

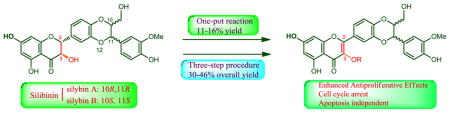
Abstract
Eight 3-O-alkyl-2,3-dehydrosilibinins have been synthesized from commercially available silibinin through two synthetic approaches. A one-pot reaction, starting with aerobic oxidation of silibinin followed by direct alkylation of the phenolic hydroxyl group in the subsequent 2,3-dehydrosilibinin, furnishes the desired derivatives in 11–16% yields. The three-step procedure employing benzyl ether to protect 7-OH in silibinin generates the desired derivatives in 30–46% overall yields. The antiproliferative activity of the 2,3-dehydrosilibinin derivatives against both androgen-sensitive and androgen-insensitive prostate cancer cells have been assessed using a WST-1 cell proliferation assay. All derivatives exhibited greater antiproliferative potency than silibinin, with 2,3-dehydrosilibinins each possessing a three- to five-carbon linear alkyl group to 3-OH (IC50 values in a range of 1.71 to 3.06 μM against PC-3 and LNCaP cells) as the optimal derivatives. The optimal potency was reached with three- to five-carbon alkyl groups. Our findings suggest that 3-O-propyl-2,3-dehydrosilibinin effectively inhibits the growth of PC-3 prostate cancer cells by arresting cell cycle in the G0/G1 phase, but not by activating PC-3 cell apoptosis.
Graphical abstract
Silibinin (1), isolated from milk thistle (Silybum marianum L. Gaertner, Asteraceae), represents the first identified and well-investigated flavonolignan. Milk thistle is a well-known traditional European medicine that has long been used for treating liver disorders and protecting the liver against a variety of xenobiotics and hepatotoxins.1 Its medicinal merits in this field were first recorded in Hieronymus Bock’s book published in 1539.2 2,3-Dehydrosilibinin (2), as the most important oxidized derivative of silibinin, was first synthesized from silibinin (1) and employed to revise the structure of silibinin by Pelter and Haensel in 1968.3 Several studies have so far confirmed that silibinin can be readily converted to 2,3-dehydrosilibinin through oxidation of the secondary aliphatic hydroxyl group to a ketone followed by enolization.2 So far, only two full reports have been published on the isolation of 2,3-dehydrosilibinin from natural sources including seeds of S. marianum subsp. anatolicum4 and the fruits of spotted milkweed (S. marianum L. Gaertn.) cultivated in Russia and CIS countries.5 Without publishing the detailed data, Gazak and co-workers pointed out that 2,3-dehydrosilibinin exists as a minor constituent in almost all crude extracts of milk thistle (silymarin) and is responsible for the yellow color of silymarin.6 It remains unclear whether 2,3-dehydrosilibinin is a naturally occurring or an artefact flavonolignan.2
Recently, 2,3-dehydrosilibinin has been reported to display significant improvements over silibinin in numerous biological activities. As compared with silibinin, 2,3-dehydrosilibinin is superior by one order of magnitude in antioxidative properties;6 it is a 25 times more potent radical scavenger; it inhibits lipid peroxidation 10 times more efficiently;6,7 it possesses more potent cytotoxicity against human prostate cancer cells;8 it exhibits better apoptotic activity in HTB cell model;8 and it exhibits a higher cytoprotective potential in hepatoma HepG2 cells.9 Additionally, C-isoprenylated or geranylated derivatives of 2,3-dehydrosilibinin were demonstrated to be effective P-glycoprotein modulators.10 Our previous studies showed that 7-O-alkyl-2,3-dehydrosilibinins with a C2–C3 double bond have better antiproliferative potency than 7-O-alkylsilibinins with a C2–C3 single bond against androgen-resistant human prostate cancer cell lines (DU145 and PC-3).11
The ultimate goal of our program on 2,3-dehydrosilibinin is to engineer new derivatives with enhanced potency and bioavailability through appropriate structure manipulations for the treatment of castration-resistant prostate cancer. At the starting point of this long standing program, our ongoing studies aim to systematically explore the appropriate structure moieties of 2,3-dehydrosilibinin for further modifications. Recently, we reported that in vitro antiproliferative potency of 2,3-dehydrosilibinin against three prostate cancer cell lines can be significantly improved through appropriate chemical modifications on 7-OH.11 This encouraged us to investigate the effects of 3-OH modifications on prostate cancer cell proliferation. However, 3-O-alkyl-2,3-dehydrosilibinins cannot be achieved by the synthetic methods employed in our previous study, which can only yield 7-O-alkyl-2,3-dehydrosilibinins and 3,7-O-dialkyl-2,3-dehydrosilibinins.11 Consequently, the present study focuses on the exploration of general methods for the synthesis of 3-O-alkyl-2,3-dehydrosilibinins and in vitro evaluation of these derivatives as anti-prostate cancer agents.
