

Abstract
Toxoplasmosis is a widespread parasitic infection by Toxoplasma gondii, a parasite with at least three distinct clonal lineages. This article reports the whole genome sequencing and de novo assembly of T. gondii RH (type I representative strain), as well as genome-wide comparison across major T. gondii lineages. Genomic DNA was extracted from tachyzoites of T. gondii RH strain and its identity was verified by PCR and LAMP. Subsequently, whole genome sequencing was performed, followed by sequence filtering, genome assembly, gene annotation assignments, clustering of gene orthologs and phylogenetic tree construction. Genome comparison was done with the already archived genomes of T. gondii. From this study, the genome size of T. gondii RH strain was found to be 69.35Mb, with a mean GC content of 52%. The genome shares high similarity to the archived genomes of T. gondii GT1, ME49 and VEG strains. Nevertheless, 111 genes were found to be unique to T. gondii RH strain. Importantly, unique genes annotated to functions that are potentially critical for T. gondii virulence were found, which may explain the unique phenotypes of this particular strain. This report complements the genomic archive of T. gondii. Data obtained from this study contribute to better understanding of T. gondii and serve as a reference for future studies on this parasite.
Introduction
Toxoplasma gondii is a globally widespread zoonotic parasite that infects approximately one-third of the world's population [1, 2]. Although most T. gondii infections are subclinical, infection by this parasite in immuno-compromised groups and pregnant women can result in severe outcomes [3, 4]. As T. gondii can persist within the host via latent infection, it may cause catastrophic consequences to those infected with HIV following reactivation of the parasite. This parasite is incriminated as one of the most fatal foodborne pathogens in USA [5].
Similar to many other members of the Apicomplexa phylum, T. gondii shows a complex life cycle, during which sexual reproduction happens within its definite host (cat). Asexual reproduction occurs within its wide range of homoeothermic intermediate hosts, including humans. Since sexual reproduction is part of its life cycle, T. gondii was expected to show high variation across the world. However, the majority of the T. gondii strains from most parts of the world belong (but are not restricted) to three distinct clonal lineages (type I, type II and type III) with minimal genetic differences among them [6–8]. However, these T. gondii lineages show varied virulence [9]. For instance, Type I T. gondii is the most virulent lineage in murine models [10]. Furthermore, type I has been persistently and disproportionately isolated from immuno-competent patients suffering from severe ocular toxoplasmosis in USA [11]. Type I is also associated with severe congenital toxoplasmosis in European countries [10, 12].
The T. gondii type I lineage is represented by GT1 (isolated from goat’s skeletal muscle) and RH (isolated from a fatal toxoplasmosis-induced encephalitis case in year 1939) strains [13, 14]. RH strain has been adapted to in vitro cultivation and commonly used in laboratory work [15]. It shows high growth and migration rates, and imparts the highest fatality in mice [9, 16]. On the other hand, type II (represented by ME49 strain) and type III (represented by VEG and CEP strains) are considered less virulent than type I [17], with type II dominating human toxoplasmosis in USA [18, 19] while type III is significantly associated with animal hosts [18, 20]. With high genetic similarities between these lineages, it is intriguing to see apparently varied virulence among these parasites [9].
The mechanisms behind the virulence and pathogenesis as well as other biological aspects of T. gondii can be understood better by performing in-depth genome analyses and comparisons on all major lineages of T. gondii. To date, whole genome sequencing has been completed on 62 T. gondii strains, including type I (GT1 and RH strains), type II (ME49 strain) and type III (VEG strain), as well as recombinant strains [21–27]. A clearer picture on genome differences between the major T. gondii lineages will contribute towards better understanding on different virulence attributed to these lineages. Here, we performed whole genome de novo sequencing and assembly on T. gondii RH strain and compared its genome with those of T. gondii GT1, ME49 and VEG strains.
Materials and Methods
Ethical approval
Experiments involving mouse model were carried out in compliance with the animal ethics approved by Institutional Animal Care and Use Committee (IACUC) of the University of Malaya, Faculty of Medicine (2014-06-03/PARA/R/CXT).
Propagation of parasites and genomic DNA extraction
Tachyzoites of T. gondii RH strain were maintained by serial intraperitoneal passage in BALB/c mice. The tachyzoites were harvested from peritoneal fluids three to four days-post- infection. The parasites were washed and re-suspended in sterile phosphate buffered saline (1x PBS) prior to usage. High molecular weight genomic DNA was extracted using DNeasy Blood and Tissue Kit (QIAGEN, Germany). The specific identity of the extracted DNA was verified by conventional nested PCR directed against the B1 gene of T. gondii (primers used: B1F1:5’-CCGTTGGTTCCGCCTCCTTC-3’; B1R1: 5’-GCAAAACAGCGGCAGCGTCT-3’ and B1F2: 5’-CCGCCTCCTTCGTCCGTCGT-3’; B1R2: 5’-GTGGGGGCGGACCTCTCTTG-3’) and Loop-Mediated Isothermal Amplification Method (LAMP) [28, 29]. The DNA yield and purity were measured spectrophotometrically (NanoDrop 2000c spectrophotometer, Thermo Scientific); DNA integrity was verified by agarose gel electrophoresis and a Bioanalyzer (2100, Agilent).
Library construction and sequencing of T. gondii
DNA samples of T. gondii were used for the constructions of five libraries with various insert sizes of 200 bp, 500 bp, 800 bp, 5 kb and 10 kb. Paired-end sequencing was performed on these five libraries through Illumina HiSeq 2000 which allows users to determine the length of the insert and to sequence both ends of the insert, generating high quality sequence data. This process generated a total of 30.89 Gb of raw sequence data (Table A in S2 File).
