked during the pairing procedure. Thus, the unpaired inputs experienced the same postsynaptic firing as the paired inputs, but were not active during the pairing procedure, so plastic changes in these inputs were heterosynaptic. For comparison, the third group of inputs shows results of intracellular tetanization experiments (data from Fig. 3C, N = 136, pale blue circle symbols).
To summarize, heterosynaptic plasticity induced by intracellular tetanization expresses properties that are well suited for serving as a robust mechanism of normalization of synaptic weights: (a) it depresses strong and potentiates weak synapses thus preventing runaway dynamics of synaptic weights, (b) it can be induced at non-active synapses, and (c) it operates on the same time scale as homosynaptic plasticity. As we will discuss below, heterosynaptic plasticity with these properties can also support synaptic competition.
Plasticity of Intrinsic Excitability and Homeostatic Scaling
Two further forms of heterosynaptic changes have been described and well documented: changes of excitability and homeostatic plasticity. Changes of global or local dendritic excitability can be both of homeostatic nature, decreasing excitability after prolonged strong activity but increasing it after activity suppression (Zhang and Linden 2003), but also may enhance or amplify effects of potentiation by increasing excitability of the dendrites at which tetanized synapses are located (Fink and O’Dell 2009; Frick and others 2004). The excitability changes may dramatically affect neuronal responses; however, as changes of excitability do not change synaptic transmission as such, this type of plasticity is not discussed further in this review.
Homeostatic plasticity mediates compensatory scaling of synaptic weights counteracting prolonged dramatic changes of activity. Synaptic weights scale up after hours/days of activity blockade by TTX (Turrigiano and others 1998) or hyperpolarization of neurons caused by expression of inwardly rectifying potassium channels (Burrone and others 2002), and scaled down after prolonged increases of activity by blockade of inhibition (Turrigiano and others 1998). Homeostatic plasticity and its effects, cell-wide or compartmentalized, have been extensively reviewed recently (Rabinowitch and Segev 2008; Turrigiano 2008; Vitureira and others 2012). Homeostatic plasticity differs from the heterosynaptic plasticity described above in two important aspects. First, homeostatic plasticity requires nonspecific dramatic changes of neuronal activity over prolonged periods, which are unlikely to happen during everyday life and learning. Second, it operates on a very long time scale, hours and days, and thus cannot counteract runaway dynamics induced within seconds and minutes by Hebbian-type learning rules (Zenke and others 2013).
Computational Properties of Plasticity Induced by Intracellular Tetanization
We used computer models to test the hypothesis that heterosynaptic plasticity observed in intracellular tetanization experiments can prevent runaway dynamics of synaptic weights and activity (Chen and others 2013b). The model neuron consisted of two compartments, axosomatic and dendritic, and received synaptic inputs from 100 simulated presynaptic neurons (Fig. 4A). Each presynaptic neuron fired action potentials at average frequency of 1 Hz, with Poisson distributed interspike intervals. Activity of presynaptic neurons was mildly correlated, with averaged cross-correlation between pairs of spike trains 0.35 ± 0.05. This input led to the firing of the postsynaptic model neuron at ~1.8 Hz. With symmetrical STDP learning rule (Fig. 4A) synaptic weights showed runaway dynamics (Fig. 4B). By the end of the simulation, all synaptic weights were saturated at the maximal value, leading to a dramatic increase of the postsynaptic firing rate from ~1.8 Hz during the first 10 seconds of simulation, to ~6.3 Hz during the last 10 s of simulation. Implementing in the model heterosynaptic plasticity with experimentally observed properties effectively prevented runaway dynamics of synaptic weights and activity (Fig. 4C). In the model with both STDP and heterosynaptic plasticity, synaptic weights slightly increased, but did not saturate and formed a normal distribution around the new mean value, within the operation range of synapses. Firing rate of the postsynaptic neuron slightly increased from ~1.8 Hz to ~2.6 Hz. Further simulations demonstrated that the stabilizing effect of heterosynaptic plasticity on synaptic weights is long-lasting and robust to variations in the patterns of presynaptic activity, changes of calcium threshold for plasticity induction, and changes of parameters of STDP learning rules over a broad range of values. The latter point is demonstrated in Figure 5. To explore effects of STDP parameters, we fixed the depression window of STDP (a- = 0.001 mS/cm2, τ- = 20 ms), but changed systematically the amplitude and the time constant of the potentiation window (Fig. 5A). The range of tested STDP rules included those strongly dominated by depression, as well as those dominated by potentiation. The STDP-only model expressed nonsaturating behavior only with a very limited set of STDP rules, specifically those dominated by depression (Fig. 5B). As soon as the potentiation window of STDP rule was ~75% of the depression window or stronger, synaptic weights and postsynaptic firing invariably expressed runaway dynamics (Fig. 5B). Addition of heterosynaptic plasticity to the model robustly prevented runaway dynamics and led to the stable and physiological synaptic weight distribution. The model equipped with heterosynaptic plasticity expressed stable behavior with all STDP windows tested—from almost exclusively depressing STDP rules, to those strongly dominated by potentiation (Fig. 5C).
Figure 4.

Heterosynaptic plasticity prevents runaway dynamics of synaptic weights. (A) A scheme of a model neuron and STDP learning rule. The model neuron consisted of axosomatic and dendritic compartments, receiving 100 synaptic inputs from 100 presynaptic neurons. Each presynaptic neuron fired action potentials at ~1 Hz, with Poisson distributed interspike intervals. In simulations shown in this figure, firing of input neurons was mildly correlated (averaged cross-correlation 0.35 ± 0.05), and STDP learning rule had symmetrical potentiation and depression windows. (B) In a model with symmetrical STDP learning rule (τ+ = τ− = 20 ms; a+ = a− = 0.001 mS/cm2) synaptic inputs express runaway dynamics, and saturate at maximal value. Color plot shows dynamics of changes of synaptic weights (color coded). Synapses were sorted by their weights at the beginning of experiment. Bottom: Time course of weight changes of synapses #10 and #90. Inset on the right shows distributions of synaptic weights at the beginning (red) and at the end of the simulation. At the end of the simulation, all inputs were potentiated to the maximum (red bar length is out of scale). (C) In a model with STDP and heterosynaptic plasticity implemented according to experimental data, same pattern of input activity did not lead to saturation of synaptic weights. Instead, synaptic weights reached a new balance and formed a normal distribution around a new mean. Conventions as in B (Modified, with permission, from Chen and others 2013b).
Figure 5.