3-O-Methyl-2,3-dehydrosilibinin was reported by Dzubak and co-workers to be capable of improving in vitro antiproliferative potency against K562 human myeloid leukemia cancer cells and of blocking functional activity of P-glycoprotein.12 No other 3-O-alkyl-2,3-dehydrosilibinins have been reported. The challenge for the synthesis of 3-O-alkyl-2,3-dehydrosilibinins lies in the competitive reactivity of the four phenolic hydroxyl groups at C-3, C-5, C-7, and C-20 in 2,3-dehydrosilibinin. The relative reactivity of the phenolic hydroxyl groups in silibinin toward the etherification reaction is approximately 7-OH > 20-OH ≫ 5-OH.2 The only known 3-O-alkyl-2,3-dehydrosilibinin reported in the literature12 is the methyl derivative (3). It was synthesized in 45% yield by direct alkylation of 2,3-dehydrosilibinin, prepared by oxidation of silibinin in 13–90%, using sodium hydride as base and DMF as solvent.2,12 This indicated that the 3-OH in 2,3-dehydrosilibinin is more reactive than 7-OH toward the etherification reaction.
Two synthetic approaches to a group of 3-O-alkyl-2,3-dehydrosilibinns have been developed in this paper. Our first synthetic approach to the 3-O-alkyl-2,3-dehydrosilibins is illustrated in Scheme 1. Specifically, the one-pot reaction starts from potassium acetate-mediated aerobic oxidation of silibinin followed by selective alkylation of 3-OH of the subsequent 2,3-dehydrosilibinin. In our hands, oxidation of silibinin to 2,3-dehydrosilibinin can be achieved under aerobic conditions using either potassium carbonate or potassium acetate as base and DMF as solvent. Using potassium carbonate to mediate the oxidation in the one-pot reaction led to decreased yields. This is probably due to the simultaneous deprotonation of 7-OH during oxidation, resulting in low selectivity of alkylation on 3-OH of 2,3-dehydrosilibinin. Prolonging the reaction time led to no significant change in yields. The one-pot reaction under the optimal conditions furnishes the desired derivatives in 11–16% yields (Table 1). Through this method, we could eliminate two steps required for the temporary protection/deprotection of other phenolic hydroxyl groups. However, it is challenging to further improve the yield due to the competitive reactivity of two phenolic hydroxyl groups at C-3 and C-7. The products from this reaction as determined by TLC analysis include the corresponding 7-O-alkyl-2,3-dehydrosilibinins and 3,7-O-dialkyl-2,3-dehydrosilibinins in addition to the desired 3-O-alkyl-2,3-dehydrosilibinins (3–10).
Scheme 1.

One-pot synthesis of 3-O-alkyl-2,3-dehydrosilibinins (3–10)
Table 1.
Yields for the two alternative syntheses of 3-O-alkyl-2,3-dehydrosilibinins (3–9)
| Derivative | One-pot method | Three-step method |
|---|---|---|
| 3 (methyl) | 13% | 35% |
| 4 (ethyl) | 14% | 30% |
| 5 (propyl) | 12% | 38% |
| 6 (butyl) | 15% | 46% |
| 7 (pentyl) | 11% | 37% |
| 8 (hexyl) | 16% | 32% |
| 9 (heptyl) | 11% | 30% |
As shown in Scheme 2, the three-step procedure includes benzyl ether protection of 7-OH in silibinin to yield derivative 11, oxidation of 11 followed by selective alkylation on 3-OH generates derivatives 12–18, and debenzylation of 12–18 in the presence of ammonium formate catalyzed by palladium carbon provides the desired derivatives 3–9 in 30–46% overall yields for three steps (Table 1). The two- to three-fold improvement in overall yields is primarily attributed to higher efficiency of oxidation of 7-O-benzylsilibinin to 7-O-benzyl-2,3-dehydrosilibinin and higher selectivity of alkylation on 3-OH.