Sequence filtering
Artificial reads or low quality paired reads derived mainly from base-calling duplicates and adapter contamination were filtered to obtain a clean read set. Base-calling duplicates, which are pseudo-sequences caused by the SOLEXA pipeline, were filtered at a threshold of Euclidean distance ≤ 3 and a mismatch rate of ≤ 0.1. Duplicated reads from PCR amplification during library construction (both long inserts of ≥ 2 kb and short inserts of 150–500 bp) were filtered to ensure high accuracy in scaffold construction. Besides, removal of low quality sequences such as those with an excess of “N” bases (> 10%) was done. Different bacterial and viral genomes were used for similarity mapping with Burrows-Wheeler Aligner (BWA). Reads mapped to the bacterial and viral genomes were removed. From these steps, the data size of all five libraries (post-filtered) was reduced significantly (Table B in S2 File).
Genome assembly
The sequencing errors were corrected based on k-mer frequency information before assembly in order to reduce the memory consumption in de Bruijn graph algorithm construction [30]. We had chosen 17-mers and corrected the sequencing errors for frequency lower than 4. The total used bases were 6.63 Gb and used reads were 66,335,501 whereas the specificity k-mer number was 231,186,702 (Table C in S2 File). The expected depth peak was 100 according to the distribution curve (Figure A in S1 File).
The corrected reads were assembled using SOAPdenovo, a software that assembles short oligonucleotide into contigs and scaffolds through de Bruijn graph algorithm [31]. During assembly, a de Bruijn graph was used to assemble all possible sequences from the Illumina reads, with a k-mer as a node and the k-1 bases overlap between two k-mers as an edge. In this T. gondii genome assembly, we chose k = 25 bp (25-mers) to construct de Bruijn graph. To avoid chimeric reads, which are responsible for misassembles by generating incorrect sequence overlap, only short insert-size (<1 kb) of single- and paired-end reads were used in this assembly. Further corrections performed during de Bruijn graph simplification encompassed errors removals (tips, low coverage linkages and bubbles removal) and tiny repeats resolving. The graph was transformed to a contig graph by transforming linearly connected k-mers into a pre-contig node. Dijkstra’s algorithm (Skiena) was used to detect bubbles, which were then merged into a single pathway when the branches sequences were identical. Using this method, repetitive sequences were collapsed and consensus sequences were obtained.
The contig sequences were obtained by conjoining the k-mers in an unambiguous path. These contigs were broken into fragments at the boundaries of repeat ambiguous connections. All reads were realigned onto the contig sequences to obtain aligned paired-end reads. Contigs were linked to a scaffolding graph with paired-end reads. Connections between contigs comprised the edges in this graph, and the branch length demonstrated the gap size, which was calculated from the insert size of the paired-end reads. Subsequently, sub-graph linearization was applied to transform interleaving contigs into a linear structure. Repeat masking was used to mask complicated connections for repeat contigs. Following this, the amount of shared paired-end relationships between contig pairs were calculated, the ratio of consistent and conflicting paired-ends were determined, and scaffolds construction was done from the shortest to the longest insert size paired-ends in a step by step manner.
Gaps, which composed mainly of masked repeats during scaffold construction, were found. Through the paired-end information, we retrieved read pairs that had one read well-aligned on the contigs and another read located in the gap region. Local assembly was performed on these reads to fill in small gaps within the scaffolds. We constructed a 27-mers de Bruijn graph with reads in the gaps and the contig ends on both sides using approach similar to that of contig construction. Gaps were filled with path sequences if an unambiguous path was found between those two contig ends. Each read in the gaps was checked to find one that had unambiguously mapped ends on both sides of the contigs and filled the gaps with read sequences. Most of the small tandem-repeat gaps were correctly filled by this way. However, longer tandem-repeat gaps could not be resolved by read sequences. Hence, we filled them with two repeat units along with a string of “N”. The GC-depth content difference is a primary factor for non-random sequencing-depth distribution [32]. Therefore it was examined to analyse the nucleotide distribution, to assess the randomness of sequencing and to inspect for possible sample contamination. Sliding of 500 bp bins (with 250 bp overlap) was applied for the examination.
Repeats
Many assembled genomes consist of repeated sequences which have to be reduced to avoid misassembly and to minimize the error rate. A combination of homology-based and de novo approach was employed in this study to identify the interspersed repeated sequences. Known transposable elements (TEs) were identified using RepeatMasker and RepeatProteinMask against the Repbase library of TEs (Table D in S2 File) [33–35]. The RepeatProteinMask was used for identifying highly diverged TEs. On the other hand, we constructed a repeat library with RepeatModeler using two programs; RECON and RepeatScout for the de novo approach [36, 37]. The outputs were consensus sequences and classification information for each repeat family. Number of repeats was further identified through several tools such as DNA, SINE, LINE and LTR (Table E in S2 File).
Gene annotation
Both homology-based and de novo approaches were employed in the predictions of gene structures in the draft genome assembly. Four gene sets from database ToxoDB (version 13.0) were used in homology-based approach [38]; Neospora caninum, T. gondii GT1, T. gondii VEG and T. gondii ME49, whereas de novo approach involved usage of AUGUSTUS, Genscan and SNAP softwares to filter partial and small genes with coding length shorter than 150 bp in order to reduce false positives. The predictions were aligned to a TE protein database using BlastP with E-value of 10−5 and filtered TE-derived genes that had more than 50% aligning rate. Different gene sources were then integrated to form a consensus gene set using GLEAN [39] by combining results from different gene predictions into a single set of gene prediction (Tables F and G in S2 File). Subsequently, individual genes were assigned functional annotation based on clustering Method. Functional annotation of the unique T. gondii genes has revealed key data that can further our understanding of differences between different strains. Uniprot database [40] was referred to understand the structure, subcellular location and functions of annotated proteins.
Besides, whole genome synteny analysis was performed. Based on the chromosomes of T. gondii GT1 strain, contigs reordering was done on the assembled scaffolds of T. gondii RH strain, where all the 14 chromosomes were reordered and reassembled with Mauve Aligner [41]. Similarities in chromosomes across different strains of T. gondii were studied using BLASTn (E value < 1e-05). Synteny blocks were retrived from the blast results to constitute the synteny plot with Circos v0.69 [42].
