

Abstract
DNA polymerases exhibit a surprising tolerance for analogs of deoxyribonucleoside triphosphates (dNTPs), despite the enzymes’ highly evolved mechanisms for the specific recognition and discrimination of native dNTPs. Here, individual DNA Polymerase I Klenow Fragment (KF) molecules were tethered to a single-walled carbon nanotube field-effect transistor (SWCNT-FET) to investigate accommodation of dNTP analogs with single-molecule resolution. Each base incorporation accompanied a change in current with its duration defined by τclosed. Under Vmax conditions, the average time of τclosed was similar for all analog and native dNTPs (0.2 to 0.4 ms), indicating no kinetic impact on this step due to analog structure. Accordingly, the average rates of dNTP analog incorporation were largely determined by durations with no change in current defined by τopen, which includes molecular recognition of the incoming dNTP. All α-thio-dNTPs were incorporated more slowly, at 40 to 65% of the rate for the corresponding native dNTPs. During polymerization with 6-Cl-2APTP, 2-thio-dTTP, or 2-thio-dCTP, the nanocircuit uncovered an alternative conformation represented by positive current excursions that do not occur with native dNTPs. A model consistent with these results invokes rotations by the enzyme’s O-helix; this motion can test the stability of nascent base pairs using non-hydrophilic interactions, and is allosterically coupled to charged residues near the site of SWCNT attachment. This model with two opposing O-helix motions differs from the previous report in which all current excursions were solely attributed to global enzyme closure and covalent bond formation. The results suggest the enzyme applies a dynamic stability-checking mechanism for each nascent base pair.
Graphical Abstract
INTRODUCTION
To ensure their survival, all organisms rely on DNA polymerases to correctly recognize deoxynucleoside triphosphates (dNTPs) and successfully catalyze their incorporation into new strands of DNA. The fidelity of DNA polymerases relies predominantly on the correct geometry of the nascent base pair.1–3 By forming base pairs with a shape and size similar to the canonical A•T and G•C base pairs, isosteric and hydrophobic dNTP analogs incapable of hydrogen bonding with native, complementary bases have been successfully incorporated by DNA polymerases.4–6 Studies with other dNTP analogs substantiate a requirement for shape complementarity7–9 and demonstrate that additional factors, including stereochemistry, sterics, electronic effects, base stacking, and hydrophobic interactions contribute to the fidelity of DNA polymerases.8,10–15
Experiments with dNTP analogs can also illustrate the prerequisite for snug fit in the DNA polymerase active site.8,12,14 For example, recent crystal structures of the A-family DNA polymerase fragment KlenTaq demonstrate a mutually induced fit to a Watson-Crick geometry by both the nascent base pair and the polymerase during accommodation of unnatural or non-native pairing,16 illustrating plasticity in the enzyme’s molecular recognition. Specifically, the O-helix of A-family DNA polymerases adopts multiple distinct conformations in the active site to discriminate against imperfect substrates.17 For example, O-helix residues F762 and Y766 in the DNA Polymerase I Klenow Fragment (KF) active site discriminate amongst dNTPs through base specificity to maintain fidelity,18–20 and are involved in the active site tightness around the nascent base pair.6,21
Detailed evaluation of unnatural dNTP polymerization beyond a single base pair could provide accurate kinetic information about this non-native polymerase activity, and uncover general insights into enzyme molecular recognition and specificity. As reported here, such information can be elucidated by single-molecule techniques; observation of intermediate steps and transition states are otherwise lost through averaging in ensemble populations.22,23 DNA polymerization experiments employing single-molecule Forster resonance energy transfer (smFRET) have revealed conformational flexibility and insights into the fidelity mechanism of KF.24–27 Additionally, the observed kinetic and thermodynamic differences between correct and mismatched dNTP-DNA-polymerase complexes quantify the driving forces for correct nucleotide incorporation into DNA templates.26–28
The experiments reported here differ from previous fluorescence-based studies of DNA polymerases. Despite its power to capture new information about enzyme dynamics, FRET requires a fluorescently labeled protein and/or substrate. Photobleaching and the flux of photons between fluorophores limit both the duration and time resolution, respectively, of FRET and smFRET experiments. Furthermore, smFRET experiments typically examine single dNTP or dideoxy-NTP (ddNTP) incorporation events. Such experiments offer the important advantage of avoiding the potentially distorted homopolymeric dsDNA products. In the experiments reported here, however, homopolymeric templates must be used to determine the kinetics of individual dNTPs and their analogs in multiple turnover events. The approach enables these first single-molecule studies to explore unnatural dNTP analog incorporation beyond the base recognition step, expanding this evaluation to processive incorporation events.
Recently, we described a new approach to single-molecule enzymology and applied it to three enzymes. In this technique, an individual protein is bioconjugated to a single-walled carbon nanotube field effect transistor (SWCNT-FET; Figure 1a). The approach uncovered new insights into the number of steps, kinetic parameters, and processivity of the well-studied T4 lysozyme.29,30 Engineered lysozyme variants revealed that significant differences in electronic signal resulted from the electrostatic charges of side chains close to the attachment site.31 Tremendously dynamic rates of cAMP-dependent protein kinase A (PKA) demonstrated the role of protein kinases as a highly variable molecular switch.32 KF conjugated to SWCNT-FETs uncovered significant kinetic and conformational differences between the enzyme-catalyzed formation of A•T/T•A and G•C/C•G sets of base pairs.33
Figure 1.
