

Abstract
Background:
Calcium homeostasis is considered to be important in antineoplastic as well as in neurotoxic adverse effects of cisplatin.
Aims:
This study aimed to investigate the role of Ca2+ in cisplatin neurotoxicity in cultured rat dorsal root ganglia (DRG) cells.
Study Design:
Cell culture study.
Methods:
DRG cells prepared from 1-day old Sprague-Dawley rats were used to determine the role of Ca2+ in the cisplatin (10–600 μM) neurotoxicity. The cells were incubated with cisplatin plus nimodipine (1–3 μM), dizocilpine (MK-801) (1–3 μM) or thapsigargin (100–300 nM). Toxicity of cisplatinon DRG cells was determined by the MTT assay.
Results:
The neurotoxicity of cisplatin was significant when used in high concentrations (100–600 μM). Nimodipine (1 μM) but not MK-801 or thapsigargin prevented the neurotoxic effects of 200 μM of cisplatin.
Conclusion:
Voltage-dependent calcium channels may play a role in cisplatin neurotoxicity.
Keywords: Calcium, cisplatin, dorsal root ganglia, primary cell culture
Platinum drugs are used in the treatment of various types of cancers including ovarian, testicular, prostate, and colon cancer. Their well-known dose limiting adverse effect is peripheral neuropathy, which occurs in about 30–40% of patients (1,2) and usually forces patients to discontinue the treatment. After the discontinuation of treatment, the neurological dysfunction may gradually improve, but sometimes it may persist for a certain period, or be permanent (3). Cisplatin therapy and cytotoxicity are known to include interstrand and intrastrand adducts formation in DNA (4), which has been suggested to be a major mechanism in cisplatin-induced neuropathy (5). Although the mechanism of neuropathy is largely unclear, it has been reported that platinum compounds induce sensory deterioration and damage in large myelinated sensory fiber secondary to their accumulation in the dorsal root ganglia (DRG) (5,6). Cisplatin is not capable of crossing the blood-brain barrier. Thus, no direct exposure to toxic levels of the drug occurs in central nervous system neurons. However, it is expected that sensory neurons are exposed to high levels of the drug during chemotherapy due to the supply of dorsal root ganglia by fenestrated capillaries (7). Although glutation (8) and other thiol compounds (9), ACTH-like peptides (10), neurotrophic agents (11), paclitaxel, which is a microtubule stabilizer (12), and calcium channel antagonists (13) have been tested in vivo and in vitro to prevent cisplatin-induced neurotoxicity, none of them have been found to be effective. Screnci and McKeage (13) have previously reviewed nimodipine and some other agents for use against platinum toxicity and found no significant preventive effect. However, there are few studies on cisplatin toxicity investigating the effect of the inhibition of different systems such as channels or pumps, binding to sulfhydryl groups of enzymes, second messengers and signaling systems involved in calcium homeostasis (14,15).
Calcium homeostasis is considered to be important in both antineoplastic as well as neurotoxic adverse effects of cisplatin. Voltage-activating calcium channels are thought to be responsible for these neurotoxic side effects of cisplatin. There are several studies revealing that voltage-activating calcium channels are responsible from these neurotoxic effects (16–18). It has been reported that calcium channel blockers, in combination with the absence of normal extracellular calcium, increase the toxicity of cisplatin (19). All of these data suggest that calcium channels may be responsible for the neurotoxicity of the drug.
This study aimed to investigate the role of calcium in cisplatin neurotoxicity by using nimodipine (an L voltage-sensitive calcium channel antagonist), MK-801 (an NMDA channel antagonist) and thapsigargin (an inhibitor of efflux of calcium from intracellular stores) and also Ca2+ and Mg2+ in cultured rat dorsal root ganglion (DRG) neurons in order to assess the role of different systems involved in calcium homeostasis.
MATERIALS AND METHODS
The experiments were carried out according to the guidelines for the care and use of laboratory animals and consent was obtained from the local ethics committee (date, 24/05/2012 and protocol number, 273-A). Dorsal root ganglia (DRG) cells were used for the determination of cisplatin (at the concentrations of 10–600 μM) (Sigma Aldrich, St. Louis, Missouri, USA) neurotoxicity. Primary cultures of DRG were prepared from 1-day old Sprague-Dawley rats by using Dulbecco’s Modified-Eagle Medium (DMEM) (Sigma Aldrich, St. Louis, Missouri, USA). The toxic effects of cisplatin were evaluated by incubating the cells with cisplatin alone and with cisplatin plus nimodipine (1–3 μM), MK-801 (1–3 μM) (Sigma Aldrich, St. Louis, Missouri, USA) or thapsigargin (100–300 nM) (Sigma Aldrich, St. Louis, Missouri, USA) and Ca2+ and Mg2+ (1–100 μM). The MTT assay, which detects only the living but not dead cells, was used to detect the toxicity of DRG cells. This is a colorimetric assay for assessing cell viability and the signal generated in this assay is dependent on the degree of activation of the cells.