Clustering of gene orthologs
All protein sequences were compared against a database containing a protein dataset of all the species studied (T. gondii RH, T. gondii GT1, T. gondii ME49, T. gondii VEG, Neospora caninum) (Table H in S2 File) (BlastP; using an E-value < 10−7), and con-joined fragment alignments for every gene were used through program Solar (Skiena). A connection (edge) was assigned between two nodes (genes) if more than one third of the region aligned to both genes. A h-score (0 to 100) was used to weight the similarity (edge). For two genes, G1 and G2, the h-score was defined as score (G1xG2) / max score (G1G1), score (G2G2), whereby the score was actually the BLAST raw score. Extracting gene families using clustering by Hcluster_sg, the average distance were used for the hierarchical clustering algorithm, requiring the minimum edge weight (h-score) to be larger than five, and minimum edge density (total number of edges / theoretical number of edges) to be larger than one third. The clustering of gene families was terminated if out-group genes were identified.
Phylogenetic reconstruction
Protein sequences of parasite gene families between T. gondii strains were retrieved from previous work on T. gondii genomes [27] to run multiple sequence alignment using MAFFT v7 program (default option) [43]. Phylogeny tree editing and annotations were performed on iTOL webserver [44]. Phylogeny tree was constructed using Neighbour joining algorithm in Mafft online server [45].
Data Access
This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the project accession LLKL01000000 (BioProject number, PRJNA294483; BioSample, SAMN04026592), consists of sequences LLKL01000001-LLKL01000441.
Results
Genome assembly and repeat content
The genome of T. gondii RH strain (TOXRH) was sequenced at 171-fold coverage (Table 1). The total k-mer number was 5,402,162,948 with the calculated depth peak of 86 (Table C in S2 File). A final draft assembly of 441 scaffolds totalling 69.35 Mb in length was produced (Table 1). All gaps in RH strain assembled genome were filled similarly to GT1 and VEG strains. Scaffolds with length shorter than 200 bp were excluded (Table I in S2 File). Besides, chromosome assemblies were conducted (Table J in S2 File).
Table 1. Comparison of assembly statistics.
ToxoDB version 13.0 was referred.
| Details | T. gondii RH | T. gondii GT1 | T. gondii ME49 | T. gondii VEG | Neospora caninum |
|---|---|---|---|---|---|
| Coverage (x) | 171 | 67.44 | 26.5 | 77.38 | N/A |
| Genome size (Mb) | 69.35 | 63.95 | 65.67 | 64.52 | 59.1 |
| Number of Scaffolds | 441 | 2063 | 2263 | 1323 | 66 |
| Contig | 7797 | 2337 | 1243 | 1340 | 52 |
| Scaffolds N50 (kb) | 2839.2 | 6105.4 | 6327.7 | 6372.9 | 5490.9 |
| GC (%) | 52.0 | 52.4 | 52.3 | 52.4 | 54.8 |
| Coding genes (n) | 7860 | 8460 | 8322 | 8410 | 7122 |
| Chromosomes | 14 | 14 | 14 | 14 | 14 |
The majority of the sequences have a GC content between 40–60% (mean = 52%) and almost all have a sequencing depth above 60X (Figures B-D in S1 File). This is indeed similar to T. gondii GT1 (Type I), T. gondii ME49 (Type II) and T. gondii VEG (Type III) (Figure C in S1 File). We detected 190 (76.61%) core essential genes by CEGMA, indicating that the assembly represents a substantial proportion of the entire genome. We also estimated a repeat content of 15.98% (equating to 11.15 Mb of DNA) in this draft genome (Table D in S2 File), comprised of 5.136% (3,583,811 bp) DNA transposons, 1.735% (1,210,317 bp) LINE, 0.037% (25,965 bp) SINE, 4.858% (3,389,530 bp) LTR and 2.914% (2,033,436 bp) unclassified dispersed elements (Table E in S2 File). Additionally, we have estimated segmental duplication of 1.69 Mb.
Gene set comparison of T. gondii RH strain with T. gondii GT1, ME49 and VEG strains
GLEAN analysis predicted 7860 protein-encoding genes and 216 non-coding DNAs. The average length of exons and introns are 419 bp and 548 bp, respectively, with an average of 5.9 exons per gene (Table F in S2 File). This finding is similar to genomes of other T. gondii strains. Based on comparison with the draft genomes of N. caninum, T. gondii GT1, T. gondii ME49 and T. gondii VEG [38], T. gondii RH strain shows high sequence similarity to all three distinct strains of T. gondii. By applying the clustering method, gene families (homologous genes clustered together from different genomes) were analyzed. Of the predicted T. gondii RH gene families, 97.8% (5246 gene families) have orthologs (BLASTp cut-off: 10−5) in T. gondii GT1 compared to that in T. gondii ME49 (n = 5237; 97.6%) and T. gondii VEG (n = 5232; 97.5%) (Fig 1). From phylogenetic analysis (Fig 2), close evolutionary relationship between T. gondii RH and T. gondii GT1 was noted. This was in parallel with the results from the clustering method mentioned earlier. Both sets of results reflected sharing of a more recent common ancestor between T. gondii RH and T. gondii GT1. Between T. gondii ME49 and VEG strains, T. gondii RH showed closer relationship with T. gondii ME49. Meanwhile, 97 gene families were predicted to be unique to the T. gondii RH genome (Fig 1; Table H in S2 File). From the synteny analysis, we found that all three genomes of T. gondii GT1, ME49 and VEG showed high level of synteny with the genome of T. gondii RH. The generated synteny blocks reflected high chromosomal conservation between T. gondii RH and T. gondii GT1 whereas both T. gondii ME49 and T. gondii VEG showed smaller length chromosomal rearrangements. Generally, T. gondii RH has 99% similarity with all strains of T. gondii at chromosomal synteny level (Fig 3).
Fig 1. Genome comparison of T. gondii RH, GT1, ME49 and VEG strains.