Heterosynaptic plasticity makes a broad range of STDP parameters compatible with stable operation of neurons. (A) Examples of STDP rules with a constant depression window (a− = 0.001 mS/cm2, τ −=20 ms), but changing the potentiation window time constant (top, τ+ = 5, 10, 20, 30, 40 ms) and amplitude (bottom, a+ = 0.2–2.5 × 10−3 mS/cm2). (B, C) Each box in the grids shows the D’Agostino-Pearson’s K2 test for normality of synaptic weight distribution after 100 seconds of simulations with different STDP potentiation windows, with a+ and τ+ as indicated on the X and Y axes. White square indicates symmetrical STDP learning rule. Synaptic weight distributions with high K2 test values (>50) indicating deviation from normality, typically contain most of the weights saturated at maximal or minimal values. Note that in simulations with STDP only model (B), only few STDP rules, with strong bias toward depression, did not lead to runaway dynamics. Most STDP rules, including examples shown in the bottom, led to runaway dynamics of synaptic weights. In contrast, model with STDP and heterosynaptic plasticity (C) did not express runaway dynamics over the whole range of tested STDP rules, including those extremely unbalanced (insets, bottom) (Modified, with permission, from Chen and others 2013b).
Thus, heterosynaptic plasticity makes a broad range of STDP parameters compatible with stable operation of neurons and neuronal networks. This is an important feature because experimental evidence indeed shows wide variations of STDP windows for potentiation and depression and of their relative strength in neurons of different types (Abbott and Nelson 2000; Caporale and Dan 2008; Feldman 2009; Froemke and others 2005; Haas and others 2006; Nishiyama and others 2000; Sjöström and others 2001; Zhou and others 2005).
Notably, despite its strong stabilizing effect on synaptic weights, heterosynaptic plasticity does not prevent synaptic competition and segregation of synaptic weights. Figure 6 shows results of simulations in which inputs to the model neuron were segregated in two groups. In one group of synapses, presynaptic firing was weakly correlated, with averaged correlation between spike trains 0.34 ± 0.02. In the other, smaller group, correlation of presynaptic firing was higher (0.61 ± 0.05). All presynaptic neurons fired at averaged frequency of ~1 Hz. In STDP-only model, strongly correlated inputs were rapidly potentiated and saturated at the maximum value, whereas distribution of the weakly correlated inputs changed little (Fig. 6A, B). In the model with both STDP and heterosynaptic plasticity, no inputs were saturated, but the groups of weakly and strongly correlated inputs formed two clearly separate distributions, both within the operation range of synaptic weights (Fig. 6C, D). Similarly, segregation was observed when the two groups of synaptic inputs differed by their average firing frequency rather than by the level of correlation. Results from Figure 6D illustrate one further notable feature introduced by heterosynaptic plasticity: the final distribution of synaptic weights is determined by the balance between STDP, or any other mechanisms that govern homosynaptic changes, and heterosynaptic plasticity.
Figure 6.

Synaptic competition and segregation of synaptic weights of strongly versus weakly correlated inputs in the model with heterosynaptic plasticity. A model neuron received input from N = 100 presynaptic neurons firing at average frequency of 1 Hz. Spike trains of 66 presynaptic neurons (inputs ## 1 to 66) were weakly correlated (averaged cross-correlation 0.34 ± 0.02), spike trains of 34 presynaptic neurons (inputs ## 67 to 100) were strongly correlated (averaged cross-correlation 0.61 ± 0.05). Symmetrical STDP rule was used in the simulations, with τ+ = τ− = 20 ms; a+ = a− = 0.001 mS/cm2. (A, C) Dynamics of synaptic weights of weakly correlated inputs (synapses ## 1 … 66) and strongly correlated inputs (synapses ## 67 … 100) in the model with STDP only (A) and the model with STDP and heterosynaptic plasticity (C). (B. D) Distributions of synaptic weights at the beginning (blue bars) and at the end (red) of simulations from A and C, respectively. Note runaway dynamics of synaptic weights and their saturation at the highest value (0.03 mS/cm2) for the group of strongly correlated inputs in STDP-only simulation (Modified, with permission, from Chen and others 2013b).
Thus, heterosynaptic plasticity does not prevent segregation of groups of synaptic inputs exhibiting activity patterns that differ by the degree of correlation or by firing frequency. Moreover, it helps preserve the ability of a neuron with plastic synapses for further learning: unsaturated synapses have higher potential for further changes than those potentiated to the maximum or depressed to zero by STDP-only learning rules.
Functional Roles for Heterosynaptic Plasticity
Heterosynaptic plasticity with experimentally observed properties can play several important roles in learning systems equipped with plastic synapses.
First, it can play a stabilizing role by effectively counteracting positive feedback introduced by Hebbian-type learning rules, and thus prevent runaway dynamics of synaptic weight and neuronal firing. One consequence of this robust stabilization is that it makes a broad variety of learning rules and input activity patterns compatible with normal, unsaturated operation of learning networks. Another consequence is that weight-dependent heterosynaptic plasticity endorses any excessive activity in the system with stabilizing effect. Because only a fraction of synaptic inputs to a cell needs to be activated to induce firing, only this fraction of inputs will be subject to the positive feedback of the Hebbian learning. All other synapses, actually the majority of them, will be subject to stabilizing effect of heterosynaptic plasticity. Finally, by preventing saturation of synaptic weights, heterosynaptic plasticity keeps synapses within their operation range, susceptible to further changes, and thus keeps the system susceptible to new learning.
Second, heterosynaptic plasticity can support and facilitate synaptic competition by introducing an additional force driving synaptic weights toward equilibrium. Indeed, the weight of a synapse is determined by the balance between homosynaptic plasticity, for example, STDP, and heterosynaptic plasticity, with STDP pressing toward one of the extremes, and heterosynaptic plasticity pressing toward its own equilibrium. If STDP is not operating at a synapse, say, because it is not activated presynaptically, its weight will be shifted toward the heterosynaptic plasticity equilibrium and thus away from the STDP-extreme. This will result in a more robust segregation of synaptic weights of competing synapses. Moreover, such a mechanism could underlie refinement of already learned patterns by decreasing weights of erroneously potentiated synapses, and in a limit would allow re-learning.
Induction of Input-Specific and Heterosynaptic Plasticity: Similarity of Mechanisms
Properties of heterosynaptic plasticity described above were derived from results of intracellular tetanization experiments. However, intracellular tetanization, though a useful tool to study plasticity that does not require presynaptic stimulation for its induction, is artificial in a way that in vivo, strong depolarization and firing of a neuron necessary for plasticity induction are produced by synaptic activity. This poses a question, whether a pairing procedure, conventionally used to induce STDP, can also induce heterosynaptic plasticity with the above properties? To address this question, we recorded synaptic responses in layer 2/3 pyramidal neurons from rat neocortex and induced STDP by pairing one of the test inputs with bursts of 5 postsynaptic action potentials. During pairing, the first spike in a burst was generated ~10 ms after the EPSP onset. Fifty pairings induced robust potentiation of EPSP amplitude to 137 ± 5.5% of the control (mean ± SEM, N = 81). The magnitude of EPSP change was significantly correlated with initial paired-pulse ratio (Fig. 7, magenta, diamond symbols; r = 0.40, P < 0.001). Importantly, the pairing procedure also induced plasticity in the inputs that were recorded in the same experiments but were not stimulated during the pairing procedure. The properties of this heterosynaptic plasticity were very similar to plasticity induced by intracellular tetanization. First, heterosynaptic changes in unpaired inputs occurred in both directions: 15 out of 50 inputs (30%) showed significant potentiation, and 12 more (24%) showed significant depression. Intracellular tetanization (data from Fig. 3) induced significant potentiation in 44 out of 136 inputs (32%) and depression in 49 inputs (36%). Second, the amplitude of EPSP changes was correlated with initial paired-pulse ratio (Fig. 7, cyan, square symbols, r = 0.39, P < 0.001; compare to r = 0.43, P < 0.001 for intracellular tetanization, pale blue). Third, although individual inputs did express significant potentiation and depression, there was no significant change of the averaged response amplitude (105.7 ± 4.7% of control, N = 50, compare to 106.8 ± 4.0% of control, N = 136, after intracellular tetanization).