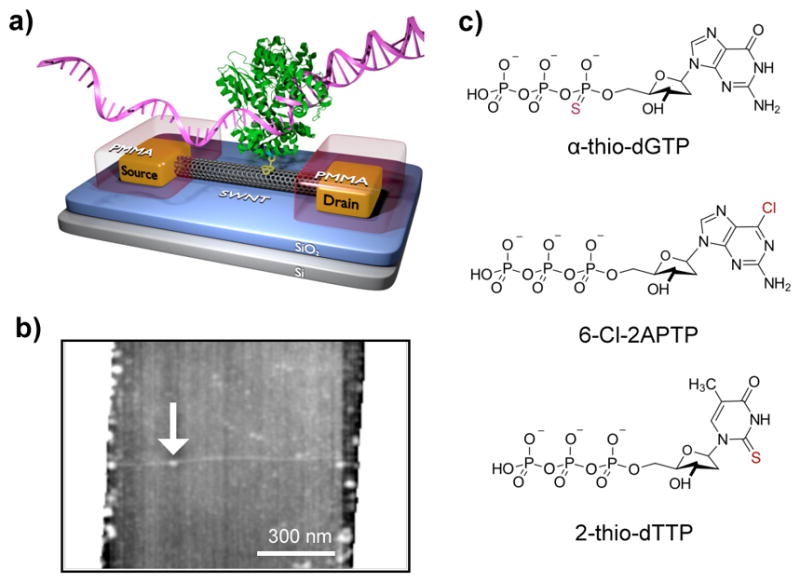
A single KF nanocircuit and the chemically modified dNTPs tested for their incorporation by KF. (a) A schematic diagram of a single-walled carbon nanotube field effect transistor (SWCNT-FET) non-covalently bio-conjugated to a single molecule of DNA polymerase I (KF) through a single cysteine introduced in the “fingers” subdomain. A pyrene-maleimide linker (yellow) adhered to the SWCNT-FET through π-π stacking and covalently attached to the single cysteine to immobilize the KF. The SWCNT-FET was grown on SiO2, connected to source and drain metal electrodes, and passivated with a polymer (PMMA, red). (b) Atomic force microscopy shows the 1–2 nm diameter of the SWCNT FET with a single KF attachment (7 nm, arrow). (c) Chemical structures of representative dNTP analogs with chemical modifications from the native dNTPs highlighted in red.
The sensitivity of the KF nanocircuit to small differences in base pair recognition is surprising since the native Watson-Crick base pairs have similar sizes. By coupling to key active site residues, the location of attachment at position 790 in the “fingers” subdomain can allow transfer of active site dynamics to charged surface residues near the SWCNT; though the approach reported here requires attachment to the SWCNT, conjugation at this position causes only minimal perturbation of KF’s activity compared to solution-phase DNA polymerization assays.33 Combining the SWCNT-FET sensitivity with KF’s malleable active site recognition, we hypothesized that the KF nanocircuit technique might also be responsive to the unnatural structures of dNTP analogs. Translation of the associated small conformational changes during base recognition and subsequent incorporation into a reliable measurement could reveal new aspects of the roles for base pair structure, both steric and electronic, during DNA polymerization.
Here, the SWCNT-FET technique was used to examine KF kinetic and conformational dynamics during native and analog dNTP incorporation. Using dNTP analogs with phosphodiester or nucleobase modifications, DNA polymerization was monitored with single base pair resolution and then statistically analyzed to reveal differences in incorporation kinetics and conformations. Analogs significantly altered the time required for nucleotide recognition, but not the kinetics of KF’s closed conformation. However, signals revealed a highly plastic aspect of the closure as the enzyme accommodated various analog base pairs with different structures, hydrogen bonding patterns, and electron densities.
EXPERIMENTAL SECTION
SWCNT-FETs were fabricated29 and functionalized with a single-cysteine variant of exonuclease-deficient KF (D355A/E357A/L790C/C907S).33,34 Purification of KF to >95% ensured its homogeneity (Figure S1). A fluorescence-based assay confirmed activity of the bulk enzyme prior to attachment (Figure S2).33,35 Attachment of KF to SWCNT-FETs was accomplished by soaking the devices in a solution of N-(1-pyrenyl)maleimide (1 mM in ethanol, 30 min), followed by incubation with KF (300 nM KF in a standard KF activity buffer of 20 mM Tris, 50 mM NaCl, 10 mM MgCl2, 100 μM TCEP, pH 8.0). Atomic force microscopy after data collection confirmed attachment of a single KF molecule to each device (Figure 1b). Such devices are referred to simply as KF nanocircuits.
Experiments used the homopolymeric templates poly(dA)42, poly(dT)42, poly(dG)42, or poly(dC)42 mixed with complementary dNTP analogs. Each template was fused to an M13 priming site and mixed with an M13 forward primer in a 1:1 stoichiometric ratio; for hybridization, the mixture was heated in a thermal cycler to 95 °C for 5 to 10 min followed by cooling to 65 °C then further cooling with a gradient of 5 °C every five min until reaching room temperature. KF nanocircuits were immersed in activity buffer with the annealed template-primer at 100 nM concentrations. Native or analog dNTPs were added to the buffer in excess, ensuring Vmax conditions for KF catalysis. To compensate for possibly reduced affinity of dNTP analogs, the experiments applied higher concentrations of analogs (Figure 1c, 100 μM, Trilink Biotechnologies) than the native dNTPs (10 μM, Fisher). Structural characterizations of the dNTP analogs were provided by the supplier.
Measurements consisted of monitoring the source-drain current, I(t), through the SWCNT-FET while the attached KF molecule interacted with its surrounding environment. The drain electrode was biased at 100 mV, and the electrolyte, which served as a gate electrode, was held at or near 0 V. Incubation of the device with any template-primer and its complementary dNTPs transduced fluctuations, ΔI(t), whereas these fluctuations were absent with non-complementary dNTPs or in control measurements missing the template-primer or KF attachment.33 I(t) fluctuations were amplified (Keithley 428), digitized at 100 kHz, and stored as uninterrupted, 600 s data sets for later analysis. Between measurements, the KF nanocircuits were rinsed twice with activity buffer, incubated in buffer for 5 min, then rinsed twice with buffer before introducing another nucleotide and template-primer. Each KF molecule was monitored with multiple analogs, their corresponding native dNTPs, and nucleotide-free buffer in order to collect directly comparable data sets, confirm typical KF activities, and reproduce the types of ΔI(t) excursions reported previously.33
RESULTS
Figures 2a and 2b show representative ΔI(t) signals produced by a KF nanocircuit processing a poly(dC)42 template in the presence of dGTP. The device produced uninterrupted sequences of negative ΔI(t) excursions, shown at three different magnifications. Each ΔI(t) excursion indicated the formation of one base pair, and the kinetic parameters derived from ΔI(t) data sets were consistent with previous single-molecule analysis of KF motions33 and ensemble KF incorporation rates.36,37 As observed previously, results with G•C or C•G base pair formation were essentially identical to one another; A•T/T•A base pair formation also provided very similar polymerization kinetics, dynamics and ΔI(t) values compared to each other. Measurements with the native dNTPs provided baseline values for comparison with dNTP analogs.