Primary cultures of DRG were prepared from 1-day old Sprague Dawley rats by modifying the methods used by Jong et al. (20) and Ulupinar et al. (21). Briefly, rats were decapitated and DRG were collected by microdissection into sterile calcium- and magnesium-free modified Hank’s balanced salt solution (HBSS) (Sigma Aldrich; Lonza, Belgium) (7,20,22). The dorsal root ganglia from each rat were digested with trypsin (0.25% trypsin-0.02% EDTA) (Gibco; OK, US) at 37°C for 10 min. Cells were dissociated by triturating with a Pasteur pipette and plated in culture plates coated with poly-D-lysine (Sigma Aldrich, St. Louis, Missouri, USA). Bases of the 25 cm3 polypropylene tissue culture flasks were covered with poly-D-lysine. For this procedure, 30000–70000 MW, 0.1 mg/mL poly-D-lysine was dissolved in phosphate buffered saline (PBS) (Hyclone; Logan, Utah, USA) and the flask bases were filled with the same solution. After incubation for 5 min at room temperature, the solution was removed and the flasks were washed with bidistilled water and then dried (23). The cells were incubated with DMEM solution supplemented with 10% fetal bovine serum, 2 mM glutamine, 50 μg/mL penicillin and streptomycin (Sigma Aldrich; St. Louis, Missouri, USA). The cultures were maintained at 37°C in an atmosphere of 5% CO2 for 1–2 days. Then, 10 μM of cytosine arabinoside (Cayman Chemicals, Ann Arbor; MI, USA) was added to the culture medium to prevent the replication of non-neuronal cells. Culture media was changed with DMEM solution twice a week and the neurons were used for neurotoxicity experiments following in vitro incubation for 8–10 days.
Cytotoxicity assay
The DRG cells were used for the determination of cisplatin neurotoxicity. Cells (5×103 cells/well) were incubated overnight in 96-well culture plates in drug-free DMEM medium and cisplatin was added to the wells with gradually increasing concentrations (10–600 μM) and incubated for 24 h. The toxic effects of cisplatin were evaluated by incubating the cells with the drug alone or in combination with nimodipine, MK-801 (1–3 μM) or thapsigargin (100–300 μM) (24) as well as with Ca2+ and Mg2+ (1–100 mM) for 24 hours. MTT assay was used for detection of the toxicity. Briefly, at the end of the incubation period, cells were further incubated with MTT solution (1 mg/ml) for 4 h at 37°C. After removing the incubation solution, cells were lysed with 150 μL of dimethylsulfoxide and optical density was measured at 540 nm as reference on an ELISA test system Multiscan EX; Franklin, Massachusetts, USA) (25,26). Each experimental group was tested at least three times. Inhibition of living cells (%) values were calculated by using the absorbance values on the formula of % inhibition=100 × (mean control-sample)/mean control. In this formula, mean (%) viability of the control group was accepted as 100% and cytotoxic effects of the different doses of cisplatin were determined to be statistically different in (%) inhibition values. On the other hand, because 10 μM of cisplatin did not result in a significant difference in (%) inhibition values, only the results for higher doses (50–600 μM) were included in the statistical analysis.
Data were analyzed by using SPSS 15.0 statistical package program (IBM SPSS Inc.; Chicago, IL, USA). The results were expressed as mean±SEM. Toxicity results for the drugs used were compared by unpaired Student’s t test. Concentration-dependent study of cisplatin was analyzed by one-way ANOVA test followed by post-hoc Dunnett test.
RESULTS
Cisplatin neurotoxicity on dorsal root ganglion neurons was concentration-dependent (Figure 1). We only included the results for 50–600 μM of cisplatin in the statistical analysis as submaximal concentrations to determine the effects of the agents involved in calcium homeostasis.
FIG. 1.
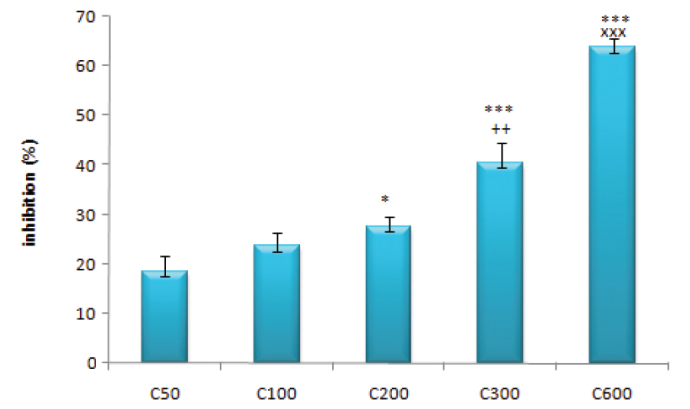
The concentration-dependent inhibitory effects of cisplatin (50–600 μM) on dorsal root ganglia
(*: p<0.05 compared with the C50 group; ***: p<0.001 compared with the C50 group; ++: p<0.01 compared with the C200 group; xxx: p<0.001 compared with the C300 group)
Nimodipine (1 μM) alone had an inhibitory effect similar to that observed with 50 μM of cisplatin, whereas the inhibitory effects of MK-801 (1 μM) and thapsigargin (100 nM) were higher than nimodipine (p<0.001) (Figure 2). When nimodipine was used with 50 μM of cisplatin, the inhibitory effect increased significantly (p<0.05). However, there were no significant changes when MK-801 or thapsigargin was used with or without cisplatin (p>0.05).
FIG. 2.