Thus, induction of homosynaptic plasticity by a pairing procedure typically used in STDP studies is accompanied by the induction of heterosynaptic plasticity in unpaired inputs. This heterosynaptic plasticity has properties similar to those of plasticity induced by intracellular tetanization, and thus can play a role in stabilizing synaptic weights and supporting synaptic competition.
This conclusion stays in apparent contradiction to the notion of input specificity of pairing-induced plasticity and to the wealth of publications reporting no changes in unpaired or control inputs. A solution to this contradiction may be the fact that heterosynaptic changes are bidirectional but balanced, as the results from Figure 7 show. To test this conjecture, we reanalyzed results published in several STDP studies. For this analysis we have selected results of 35 experimental series from 8 studies of STDP, which stated the mean, the SD or SEM, and the number of observations contributing to the reported changes of response amplitude (Birtoli and Ulrich 2004; Feldman 2000; Hardingham and others 2007; Letzkus and others 2006; Nevian and Sakmann 2006; Sjöström and others 2001; Watt and others 2004; our data from Fig. 7). All these papers present clear cases of STDP of excitatory inputs to layer 2/3 or layer 5 pyramidal neurons in slices from somatosensory, visual, or auditory areas of rat neocortex. Figure 8 shows results of this analysis. Each bar shows the averaged after-pairing change of response amplitude (diamond symbol) and the range covered by ±2 SD. This range includes 95% of normally distributed values. Note that number of inputs contributing to each experimental series from published papers (Fig. 8, a-g) ranged between N = 4 and N = 20, and thus actually measured values have not necessary covered the whole ±2 SD range. Figure 8 illustrates several important points. First, ranges of response amplitude changes after most LTP and LTD protocols overlap, and typically include changes of the opposite signs (Fig. 8, magenta and green). LTP protocols, alongside with—by design—increase of the averaged amplitude, lead to highly variable effects, typically including amplitude decreases (in 8 out of 11 LTP protocols shown). This indicates that factors other than timing, such as synaptic predispositions for plasticity, contributed to the final change of response amplitude. Second, the ranges of response amplitudes in “No change or unpaired” groups (Fig. 8, blue), typically express substantial overlap with LTP and LTD ranges, and thus show evidence for heterosynaptic changes. However, most probably because heterosynaptic potentiation and depression were balanced, the average was not significantly different from zero. Third, the ranges of amplitude changes after AP bursts only protocols (Fig. 8, d, e, and our data in h) clearly include cases of potentiation and depression, though the average is not significantly different from zero, probably because of balanced LTP and LTD. Notably, Hardingham and others (2007) found that potentiation and depression can be induced by the same protocol, whereby the direction of the EPSP amplitude change was correlated with initial release probability. The LTP and LTD data from this article represent cases selected by the direction of the change. The third bar in this group (Fig. 8, g) shows LTP and LTD data pooled together. Finally, the range of amplitude changes of unpaired inputs is typically smaller than the range of amplitude changes induced by spike burst-only protocols (Fig. 8, d, e, and h). This may indicate that plastic synapses compete for limited resources. In this scenario, pairing may facilitate access to resources for paired inputs via mechanism of synaptic tagging (Fonseca and others 2004; Frey and Morris 1997) or a similar process, which would leave fewer resources available for heterosynaptic changes at unpaired inputs. Spikes-only protocols leave more resources available for heterosynaptic changes, and thus induce heterosynaptic plasticity of larger amplitude.
Figure 8.

Comparison of reported changes of response amplitude at inputs that were active during the induction (homosynaptic, input-specific) and those not active during the induction (heterosynaptic). The plot shows results of 35 experimental series (bars) from 8 articles (groups of bars) on pairing-induced long-term plasticity (STDP), in which the mean amplitude changes were reported together with the SD (or SEM) and number of observations. Each bar shows an average (diamond symbol) change of EPSP amplitude after pairing procedure ±2 SD. This range includes 95% of normally distributed values. Magenta: changes after potentiation-inducing protocols (post after pre). Green: changes after depression inducing protocols (pre after post). Blue: range of EPSP amplitudes after protocols that did not lead to significant changes of the averaged response (such as interval between pre and post spikes outside plasticity windows). Gray: range of EPSP amplitudes after presynaptic only stimulation without postsynaptic spikes. Black, bars from cyan to pink (in d, e, h): range of EPSP amplitudes after bursts of postsynaptic spikes only, without presynaptic stimulation. Data for excitatory inputs to L2/3 or L5 pyramidal neurons from somatosensory, visual, or auditory cortex, from the following articles: Feldman 2000 (a); Sjöström and others 2001 (b); Watt and others 2004 (c); Birtoli and Ulrich 2004 (d); Nevian and Sakmann 2006 (e); Letzkus and others 2006 (f); Hardingham and others 2007 (g); Our data from Figure 7 (h).
The above analysis demonstrates that although amplitude changes induced in unpaired inputs or by spike bursts-only protocols were not significant when averaged across the inputs, these groups might have included cases of potentiation and depression of individual inputs in most of the data shown in Figure 8. The reason for the absence of significant change of group-averages might be the balanced nature of heterosynaptic plasticity. It is also important to note that in articles specifically aimed at investigating heterosynaptic plasticity, it was readily induced by regular pairing (Arami and others 2013; Huang and others 2008; Nishiyama and others 2000), afferent tetanization (Bauer and LeDoux 2004; Chevalyre and others 2003; Cummings and others 1996; Nugent and others 2007; Pascual and others 2005; Royer and Paré 2003; Staubli and Ji 1996; Wöhrl and others 2007), or purely postsynaptic protocols (e.g., Christofi and others 1993; Cummings and others 1996; Pockett and others 1990). This analysis substantiates our conclusion that induction of homosynaptic plasticity by a typical pairing procedure used in STDP studies is accompanied by induction of heterosynaptic plasticity in unpaired inputs.