Scheme 2.

Three-step synthetic procedure for 3-O-alkyl-2,3-dehydrosilibinins (2–9)
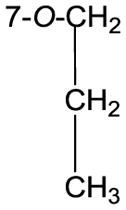
The structures of the eight 3-O-alkyl-2,3-dehydrosilibinins were characterized by interpreting their NMR, HRMS, and FTIR data. The 1H and 13C NMR data for compound 5 (Table 2) were fully assigned based on the interpretation of their COSY, HMQC, and HMBC data. The propyl group in compound 5 was assigned to 3-OH based on the key HMBC correlations from the triplet signal at δH 4.02 (CH2 in propyl) to the signal at δC 138.8 (C-3, Fig. 1). This assignment is also supported by the absence of a broad singlet signal at around δH 6.5 for the proton of 3-OH in 2,3-dehydrosilibinin (2).
Table 2.
NMR Data for 3-O-propyl-2,3-dehydrosilibinin (5) (1H NMR: 300 MHz; 13C NMR: 75 MHz).
| Position | 3-O-propyl-2,3-dehydrosilibinin (acetone-d6) | |
|---|---|---|
| δC, type | δH, (J in Hz) | |
|
|
|
|
| 2 | 147.1, C | - |
| 3 | 138.8, C | - |
| 4 | 175.4, C | - |
| 4a | 105.9, C | - |
| 5 | 163.2, C | - |
| 6 | 99.4, CH | 6.25, s |
| 7 | 165.0, C | - |
| 8 | 94.5, CH | 6.52, s |
| 8a | 157.8, C | - |
| 10 | 80.0, CH | 4.25–4.23, m |
| 11 | 77.2, CH | 5.05, d (7.8) |
| 12a | 144.8, C | - |
| 13 | 118.0, CH | 7.70, s |
| 14 | 124.4, C | - |
| 15 | 123.2, CH | 7.74, d (8.1) |
| 16 | 117.7, CH | 7.06, d (8.1) |
| 16a | 156.2, C | - |
| 17 | 128.9, C | - |
| 18 | 111.9, CH | 7.16, s |
| 19 | 148.6, C | - |
| 20 | 148.1, C | - |
| 21 | 115.8, CH | 6.90, d (8.1) |
| 22 | 121.7, CH | 7.00, d (8.1) |
| 23 | 61.7, CH2 | 3.80, br.d (12.3) |
| 3.55, br.d (12.3) | ||
|
| ||
|
74.8, CH2 | 4.02, t (6.6) |
| 24.0, CH2 | 1.73, Hex (7.2) | |
| 10.8, CH3 | 0.96, t (7.5) | |
|
| ||
| 19-OMe | 56.3, CH3 | 3.95, s |
| 5-OH | - | 11.67, s |
| 20-OH | - | 5.79, s |
| 23-OH | - | |
Figure 1.
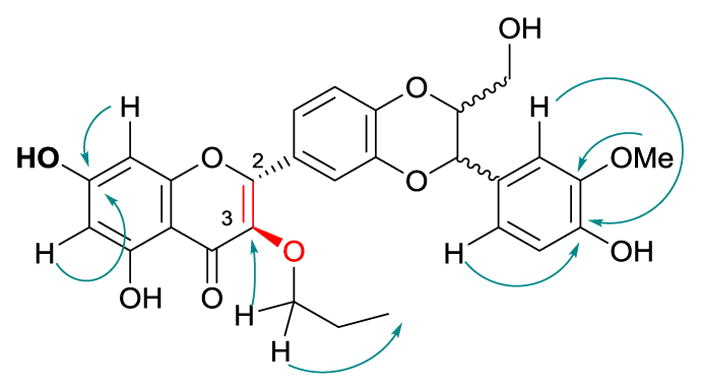
Diagnostic HMBC correlations in 3-O-propyl-2,3-dehydrosilibinin (5)
The in vitro anti-proliferative activities of the dehydrosilibinin derivatives were evaluated using a WST-1 cell proliferation assay in both androgen-sensitive (LNCaP) and androgen-insensitive (PC-3 and DU145) human prostate cancer cell lines. The detailed procedure is described in the Experimental Section in Supplementary Data. Silibinin was used as a positive control for comparison in the parallel experiments and the IC50 values are listed in Table 3. The cytotoxicity of 2,3-dehydrosilibinin at 30 μM and 60 μM against PC-3 human prostate cancer cells has been reported, but without an IC50 value in the literature.8 Here, the antiproliferative activity of 2,3-dehydrosilibinin (2) is first reported with IC50 values against three human prostate cancer cell lines. All eight 3-O-alkyl-2,3-dehydrosilibinins (3–10) as well as 2,3-dehydrosilibinin exhibit significantly greater anti-proliferative potency by comparing their IC50 values with that of silibinin (Table 3). The potency is slightly enhanced with increasing length of the alkyl group, reaching the maximum with three- to five-carbon alkyl groups. Consequently, 2,3-dehydrosilibinins 5–7 each with a three- to five-carbon linear alkyl group attached to 3-OH were identified as the optimal derivatives with IC50 values in a range of 1.71–3.06 μM and 1.99–2.07 μM against PC-3 and LNCaP cells, respectively. All synthesized 3-O-alkyl-2,3-dehydrosilibinins (3–10) as well as 2,3-dehydrosilibinin (2) are more effective in inhibiting proliferation of LNCaP and PC-3 cells than of DU145 cells. Specifically, they are 5–42 times more potent toward LNCaP and PC-3 cell lines, but only 4–8 folds more potent against the DU145 cell line, as compared with silibinin.