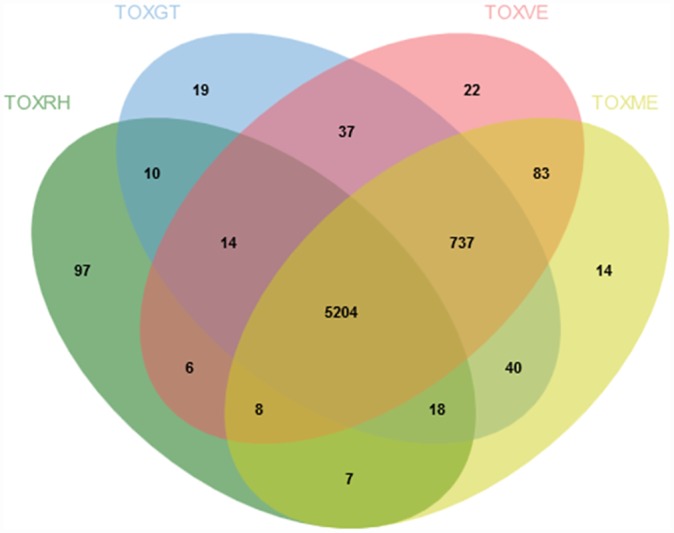
Abbreviation: TOXME: T. gondii ME49 strain; TOXVE: T. gondii VEG strain; TOXGT: T. gondii GT1 strain; TOXRH: T. gondii RH strain.
Fig 2. Phylogeny tree in circular format representing genes related to the parasite gene families of T. gondii.

Different colors represent the various parasite gene families.
Fig 3. Circos plot illustrating the levels of synteny among genomes of different T. gondii strains.

Outer ring represents chromosomes of each strain, with the labelling of respective chromosome numbering. The scale marks on the chromosome represent 1Mb. The color bands represent syntenic blocks between the chromosomes under comparison with the chromosomes of T. gondii RH. The blue lines within the chromosome blocks of T. gondii RH indicate the T. gondii RH-unique genes identified as part of the orthologous gene study.
Annotation and protein classifications
Three types of noncoding RNA were found, i.e., 181 copies of tRNA, 18 copies of rRNA and 4 copies of miRNA (Table K in S2 File). In all, 7563 genes were clustered in 5364 families with 97 unique families encoding a total of 111 unique genes (Tables H and L in S2 File). The unique genes found were indicated in the circos plot of synteny study as well (Fig 3). These unique genes were found to be distributed unevenly across all chromosomes of T. gondii RH except chromosomes 11 and 12 (Table M in S2 File). The T. gondii RH-unique genes were “concentrated” in chromosomes 8, 6, 10, 7A, and 9.
To understand more about evolutionary origin of the 111 T. gondii RH unique genes, we decided to conduct a protein-protein BLAST (BLASTp) screening against genomes of other T. gondii strains available from ToxoDB. From this local alignment, 12 of the 111 genes showed good BLAST matches with genes of other T. gondii strains, validating the presence of similar homologs within the genomes of other T. gondii strains (Table N in S2 File). BLASTX of T. gondii ME49 unique genes found previously [27] against the unique genes found in T. gondii RH draft genome was performed. There were nine genes with unknown annotations showing good matches (Table O in S2 File). Besides, unique gene synteny similarity search between the T. gondii RH scaffold and T. gondii GT1 chromosomes were conducted. From this investigation, only eight of the T. gondii RH unique genes showed overlaps of synteny and the remaining 103 genes were specific to T. gondii RH strain (Table P in S2 File). These findings suggest divergence evolution of T. gondii strains from a common ancestor, giving rise to the many T. gondii RH genes that are not recognizable as homologs under comparisons with genomes of other T. gondii strains.
Most of the unique genes were annotated to unknown functions. Nevertheless, 17 unique genes were annotated to proteins with vital functions (Table Q in S2 File). Functions annotated by the 11 genes involved in gene expression regulation could be classified into pre-translational regulation (TOXaeaD_GLEAN_10004604, TOXaeaD_GLEAN_10007767, TOXaeaD_GLEAN_10006721, TOXaeaD_GLEAN_10006347), translation “patrolling” (TOXaeaD_GLEAN_10005699, TOXaeaD_GLEAN_10005558), and post-translational modifications (TOXaeaD_GLEAN_10000645, TOXaeaD_GLEAN_10000683, TOXaeaD_GLEAN_10003567, TOXaeaD_GLEAN_10005019, TOXaeaD_GLEAN_10005632). Differences in regulation of gene expression may lead to different and unique phenotypes. Importantly, these genes may play critical roles in epigenetics of the T. gondii RH strain, which may contribute to distinct phenotypes among different strains despite high similarity at the genomic level. Indeed, epigenetics of T. gondii have received more research attention in the field of parasitology [46]. In addition, T. gondii is known for its temporal regulation of gene expression in conjunction with stage transition [47]. When compared with the type I and type II parasites, T. gondii RH strain has relatively lower tendency to form bradyzoites. These T. gondii RH-unique genes may involve in its regulation of stage transformation (i.e., tachyzoite to bradyzoite) that is different from other T. gondii clonal lineages.
Four T. gondii RH unique genes were annotated to regulation of replication, encompassing DNA repairing (TOXaeaD_GLEAN_10004705), mitotic cell cycle regulation (TOXaeaD_GLEAN_10005699, TOXaeaD_GLEAN_10006721, TOXaeaD_GLEAN_10004705), and meiosis regulation (TOXaeaD_GLEAN_10004604). It is noteworthy that the unique genes responsible for replication regulation were found to be annotated to functions that are closely associated with gene expression regulation as well. After all, the speed of replication is linked to the tachyzoite-bradyzoite stage conversion [48], which implies different protein expressions.
Two unique genes were annotated to exosomal proteins carrying calcium-related functions. Gene TOXaeaD_GLEAN_10001862 was annotated to hippocalcin-like protein 1 (HPCAL1). As indicated by the name, this protein is involved in calcium ion binding. Another unique gene (TOXaeaD_GLEAN_10003157) is annotated to calmodulin-like protein 3 (CALML3), which is also known as protein NB-1. CALML3 is an exosomal protein involved in calcium binding and release [49]. We believe that this protein may be involved in regulation of locomotion, invasion and egression [50, 51]. We have verified the presence of this gene within the T. gondii RH genome with PCR and sequencing as well (Figure E in S1 File). Needless to say, further work is needed to verify this postulation.