Mechanisms of Heterosynaptic Plasticity
Rise of intracellular calcium concentration is an obvious candidate for triggering heterosynaptic plasticity. Indeed, the rise of [Ca2+]in is sufficient to induce plasticity (Neveu and Zucker 1996; Yang and others 1999). Induction of long-term heterosynaptic plasticity by intracellular tetanization and other protocols was impaired or blocked by chelation of intracellular calcium with EGTA (Arami and others 2013; Bright and Brickley 2008; Han and Heinemann 2013; Lee and others 2012; Nugent and others 2007). Substantial rise of [Ca2+]in is produced by bursts of back-propagating action potentials activating voltage-dependent calcium channels (Miyakawa and others 1992; Petrozzino and Connor 1994; Schiller and others 1995; Schiller and others 1998; Yuste and Denk 1995), and can be further amplified by calcium release from internal stores (Berridge 1998; Fagni and others 2000; Nakamura and others 1999). Because this [Ca2+]in rise depends on backpropagating spikes, it can be induced cell-wide, including dendrites with no recently activated synapses. The profile of the [Ca2+]in rise in nonactive dendrites will be determined by their ability to support backpropagation and by the number and frequency of action potentials (Golding and others 2002; Larkum and others 1999; Lisman and Spruston 2005; Spruston and others 1995). For example, bursts of action potentials can invade dendrites of layer 5 pyramidal neurons more reliably than single spikes (Kampa and others 2006; Sjöström and Häusser 2006). The resulting profile of [Ca2+]in may promote induction of heterosynaptic LTP at sites of strong calcium rise, and LTD at sites of more moderate rise. However, the final outcome, LTP or LTD, might be also strongly influenced by predispositions of synapses for plasticity, because both potentiation and depression could be induced at synapses presumably located at similar distances and thus experiencing similar calcium rises (Lee and others 2012).
Strong local activation produces [Ca2+]in rise that is not restricted to the activated synapses, and thus can trigger local heterosynaptic plasticity at nearby sites. Indeed, profile of [Ca2+]in rise around the activation site can explain the Mexican-hat type profile of potentiation and depression induced along the activated dendrite (Royer and Paré 2003). Calcium release from internal stores may facilitate induction of this local heterosynaptic plasticity (Daw and others 2002; Dudman and others 2007; Nagase and others 2013; Nishiyama and others 2000; Royer and Paré 2003).
Plasticity induced by intracellular tetanization involves presynaptic components, implying retrograde signaling (Lee and others 2012; Volgushev and others 1997; Volgushev and others 2000). One candidate retrograde messenger is nitric oxide (NO). It is involved in mediating local heterosynaptic plasticity of both excitatory and inhibitory transmission (Lange and others 2012; Nugent and others 2007; Schuman and Madison 1994). These effects could be due to diffusion of NO produced at activated synapses (Böhme and others 1991; Gally and others 1990; Hardingham and others 2013; Hölscher and others 1997; O’Dell and Alger, 1991). Disrupting NO signaling by NO-synthase inhibitors and NO-scavengers abolished the dependence of plasticity induced by intracellular tetanization on initial paired-pulse ratio (Lee and others 2012; Volgushev and others 2000). Weaker intracellular tetanization also induced plasticity that was not correlated with initial paired-pulse ratio, suggesting that NO signaling required strong postsynaptic activity (Volgushev and others 2000). Consistent with this suggestion are results demonstrating that the NO-dependent presynaptic component of homosynaptic plasticity was observed only after strong induction protocol, but not after a weaker protocol (Phillips and others 2008).
Disruption of NO signaling in intracellular tetanization experiments did not completely abolish presynaptic changes, implying involvement of additional retrograde signaling systems (Lee and others 2012; Volgushev and others 2000). Activation of metabotropic glutamate receptors (mGluRs) can trigger the production, release, and retrograde action of endocannabinoids, including heterosynaptic LTD at inhibitory synapses (Chevaleyre and Castillo 2003; Chevaleyre and others 2006; Chiu and others 2010; Maejima and others 2001; Pan and others 2008). The spread of retrograde signal to heterosynaptic sites was suggested to be due either to spillover of glutamate acting on local mGluR1/5-Rs (Chevaleyre and Castillo 2003; Yang and Calakos 2013), or the diffusion of endocannabinoids produced at active synapses (Yasuda and others 2008).
Heterosynaptic depression can be also mediated by activity-dependent release of ATP and adenosine from neurons (Lovatt and others 2012) and astrocytes (Chen and others 2013a; Pascual and others 2005; Serrano and others 2006; Zhang and others 2003). This depression was A1 receptor dependent: it was abolished by the A1 receptor antagonist DPCPX (Lovatt and others 2012; Pascual and others 2005) and was absent in mice with impaired adenosine tone and diminished A1 receptor activation (Pascual and others 2005).
Thus, multiple mechanisms mediate heterosynaptic plasticity. Most of these mechanisms overlap with those mediating homosynaptic plasticity, as indicated by at least partial occlusion between homo- and heterosynaptic plastic changes (Cummings and others 1996; Kuhnt and others 1994; Neveu and Zucker 1996; Volgushev and others 1999; Yang and others 1999). Heterosynaptic changes could be local, mediated by the spread of plasticity inducing factors, such as intracellular or extracellular messengers including retrograde signals, around the activated synapses. The spread involving glial cells may cover glial islets and thus reach several hundreds of dendrites (up to 600; Halassa and others 2007) arising from potentially large pool of distinct neurons, with thousands of synapses. In addition, heterosynaptic changes can be induced by cell-wide signals, such as [Ca2+]in rises produced by backpropagating action potentials. Although heterosynaptic plasticity in its simplest sense does not demand associative activation of presynaptic sites, the physiological induction of heterosynaptic plasticity is almost certainly shaped by these signals.
Conclusion: A Multifaceted Picture of Plasticity
The emerging picture of long-term regulation of synaptic transmission includes an increasing number of forms and mechanisms of plasticity mediated by a variety of biochemical cascades. This multifaceted picture includes both mechanisms that mediate changes necessary for learning new behaviors, as well as those securing stability of the system by mediating compensatory changes to counteract positive feedback imposed by associative learning rules. The first task is best served by input-specific, associative Hebbian-type homosynaptic plasticity. It is mediated by mechanisms that require local synaptic activity and modify efficacy of activated synapses, such as canonical NMDA-receptor mediated plasticity. The second task, of achieving stability, requires balancing of synaptic weights within populations of synapses and depends on modifications at nonactivated synapses—heterosynaptic plasticity. Mechanisms mediating heterosynaptic plasticity overlap with those of homosynaptic plasticity, as indicated by the partial occlusion of effects of these two plasticity forms. Heterosynaptic plasticity can be local, mediated by the spread of a signal that induces changes at nearby active synapses. For example, short-range spread of NO may mediate distributed potentiation, which actually amplifies the effect of input-specific potentiation. The profile of [Ca2+]in rise along a dendrite after a strong local stimulation may underlie the Mexican-hat profile of potentiation and depression, which balances the effect of the input-specific LTP or LTD. The spread of that kind of local heterosynaptic plasticity may be enhanced by intracellular mechanisms, such as activation of Ca2+ release from endoplasmic reticulum, or extracellular pathways, for example, involving glial cells. The latter pathway may cover glial islets and thus reach several hundreds of dendrites with thousands of synapses. Candidate trigger for inducing cell-wide heterosynaptic plasticity is [Ca2+]in rise produced by back-propagating action potentials, eventually amplified by calcium-dependent calcium release from internal stores. At dendritic locations far away from the activity site, pre-dispositions of synapses for potentiation or depression may play a stronger role in determining plastic changes because local resources are not depleted by privileged-access activated synapses.