Table 3.
In vitro anti-proliferative activity (IC50, μM)a of the compounds against prostate cancer cell lines
| Comp. No | IC50 (μM) | IC50 (silibinin)/IC50 (derivitive) | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| LNCaP b | DU145 c | PC-3d | LNCaP | DU145 | PC-3 | |
| Silibinin (1) | 43.03 ± 7.84 | 93.34 ± 13.76 | 72.65 ± 3.15 | 1 | 1 | 1 |
| 2 | 3.09 ± 1.30 | 11.48 ± 1.42 | 9.45 ± 0.56 | 14 | 8 | 8 |
| 3 | 8.14 ± 2.35 | 21.64 ± 0.53 | 12.58 ± 1.28 | 5 | 4 | 6 |
| 4 | 3.22 ± 0.59 | 16.44 ± 0.49 | 7.52 ± 0.22 | 14 | 6 | 10 |
| 5 | 2.07 ± 0.18 | 11.04 ± 0.68 | 1.71 ± 0.45 | 21 | 8 | 42 |
| 6 | 1.99 ± 0.10 | 14.36 ± 0.40 | 2.29 ± 0.12 | 22 | 7 | 32 |
| 7 | 2.07 ± 0.35 | 14.03 ± 0.66 | 3.06 ± 0.48 | 21 | 7 | 24 |
| 8 | 3.50 ± 0.21 | 21.11 ± 0.76 | 6.04 ± 0.80 | 12 | 4 | 12 |
| 9 | 3.96 ± 0.38 | 19.24 ± 0.88 | 10.66 ± 1.62 | 11 | 5 | 7 |
| 10 | 3.77 ± 0.40 | 17.76 ± 1.98 | 4.46 ± 2.24 | 11 | 6 | 16 |
IC50 is the drug concentration effective in inhibiting 50% of the cell viability measured by the WST-1 cell proliferation Assay after 3 days exposure.
Human androgen-sensitive prostate cancer cell line
Human androgen-independent prostate cancer cell line
Human androgen-independent prostate cancer cell line
Our data further corroborate that 2,3-dehydrosilibinin has greater anti-proliferative potency than silibinin against prostate cancer cells. Additionally, we found for the first time that 3-O-alkyl-2,3-dehydrosilibinins possess greater anti-proliferative potency than silibinin toward both androgen-sensitive and androgen-resistant human prostate cancer cell lines (LNCaP, DU145 and PC-3). Three- to five-carbon alkyl groups attached to 3-OH of 2,3-dehydrosilibinin maximize the in vitro antiproliferative potency. However, 7-O-methyl-2,3-dehydrosilibinin and 7-O-ethyl-2,3-dehydrosilibinin represent the most potent derivatives among the series of 7-O-alkyl-2,3-dehydrosilibinins.11
Silibinin has been demonstrated to arrest cell cycle at G1 phase in various prostate cancer cell models. 13–15 The effect of 3-O-propyl-2,3-dehydrosilibinin (5) on the PC-3 cell cycle was evaluated using flow cytometric analysis with propidium iodide DNA staining. Derivative 5 increased the population of PC-3 cells in a G0/G1 phase (Fig. 2), while fewer cells were observed in the G2 phase. Specifically, the G0/G1 PC-3 cells were increased from 48% and 60% in control cells at 16 hours and 24 hours, respectively, to 68% in derivative 5-treated cells at both time points (Table 4). The cell population in G2 phase slightly decreased from 31% in control cells to 18% at 16 hours, and from 21% in control cells to 18% at 24 hours. Similarly, 2,3-dehydrosilibinin (2) also induces the PC-3 cell cycle arrest at the G0/G1 phase (Fig. 2). It increased the population of PC-3 cells in the G0/G1 phase from 48% and 60% (control cells) to 65% and 63% at 16 hours and 24 hours, respectively (Table 4).