Figure 2.

ΔI(t) excursions during native and analog dNTP incorporation. High and low current states correspond to conformational dynamics of the enzyme during processive nucleotide incorporation. (a) In the presence of poly(dC)42 template and its complementary native dGTP, ΔI(t) excursions occur during each base incorporation. (b) Magnification of the highlighted region in (a) illustrates the ΔI(t) events corresponding to single base incorporations. For comparison, representative 1 s data sets are shown for the same KF nanocircuit incorporating the dNTP analogs (c) αthio-dGTP, (d) 6-Cl-2APTP, and (e,f) 2-thio-dCTP. To the right of each data set, the magnified view depicts one typical ΔI(t) excursion.
Commercially available dNTP analogs were incorporated into DNA through KF polymerization in both ensemble (Figure S3) and single-molecule assays. Measurements with α-thio-dNTP, or dNTPαS, analogs produced ΔI(t) data sets that appeared quite similar to the native dNTPs, but with different incorporation rates (Figure 2c). When measured with KF nanocircuits, incorporation of 6-chloro-2-aminopurine-drTP, or 6-Cl-2APTP, opposite both poly(dC)42 and poly(dT)42 templates caused ΔI(t) signals with inverted amplitude reflecting a different KF conformation (Figure 2d). This analog incorporated more slowly; for example, opposite poly(dC)42, 6-Cl-2APTP produced ΔI(t) excursions at 80% of the rate of dGTP. ΔI(t) records with the 2-thio-dNTP analogs produced mixed behaviors in which KF activity produced negative ΔI(t) excursions during one minute, positive ΔI(t) excursions during another minute, and, more rarely, mixtures of both behaviors along a single template strand (Figures 2e,f).
In our previous report with native dNTPs, the time constants for the experimental baseline current, τopen here, were referred to as τhi. Time constants representing a native dNTP incorporation event all occurred with lower current, and were referred to as τlo.33 Since positive, negative, or mixtures of both positive and negative ΔI(t) excursions are reported here, time constants for either direction of excursions are termed τclosed. Distributions of τopen and τclosed were derived from each record of polymerization data.
Figure 3 shows example distributions for incorporation of dGTP substrates into poly(dC)42 templates. The distributions from native and analog dGTP τclosed events were nearly indistinguishable except for rare events in the tails, for which we have the poorest statistics (Figure 3a). To draw comparisons between native and analog dNTPs, we focused on the mean time constant <τ> of the primary, Poissonian component of these distributions. All of the mean values for <τclosed> were in close agreement around 0.3±0.1 ms. By comparison, the distributions and mean values of <τopen> were clearly different. For example, KF spent 63.6±2.8 ms in its open conformation when processing α-thio-dGTP, which is 56% longer than the 40.8±0.6 ms observed for native dGTP (Figure 3b).
Figure 3.

Direct comparison of the probability distributions of <τopen> and <τclosed> durations during incorporation of the indicated dNTPs from >50 s data sets. For both (a) τclosed and (b) τopen, the homopolymeric poly(dC)42 was the template used. Single-exponential fits for each nucleotide are shown as solid lines.
The kinetic parameters <τclosed>, <τopen>, and the average rate of incorporation k were analyzed for the four homopolymeric templates with native and analog dNTPs (Table 1). As with the case described above, every combination produced identical τclosed distributions with <τclosed> values in the range of 0.3±0.1 ms. While a similar effect was previously observed for the four native dNTPs,33 the extension of this result to dNTP analogs having different nucleobase sizes, electronic properties, hydrogen bonding, or substitution at the α-phosphodiester was unexpected.
Table 1.
Kinetics of native and analog dNTP incorporation by KF.a
| Template | Nucleotide | <τopen (ms)> | <τclosed (ms)> | k (1/s) |
|---|---|---|---|---|
| poly(dT)42 | dATP | 58.9 ± 1.2 | 0.34 ± 0.18 | 16.8 ± 0.4 |
| α-thio-dATP | 145.9 ± 8.4 | 0.38 ± 0.21 | 6.8 ± 0.4 | |
|
| ||||
| poly(dA)42 | dTTP | 69.6 ± 2.3 | 0.33 ± 0.12 | 14.3 ± 0.5 |
| α-thio-dTTP | 152.1 ± 6.6 | 0.29 ± 0.13 | 6.6 ± 0.3 | |
| 2-thio-dTTP | 61.1 ± 3.2b | 0.23 ± 0.14b | 16.3 ± 0.9b | |
|
| ||||
| poly(dG)42 | dCTP | 42.8 ± 5.0 | 0.35 ± 0.20 | 23.2 ± 3.2 |
| α-thio-dCTP | 68.8 ± 4.6 | 0.33 ± 0.19 | 14.5 ± 1.0 | |
| 2-thio-dCTP | 32.3 ± 1.1b | 0.41 ± 0.15b | 30.6 ± 1.2b | |
|
| ||||
| poly(dC)42 | dGTP | 40.8 ± 6.0 | 0.40 ± 0.20 | 24.3 ± 4.3 |
| α-thio-dGTP | 63.6 ± 2.8 | 0.21 ± 0.15 | 15.7 ± 0.7 | |
| 6-Cl-2APTP | 50.5 ± 1.4 | 0.20 ± 0.12 | 19.7 ± 0.6 | |
Average values ± standard deviation
Similar values were observed for both upor down-switching events.