This interacting network of complementary plasticity mechanisms introduces several forces that drive synaptic weight changes in opposite directions (Fig. 9). Homosynaptic plasticity is induced by specific patterns of input activity such as high-frequency tetanization for LTP or prolonged low-frequency stimulation for LTD, or specific timing relative to postsynaptic spikes for STDP. Homosynaptic changes mediate associative learning, but they also drive synaptic weights to the extreme values, bringing the system out of balance. Heterosynaptic plasticity is not restricted to activated synapses, thus making any synapse a potential target for modification by episodes of strong postsynaptic activity. Because of the weight-dependence of the direction of heterosynaptic changes, LTP for weak synapses but LTD for strong synapses, it introduces two forces that drive synaptic weights away from the extremes, toward an equilibrium within the operation range. Relative effectiveness of these opposite forces can possibly change as a function of brain state (e.g., sleep vs. wakefulness), thus opening possibility for promoting either local stimulus specific changes or global homeostatic scaling. Because all four forces operate on the same time scale, they can effectively balance each other. The weight of a synapse is determined by the balance between the homosynaptic and heterosynaptic plasticity forces. The combined action of these forces on synaptic weights allows neuronal networks to sustain the ability for associative synaptic changes while preserving secure balance around a stable operating point.
Figure 9.
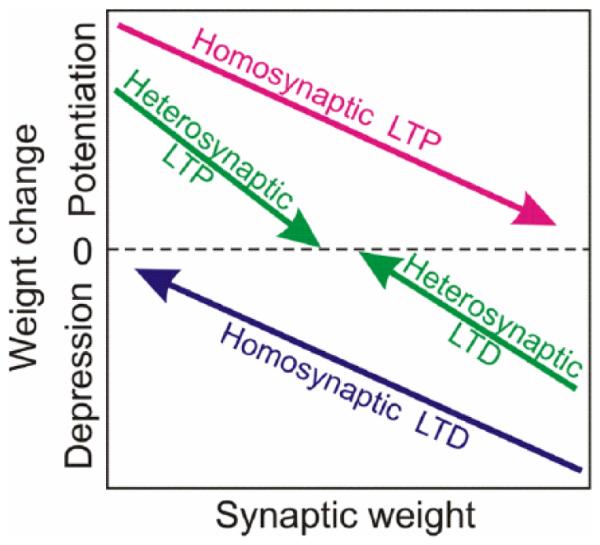
Driving forces for homosynaptic and heterosynaptic weight changes. Homosynaptic plasticity, LTP or LTD, is induced only at synapses active during the induction, and requires specific activity patterns. Homosynaptic LTP leads to weight-dependent increase of synaptic weights: potentiation is stronger at initially weak than at initially strong synapses. Homosynaptic LTD leads to weight-dependent decrease of synaptic weights: depression of strong synapses is stronger than depression of the weak synapses. Heterosynaptic plasticity can be induced at any synapse during episodes of strong postsynaptic activity. Initially weak synapses are subject to weight-dependent heterosynaptic LTP. Initially strong synapses are subject to weight-dependent heterosynaptic LTD.
Outlook and Open Questions
The complementary nature of homosynaptic and heterosynaptic plasticity stresses the need for understanding the interaction between these forms of plasticity in governing synaptic changes. What determines the balance between homosynaptic and heterosynaptic plasticity at individual synapses and in neuronal networks? What are synapse-specific, cell type-specific, and structure-specific mechanisms of maintaining this balance? How is the balance between homosynaptic and heterosynaptic plasticity regulated, and what factors may disturb it? Revealing these factors may have important implications to understanding emergence of pathological states of the brain, such as posttraumatic stress disorder. Shifting the balance toward homosynaptic plasticity, for example, during traumatizing experiences, may lead to runaway potentiation and/or depression of selected populations of synapses, resulting in the restriction of future activity routing to the few deeply carved pathways.
How is the balance between homosynaptic and heterosynaptic plasticity regulated during development? During formation of sensory representations, runaway dynamics may be a useful process contributing to elimination of excessive connections while preserving the “correct” synapses. After the representations have been established, however, the balance might shift so that the stabilizing effect of heterosynaptic plasticity becomes capable to counteract runaway dynamics imposed by associative learning rules.
What determines the predispositions of synapses for potentiation and depression, and what are mechanisms of weight-dependence of both homosynaptic and heterosynaptic plasticity? Finally, how is long-term stability of connections mediating life-long memory traces achieved in the face of the multitude of plasticity mechanisms driving homosynaptic and heterosynaptic changes? How can some synapses escape the drive for modifications imposed by these forces? Answering these questions will advance our understanding of multifacet roles of synaptic plasticity: how it mediates learning of adaptive behaviors, but also may lead to pathological brain states.