Fig. 2.

Cell cycle analysis of PC-3 cells. PC-3 cancer cells were untreated or treated with 2 and 5. Cells were harvested after 16 and 24 hours, fixed, stained, and analyzed for DNA content.
Table 4.
The distribution and percentage of PC-3 cells in G1/G0 and G2 phase of the cell cycle
| PC-3 cells | 16 hours | 24 hours | ||
|---|---|---|---|---|
| G0/G1 | G2 | G0/G1 | G2 | |
| Control cells | 48% | 31% | 60% | 21% |
| 2-treated (50 μM) | 65% | 18% | 63% | 18% |
| 5-treated (50 μM) | 68% | 18% | 68% | 18% |
Agarwal and co-workers have reported that silibinin can activate cell apoptosis in PC-3 tumor xenografts.16 F2N12S and CYTOX AADvanced double staining flow cytometry-based assay was chosen to discriminate PC-3 cells dying from apoptosis from those dying from necrosis in response to increasing concentrations of 2,3-dehydrosilibinin (2) and 3-O-propyl-2,3-dehydrosilibinin (5). PC-3 cells were incubated with 2 or 5 for 16 h. Staurosporine was used as a specific apoptotic inducer and positive apoptotic control in these experiments (not shown). As illustrated in Fig. 3 and Fig. 5, derivative 5 with a propyl group at 3-OH in 2,3-dehydrosilibinin did not induce significant levels of apoptotic cell death in the androgen-insensitive PC-3 prostate cancer cell line at a dose of up to 100 μM after a 16-hour treatment. In contrast, 2,3-dehydrosilibinin (2) induced significant levels of PC-3 apoptotic cell death after a 16-hour treatment, as illustrated in Fig. 4–5. Specifically, 60 μM of 2 could induce detectable early phase of apoptosis in PC-3 cells as compared with control cells; treatment with 100 μM of 2 led to 46% early apoptotic cells and 16% late apoptotic/necrotic cells. Both apoptotic and necrotic cell populations increased in response to increasing concentration of 2 (0–100 μM final concentration range). Interestingly, 2,3-dehydrosilibinin (2), 3-O-propyl-2,3-dehydrosilibinin (5), and 7-O-ethyl-2,3-dehydrosilibinin11 show similar inhibitory effect on PC-3 cell proliferation but different inductive effect on PC-3 cell apoptosis, indicating that incorporation of an alkyl group to 7-OH in 2,3-dehydrosilibinin promotes the apoptotic activation and that introduction of an alkyl group to 3-OH in 2,3-dehydrosilibinin reverses the apoptotic response. Recently, the inhibitory effect of silibinin on PC-3 and other cancer cell proliferation was demonstrated to be associated with both cell apoptotic and autophagic induction.17–19 Regulation of autophagy could be an important mechanism contributing to the significant anti-proliferative effect of 3-O-alkyl-2,3-dehydrosilibinins.
Fig. 3.