On the other hand, τopen was far more sensitive to dNTP identity. The mean duration of <τopen> ranged from 23 ms with native dCTP to 145 ms with α-thio-dATP. Among the four native dNTPs, <τopen> was longer for dTTP or dATP incorporation than for dGTP or dCTP incorporation. This hierarchy was preserved within longer <τopen> durations measured for all four α-thio-dNTPs. The α-thio substitution increased <τopen> by 50% in the case of dGTP and dCTP, whereas the increase was more than 100% for dTTP and dATP.
The average KF processing rate for dNTP incorporation was calculated as k = (<τopen> + <τclosed>)−1. τopen largely determines k, because it is at least 60 times longer than τclosed. At its fastest, KF incorporated 2-thio-dCTP at more than 30 s−1. The increase in τopen described above for αthio-dNTPs reduced k to 15 s−1 for α-thio-dCTP and α-thio-dGTP and 7 s−1 for α-thio-dATP and α-thio-dTTP. Rates for 6-Cl-2APTP incorporation compared most favorably to the slowest rates observed for native dGTP incorporation. Conversely, 2-thio-dTTP and 2-thio-dCTP incorporation appeared slightly faster than incorporation of their native counterparts.
Similar results were reproduced using a dozen different KF molecules. Each KF was attached to a different SWCNT-FET and measured independently. For comparison, a non-homopolymeric template measured with dNTP analogs resulted in similar kinetics (data not shown). As mentioned previously, our experiments applied 100 μM of dNTP analogs to ensure steady state conditions; for comparision, 10 μM α-thio-dATP with the poly(dT)42 template did not affect DNA polymerization. Due to static disorder, some KF molecules processed faster or slower than the ensemble average, but without any significant change to the relative comparison of analog to native dNTPs.
DISCUSSION
The dNTP analogs were chosen for their ability to be incorporated into DNA templates by DNA polymerases and variations in sizes, structures, and reactivity. We examined either substitution at the α-phosphate or nucleobase. The first type of analog, α-thio-dNTP, substituted a non-bridging, α-phosphoryl oxygen atom with sulfur to introduce a new stereocenter and alter the reactivity at this crucial site. The second category of dNTP analogs, halogen or sulfur substitution on the nucleobase, changes the size and electronic structure of the base pair; some analogs also alter the hydrogen bonding available for base pairing. For example, 6-Cl-2-APTP (Figure 1c), has two hydrogen bonding profiles, allowing its incorporation opposite both T and C bases.9,15,38 Compared to dATP, 6-Cl-2-APTP replaces the 6-amino group with chlorine, but introduces a 2-amino functionality; this configuration ultimately provides the same number of Watson-Crick hydrogen bonds complementary to T as dATP. When used as a dGTP analog, 6-Cl-2-APTP has different tautomerization, which changes the N-1 from a hydrogen bond donor to an acceptor. In this case, replacement of oxygen with chlorine dramatically decreases the strength of the hydrogen bonding.39 Like 6-Cl-2APTP, sulfur-substituted analogs 2-thio-dTTP and 2-thio-dCTP also form larger base pairs due to the increased bond length of the thiocarbonyl.40
The single-molecule experiments carried out in this study illustrate and shed new light on the well-appreciated plasticity of DNA polymerases like KF. This class of enzymes can accommodate even dramatically modified incoming dNTPs. However, we directly observe conformational motions required by the enzyme to maintain fidelity when faced with certain altered dNTPs. Reflecting the limits for such accommodations, DNA polymerases are known to exhibit strong sensitivity to minor changes in dNTP size and shape.8,12 Our analysis benefits from comparing single molecule data with native and analog dNTPs during numerous processive incorporation events. This analysis begins with the kinetics of the two observed enzyme conformations during catalysis, which were captured by τopen and τclosed.
Events taking place during τopen include the rate-limiting step of dNTP recognition, which is sensitive to both nucleobase and backbone modifications. Successful recognition and binding of the appropriate nucleotide triggers KF’s activation and closure.41 Previous FRET-based experiments with the related T7 DNA polymerase have identified a “fully open” conformational state resulting from mismatch recognition.42 However, using the L790C attachment site, the SWCNT-FET records no KF motions and no signals in the presence of mismatched dNTPs. The absence of intermediate states or mismatch-associated motions suggests that our attachment site is insensitive to this initial fidelity checkpoint. Thus, ΔI(t) excursions result from a catalytically committed conformation, and are not restricted to simply the global motion of the enzyme opening and closing.
We propose that the differences observed in τopen largely reflect the mechanisms for recognizing and binding unnatural dNTPs. Long tails in the distributions for α-thio-dGTP and 6-Cl-2APTP compared to native dGTP may have been responsible for the <τopen> increase (Figure 3b). In fact, these tails can be fit to second exponentials with time constants of 200 ms, about five times longer than <τopen> for native dGTP. Similar long tails were observed with all tested dNTP analogs, illustrating the challenges faced by the enzyme when incorporating non-natural substrates. Steps other than recognition potentially take place during the τopen reported here; covalent bond formation is one possible example that would occur too quickly, even with the slowed reactivity of α-thio-dNTPs, to be detectable as rate-limiting.43 Faster rates of incorporation observed with the 2-thio analogs can result from more stable base pair formation, effectively shortening <τopen> values. The larger size of the 2-thio-dCTP sulfur atom at the hydrogen bonding interface with the template G base does not appear to affect the ability of 2-thio-dCTP to base pair efficiently. These results agree with the previously observed increase in polymerization efficiency with 2-thio-dTTP and 4-thio-dTTP compared to dTTP incorporation.14,44
The 6-Cl-2APTP analog, with much weaker hydrogen bonding and consequent imperfect base pairing compared to dGTP, exemplifies the challenges of base pairing recognition during KF-catalyzed DNA polymerization. Longer <τopen> values for 6-Cl-2APTP versus dGTP incorporation opposite a poly(dC)42 template illustrate the willingness of DNA polymerases to accept unnatural dNTPs in part by lengthening the time allotted for recognition. The <τopen> value, and thus the rate of incorporation, observed during 6-Cl-2APTP polymerization opposite poly(dC)42 fell between the values measured for native dGTP and dATP incorporation opposite complementary, homopolymeric templates. Thus, despite its altered tautomerization and consequent loss of at least one base pairing hydrogen bond when compared to dGTP, 6-Cl-2APTP can still be incorporated more quickly opposite poly(dC)42 than native dATP opposite poly(dT)42. Notably, the base pairing hydrogen bond in the minor groove remains unchanged when 6-Cl-2APTP is considered a dGTP analog, and could govern the relatively faster rates observed for dGTP, dCTP, and 6-Cl-2APTP opposite a poly(dC)42 template.