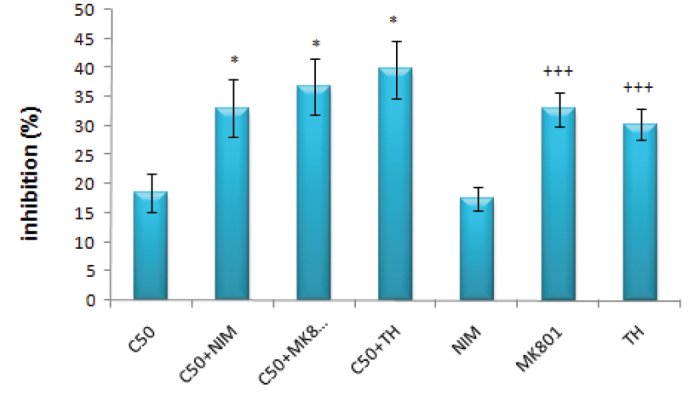
The inhibitory effects of 50 μM cisplatin and/or nimodipine, MK–801 (1 μM) and thapsigargin (100 nM) on DRG
(*: p<0.05 compared with the C50 group; +++: p<0.001 compared with the nimodipine group)
While the inhibitory effect of 1 μM of nimodipine±cisplatin was not significantly different from that of 100 μM of cisplatin, MK-801 and thapsigargin used in combination with 100 μM of cisplatin had significantly higher inhibitory effect compared to cisplatin alone (for both, p<0.05). On the other hand, the inhibitory effects of 3 μM of nimodipine (p<0.01) and MK-801 and thapsigargin used with 100 μM of cisplatin (for both; p<0.001) were significantly higher than that of 100 μM cisplatin alone. Although nimodipine alone at a concentration of 1 μM had an inhibitory effect similar to that of 100 μM of cisplatin alone (p>0.05), the inhibitory effects of MK-801 and thapsigargin were significantly higher compared to that of 100 μM of cisplatin (for both; p<0.001) (Figure 3).
FIG. 3.
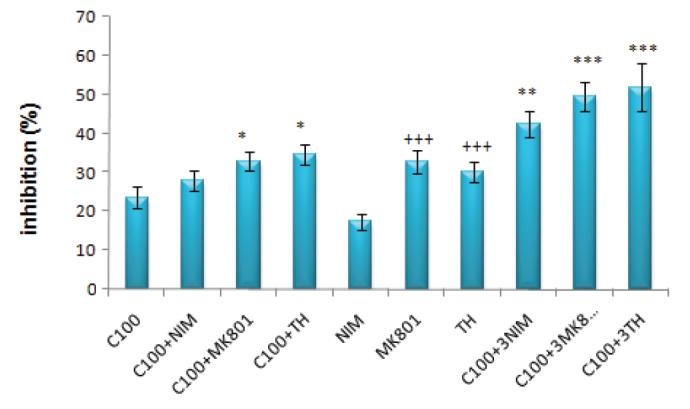
The inhibitory effects of 100 μM of cisplatin and/or NIM (nimodipin, 1 μM) & 3NIM (nimodipine, 3 μM), MK–801 (1 μM) & 3MK-801 (MK–801, 3 μM) and TH (thapsigargin, 100 nM) & 3TH (thapsigargin, 300 nM) on DRG
(*: p<0.05 compared with the C100 group; **: p<0.01 compared with the C100 group; ***: p<0.001 compared with the C100 group; +++: p<0.001 compared with the nimodipine group)
The inhibitory effect of nimodipine (1 μM) when used alone or with 200 μM of cisplatin was significantly lower than that of 200 μM of cisplatin alone (for both; p<0.05). On the other hand, MK-801 and thapsigargin used with 200 μM of cisplatin resulted in significantly higher inhibitory effects compared to 200 μM of cisplatin used alone (for both; p<0.001). When used alone, MK-801 and thapsigargin had a significantly higher inhibitory effect than nimodipine alone (for both; p<0.001), but not than cisplatin alone (p>0.05) (Figure 4).
FIG. 4.
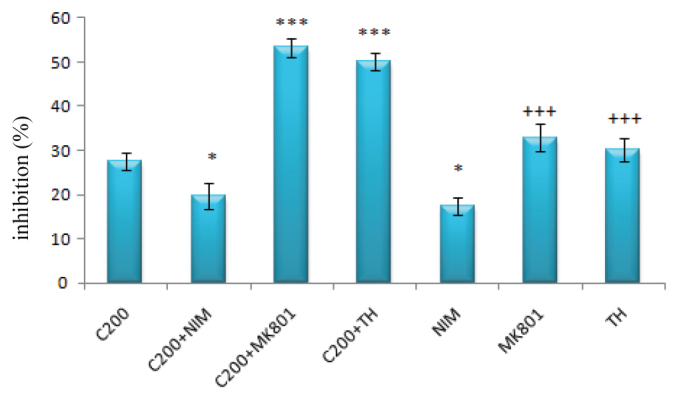
The inhibitory effects of 200 μM cisplatin and/or nimodipine, MK–801 (1 μM) and thapsigargin (100 nM) on DRG
(*: p<0.05 compared with the C200 group; ***: p<0.001 compared with the C200 group; +++: p<0.001 compared with the nimodipine group)
(NIM: nimodipine; MK-801: dizocilpine; DRG: dorsal root ganglia; TH: thapsigargin)
While the inhibitory effects of nimodipine (1 μM) were significantly lower (p<0.001) compared to the 300 μM of cisplatin group, the inhibitory effects of nimodipine (1 μM), MK-801 (1 μM) and thapsigargin (100 nM) used in combination with 300 μM of cisplatin resulted in no significant difference compared to the group treated only with 300 μM of cisplatin (Figure 5).
FIG. 5.