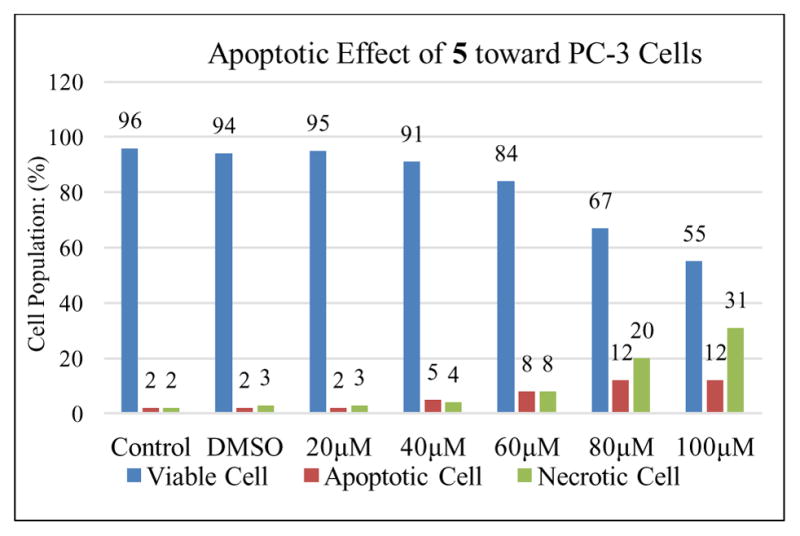
Evolution of viable, apoptotic, and necrotic PC-3 cells populations in response to increasing dosages of derivative 5
Fig. 5.

Apoptosis in PC-3 cells treated with derivatives 2 and 5 at 80 and 100 μM (by F2N12S and CYTOX AADvanced double staining)
Fig. 4.
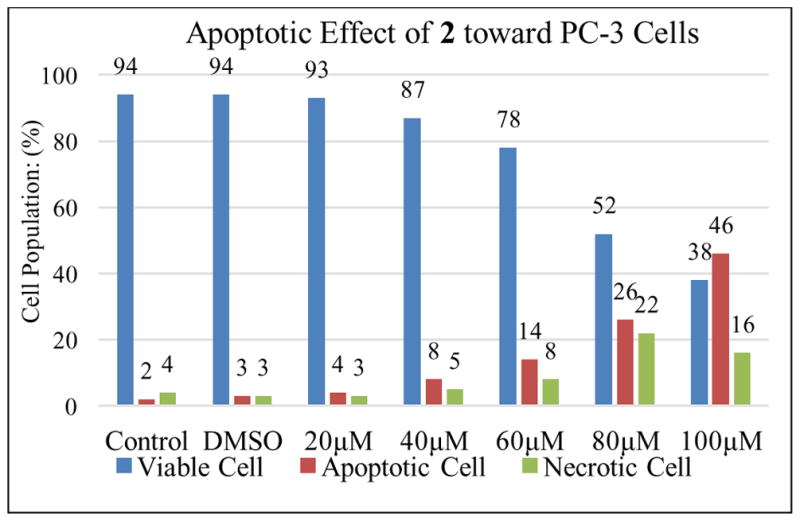
Evolution of viable, apoptotic, and necrotic PC-3 cells populations in response to increasing dosages of derivative 2
In summary, eight 3-O-alkyl-2,3-dehydrosilibinins have been successfully synthesized through one-pot reaction procedure. Seven of them have also been obtained by a three-step procedure in significantly improved yields. Their antiproliferative potency against three prostate cancer cell lines, as evaluated by WST-1 cell proliferation assay, is significantly greater than silibinin. 2,3-Dehydrosilibinins 5–7 each with a three- to five-carbon linear alkyl group attached to 3-OH were identified as the optimal derivatives with IC50 values in the range of 1.71 – 3.06 μM toward PC-3 and LNCaP prostate cancer cell lines, a 24- to 42-fold improvement in potency as compared with silibinin. Importantly, the antiproliferative potency of 3-O-propyl-2,3-dehydrosilibinin against PC-3 prostate cancer cells is not primarily associated with its capability to induce PC-3 cell apoptosis. However, 3-O-propyl-2,3-dehydrosilibinin appears to inhibit prostate cancer cell growth by confining more cells in the G0/G1 phase. Accordingly, this scaffold is worth further exploration to define the mechanism of action and to optimize the lead compounds via chemical modifications.
Supplementary Material
Acknowledgments
This work was financially supported by California State University (CSU)-Fresno. The HRMS data were supported by NIH RCMI program at Xavier University of Louisiana through Grant 2G12MD007595 (G.W.) and NIH-NIGMS through Grant 1U54GM104940 (G.W.). We are also grateful to i) the ASI and the Graduate Net Initiative at CSU-Fresno for Graduate Research Grants (to S.Z. and X.Z.) and ii) CSUPERB for Presidents’ Commission Scholar Award (to T. L.)