Acknowledgments
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Supported by NIH Grant R01 MH087631.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nature Neurosci. 2000;3:1178–83. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19(4):126–30. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- Arami MK, Sohya K, Sarihi A, Jiang B, Yanagawa Y. Reciprocal homosynaptic and heterosynaptic long-term plasticity of corticogeniculate projection neurons in layer VI of the mouse visual cortex. J Neurosci. 2013;33(18):7787–98. doi: 10.1523/JNEUROSCI.5350-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer EP, LeDoux JE. Heterosynaptic long-term potentiation of inhibitory interneurons in the lateral amygdala. J Neurosci. 2004;24:9507–12. doi: 10.1523/JNEUROSCI.3567-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birtoli B, Ulrich D. Firing mode-dependent synaptic plasticity in rat neocortical pyramidal neurons. J Neurosci. 2004;24:4935–40. doi: 10.1523/JNEUROSCI.0795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–9. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–56. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhme GA, Bon C, Stutzmann JM, Doble A, Blanchard JC. Possible involvement of nitric oxide in long-term potentiation. Eur J Pharmacol. 1991;199(3):379–81. doi: 10.1016/0014-2999(91)90505-k. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer T, Staiger V, Aersten A. Synaptic plasticity in rat hippocampal slice cultures: local “Hebbian” conjunction of pre- and postsynaptic stimulation leads to distributed synaptic enhancement. Proc Natl Acad Sci U S A. 1989;86:8113–7. doi: 10.1073/pnas.86.20.8113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, O’Byrne M, Murthy VN. Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature. 2002;420:414–8. doi: 10.1038/nature01242. [DOI] [PubMed] [Google Scholar]
- Caporale N, Dan Y. Spike timing-dependent plasticity: a Hebbian learning rule. Annu Rev Neurosci. 2008;31:25–46. doi: 10.1146/annurev.neuro.31.060407.125639. [DOI] [PubMed] [Google Scholar]
- Chen J, Tan Z, Zeng L, Zhang X, He Y, Gao W. Heterosynaptic long-term depression mediated by ATP released from astrocytes. Glia. 2013a;61(2):178–91. doi: 10.1002/glia.22425. others. [DOI] [PubMed] [Google Scholar]
- Chen JY, Lonjers P, Lee C, Chistiakova M, Volgushev M, Bazhenov M. Heterosynaptic plasticity prevents runaway synaptic dynamics. J Neurosci. 2013b;33(40):15915–29. doi: 10.1523/JNEUROSCI.5088-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38(3):461–72. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Chistiakova M, Volgushev M. Heterosynaptic plasticity in the neocortex. Exp Brain Res. 2009;199(3–4):377–90. doi: 10.1007/s00221-009-1859-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CQ, Puente N, Grandes P, Castillo PE. Dopaminergic modulation of endocannabinoid-mediated plasticity at GABAergic synapses in the prefrontal cortex. J Neurosci. 2010;30(21):7236–48. doi: 10.1523/JNEUROSCI.0736-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofi G, Nowicky AV, Bolsover SR, Bindman LJ. The postsynaptic induction of nonassociative long-term depression of excitatory synaptic transmission in rat hippocampal slices. J Neurophysiol. 1993;69:219–29. doi: 10.1152/jn.1993.69.1.219. [DOI] [PubMed] [Google Scholar]
- Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron. 1996;16(4):825–33. doi: 10.1016/s0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- Daw MI, Bortolotto ZA, Saulle E, Zaman S, Collingridge GL, Isaac JT. Phosphatidylinositol 3 kinase regulates synapse specificity of hippocampal long-term depression. Nat Neurosci. 2002;5:835–6. doi: 10.1038/nn903. [DOI] [PubMed] [Google Scholar]
- Dudman JT, Tsay D, Siegelbaum SA. A role for synaptic inputs at distal dendrites: instructive signals for hippocampal long-term plasticity. Neuron. 2007;56:866–79. doi: 10.1016/j.neuron.2007.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott T, Shadbolt NR. Multiplicative synaptic normalization and a nonlinear Hebb rule underlie a neurotrophic model of competitive synaptic plasticity. Neural Comput. 2002;14:1311–22. doi: 10.1162/089976602753712954. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Synapse specificity of long-term potentiation breaks down at short distances. Nature. 1997;388:279–84. doi: 10.1038/40870. [DOI] [PubMed] [Google Scholar]
- Fagni L, Chavis P, Ango F, Bockaert J. Complex interactions between mGluRs, intracellular Ca2+ stores and ion channels in neurons. Trends Neurosci. 2000;23(2):80–8. doi: 10.1016/s0166-2236(99)01492-7. [DOI] [PubMed] [Google Scholar]
- Feldman DE. Timing-based LTP and LTD at vertical imputs to layer II/III pyramidal cells in rat barrel cortex. Neuron. 2000;27:45–56. doi: 10.1016/s0896-6273(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Feldman DE. Synaptic mechanisms for plasticity in neocortex. Annu Rev Neurosci. 2009;32:33–55. doi: 10.1146/annurev.neuro.051508.135516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finelli LA, Haney S, Bazhenov M, Stopfer M, Sejnowski TJ. Synaptic learning rules and sparse coding in a model sensory system. PLoS Comput Biol. 2008;4(4):e1000062. doi: 10.1371/journal.pcbi.1000062. doi:10.1371/journal.pcbi.1000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink AE, O’Dell TJ. Short trains of theta frequency stimulation enhance CA1 pyramidal neuron excitability in the absence of synaptic potentiation. J Neurosci. 2009;29(36):11203–14. doi: 10.1523/JNEUROSCI.1450-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca R, Nägerl UV, Morris GM, Bonhoeffer T. Competing for memory: hippocampal LTP under regimes of reduced protein synthesis. Neuron. 2004;44:1011–20. doi: 10.1016/j.neuron.2004.10.033. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RGM. Synaptic tagging and long-term potentiation. Nature. 1997;385(6):533–6. doi: 10.1038/385533a0. [DOI] [PubMed] [Google Scholar]
- Frick A, Magee J, Johnston D. LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat Neurosci. 2004;7:126–35. doi: 10.1038/nn1178. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Mu-Ming P, Dan Y. Spike-timing-dependent synaptic plasticity depends on dendritic location. Nature. 2005;434:221–5. doi: 10.1038/nature03366. [DOI] [PubMed] [Google Scholar]
- Gally IA, Montague PR, Reeke GN, Edelman GM. The NO hypothesis: possible effects of a short-lived, rapidly diffusible signal in the development and function of the nervous system. Proc Natl Acad Sci U S A. 1990;87:3547–51. doi: 10.1073/pnas.87.9.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjorgjieva J, Clopath C, Audet J, Pfister JP. A triplet spike-timing-dependent plasticity model generalizes the Bienenstock–Cooper–Munro rule to higher-order spatiotemporal correlations. Proc Natl Acad Sci U S A. 2011;108:19383–8. doi: 10.1073/pnas.1105933108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding NL, Staff NP, Spruston N. Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature. 2002;418(6895):326–31. doi: 10.1038/nature00854. [DOI] [PubMed] [Google Scholar]