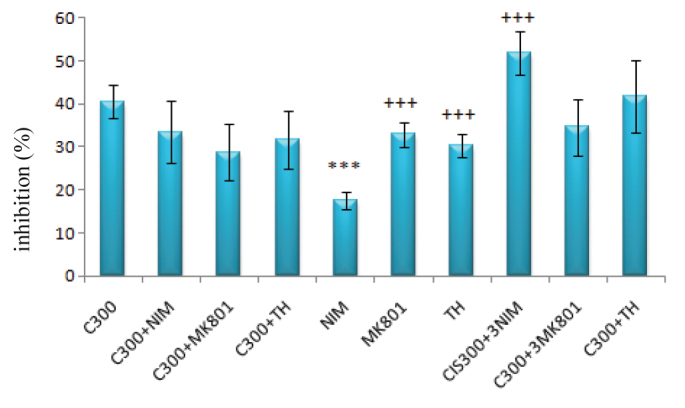
The inhibitory effects of 300 μM cisplatin and/or NIM (nimodipin, 1 μM) & 3NIM (nimodipine, 3 μM), MK–801 (1 μM) & 3MK-801 (MK–801, 3 μM) and TH (thapsigargin, 100 nM) & 3TH (thapsigargin 300 nM) on DRG
(***: p<0.001 compared with the C300 group; +++: p<0.001 compared with the nimodipine group)
Nimodipine (1 μM), MK-801 (1 μM) and thapsigargin (100 nM) alone had a significantly lower inhibitory effect compared to cisplatin alone at a concentration of 600 μM (for all; p<0.001). However, the inhibitory effects were not significantly different from that of 600 μM of cisplatin alone when all three agents were used in combination with 600 μM of cisplatin (for all; p>0.05). However, when the concentration of nimodipine was increased by three times, cisplatin toxicity increased significantly (p<0.001), whereas when thapsigargin concentration was increased three times, the neurotoxicity of cisplatin decreased significantly (p<0.05) (Figure 6).
FIG. 6.
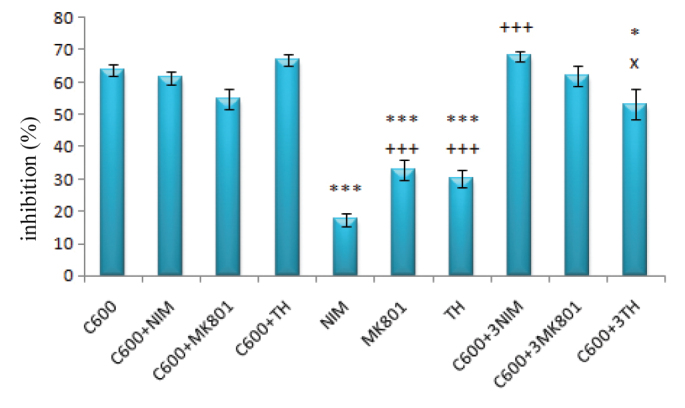
The inhibitory effects of 600 μM cisplatin and/or NIM (nimodipin, 1 μM) & 3NIM (nimodipine, 3 μM), MK–801 (1 μM) & 3MK-801 (MK–801, 3 μM) and TH (thapsigargin, 100 nM) & 3TH (thapsigargin 300 nM) on DRG
(***: p<0.001 and *: p<0.05 compared with the C600 group; +++ p<0.001 compared with the nimodipine group; x: p<0.05 compared with the C600+TH group)
As seen in Figure 7, although the absorbance values at 450 nm were significantly lower in the groups treated with 200 μM of cisplatin alone and 1 μM of Ca2+ alone compared to the control values (for both; p<0.05), no significant differences were obtained when Ca2+ was used alone at the concentrations of 10 and 100 μM or 200 μM of cisplatin was used with Ca2+ (1, 10 and 100 μM).
FIG. 7.
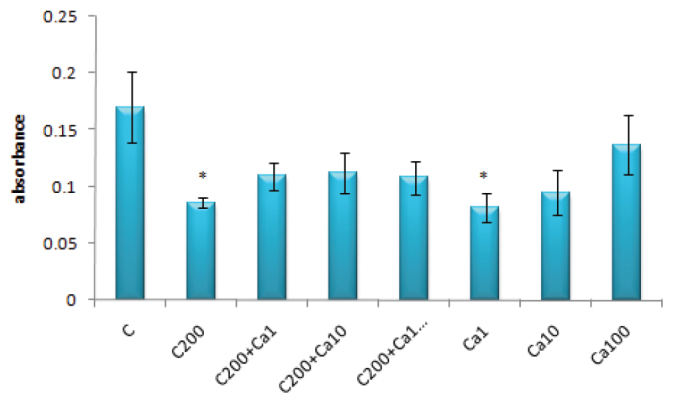
The absorbance values for 200 μM of cisplatin and/or Ca2+ (1–100 μM) on DRG
(*: p<0.05 compared with the control group)
With regard to the effects of magnesium ion, 200 μM of cisplatin alone or with Mg2+ (1, 10 and 100 μM) resulted in significantly lower absorbance values compared to the controls (for all; p<0.05). Although the absorbance value for 1 μM of Mg2+ was not significantly different to that of controls, 10 μM of Mg2+ resulted in significantly higher absorbance values than 200 μM of cisplatin (p<0.01) and 100 μM of Mg2+ had an absorbance value that was significantly higher to both controls (p<0.01) and 200 μM of cisplatin (p<0.001) (Figure 8).
FIG. 8.
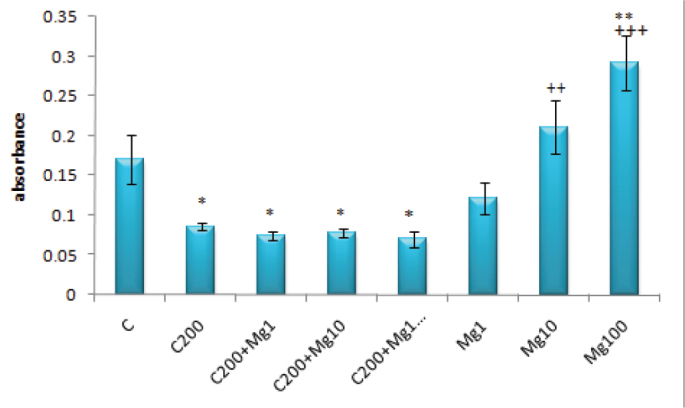
The absorbance values for 200 μM cisplatin and/or Mg2+ (1–100 μM) on DRG
(*: p<0.05 and **: p<0.01 compared with the control group; ++: p<0.01 and +++: p<0.001 compared with the C200 group)
DISCUSSION
The present study demonstrates that calcium homoeostasis is one of the important factors in cisplatin-induced neurotoxicity. The neurotoxicity of cisplatin was significant when used at high concentrations (200–600 μM), because there is no difference between 50 and 100 μM of cisplatin Due to its accumulation in DRG, we used high concentrations of cisplatin up to 600 μM. Nimodipine (1 μM) prevented the neurotoxic effects of cisplatin only at 200 μM, whereas a beneficial effect was not observed with MK-801 or thapsigargin. However, the neurotoxicity of high concentration of cisplatin (600 μM) was only decreased by high concentration of thapsigargin (300 nM). These results suggest that calcium ions may have a modulating role in cisplatin neurotoxicity.