Footnotes
Supplementary data (synthetic procedures and structural characterization) associated with this article can be found, in the online version, at http://dx.doi.org/
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Abenavoli L, Capasso R, Milic N, Capasso F. Phytother Res. 2010;24:1423. doi: 10.1002/ptr.3207. [DOI] [PubMed] [Google Scholar]
- 2.Biedermann D, Vavrikova E, Cvak L, Kren V. Nat Prod Rep. 2014;31:1138. doi: 10.1039/c3np70122k. [DOI] [PubMed] [Google Scholar]
- 3.Pelter A, Haensel R. Tetrahedron Lett. 1968:2911. [Google Scholar]
- 4.Mericli AH. Planta Medica. 1988;54:44. doi: 10.1055/s-2006-962330. [DOI] [PubMed] [Google Scholar]
- 5.Kurkin VA, Zapesochnaya GG, Volotsueva AV, Avdeeva EV, Pimenov KS. Chem Nat Comp (Translation of Khimiya Prirodnykh Soedinenii) 2002;37:315. [Google Scholar]
- 6.Gazak R, Svobodova A, Psotova J, Sedmera P, Prikrylova V, Walterova D, Kren V. Bioorg Med Chem. 2004;12:5677. doi: 10.1016/j.bmc.2004.07.064. [DOI] [PubMed] [Google Scholar]
- 7.Huber A, Thongphasuk P, Erben G, Lehmann WD, Tuma S, Stremmel W, Chamulitrat W. Biochim Biophys Acta. 2008;1780:837. doi: 10.1016/j.bbagen.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 8.Agarwal C, Wadhwa R, Deep G, Biedemann D, Gazak R, Kren V, Agarwal R. PloS One. 2013;8:e60074. doi: 10.1371/journal.pone.0060074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pyszkova M, Biler M, Biedermann D, Valentova K, Kuzma M, Vrba J, Ulrichova J, Sokolova R, Mojovic M, Popovic-Bijelic A, Kubala M, Trouillas P, Kren V, Vacek J. Free Radic Biol Med. 2016;90:114. doi: 10.1016/j.freeradbiomed.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 10.Maitrejean M, Conte G, Barron D, El Kirat K, Conseil G, Di Pietro A. Bioorg Med Chem Lett. 2000;10:157. doi: 10.1016/s0960-894x(99)00636-8. [DOI] [PubMed] [Google Scholar]
- 11.Vue B, Zhang S, Zhang X, Parisis K, Zhang Q, Zheng S, Wang G, Chen QH. Eur J Med Chem. 2016;109:36. doi: 10.1016/j.ejmech.2015.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dzubak P, Hajduch M, Gazak R, Svobodova A, Psotova J, Walterova D, Sedmera P, Kren V. Bioorg Med Chem. 2006;14:3793. doi: 10.1016/j.bmc.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 13.Tyagi A, Bhatia N, Condon MS, Bosland MC, Agarwal C, Agarwal R. Prostate. 2002;53:211. doi: 10.1002/pros.10146. [DOI] [PubMed] [Google Scholar]
- 14.Zi X, Agarwal R. Proc Natl Acad Sci USA. 1999;96:7490. doi: 10.1073/pnas.96.13.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deep G, Singh RP, Agarwal C, Kroll DJ, Agarwal R. Oncogene. 2006;25:1053. doi: 10.1038/sj.onc.1209146. [DOI] [PubMed] [Google Scholar]
- 16.Singh RP, Deep G, Blouin MJ, Pllak MN, Agarwal R. Carcinogenesis. 2007;28:2567. doi: 10.1093/carcin/bgm218. [DOI] [PubMed] [Google Scholar]
- 17.Kim SH, Kim KY, Yu SN, Park SK, Choi HD, Ji JH, Ahn SC. Biochem, Biophys Res Commun. 2015;468:151. doi: 10.1016/j.bbrc.2015.10.143. [DOI] [PubMed] [Google Scholar]
- 18.Yu Y, Fan SM, Yuan SJ, Tashiro S, Onodera S, Ikejima T. Free Radic Res. 2012;46:1346. doi: 10.3109/10715762.2012.715369. [DOI] [PubMed] [Google Scholar]
- 19.Duan W, Jin X, Li Q, Tashiro S, Onodera S, Ikejima T. J Pharmacol Sci. 2010;113:48. doi: 10.1254/jphs.09315fp. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.