In addition to recognition and binding, prolonged <τopen> values for α-thio-dNTP incorporation could result from the reduced stability of the newly synthesized DNA. KF-catalyzed processing of homopolymeric templates can result in distorted dsDNA.45 Furthermore, α-thio-dNTPs are particularly prone to form less stable binary complexes with unfavorable DNA backbone interactions, which progressively slows the catalytic rate of KF.46,47 More pronounced effects on this step, compared to experiments with the respective native dNTPs, were observed during α-thio-dATP/α-thio-dTTP versus α-thio-dGTP/α-thio-dCTP incorporation. Such results may indicate sequence-dependent DNA instability, which underscores an important caveat to these studies with homopolymeric templates. Alternatively, this difference could suggest that the α-thio substitution further interferes with the mechanism that causes <τopen> to be longer for native A•T/T•A base pairs. Some of the variation in τopen associated with α-thio-dNTP incorporation could result from the weakly inhibitory Rp stereoisomer (Ki ≈ 30 μM), present at an approximately 1:1 ratio with the Sp stereoisomer in the commercial synthesis of this analog.48 This inhibition is about an order of magnitude weaker than the Km for the native dNTP,10 and thus can be expected to affect <τopen> values only modestly.
During τclosed, KF undergoes a distinct conformational change corresponding to formation of one phosphodiester bond between the incoming nucleotide and the nascent dsDNA. In substrate-limited experiments, the number of ΔI(t) excursions matched the number of overhanging template bases;33 thus, the conformational change during τclosed must occur for each successful, processive nucleotide incorporation. Earlier, the short and equal duration of <τclosed> for native dNTPs supported a model in which τclosed results from the covalent bond-forming step itself.33 Here, we reevaluate this assignment due to three observations with dNTP analogs. First, the direction of ΔI(t) excursions was reversed for some dNTP analogs. Second, incorporation of 2-thio-dNTP analogs produced mixtures of both positive and negative ΔI(t) excursions. Third, as shown in Table 1, the invariance in <τclosed> extended to all analogs tested despite substitutions at the electrophilic α-phosphate or the likely alternative conformations needed to accommodate substitutions on the nucleobase.
In this electronic technique, the underlying SWCNT-FET is extremely sensitive to electrostatic gating by the protein’s charged surface residues within 1 nm of the attachment site.31 Previous work proved that different variants of the same enzyme can exhibit either positive or negative ΔI(t) excursions depending on the charge of the SWCNT-adjacent residues and the directions of their motion.31 KF and its charged residues electrostatically gating the SWCNT-FET remain invariant in this study. Therefore, variable ΔI(t) excursions indicate that the residues adjacent to the KF attachment site are adopting different motions in response to certain dNTP analogs during a catalytically competent cycle. Such motions are likely transmitted from the KF active site through allostery, but they are not necessarily the motions of covalent bond formation. In fact, we conclude that the covalent step could not proceed by the same mechanism and with the same <τclosed> duration but with two opposing motions. Instead, the relevant residue motions responsible for τclosed are likely independent of both initial molecular recognition and the chemical step of KF catalysis.
KF is attached to the SWCNT-FET through the protein’s L790C sidechain in the “fingers” subdomain, linking the electrostatic gating motions of relevant charged residues to catalytically committed motions during τclosed. We propose that each τclosed event results from the active site O-helix itself or a particular O-helix residue twisting in two possible directions during the observed stage of successful nucleotide incorporation. This proposed twisting is inferred by considering active site residue motions during known stages of nucleotide incorporation and their effect on the theoretical proximity of charged residues to the SWCNT-FET. For example, smFRET experiments with KF reveal an intermediate conformation of the active site O-helix between the open and closed states;27 a potentially analogous “ajar” conformation was observed in the crystal structures of the KF homolog Bst Pol I. The C-terminus of the Bst Pol I O-helix kinks on the pathway to closure such that a large shift of the KF Y766 equivalent is accompanied by a subtle rotation of the KF F762 equivalent. The rotation of the KF F762 equivalent continues until enzyme closure.49–51
By comparing crystal structures of KF and Bst Pol I, we identified charged residues adjacent to the SWCNT-FET that could move in response to rotations by Y766 and F762 in the KF active site (Figure 4). Thus, we hypothesize that the source of ΔI(t) excursions is likely additional motions of Y766 and/or F762 after enzyme closure and base incorporation that continue to propagate to charged residues near the SWCNT-FET. Indeed, an additional KF conformational change when the nascent base pair moves to the KF post-insertion site has been observed by smFRET following successful nucleotide incorporation,24 and is possibly the motion measured by τclosed. Significant interactions imparted by aromatic active site residues could include π–π stacking with the newly formed base pair. Such a motion would assess the electronic configuration of the base pair and interrogate the fidelity of the bond formation step without requiring hydrophilic interactions, which are altered by the dNTP analog’s substitutions.