Two genes were found to be annotated to enzymatic digestion. Gene TOXaeaD_GLEAN_10006826 was annotated to pepsin II-4 (pepsin A), whereas gene TOXaeaD_GLEAN_10004048 was annotated to a putative peroxisomal acyl-coenzyme A oxidase 1.2, which catalyzes the desaturation of acyl-CoAs to 2-trans-enoyl-CoAs. It regulates beta oxidation of fatty acids, which transforms lipids into acetyl CoA. The transformed acetyl CoA will then be led into Kreb’s cycle for energy production. Efficient metabolism and energy generation may enable T. gondii RH strain to stay “active” as tachyzoites instead of facing “starvation” that drives bradyzoite stage conversion [52].
One gene (TOXaeaD_GLEAN_10002720) was annotated to a transporter known as multidrug resistance-associated protein 1 (MRP1), which is also known as ATP-binding cassette sub-family C member 1 protein (ABC.C1). The ABC proteins have been linked to drug resistance in protozoan parasites [53]. This “detox” transporter acts as a multi-specific organic anion transporter, with oxidized glutathione, cysteinyl leukotrienes and activated aflatoxin B1 as substrates. It also transports glucuronides and sulfated conjugates of steroids and bile salts. Indeed, it was reported in an earlier study that three ABC transporters of T. gondii RH (which showed larger plaque formation) were significantly over-expressed when compared to T. gondii GT1, suggestive of association between the ABC transporters and the parasite’s higher growth rate [54]. Besides, a recent study found that the ABC.C1 of type I T. gondii is different from those of non-type I T. gondii parasites [55]. Although sulfadiazine resistance in T. gondii has been shown to be unrelated to ABC transporters [55], we believe that this T. gondii RH-unique ABC transporter may play important roles in detox machinery of the parasite. This protein may modulate accumulation and removal of other toxic substances or xenobiotics, which enables longer extracellular survival for the parasite.
Discussion
Whole genome alignments represent the fundamental basis for comparative analyses aimed at identifying and characterising functional related stretches of genes that are clustered. For instance, similarity across a wide range of evolutionary distance can be detected by a multiple alignments of homologous sequences from several species, whereby conserved and important biological similarities are usually revealed. Similarly, estimation of local rates of evolution on the basis of multiple alignments provides quantitative assessment of the strength of evolutionary constraints and the importance of functional elements. Collectively, these contribute to better understanding of the cellular biology of a parasite.
In this study, we generated a draft genome of T. gondii RH strain that was slightly larger than those reported by a number of studies [22–24, 27]. This may be due to a number of reasons. The performed sequencing depth of coverage is one of the possible reasons. Increased depth of coverage can compensate the possible presence of sequencing errors in the short reads, which may otherwise be filtered even if the reads are not sequencing errors. Thus, more reads are retained, resulting in a larger assembly. Besides, the larger amount of non-coding DNAs within the draft genome of T. gondii RH strain used in this study may contribute to the larger genome size as well. Interestingly, the number of coding genes in T. gondii RH draft genome of this study happened to be of the lowest under comparison with the genomes of T. gondii GT1, ME49 and VEG strains (Table 1). Nevertheless, the draft genome size predicted from this study is within the expected size range [56].
By referring to a list of pathogenesis-associated, parasite gene families presented by a recently published work [27], we constructed a phylogeny tree to study the evolutionary relationship between T. gondii RH, GT1, ME49 and VEG strains. We managed to find 23 of the 24 reported parasite-specific domains. Only one domain (para_38; with repeat motifs) was not found in T. gondii RH draft genome (Table R in S2 File). A total of 1041 genes from the genome of T. gondii RH, and the published genomes of T. gondii GT1, VEG and ME49 strains were recruited. Interestingly, T. gondii RH genome had the smallest number of genes in most of these gene families as compared with the GT1, VEG and ME49 strains. T. gondii RH draft genome assembled from this study had more genes in few of the domains (Para_101 and Para_27, both annotated to hypothetical proteins; and Para_41 and Para-44, which were annotated to rhoptry proteins) than draft genomes of other strains. Such differences may be associated with certain phenotypic variations among the different strains of T. gondii. On the whole, T. gondii RH shared the closest evolutionary relationship with T. gondii GT1, as compared with the ME49 and VEG strains.
Our genome synteny analysis revealed that T. gondii RH genome shared high level of synteny with the genomes of three strains of T. gondii (GT1, ME49 and VEG) (Fig 3). The level of synteny depends on chromosomal rearrangement throughout the process of evolution. Chromosomal rearrangement can happen via chromosome translocation, inversion, fission and fusion. These processes result in loss of synteny between chromosomes under comparison. Likewise, such rearrangement may restore the synteny between chromosomes under comparison along the course of evolution. Previously, it was suggested that crossing of type II T. gondii with other ancestral strains gave rise to type III (as the first generation offspring) and type I (likely the second generation offspring) parasites [57]. It is likely that the incidence of chromosomal rearrangement was low along the course of evolution from type II to type III and type I parasites.
Different strains of T. gondii show varied biological properties. For instance, type I strain was reported to show higher trans-epithelial migration rate than type II and type III strains [58]. Indeed, even T. gondii strains of the same clonal lineage have been reported to demonstrate different phenotypes. The laboratory-adapted RH strains are known to be incapable of forming bradyzoites that are responsible for chronic infection [59]. Furthermore, the RH strain also poses a higher extracellular survival rate than GT1 strain [54]. These biological phenotype variations contribute to the virulence differences among these parasites. Intriguingly, the three major T. gondii clonal lineages show high similarity at the genome level, with the RH strain having only 111 unique genes. Therefore, these unique genes may be responsible for the different characteristics of T. gondii RH strain compared to other strains. We managed to locate these genes at the chromosomes of the parasite (Fig 3 and Table M in S2 File). Only chromosomes 11 and 12 did not have any T. gondii RH- unique genes. Most of these 111 T. gondii RH-unique genes were annotated to proteins with unknown functions. Nonetheless, 17 genes were annotated to proteins known for functions that are vital to cellular biology, many of which were related to regulation of gene expression.