Peripheral neuropathy is a common adverse effect of several antineoplastic drugs including cisplatin. This can be extremely painful and a treatment-limiting phenomenon occurring with this potentially beneficial drug. The occurrence and severity of neuropathy is dependent on many factors related to the usage of drug such as dose, duration of infusion, co-medications, alcohol abuse, and diabetes mellitus. Although the mechanism of neuropathy is unclear, cisplatin was shown to result in the formation of intrastrand and interstrand adducts in DNA (4,5). This effect of cisplatin has been suggested to be one of the major mechanisms responsible for neurotoxicity (5,27). Additionally, the increased generation of reactive oxygen species, nitric oxide, inflammation, mitochondrial dysfunction, protein kinase C, mitogen activated protein kinase (MAPK), calcium-activated proteases such as caspases, neuropeptides, NMDA receptors, calcium, potassium and sodium channels have all been suggested to have a role in cisplatin-induced peripheral neuropathy (28). To target these mechanisms, several neuroprotective approaches such as antioxidants (29), thiol compounds, neurotrophic factors, calcium channel antagonists (13), vitamin E, calcium and magnesium infusions, anti-convulsants (30) glutamine and erythropoietin (29) have been tested in cisplatin-induced neurotoxicity in several experimental models and clinical applications. Unfortunately, there has been no development regarding the use of these applications effectively yet in human treatment and their rationales to prevent toxicity may be only empiric.
It is well-known that cisplatin affects calcium homeostasis and its desired and undesired effects involve the change of calcium homeostasis (30). It has been reported that the clinical and side effects of cisplatin could be changed by altering the calcium homeostasis. It has been shown that there is a synergistic effect between nifedipine and cisplatin in the antitumor effect of cisplatin on multidrug-resistant GB-1 cells lacking Ca2+-dependent endonuclease, and nifedipine subsequently induces apoptosis by interacting with an uncharacterized functional site other than the calcium channel on GB-1 cells (19). Supplemental treatments with calcium and ergocalciferol plus elemental calcium have also been demonstrated to protect against severe gastrointestinal toxicity and nephrotoxicity (31).
Interestingly, intracellular administration of bis-(o-aminophenoxy)-N,N,N′,N′-tetraacetic acid (BAPTA), a chelator of calcium ions, could imitate the effect of oxaliplatin and the effects of this drug on voltage-gated sodium channels may partly cause the neuronal damage induced by oxaliplatin. Therefore, it has been suggested that oxaliplatin has the capacity to change the voltage-gated sodium channels through a pathway involving calcium ions (32).
Interestingly, 1 μM nimodipine showed biphasic effect on cisplatin-induced neurotoxicity (Figures 2–6) and its significant protective activity was on 200 μM of cisplatin (Figure 4). Although 1 μM of MK-801 and 100 nM thapsigargin increased cisplatin neurotoxicity at lower concentrations of cisplatin (50–200 μM), there was no significant interaction at higher concentrations (300–600 μM). On the other hand, 300 nM of thapsigargin significantly decreased but 3 μM nimodipine significantly increased the neurotoxicity of 600 μM of cisplatin (Figure 6). In addition, 1 μM MK-801 and 200 μM cisplatin synergistically increased the neurotoxicity (Figure 4), but it was not observed with the other concentrations used. Thus, 200 μM cisplatin seems to be a critical concentration for calcium ions to be effective on neurotoxicity. Taken together, all of these results suggest that cisplatin (at lower concentrations) may activate L-type voltage-gated calcium channels and increase calcium accumulation in the cells, thus playing a role in cisplatin-induced neurotoxicity. On the other hand, at higher concentrations of cisplatin, the efflux of calcium from internal stores seems to play a role in the toxicity of the drug. Nimodipine prevents the increase in cytosolic calcium. This suggests that cisplatin initially increases the cytosolic calcium by L-type voltage-dependent calcium channels with the subsequent additional efflux from internal sources. In regard to these results and the use of calcium and magnesium infusion for preventing cisplatin neurotoxicity (2,29), we tested the effects of calcium and magnesium ions on cisplatin-induced neurotoxicity. Calcium (1–100 μM) did not significantly change cisplatin neurotoxicity and 1 μM concentration of calcium had inhibitory effects as much as that of 200 μM cisplatin (Figure 7). However, there was no significant change between control and the groups of calcium+cisplatin. Thus, increasing extracellular calcium concentration seems to have a neuroprotective effect. On the other hand, magnesium (1–100 μM) did not change cisplatin neurotoxicity, but when it was used alone at concentrations of 10 and 100 μM, cell growth was significantly increased (Figure 8). It has been previously demonstrated that increasing the concentration of extracellular calcium facilitates sodium channel closing (33). Thus, it has been suggested that this effect would decrease the peripheral neurotoxicity of platinum compounds without affecting the antitumor efficacy.