Figure 4.
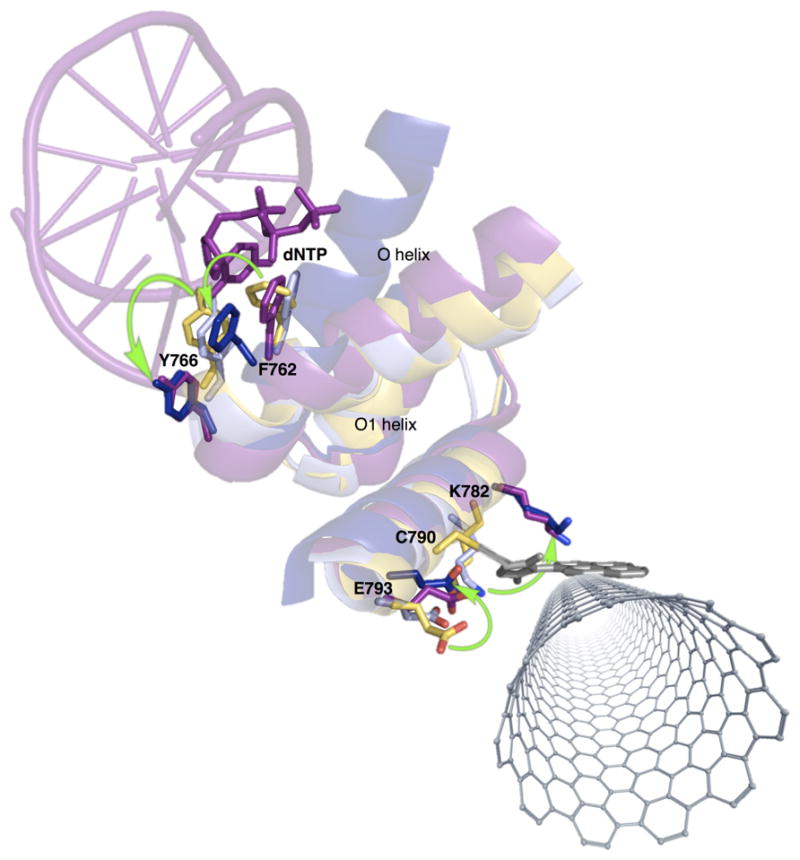
Model of a KF-functionalized nanocircuit showing various stages of dNTP incorporation. The open conformation of KF (yellow, PDB ID: 1KFD) can be structurally aligned with the following structures: the homologous Bst Pol I fragment in its open conformation without dNTP bound (light blue, PDB ID: 1L3T), its closed conformation (dark blue, PDB ID: 1LV5), and its ajar conformation with a matched base pair (magenta, PDB ID: 3HT3). The equivalent of KF F762 makes small rotations during each stage of nucleotide incorporation shown, while the equivalent of KF Y766 moves dramatically only from the open to “ajar” state. Representative charged residues near the attachment site show variable movement that correspond to the active site residues, and likely continue to move in concert with F762 and Y766 as the enzyme closes and checks the nascent base pair’s electronic structure. Green curved arrows show the direction of movement for each labeled residue from the open to closed states. Negative and positive ΔI(t) excursions likely result from these rotations during incorporation of native and non-native dNTPs, respectively.
The proposed O-helix twisting mechanism explains key observations of DNA polymerases, including results from our previous single-molecule work. The KF nanocircuit reveals larger ΔI(t) excursions for the A•T/T•A set than the G•C/C•G set of base pairs. Structural results have suggested that A and T template bases are most deeply buried in the DNA polymerase active site,21 and, therefore, the swiveling of the O-helix could be maximized. KF E710 and Y766 or homologous, active site glutamate and tyrosine residues have been implicated in a mechanism for stabilization of A•T/T•A base pairs over G•C/C•G base pairs.52 Thus, the hydrogen bonding interaction between KF E710 and KF Y766 prior to nucleotide incorporation could influence the size and shape of the active site and may play an important role in the τclosed step of dNTP analog recognition. Further insights will require structural analysis, mutagenesis, and modeling.
Similar results during incorporation of 2-thio-dTTP and 2-thio-dCTP illustrate KF’s preferential recognition of the base pair’s electronic structure. Although the sulfur substitution only affects a Watson-Crick hydrogen bond acceptor in the 2-thio-dCTP analog, both 2-thio-dNTP analogs result in mixtures of positive and negative ΔI(t) excursions and thus both cause similar KF motions during incorporation. The sulfur substitution for the 2-thio-dNTP analogs is minor compared to the more dramatic electronic variations introduced into 6-Cl-2APTP, but the enzyme responds in a similar, although non-exclusive, manner. The observed mixtures of both negative and positive ΔI(t) excursions suggest that KF accesses both native and alternative motions, respectively, during incorporation of the 2-thio-substituted dNTPs. An apparent memory effect locks the enzyme into one motion or the other for tens of seconds, implicating an additional conformational change that is energetically bistable in the special case of 2-thio-dNTPs.
Finally, we have also considered shuttling of the nascent DNA to the inactive exonuclease (exo) domain as a possible source of positive ΔI(t) excursions. Upon melting of an unstable primer terminus due to imperfect base pairing, DNA shuttles to and from an inactive exo domain, and KF undergoes distinct conformational changes.24,53–55 However, such transitions occur distant from the attachment site and positive ΔI(t) excursions observed here do not change durations of <τclosed>. Accordingly, shuttling to the exo domain seems inconsistent with the observation of positive ΔI(t) excursions. Similar to the conformational steps known to occur during mismatched dNTP recognition, shuttling to the exo domain must take place during τopen. In summary, the ΔI(t) excursions reported here only occur during a committed catalytic cycle, and likely represent an adaptable KF motion consistent with a swiveling O-helix testing the electronic integrity of the newly formed DNA base pair.