By analyzing T. gondii RH draft genome, we managed to understand better the genomic structure and evolution of this strain. The differences between the closely related T. gondii genomes were studied. We unraveled a short list of genes unique to T. gondii RH strain, where annotation of a number of unique genes fits in elegantly with findings from previous T. gondii RH genome-wise studies.
The genome of T. gondii RH was recently archived [27]. Nevertheless, we did not do comparison between the published T. gondii RH draft genome and the T. gondii RH draft genome assembled from this study. As the recently published T. gondii RH genome is a raw data without assemblies, any comparison done with such raw data would be incomplete. Needless to say, the availability of a richer T. gondii genomic database contributed by different parties will definitely benefit researchers in future studies.
The unique genes described above may be the genetic markers responsible for phenotypic differences between T. gondii RH strain and other strains of T. gondii. Nevertheless, it is important to acknowledge the difficulty of drawing a solid conclusion with only data garnered from draft genomes. Genomic comparison would be more powerful with downstream experiments such as transcriptome comparisons, gene silencing, gene knock-out and knock-in experiments. However, our findings have highlighted a short list of potential gene candidates, which may serve as a reference for future in-depth studies. Further investigations on the T. gondii RH unique genes with hypothetical protein annotations may complement our understandings on the virulence difference among the different strains of T. gondii. This genome is useful as a resource to study gene rearrangements of T. gondii as well.
Conclusions
We have performed full genome sequencing and de novo assembly on the RH strain of T. gondii, which complement the genomic archive of T. gondii. High genome similarity between the different strains of T. gondii was seen, and 111 T. gondii RH strain-unique genes were found, some of which may be related to the distinct phenotypes of this particular strain. Data obtained from this study contribute to better understanding on T. gondii and serve as a reference for future studies on this parasite.
Supporting Information
Figure A. Estimation of genome size of T. gondii RH strain using K-mer analysis. Figure B. GC content and sequencing depth of the T. gondii RH strain recruited in this project. Figure C. GC content distributions for the genome. The x-axis represents GC content and the y-axis represents the proportion of the bins number divided by the total windows. Figure D. Sequence depth distribution curve. Figure E. Primer design, PCR and sequencing of a randomly selected T. gondii RH-unique gene (TOXaeaD_GLEAN_10003157) found in this study.
(DOC)
Table A. Statistic of pre-filter data. Table B. Statistic of post-filter data. Table C. 17-mer statistics information. Table D. General statistics of repeats in genome. Table E. TEs content in the assembled T. gondii RH genome. Table F. General statistics of predicted protein-coding genes. Table G. Summary of evidence for the GLEAN gene models. Table H. Statistical analysis of gene families. Table I. Statistics of the assembled sequence length. Table J. Chromosome assembly. Table K. Non-coding RNA genes in the genome. Table L. The 111 genes were predicted to be unique to the T. gondii RH genome based on Clustering Method. Table M. Chromosomal loci of the 111 T. gondii RH-unique genes. Table N. BLASTp result of 111 unique genes to the latest release from Toxodb.org. Table O. BLASTx of T. gondii ME49 unique genes vs T.gondii RH (external data retrieved: supplementary table G of Lorenzi et al. 2016[27]). Table P. Synteny overlaps of the T. gondii RH unique gene loci on scaffolds with the T. gondii GT1 genome. Table Q. The 17 unique genes were annotated to proteins with vital functions. Table R. Single Copy clusters from T. gondii RH comparad to recently published gene families of T. gondii containing unique parasite-specific (para) domains. (external data retrieved: supplementary table G of Lorenzi et al. 2016[27]).
(XLS)
Data Availability
All relevant data are within the paper and its Supporting Information files. In addition, this Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession SUB1075932 (BioProject number, PRJNA294483; BioSample, SAMN04026592).
Funding Statement
This research project was supported by University of Malaya High Impact Research (HIR) Grant UM-MOHE (UM.C/HIR/MOHE/MED/16) from the Ministry of Higher Education, Malaysia.