It is well-known that the induction of neurotoxicity by cisplatin occurs through multiple actions, including aberrant re-entry into the cell cycle, apoptosis through binding to mitochondrial DNA, inhibition of transcription and synthesis of mitochondrial proteins and loss of mitochondrial functions (5,7,27). It is also known that cisplatin causes both apoptotic cell death and axonal damage by inducing early mitochondrial dysfunction with a loss of membrane potential (34). This suggests that mitochondria may play a crucial role in neurodegenerative processes such as diabetic neuropathy (35). Frataxin is a fundamental mitochondrial protein with antioxidant and chaperone features and increases mitochondrial membrane potential (36). Overexpression of this protein induces antioxidant cellular effects by activating the glutathione peroxidase, provides contribution to cell resistance against oxidative stress and supports the cell survival (19). It has been demonstrated that cisplatin decreases the expression of frataxin and it is suggested that frataxin may play a key role in neuroprotective mechanisms to cisplatin toxicity (34). Recently, it has been indicated that cisplatin forms adducts with mitochondrial DNA similarly as nuclear DNA and that these adducts inhibit mitochondrial DNA replication and transcription and evoke morphological alterations within mitochondria (37). Thus, this leads to energy failure and provides a different pathway for neuronal dysfunction.
The certain mechanism of cisplatin-induced neurotoxicity is still not fully understood. Modulation of the intracellular calcium concentration plays an important role in cell proliferation. It has been reported that cisplatin causes a time- and concentration-dependent apoptotic effect, which could be one reason for the results of increased calcium concentration (16). Mitochondria play an essential role in sustaining the homeostasis of intracellular calcium with their extensive buffering capability. Impairment of mitochondrial calcium homeostasis may also be responsible for cisplatin-induced neuropathy. Besides, oxaliplatin has been demonstrated to increase the Na+ current (38) and mexiletine, the sodium channel blocker, has been showed to relieve the oxaliplatin-induced neuropathic pain (39). Moreover, it has been reported that oxaliplatin causes the reduced expression of TREK1, TRAAK type of potassium channels and enhanced expression of hyperpolarization-activated channels, leading to over-excitability of the nerve fibers.
In conclusion, the outcomes of this study suggest that calcium homeostasis is likely to play a role in cisplatin neurotoxicity. It seems that calcium is an important ion for cell growth, because the three agents involved in calcium homeostasis had significant inhibitory effects on DRG. Further research is required to explore the role of ion channels on cisplatin-induced neurotoxicity.
Acknowledgments
The authors are alone responsible for the content and writing of the paper.
Footnotes
Ethics Committee Approval: Ethics committee approval was received for this study from the ethics committee of Eskişehir Osmangazi University School of Medicine. (24/05/2012 and protocol number, 273-A9).
Informed Consent: N/A.
Peer-review: Externally peer-reviewed.
Author contributions: Concept - K.E.; Design - K.E. Supervision - K.E. Resource - K.E. S.Y.; Materials - K.E. S.Y., Ç.Ü.; Data Collection and/or Processing - S.Y., Ç.Ü.; Analysis and/or Interpretation - B.K., E.Y.; Literature Search - K.E., S.Y.; Writing - K.E., S.Y.; Critical Reviews - K.E.
Conflict of Interest: There are no conflict of interest to declare.
Financial Disclosure: This study was financially supported by the Scientific Research Projects Unit of Eskişehir Osmangazi University under the project number of 2011/11003.
REFERENCES
- 1.Windebank AJ, Grisold W. Chemotherapy-induced neuropathy. J Peripher Nerv Syst. 2008;13:27–46. doi: 10.1111/j.1529-8027.2008.00156.x. http://dx.doi.org/10.1111/j.1529-8027.2008.00156.x. [DOI] [PubMed] [Google Scholar]
- 2.Wolf S, Barton D, Kottschade L, Grothey A, Loprinzi C. Chemotherapy-induced peripheral neuropathy: prevention and treatment strategies. Eur J Cancer. 2008;44:1507–15. doi: 10.1016/j.ejca.2008.04.018. http://dx.doi.org/10.1016/j.ejca.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 3.von Schlippe M, Fowler CJ, Harland SJ. Cisplatin neurotoxicity in the treatment of metastatic germ cell tumour: time course and prognosis. Br J Cancer. 2001;85:823–6. doi: 10.1054/bjoc.2001.2006. http://dx.doi.org/10.1054/bjoc.2001.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu F, Lin X, Okuda T, Howell SB. DNA polymerase zeta regulates cisplatin cytotoxicity, mutagenicity, and the rate of development of cisplatin resistance. Cancer Res. 2004;64:8029–35. doi: 10.1158/0008-5472.CAN-03-3942. http://dx.doi.org/10.1158/0008-5472.CAN-03-3942. [DOI] [PubMed] [Google Scholar]