CONCLUSION
The experiments reported here with dNTP analogs challenge the limits of nucleotide incorporation by DNA polymerases, including the stereochemistry at the electrophilic phosphate, the hydrogen bonding capability of the incoming base, and the mechanisms of fidelity checking. Since most dNTP analogs increase average <τopen> and the broadness of its kinetic distributions, the rate-determining dNTP recognition step appears highly sensitive to even minor variation in substrate structure. However, dramatic substitutions at the reactive site of bond formation fail to impact the durations of <τclosed>. The direction of the ΔI(t) excursions, on the other hand, switches to positive or a mixture of both negative and positive signals with base-modified dNTP analogs. Since these dNTP analogs have functionalities at the bond formation center identical to native substrates, we interpret the dramatic changes in ΔI(t) direction to result from fidelity checking by KF before opening to process the next substrate. Such events can be readily distinguished from native dNTP incorporation events and provide direct observation of the enzyme accommodating unnatural dNTPs by reversing the direction of its dynamic error checking. Such insights could lay the groundwork for a range of applications from DNA sequencing to drug design.
Supplementary Material
Acknowledgments
We thank S. Benkovic and L. Romano for helpful suggestions and B. Corso, D. Pan, and A. Rajapakse for assistance with device fabrication. This research was supported financially by the NIH NCI (R01 CA133592-01), the NIH NIGMS (1R01GM106957-01) and the NSF (DMR-1104629 and ECCS1231910).
ABBREVIATIONS
- 2-thio-dTTP
2-thiothymidine-5′-triphosphate
- 2-thio-dCTP
2-thiocytidine-5′-triphosphate
- 6-Cl-2APTP
2-amino-6-Clpurine-2′-deoxyriboside-triphosphate
- α-thio-dNTP
2′deoxynucleoside-5′-O-(1-thiotriphosphate)
Footnotes
Supporting Information. Materials and methods, SDSPAGE of L790C, and bulk measurements of KF catalytic activity with various dNTPs. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Echols H, Goodman MF. Annu Rev Biochem. 1991;60:477. doi: 10.1146/annurev.bi.60.070191.002401. [DOI] [PubMed] [Google Scholar]
- 2.Kool ET. Annu Rev Biochem. 2002;71:191. doi: 10.1146/annurev.biochem.71.110601.135453. [DOI] [PubMed] [Google Scholar]
- 3.Kunkel TA. J Biol Chem. 2004;279:16895. doi: 10.1074/jbc.R400006200. [DOI] [PubMed] [Google Scholar]
- 4.Schweitzer BA, Kool ET. J Am Chem Soc. 1995;117:1863. doi: 10.1021/ja00112a001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morales JC, Kool ET. Nat Struct Biol. 1998;5:950. doi: 10.1038/2925. [DOI] [PubMed] [Google Scholar]
- 6.Potapova O, Chan C, DeLucia AM, Helquist SA, Kool ET, Grindley NDF, Joyce CM. Biochemistry. 2006;45:890. doi: 10.1021/bi051792i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiaramonte M, Moore CL, Kincaid K, Kuchta RD. Biochemistry. 2003;42:10472. doi: 10.1021/bi034763l. [DOI] [PubMed] [Google Scholar]
- 8.Sintim HO, Kool ET. Angew Chem Int Ed Engl. 2006;45:1974. doi: 10.1002/anie.200504296. [DOI] [PubMed] [Google Scholar]
- 9.Devadoss B, Lee I, Berdis AJ. Biochemistry. 2007;46:13752. doi: 10.1021/bi701328h. [DOI] [PubMed] [Google Scholar]
- 10.Burgers PM, Eckstein F. J Biol Chem. 1979;254:6889. [PubMed] [Google Scholar]
- 11.Gao J, Liu H, Kool ET. J Am Chem Soc. 2004;126:11826. doi: 10.1021/ja048499a. [DOI] [PubMed] [Google Scholar]
- 12.Kim TW, Delaney JC, Essigmann JM, Kool ET. Proc Natl Acad Sci U S A. 2005;102:15803. doi: 10.1073/pnas.0505113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kincaid K, Beckman J, Zivkovic A, Halcomb RL, Engels JW, Kuchta RD. Nucleic Acids Res. 2005;33:2620. doi: 10.1093/nar/gki563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sintim HO, Kool ET. J Am Chem Soc. 2006;128:396. doi: 10.1021/ja0562447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X, Motea E, Lee I, Berdis AJ. Biochemistry. 2010;49:3009. doi: 10.1021/bi901523y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Betz K, Malyshev DA, Lavergne T, Welte W, Diederichs K, Dwyer TJ, Ordoukhanian P, Romesberg FE, Marx A. Nat Chem Biol. 2012;8:612. doi: 10.1038/nchembio.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang W, Wu EY, Hellinga HW, Beese LS. J Biol Chem. 2012;287:28215. doi: 10.1074/jbc.M112.366609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carroll SS, Cowart M, Benkovic SJ. Biochemistry. 1991;30:804. doi: 10.1021/bi00217a034. [DOI] [PubMed] [Google Scholar]
- 19.Astatke M, Ng K, Grindley NDF, Joyce CM. Proc Natl Acad Sci. 1998;95:3402. doi: 10.1073/pnas.95.7.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hohlbein J, Aigrain L, Craggs TD, Bermek O, Potapova O, Shoolizadeh P, Grindley NDF, Joyce CM, Kapanidis AN. Nat Commun. 2013;4:2131. doi: 10.1038/ncomms3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bermek O, Grindley NDF, Joyce CM. Biochemistry. 2013;52:6258. doi: 10.1021/bi400837k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Min W, English BP, Luo G, Cherayil BJ, Kou SC, Xie XS. Acc Chem Res. 2005;38:923. doi: 10.1021/ar040133f. [DOI] [PubMed] [Google Scholar]