References
- 1.Pappas G, Roussos N, Falagas ME. Toxoplasmosis snapshots: global status of Toxoplasma gondii seroprevalence and implications for pregnancy and congenital toxoplasmosis. Int J Parasitol. 2009; 39: 1385–94. 10.1016/j.ijpara.2009.04.003 [DOI] [PubMed] [Google Scholar]
- 2.Wasmuth JD, Pszenny V, Haile S, Jansen EM, Gast AT, Sher A, et al. Integrated bioinformatics and targeted deletion analyses of the SRS gene superfamily identify SRS29C as a negative regulator of Toxoplasma virulence. MBio. 2012; 3: e00321–12. 10.1128/mBio.00321-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Contini C. Clinical and diagnostic management of toxoplasmosis in the immunocompromised patient. Parassitologia. 2008; 50: 45–50. [PubMed] [Google Scholar]
- 4.Fatoohi AF, Cozon GJ, Greenland T, Ferrandiz J, Bienvenu J, Picot S, et al. Cellular immune responses to recombinant antigens in pregnant women chronically infected with Toxoplasma gondii. Clin Diagn Lab Immunol. 2002; 9: 704–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, et al. Foodborne illness acquired in the United States- major pathogens. Emerg Infect Dis. 2011; 17: 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bülow R, Boothroyd JC. Protection of mice from fatal Toxoplasma gondii infection by immunization with p30 antigen in liposomes. J Immunol. 1991; 147: 3496–500. [PubMed] [Google Scholar]
- 7.Dardé ML, Bouteille B, Pestre-Alexandre M. Isoenzyme analysis of 35 Toxoplasma gondii isolates and the biological and epidemiological implications. J Parasitol. 1992; 78: 786–94. [PubMed] [Google Scholar]
- 8.Su C, Evans D, Cole RH, Kissinger JC, Ajioka JW, Sibley LD. Recent expansion of Toxoplasma through enhanced oral transmission. Science. 2003; 299: 414–16. [DOI] [PubMed] [Google Scholar]
- 9.Saeij JP, Boyle JP, Boothroyd JC. Differences among the three major strains of Toxoplasma gondii and their specific interactions with the infected host. Trends Parasitol. 2005; 21: 476–81. [DOI] [PubMed] [Google Scholar]
- 10.Howe DK, Honoré S, Derouin F, Sibley LD. Determination of genotypes of Toxoplasma gondii strains isolated from patients with toxoplasmosis. J Clin Microbiol. 1997; 35: 1411–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grigg ME, Ganatra J, Boothroyd JC, Margolis TP. Unusual abundance of atypical strains associated with human ocular toxoplasmosis. J Infect Dis. 2001; 184: 633–9. [DOI] [PubMed] [Google Scholar]
- 12.Fuentes I, Rubio JM, Ramírez C, Alvar J. Genotypic characterization of Toxoplasma gondii strains associated with human toxoplasmosis in Spain: direct analysis from clinical samples. J Clin Microbiol. 2001; 39: 1566–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sabin AB. Toxoplasmic encephalitis in children. J Am Med Assoc. 1941; 116: 801–7. [Google Scholar]
- 14.Dubey JP. Mouse pathogenicity of Toxoplasma gondii isolated from a goat. Am J Vet Res. 1980; 41: 427–9. [PubMed] [Google Scholar]
- 15.Pfefferkorn ER, Pfefferkorn LC. Toxoplasma gondii: isolation and preliminary characterization of temperature-sensitive mutants. Exp Parasitol. 1976; 39: 365–76. [DOI] [PubMed] [Google Scholar]
- 16.Kaufman HE, Remington JS, Jacobs L. Toxoplasmosis: the nature of virulence. Am J Ophthalmol. 1958; 46: 255–60. [DOI] [PubMed] [Google Scholar]
- 17.Sibley LD, Boothroyd JC. Construction of a molecular karyotype for Toxoplasma gondii. Mol Biochem Parasitol. 1992; 51: 291–300. [DOI] [PubMed] [Google Scholar]
- 18.Howe DK, Sibley LD. Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. J Infect Dis. 1995; 172: 1561–6. [DOI] [PubMed] [Google Scholar]
- 19.Dardé ML. Genetic analysis of the diversity in Toxoplasma gondii. Ann Ist Super Sanita. 2004; 40: 57–63. [PubMed] [Google Scholar]
- 20.Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004; 363: 1965–76. [DOI] [PubMed] [Google Scholar]
- 21.Kissinger JC, Gajria B, Li L, Paulsen IT, Roos DS. ToxoDB: accessing the Toxoplasma gondii genome. Nucleic Acids Res. 2003; 31: 234–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gajria B, Bahl A, Brestelli J, Dommer J, Fischer S, Gao X, et al. ToxoDB: an integrated Toxoplasma gondii database resource. Nucleic Acids Res. 2008; 36: D553–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paulsen I. Nucleotide sequences [large scale genomic DNA] of Toxoplasma gondii VEG strain [ECO:0000313 EMBL:ESS29589.1]. Submitted (March 2007) to: EMBL/GenBank/DDBJ databases.
- 24.Bontell IL, Hall N, Ashelford KE, Dubey JP, Boyle JP, Lindh J, et al. Whole genome sequencing of a natural recombinant Toxoplasma gondii strain reveals chromosome sorting and local allelic variants. Genome Biol. 2009; 10: R53 10.1186/gb-2009-10-5-r53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farrell A, Thirugnanam S, Lorestani A, Dvorin JD, Eidell KP, Ferguson DJ, et al. A DOC2 protein identified by mutational profiling is essential for apicomplexan parasite exocytosis. Science. 2012; 335(6065): 218–21. 10.1126/science.1210829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang N, Farrell A, Niedelman W, Melo M, Lu D, Julien L, et al. Genetic basis for phenotypic differences between different Toxoplasma gondii type I strains. Bmc Genomics. 2013; 14(1): 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lorenzi H, Khan A, Behnke MS, Namasivayam S, Swapna LS, Hadjithomas M, et al. Local admixture of amplified and diversified secreted pathogenesis determinants shapes mosaic Toxoplasma gondii genomes. Nat Commun. 2006; 7: 10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burg JL, Grover CM, Pouletty P, Boothroyd JC. Direct and sensitive detection of a pathogenic protozoan, Toxoplasma gondii, by polymerase chain reaction. J Clin Microbiol. 1989; 27: 1787–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lau YL, Meganathan P, Sonaimuthu P, Thiruvengadam G, Nissapatorn V, Chen Y. Specific, sensitive, and rapid diagnosis of active toxoplasmosis by a loop-mediated isothermal amplification method using blood samples from patients. J Clin Microbiol. 2010; 48: 3698–3702. 10.1128/JCM.00462-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Bruijn NG. A combinatorial problem. Koninklijke Nederlandse Akademie v. Wetenschappen. 1946; 49: 758–64. [Google Scholar]
- 31.Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010; 20: 265–72. 10.1101/gr.097261.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008; 456: 53–59. 10.1038/nature07517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.RepeatMasker [http://www.repeatmasker.org]. Accessed 20 July 2014.
- 34.Repeat Protein Mask [http://www.repeatmasker.org]. Accessed 20 July 2014.
- 35.Repbase [www.girinst.org/repbase/]. Accessed 20 July 2014.
- 36.Bao Z, Eddy SR. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002; 12: 1269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005; 21 Suppl 1:i351–8. [DOI] [PubMed] [Google Scholar]
- 38.ToxoDB [www.toxodb.org/toxo/]. Accessed 25 march 2016.