- Haas JS, Nowotny T, Abarbanel HDI. Spike-timing-dependent plasticity of inhibitory synapses in the entorhinal cortex. J Neurophysiol. 2006;96:3305–13. doi: 10.1152/jn.00551.2006. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG. Synaptic islands defined by the territory of a single astrocyte. J Neurosci. 2007;27(24):6473–7. doi: 10.1523/JNEUROSCI.1419-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han EB, Heinemann SF. Distal dendritic inputs control neuronal activity by heterosynaptic potentiation of proximal inputs. J Neurosci. 2013;33:1314–25. doi: 10.1523/JNEUROSCI.3219-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham N, Dachtler J, Fox K. The role of nitric oxide in pre-synaptic plasticity and homeostasis. Front Cell Neurosci. 2013;7:190. doi: 10.3389/fncel.2013.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham NR, Hardingham GF, Fox KD, Jack JB. Presynaptic efficacy directs normalization of synaptic strength in layer 2/3 rat neocortex after paired activity. J Neurophysiol. 2007;97:2965–75. doi: 10.1152/jn.01352.2006. [DOI] [PubMed] [Google Scholar]
- Hebb DO. The organisation of behavior. Wiley; New York, NY: 1949. [Google Scholar]
- Hölscher C, Anwyl R, Rowan MJ. Stimulation on the positive phase of hippocampal theta rhythm induces long-term potentiation that can be depotentiated by stimulation on the negative phase in area CA1 in vivo. J Neurosci. 1997;17(16):6470–7. doi: 10.1523/JNEUROSCI.17-16-06470.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Yasuda H, Sarihi A, Tsumoto T. Roles of endocannabinoids in heterosynaptic long-term depression of excitatory synaptic transmission in visual cortex of young mice. J Neurosci. 2008;28(28):7074–83. doi: 10.1523/JNEUROSCI.0899-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa BM, Letzkus JJ, Stuart GJ. Requirement of dendritic calcium spikes for induction of spike-timing-dependent synaptic plasticity. J Physiol. 2006;574(1):283–90. doi: 10.1113/jphysiol.2006.111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempter R, Gerstner W, van Hemmen JL. Intrinsic stabilization of output rates by spike-based Hebbian learning. Neural Comp. 2001;13:2709–41. doi: 10.1162/089976601317098501. [DOI] [PubMed] [Google Scholar]
- Kossel A, Bonhoeffer T, Bolz J. Non-Hebbian synapses in rat visual cortex. Neuroreport. 1990;1:115–8. doi: 10.1097/00001756-199010000-00008. [DOI] [PubMed] [Google Scholar]
- Kuhnt U, Voronin L. Interaction between paired-pulse facilitation and long-term potentiation in area CA1 of guinea-pig hippocampal slices: application of quantal analysis. Neuroscience. 1994;62:391–7. doi: 10.1016/0306-4522(94)90374-3. [DOI] [PubMed] [Google Scholar]
- Lange MD, Doengi M, Lesting J, Pape HC, Jüngling K. Heterosynaptic long-term potentiation at interneuron–principal neuron synapses in the amygdala requires nitric oxide signalling. J Physiol. 2012;590(1):131–43. doi: 10.1113/jphysiol.2011.221317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Kaiser KMM, Sakmann B. Calcium electrogenesis in distal apical dendrites of layer 5 pyramidal cells at a critical frequency of back-propagating action potentials. Proc Natl Acad Sci U S A. 1999;96(25):14600–4. doi: 10.1073/pnas.96.25.14600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CM, Stoelzel C, Chistiakova M, Volgushev M. Heterosynaptic plasticity induced by intracellular tetanization in layer 2/3 pyramidal neurons in rat auditory cortex. J Physiol. 2012;590(10):2253–71. doi: 10.1113/jphysiol.2012.228247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzkus JJ, Kampa BM, Stuart GJ. Learning rules for spike timing-dependent plasticity depend on dendritic synapse location. J Neurosci. 2006;26:10420–9. doi: 10.1523/JNEUROSCI.2650-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Spruston N. Postsynaptic depolarization requirements for LTP and LTD: a critique of spike timing-dependent plasticity. Nat Neurosci. 2005;8(7):839–41. doi: 10.1038/nn0705-839. [DOI] [PubMed] [Google Scholar]
- Lovatt D, Xu Q, Liu W, Takano T, Smith NA, Schnermann J. Neuronal adenosine release, and not astrocytic ATP release, mediates feedback inhibition of excitatory activity. Proc Natl Acad Sci. 2012;109:6265–70. doi: 10.1073/pnas.1120997109. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch GS, Dunwiddie T, Gribkoff V. Heterosynaptic depression: a postsynaptic correlate of long-term potentiation. Nature. 1977;266:737–9. doi: 10.1038/266737a0. [DOI] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31(3):463–75. doi: 10.1016/s0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275(5297):209–13. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Markram H, Luebke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–5. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Miller KD. Synaptic economics: competition and cooperation in synaptic plasticity. Neuron. 1996;17:371–4. doi: 10.1016/s0896-6273(00)80169-5. [DOI] [PubMed] [Google Scholar]
- Miller KD, MacKay DJC. The role of constraints in Hebbian learning. Neural Comput. 1993;6:100–26. [Google Scholar]
- Miyakawa H, Ross WH, Jaffe D, Callaway JC, Lasser-Ross N, Lisman JE. Synaptically activated increases in Ca2+ concentration in hippocampal CA1 pyramidal cells are primarily due to voltage-gated Ca2+ channels. Neuron. 1992;9:1163–73. doi: 10.1016/0896-6273(92)90074-n. others. [DOI] [PubMed] [Google Scholar]
- Nagase T, Ito KI, Kato K, Kaneko K, Kohda K, Matsumoto M. Long-term potentiation and long-term depression in hippocampal CA1 neurons of mice lacking the IP3 type 1 receptor. Neuroscience. 2003;117:821–30. doi: 10.1016/s0306-4522(02)00803-5. others. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Barbara JG, Nakamura K, Ross WN. Synergistic Release of Ca2+ from IP3 sensitive stores evoked by synaptic activation of mGluRs paired with back-propagating action potentials. Neuron. 1999;24(3):727–37. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- Neveu D, Zucker RS. Long-lasting potentiation and depression without presynaptic activity. J Neurophysiol. 1996;75:2157–60. doi: 10.1152/jn.1996.75.5.2157. [DOI] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Single spine Ca2+ signals evoked by coincident EPSPs and backpropagating action potentials in spiny stellate cells of layer 4 in the juvenile rat somatosensory barrel cortex. J Neurosci. 2006;26:11001–13. doi: 10.1523/JNEUROSCI.3332-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama M, Hong K, Mikoshiba K, Poo M, Kato K. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature. 2000;408:584–8. doi: 10.1038/35046067. [DOI] [PubMed] [Google Scholar]
- Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature. 2007;446(7139):1086–90. doi: 10.1038/nature05726. [DOI] [PubMed] [Google Scholar]
- O’Dell TJ, Alger BE. Single calcium channels in rat and guinea-pig hippocampal neurons. J Physiol. 1991;436:739–67. doi: 10.1113/jphysiol.1991.sp018577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oja E. A simplified neuron model as a principal component analyzer. J Math Biol. 1982;15:267–73. doi: 10.1007/BF00275687. [DOI] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu QS. Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J Neurosci. 2008;28(6):1385–97. doi: 10.1523/JNEUROSCI.4033-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–6. doi: 10.1126/science.1116916. others. [DOI] [PubMed] [Google Scholar]
- Petrozzino JJ, Connor JA. Dendritic Ca2+ accumulations and metabotropic glutamate receptor activation associated with an NMDA receptor-independent long-term potentiation in hippocampal CA1 neurons. Hippocampus. 1994;4:546–58. doi: 10.1002/hipo.450040504. [DOI] [PubMed] [Google Scholar]