- 23.Lu HP. Chem Soc Rev. 2014;43:1118. doi: 10.1039/c3cs60191a. [DOI] [PubMed] [Google Scholar]
- 24.Christian TD, Romano LJ, Rueda D. Proc Natl Acad Sci U S A. 2009;106:21109. doi: 10.1073/pnas.0908640106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santoso Y, Joyce CM, Potapova O, Le Reste L, Hohlbein J, Torella JP, Grindley NDF, Kapanidis AN. Proc Natl Acad Sci U S A. 2010;107:715. doi: 10.1073/pnas.0910909107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markiewicz RP, Vrtis KB, Rueda D, Romano LJ. Nucleic Acids Res. 2012;40:7975. doi: 10.1093/nar/gks523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berezhna SY, Gill JP, Lamichhane R, Millar DP. J Am Chem Soc. 2012;134:11261. doi: 10.1021/ja3038273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garalde DR, Simon CA, Dahl JM, Wang H, Akeson M, Lieberman KR. J Biol Chem. 2011;286:14480. doi: 10.1074/jbc.M111.218750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi Y, Moody IS, Sims PC, Hunt SR, Corso BL, Perez I, Weiss GA, Collins PG. Science. 2012;335:319. doi: 10.1126/science.1214824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi Y, Moody IS, Sims PC, Hunt SR, Corso BL, Seitz DE, Blaszczak LC, Blaszcazk LC, Collins PG, Weiss GA. J Am Chem Soc. 2012;134:2032. doi: 10.1021/ja211540z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi Y, Olsen TJ, Sims PC, Moody IS, Corso BL, Dang MN, Weiss GA, Collins PG. Nano Lett. 2013;13:625. doi: 10.1021/nl304209p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sims PC, Moody IS, Choi Y, Dong C, Iftikhar M, Corso BL, Gul OT, Collins PG, Weiss GA. 2013 doi: 10.1021/ja311604j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olsen TJ, Choi Y, Sims PC, Gul OT, Corso BL, Dong C, Brown WA, Collins PG, Weiss GA. J Am Chem Soc. 2013;135:7855. doi: 10.1021/ja311603r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Derbyshire V, Freemont P, Sanderson M, Beese L, Friedman J, Joyce C, Steitz T. Science. 1988;240:199. doi: 10.1126/science.2832946. [DOI] [PubMed] [Google Scholar]
- 35.Frey MW, Sowers LC, Millar DP, Benkovic SJ. Biochemistry. 1995;34:9185. doi: 10.1021/bi00028a031. [DOI] [PubMed] [Google Scholar]
- 36.Kuchta RD, Mizrahi V, Benkovic PA, Johnson KA, Benkovic SJ. Biochemistry. 1987;26:8410. doi: 10.1021/bi00399a057. [DOI] [PubMed] [Google Scholar]
- 37.Dahlberg ME, Benkovic SJ. Biochemistry. 1991;30:4835. doi: 10.1021/bi00234a002. [DOI] [PubMed] [Google Scholar]
- 38.Patro JN, Urban M, Kuchta RD. Biochemistry. 2009;48:180. doi: 10.1021/bi801823z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Politzer P, Lane P, Concha MC, Ma Y, Murray JS. J Mol Model. 2007;13:305. doi: 10.1007/s00894-006-0154-7. [DOI] [PubMed] [Google Scholar]
- 40.Biedermann M, Hartung H, Dolling W, Verjus P. Acta Crystallogr Sect C Cryst Struct Commun. 1998;54:507. [Google Scholar]
- 41.Dzantiev L, Romano LJ. Biochemistry. 2000;39:356. doi: 10.1021/bi991952o. [DOI] [PubMed] [Google Scholar]
- 42.Tsai YC, Johnson KA. Biochemistry. 2006;45:9675. doi: 10.1021/bi060993z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bryant FR, Johnson KA, Benkovic SJ. Biochemistry. 1983;22:3537. doi: 10.1021/bi00284a001. [DOI] [PubMed] [Google Scholar]
- 44.Sismour AM, Benner SA. Nucleic Acids Res. 2005;33:5640. doi: 10.1093/nar/gki873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leslie AGW, Arnott S, Chandrasekaran R, Ratliff RL. J Mol Biol. 1980;143:49. doi: 10.1016/0022-2836(80)90124-2. [DOI] [PubMed] [Google Scholar]
- 46.Eckstein F, Jovin TM. Biochemistry. 1983;22:4546. doi: 10.1021/bi00288a030. [DOI] [PubMed] [Google Scholar]
- 47.Mizrahi V, Henrie RN, Marlier JF, Johnson KA, Benkovic SJ. Biochemistry. 1985;24:4010. doi: 10.1021/bi00336a031. [DOI] [PubMed] [Google Scholar]
- 48.Ludwig J, Eckstein F. J Org Chem. 1989;54:631. [Google Scholar]
- 49.Johnson SJ, Taylor JS, Beese LS. Proc Natl Acad Sci U S A. 2003;100:3895. doi: 10.1073/pnas.0630532100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trostler M, Delier A, Beckman J, Urban M, Patro JN, Spratt TE, Beese LS, Kuchta RD. Biochemistry. 2009;48:4633. doi: 10.1021/bi900104n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu EY, Beese LS. J Biol Chem. 2011;286:19758. doi: 10.1074/jbc.M110.191130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Donlin MJ, Johnson KA. Biochemistry. 1994;33:14908. doi: 10.1021/bi00253a030. [DOI] [PubMed] [Google Scholar]
- 53.Joyce C. J Biol Chem. 1989;264:10858. [PubMed] [Google Scholar]
- 54.Tuske S, Singh K, Kaushik N, Modak MJ. J Biol Chem. 2000;275:23759. doi: 10.1074/jbc.M001804200. [DOI] [PubMed] [Google Scholar]
- 55.Lamichhane R, Berezhna SY, Gill JP, Van der Schans E, Millar DP. J Am Chem Soc. 2013;135:4735. doi: 10.1021/ja311641b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.