- 5.McDonald ES, Randon KR, Knight A, Windebank AJ. Cisplatin preferentially binds to DNA in dorsal root ganglion neurons in vitro and in vivo: a potential mechanism for neurotoxicity. Neurobiol Dis. 2005;18:305–13. doi: 10.1016/j.nbd.2004.09.013. http://dx.doi.org/10.1016/j.nbd.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 6.Ocean AJ, Vahdat LT. Chemotherapy-induced peripheral neuropathy: pathogenesis and emerging therapies. Support Care Cancer. 2004;12:619–25. doi: 10.1007/s00520-004-0657-7. http://dx.doi.org/10.1007/s00520-004-0657-7. [DOI] [PubMed] [Google Scholar]
- 7.Gill JS, Windebank AJ. Cisplatin-induced apoptosis in rat dorsal root ganglion neurons is associated with attempted entry into the cell cycle. J Clin Invest. 1998;101:2842–50. doi: 10.1172/JCI1130. http://dx.doi.org/10.1172/JCI1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavaletti G, Minoia C, Schieppati M, Tredici G. Protective effects of glutathione on cisplatin neurotoxicity in rats. Int J Radiat Oncol Biol Phys. 1994;29:771–6. doi: 10.1016/0360-3016(94)90565-7. http://dx.doi.org/10.1016/0360-3016(94)90565-7. [DOI] [PubMed] [Google Scholar]
- 9.Capizzi RL. Protection of normal tissues from the cytotoxic effects of chemotherapy by amifostine (Ethyol): clinical experiences. Semin Oncol. 1994;21:8–15. [PubMed] [Google Scholar]
- 10.Hol EM, Gispen WH, Bar PR. ACTH-related peptides: receptors and signal transduction systems involved in their neurotrophic and neuroprotective actions. Peptides. 1995;16:979–93. doi: 10.1016/0196-9781(95)00017-e. http://dx.doi.org/10.1016/0196-9781(95)00017-E. [DOI] [PubMed] [Google Scholar]
- 11.Konings PN, Makkink WK, van Delft AM, Ruigt GS. Reversal by NGF of cytostatic drug-induced reduction of neurite outgrowth in rat dorsal root ganglia in vitro. Brain Res. 1994;640:195–204. doi: 10.1016/0006-8993(94)91873-2. http://dx.doi.org/10.1016/0006-8993(94)91873-2. [DOI] [PubMed] [Google Scholar]
- 12.McKeage MJ, Haddad GG, Ding L, Galettis P, Screnci D, Zhuang L, et al. Neuroprotective interactions in rats between paclitaxel and cisplatin. Oncol Res. 1999;11:287–93. [PubMed] [Google Scholar]
- 13.Screnci D, McKeage MJ. Platinum neurotoxicity: clinical profiles, experimental models and neuroprotective approaches. Journal of inorganic biochemistry. 1999;77:105–10. doi: 10.1016/s0162-0134(99)00135-x. http://dx.doi.org/10.1016/S0162-0134(99)00135-X. [DOI] [PubMed] [Google Scholar]
- 14.Piccolini VM, Bottone MG, Bottiroli G, De Pascali SA, Fanizzi FP, Bernocchi G. Platinum drugs and neurotoxicity: effects on intracellular calcium homeostasis. Cell Biol Toxicol. 2013;29:339–53. doi: 10.1007/s10565-013-9252-3. http://dx.doi.org/10.1007/s10565-013-9252-3. [DOI] [PubMed] [Google Scholar]
- 15.Avan A, Postma TJ, Ceresa C, Avan A, Cavaletti G, Giovannetti E, et al. Platinum-Induced Neurotoxicity and Preventive Strategies: Past, Present, and Future. Oncologist. 2015;20:411–32. doi: 10.1634/theoncologist.2014-0044. http://dx.doi.org/10.1634/theoncologist.2014-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cummings BS, Gelasco AK, Kinsey GR, McHowat J, Schnellmann RG. Inactivation of endoplasmic reticulum bound Ca2+-independent phospholipase A2 in renal cells during oxidative stress. J Am Soc Nephrol. 2004;15:1441–51. doi: 10.1097/01.asn.0000127923.57438.ec. http://dx.doi.org/10.1097/01.ASN.0000127923.57438.EC. [DOI] [PubMed] [Google Scholar]
- 17.Tomaszewski A, Busselberg D. Cisplatin modulates voltage gated channel currents of dorsal root ganglion neurons of rats. Neurotoxicology. 2007;28:49–58. doi: 10.1016/j.neuro.2006.07.005. http://dx.doi.org/10.1016/j.neuro.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 18.Jaffe LF. A calcium-based theory of carcinogenesis. Advances in cancer research. 2005;94:231–63. doi: 10.1016/S0065-230X(05)94006-2. http://dx.doi.org/10.1016/S0065-230X(05)94006-2. [DOI] [PubMed] [Google Scholar]
- 19.Kondo S, Yin D, Morimura T, Kubo H, Nakatsu S, Takeuchi J. Combination therapy with cisplatin and nifedipine induces apoptosis in cisplatin-sensitive and cisplatin-resistant human glioblastoma cells. Br J Cancer. 1995;71:282–9. doi: 10.1038/bjc.1995.57. http://dx.doi.org/10.1038/bjc.1995.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jong NN, Nakanishi T, Liu JJ, Tamai I, McKeage MJ. Oxaliplatin transport mediated by organic cation/carnitine transporters OCTN1 and OCTN2 in overexpressing human embryonic kidney 293 cells and rat dorsal root ganglion neurons. J Pharmacol Exp Ther. 2011;338:537–47. doi: 10.1124/jpet.111.181297. http://dx.doi.org/10.1124/jpet.111.181297. [DOI] [PubMed] [Google Scholar]