- Pfister JP, Gerstner W. Triplets of spikes in a model of spike timing-dependent plasticity. J Neurosci. 2006;26:9673–82. doi: 10.1523/JNEUROSCI.1425-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postsynaptic action potentials are required for nitric-oxide-dependent long-term potentiation in CA1 neurons of adult GluR1 knock-out and wild-type mice. J Neurosci. 2008;28(52):14031–41. doi: 10.1523/JNEUROSCI.3984-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pockett S, Brookes NH, Bindman LJ. Long-term depression at synapses in slices of rat hippocampus can be induced by bursts of postsynaptic activity. Exp Brain Res. 1990;80:196–200. doi: 10.1007/BF00228861. [DOI] [PubMed] [Google Scholar]
- Rabinowitch I, Segev I. Two opposing plasticity mechanisms pulling a single synapse. Trends Neurosci. 2008;31:377–83. doi: 10.1016/j.tins.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Royer S, Paré D. Conservation of total synaptic weight through balanced synaptic depression and potentiation. Nature. 2003;422:518–22. doi: 10.1038/nature01530. [DOI] [PubMed] [Google Scholar]
- Schiller J, Helmchen F, Sakmann B. Spatial profile of dendritic calcium transients evoked by action potentials in rat neocortical pyramidal neurones. J Physiol. 1995;487(3):583–600. doi: 10.1113/jphysiol.1995.sp020902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Clapham DE. NMDA receptors amplify calcium influx into dendritic spines during associative pre- and postsynaptic activation. Nat Neurosci. 1998;1(2):114–8. doi: 10.1038/363. [DOI] [PubMed] [Google Scholar]
- Schuman EM, Madison DV. Locally distributed synaptic potentiation in the hippocampus. Science. 1994;263:532–6. doi: 10.1126/science.8290963. [DOI] [PubMed] [Google Scholar]
- Serrano A, Haddjeri N, Lacaille JC, Robitaille R. GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J Neurosci. 2006;26(20):5370–82. doi: 10.1523/JNEUROSCI.5255-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Häusser M. A cooperative switch determines the sign of synaptic plasticity in distal dendrites of neocortical pyramidal neurons. Neuron. 2006;51(2):227–38. doi: 10.1016/j.neuron.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB. Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron. 2001;32:1149–64. doi: 10.1016/s0896-6273(01)00542-6. [DOI] [PubMed] [Google Scholar]
- Skorheim S, Lonjers P, Bazhenov M. A spiking network model of decision making employing rewarded STDP. Plos One. 2014 doi: 10.1371/journal.pone.0090821. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruston N, Schiller Y, Stuart G, Sakmann B. Activity-dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science. 1995;268(5208):297–300. doi: 10.1126/science.7716524. [DOI] [PubMed] [Google Scholar]
- Staubli UV, Ji ZX. The induction of homo- vs. heterosynaptic LTD in area CA1 of hippocampal slices from adult rats. Brain Res. 1996;714(1):169–76. doi: 10.1016/0006-8993(95)01523-x. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–35. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–6. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Van Rossum MC, Bi GQ, Turrigiano GG. Stable Hebbian learning from spike timing-dependent plasticity. J Neurosci. 2000;20:8812–21. doi: 10.1523/JNEUROSCI.20-23-08812.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitureira N, Letellier M, Goda Y. Homeostatic synaptic plasticity: from single synapses to neural circuits Curr Opin Neurobiol. 2012;22:516–21. doi: 10.1016/j.conb.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volgushev M, Chistiakova M, Balaban P, Eysel UT. Retrograde signalling with nitric oxide at neocortical synapses. Eur J Neurosci. 2000;12:4255–67. doi: 10.1046/j.0953-816x.2000.01322.x. [DOI] [PubMed] [Google Scholar]
- Volgushev M, Mittmann T, Chistiakova M, Balaban P, Eysel UT. Interaction between intracellular tetanization and pairing-induced long-term synaptic plasticity in the rat visual cortex. Neuroscience. 1999;93:1227–32. doi: 10.1016/s0306-4522(99)00265-1. [DOI] [PubMed] [Google Scholar]
- Volgushev M, Voronin LL, Chistiakova M, Singer W. Induction of LTP and LTD in visual cortex neurons by intracellular tetanization. NeuroReport. 1994;5:2069–72. doi: 10.1097/00001756-199410270-00020. [DOI] [PubMed] [Google Scholar]
- Volgushev M, Voronin LL, Chistiakova M, Singer W. Relations between long-term synaptic modifications and paired-pulse interactions in the rat neocortex. Eur J Neurosci. 1997;9:1656–65. doi: 10.1111/j.1460-9568.1997.tb01523.x. [DOI] [PubMed] [Google Scholar]
- Von der Malsburg C. Self-organization of orientation sensitive cells in the striate cortex. Kybernetik. 1973;14:85–100. doi: 10.1007/BF00288907. [DOI] [PubMed] [Google Scholar]
- Watt AJ, Sjöström PJ, Häusser M, Nelson SB, Turrigiano GG. A proportional but slower NMDA potentiation follows AMPA potentiation in LTP. Nat Neurosci. 2004;7:518–24. doi: 10.1038/nn1220. [DOI] [PubMed] [Google Scholar]
- White G, Levy WB, Steward O. Spatial overlap between populations of synapses determines the extent of their associative interaction during the induction of long-term potentiation and depression. J Neurophysiol. 1990;64(4):1186–98. doi: 10.1152/jn.1990.64.4.1186. [DOI] [PubMed] [Google Scholar]
- Wöhrl R, von Haebler D, Heinemann U. Low-frequency stimulation of the direct cortical input to area CA1 induces homosynaptic LTD and heterosynaptic LTP in the rat hippocampal-entorhinal cortex slice preparation. Eur J Neurosci. 2007;25(1):251–8. doi: 10.1111/j.1460-9568.2006.05274.x. [DOI] [PubMed] [Google Scholar]
- Wu Z, Yamaguchi Y. Conserving total synaptic weight ensures one-trial sequence learning of place fields in the hippocampus. Neural Networks. 2006;19:547–63. doi: 10.1016/j.neunet.2005.06.048. [DOI] [PubMed] [Google Scholar]
- Yang SN, Tang YG, Zucker RS. Selective induction of LTP and LTD by postsynaptic [Ca2+]i elevation. J Neurophysiol. 1999;81:781–7. doi: 10.1152/jn.1999.81.2.781. [DOI] [PubMed] [Google Scholar]
- Yang Y, Calakos N. Presynaptic long-term plasticity. Front Synaptic Neurosci. 2013;5:8. doi: 10.3389/fnsyn.2013.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda H, Huang Y, Tsumoto T. Regulation of excitability and plasticity by endocannabinoids and PKA in developing hippocampus. Proc Natl Acad Sci. 2008;105(8):3106–11. doi: 10.1073/pnas.0708349105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung LC, Shouval HZ, Blais BS, Cooper LN. Synaptic homeostasis and input selectivity follow from a calcium-dependent plasticity model. Proc Natl Acad Sci U S A. 2004;101:14943–8. doi: 10.1073/pnas.0405555101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Denk W. Dendritic spines as basic functional units of neuronal integration. Nature. 1995;375:682–4. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]
- Zenke F, Hennequin G, Gerstner W. Synaptic plasticity in neural networks needs homeostasis with a fast rate detector. PLoS Comput Biol. 2013;9:e1003330. doi: 10.1371/journal.pcbi.1003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Linden DJ. The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci. 2003;4:885–900. doi: 10.1038/nrn1248. [DOI] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;40(5):971–82. doi: 10.1016/s0896-6273(03)00717-7. others. [DOI] [PubMed] [Google Scholar]
- Zhou YD, Acker CD, Netoff TI, Sen K, White JA. Increasing Ca2+ transients by broadening postsynaptic action potentials enhances timing-dependent synaptic depression. Proc Natl Acad Sci U S A. 2005;102:19121–5. doi: 10.1073/pnas.0509856103. [DOI] [PMC free article] [PubMed] [Google Scholar]