- 39.GLEAN [http://glean-gene.sourceforge.net/]. Accessed 20 July 2014.
- 40.Uniprot [www.uniprot.org.]. Accessed 21 September 2015.
- 41.Darling AE, Mau B, Perna NT. Progressive Mauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010; 5: e11147 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009; 19: 1639–1645. 10.1101/gr.092759.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002; 30: 3059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Letunic I, Bork P. Interative Tree of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007; 23: 127–8. [DOI] [PubMed] [Google Scholar]
- 45.MAFFT [http://mafft.cbrc.jp/alignment/server/]. Accessed 30 May 2016.
- 46.Dixon SE, Stilger KL, Elias EV, Naguleswaran A, Sullivan WJ Jr. A decade of epigenetic research in Toxoplasma gondii. Mol Biochem Parasitol. 2010; 173: 1–9. 10.1016/j.molbiopara.2010.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Radke JR, Behnke MS, Mackey AJ, Radke JB, Roos DS, White MW. The transcriptome of Toxoplasma gondii. BMC Biol. 2005; 3: 26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bohne W, Heesemann J, Gross U. Reduced replication of Toxoplasma gondii is necessary for induction of bradyzoite-specific antigens: a possible role for nitric oxide in triggering stage conversion. Infect Immun. 1994; 62: 1761–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yaswen P, Smoll A, Peehl DM, Trask DK, Sager R, Stampfer MR. Down-regulation of a calmodulin-related gene during transformation of human mammary epithelial cells. Proc Natl Acad Sci U S A. 1990; 87: 7360–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arrizabalaga G, Boothroyd JC. Role of Calcium during Toxoplasma gondii invasion and egress. Int J Parasitol. 2004; 34: 361–8. [DOI] [PubMed] [Google Scholar]
- 51.Pezzella-D’Alessandro N, Le Moal H, Bonhomme A, Valere A, Klein C, Gomez-Marin J, et al. Calmodulin distribution and the actomyosin cytoskeleton in Toxoplasma gondii. J Histochem Cytochem. 2001; 49: 445–54. [DOI] [PubMed] [Google Scholar]
- 52.Ihara F, Nishikawa Y. Starvation of low-density lipoprotein-derived cholesterol induces bradyzoite conversion in Toxoplasma gondii. Parasit Vectors. 2014; 7: 248 10.1186/1756-3305-7-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sauvage V, Aubert D, Escotte-Binet S, Villena I. The role of ATP-binding cassette (ABC) proteins in protozoan parasites. Mol Biochem Parasitol. 2009; 167: 81–94. 10.1016/j.molbiopara.2009.05.005 [DOI] [PubMed] [Google Scholar]
- 54.Khan A, Behnke MS, Dunay IR, White MW, Sibley LD. Phenotypic and gene expression changes among clonal type I strains of Toxoplasma gondii. Eukaryot Cell. 2009; 8: 1828–36. 10.1128/EC.00150-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doliwa C, Escotte-Binet S, Aubert D, Sauvage V, Velard F, Schmid A, et al. Sulfadiazine resistance in Toxoplasma gondii: no involvement of overexpression or polymorphisms in genes of therapeutic targets and ABC transporters. Parasite. 2013; 20: 19 10.1051/parasite/2013020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boothroyd JC. Toxoplasma gondii: 25 years and 25 major advances for the field. Int J Parasitol. 2009; 39: 935–46. 10.1016/j.ijpara.2009.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boyle JP, Rajasekar B, Saeij AP, Ajioka JW, Berriman M, Paulsen I, et al. Just one cross appears capable of dramatically altering the population biology of a eukaryotic pathogen like Toxoplasma gondii. Proc Natl Acad Sci U S A. 2006; 103: 10514–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barragan A, Sibley LD. Transepithelial migration of Toxoplasma gondii is linked to parasite motility and virulence. J Exp Med. 2002; 195: 1625–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Villard O, Candolfi E, Ferguson DJ, Marcellin L, Kien T. Loss of oral infectivity of tissue cysts of Toxoplasma gondii RH strain to outbred Swiss Webster mice. Int J Parasitol. 1997; 27: 1555–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure A. Estimation of genome size of T. gondii RH strain using K-mer analysis. Figure B. GC content and sequencing depth of the T. gondii RH strain recruited in this project. Figure C. GC content distributions for the genome. The x-axis represents GC content and the y-axis represents the proportion of the bins number divided by the total windows. Figure D. Sequence depth distribution curve. Figure E. Primer design, PCR and sequencing of a randomly selected T. gondii RH-unique gene (TOXaeaD_GLEAN_10003157) found in this study.
(DOC)
Table A. Statistic of pre-filter data. Table B. Statistic of post-filter data. Table C. 17-mer statistics information. Table D. General statistics of repeats in genome. Table E. TEs content in the assembled T. gondii RH genome. Table F. General statistics of predicted protein-coding genes. Table G. Summary of evidence for the GLEAN gene models. Table H. Statistical analysis of gene families. Table I. Statistics of the assembled sequence length. Table J. Chromosome assembly. Table K. Non-coding RNA genes in the genome. Table L. The 111 genes were predicted to be unique to the T. gondii RH genome based on Clustering Method. Table M. Chromosomal loci of the 111 T. gondii RH-unique genes. Table N. BLASTp result of 111 unique genes to the latest release from Toxodb.org. Table O. BLASTx of T. gondii ME49 unique genes vs T.gondii RH (external data retrieved: supplementary table G of Lorenzi et al. 2016[27]). Table P. Synteny overlaps of the T. gondii RH unique gene loci on scaffolds with the T. gondii GT1 genome. Table Q. The 17 unique genes were annotated to proteins with vital functions. Table R. Single Copy clusters from T. gondii RH comparad to recently published gene families of T. gondii containing unique parasite-specific (para) domains. (external data retrieved: supplementary table G of Lorenzi et al. 2016[27]).
(XLS)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files. In addition, this Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession SUB1075932 (BioProject number, PRJNA294483; BioSample, SAMN04026592).