- 21.Ulupinar E, Erzurumlu RS. Peripheral target-specific influences on embryonic neurite growth vigor and patterns. J Comp Neurol. 1998;399:427–39. doi: 10.1002/(sici)1096-9861(19981005)399:4<427::aid-cne1>3.0.co;2-3. http://dx.doi.org/10.1002/(SICI)1096-9861(19981005)399:4<3c427::AID-CNE1>3e3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 22.Leach MK, Feng ZQ, Gertz CC, Tuck SJ, Regan TM, Naim Y, et al. The culture of primary motor and sensory neurons in defined media on electrospun poly-L-lactide nanofiber scaffolds. J Vis Exp. 2011 doi: 10.3791/2389. http://dx.doi.org/10.3791/2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duzenli S, Bakuridze K, Gepdiremen A. The effects of ruthenium red, dantrolene and nimodipine, alone or in combination, in NMDA induced neurotoxicity of cerebellar granular cell culture of rats. Toxicol In Vitro. 2005;19:589–94. doi: 10.1016/j.tiv.2005.03.007. http://dx.doi.org/10.1016/j.tiv.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 24.Tjiattas L, Ortiz DO, Dhivant S, Mitton K, Rogers E, Shea TB. Folate deficiency and homocysteine induce toxicity in cultured dorsal root ganglion neurons via cytosolic calcium accumulation. Aging Cell. 2004;3:71–6. doi: 10.1111/j.1474-9728.2004.00086.x. http://dx.doi.org/10.1111/j.1474-9728.2004.00086.x. [DOI] [PubMed] [Google Scholar]
- 25.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. http://dx.doi.org/10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 26.Scudiero DA, Shoemaker RH, Paull KD, Monks A, Tierney S, Nofziger TH, et al. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res. 1988;48:4827–33. [PubMed] [Google Scholar]
- 27.Ta LE, Espeset L, Podratz J, Windebank AJ. Neurotoxicity of oxaliplatin and cisplatin for dorsal root ganglion neurons correlates with platinum-DNA binding. Neurotoxicology. 2006;27:992–1002. doi: 10.1016/j.neuro.2006.04.010. http://dx.doi.org/10.1016/j.neuro.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 28.Jaggi AS, Singh N. Mechanisms in cancer-chemotherapeutic drugs-induced peripheral neuropathy. Toxicology. 2012;291:1–9. doi: 10.1016/j.tox.2011.10.019. http://dx.doi.org/10.1016/j.tox.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 29.Beijers AJ, Jongen JL, Vreugdenhil G. Chemotherapy-induced neurotoxicity: the value of neuroprotective strategies. Neths j med. 2012;70:18–25. [PubMed] [Google Scholar]
- 30.Amptoulach S, Tsavaris N. Neurotoxicity caused by the treatment with platinum analogues. Chemother Res Pract. 2011;2011:843019. doi: 10.1155/2011/843019. http://dx.doi.org/10.1155/2011/843019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meara DJ, Johnson B, Wang Y, Aggarwal SK. Role of calcium in modulation of toxicities due to cisplatin and its analogs: a histochemical approach. Anticancer Drugs. 1997;8:988–99. doi: 10.1097/00001813-199711000-00010. http://dx.doi.org/10.1097/00001813-199711000-00010. [DOI] [PubMed] [Google Scholar]
- 32.Grolleau F, Gamelin L, Boisdron-Celle M, Lapied B, Pelhate M, Gamelin E. A possible explanation for a neurotoxic effect of the anticancer agent oxaliplatin on neuronal voltage-gated sodium channels. J Neurophysiol. 2001;85:2293–7. doi: 10.1152/jn.2001.85.5.2293. [DOI] [PubMed] [Google Scholar]
- 33.Armstrong CM, Cota G. Calcium block of Na+ channels and its effect on closing rate. Proc Natl Acad Sci USA. 1999;96:4154–7. doi: 10.1073/pnas.96.7.4154. http://dx.doi.org/10.1073/pnas.96.7.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melli G, Taiana M, Camozzi F, Triolo D, Podini P, Quattrini A, et al. Alpha-lipoic acid prevents mitochondrial damage and neurotoxicity in experimental chemotherapy neuropathy. Exp Neurol. 2008;214:276–84. doi: 10.1016/j.expneurol.2008.08.013. http://dx.doi.org/10.1016/j.expneurol.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 35.Leinninger GM, Edwards JL, Lipshaw MJ, Feldman EL. Mechanisms of disease: mitochondria as new therapeutic targets in diabetic neuropathy. Nat Clin Pract Neurol. 2006;2:620–8. doi: 10.1038/ncpneuro0320. http://dx.doi.org/10.1038/ncpneuro0320. [DOI] [PubMed] [Google Scholar]
- 36.Shoichet SA, Baumer AT, Stamenkovic D, Sauer H, Pfeiffer AF, Kahn CR, et al. Frataxin promotes antioxidant defense in a thiol-dependent manner resulting in diminished malignant transformation in vitro. Hum Mol Genet. 2002;11:815–21. doi: 10.1093/hmg/11.7.815. http://dx.doi.org/10.1093/hmg/11.7.815. [DOI] [PubMed] [Google Scholar]
- 37.Podratz JL, Knight AM, Ta LE, Staff NP, Gass JM, Genelin K, et al. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol Dis. 2011;41:661–8. doi: 10.1016/j.nbd.2010.11.017. http://dx.doi.org/10.1016/j.nbd.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adelsberger H, Quasthoff S, Grosskreutz J, Lepier A, Eckel F, Lersch C. The chemotherapeutic oxaliplatin alters voltage-gated Na(+) channel kinetics on rat sensory neurons. Eur J Pharmacol. 2000;406:25–32. doi: 10.1016/s0014-2999(00)00667-1. http://dx.doi.org/10.1016/S0014-2999(00)00667-1. [DOI] [PubMed] [Google Scholar]
- 39.Egashira N, Hirakawa S, Kawashiri T, Yano T, Ikesue H, Oishi R. Mexiletine reverses oxaliplatin-induced neuropathic pain in rats. J Pharmacol Sci. 2010;112:473–6. doi: 10.1254/jphs.10012sc. http://dx.doi.org/10.1254/jphs.10012SC. [DOI] [PubMed] [Google Scholar]