

Abstract
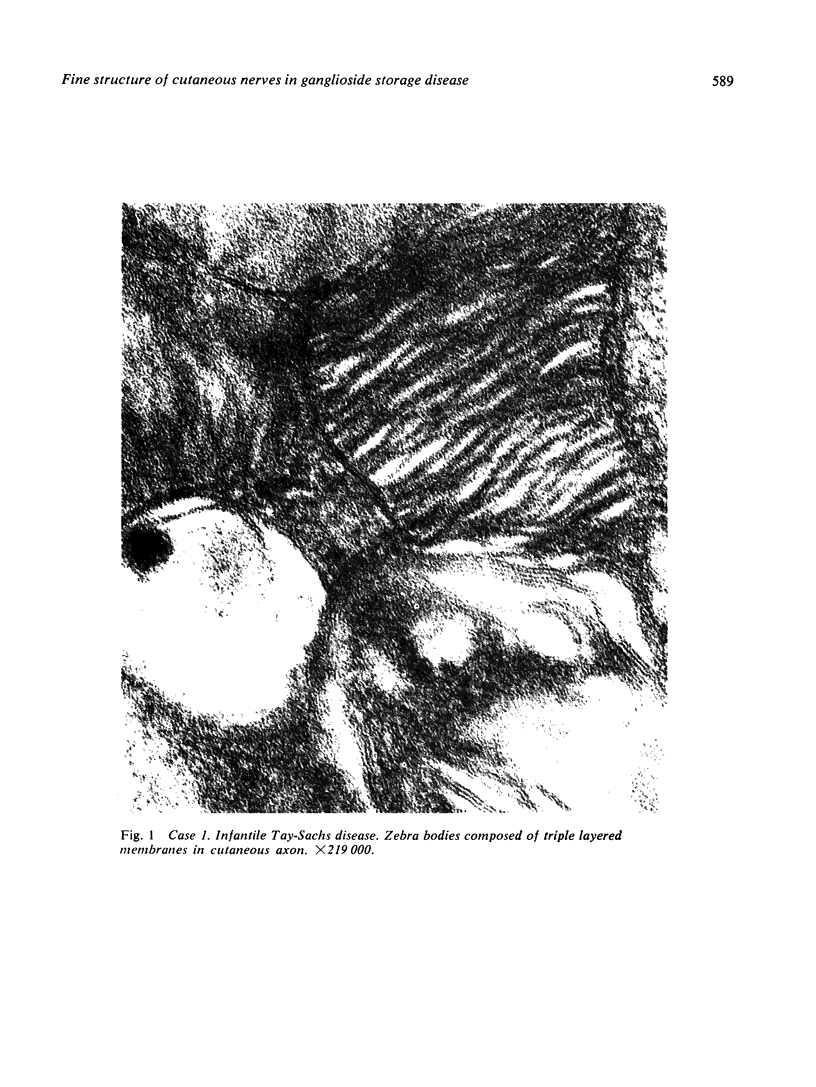
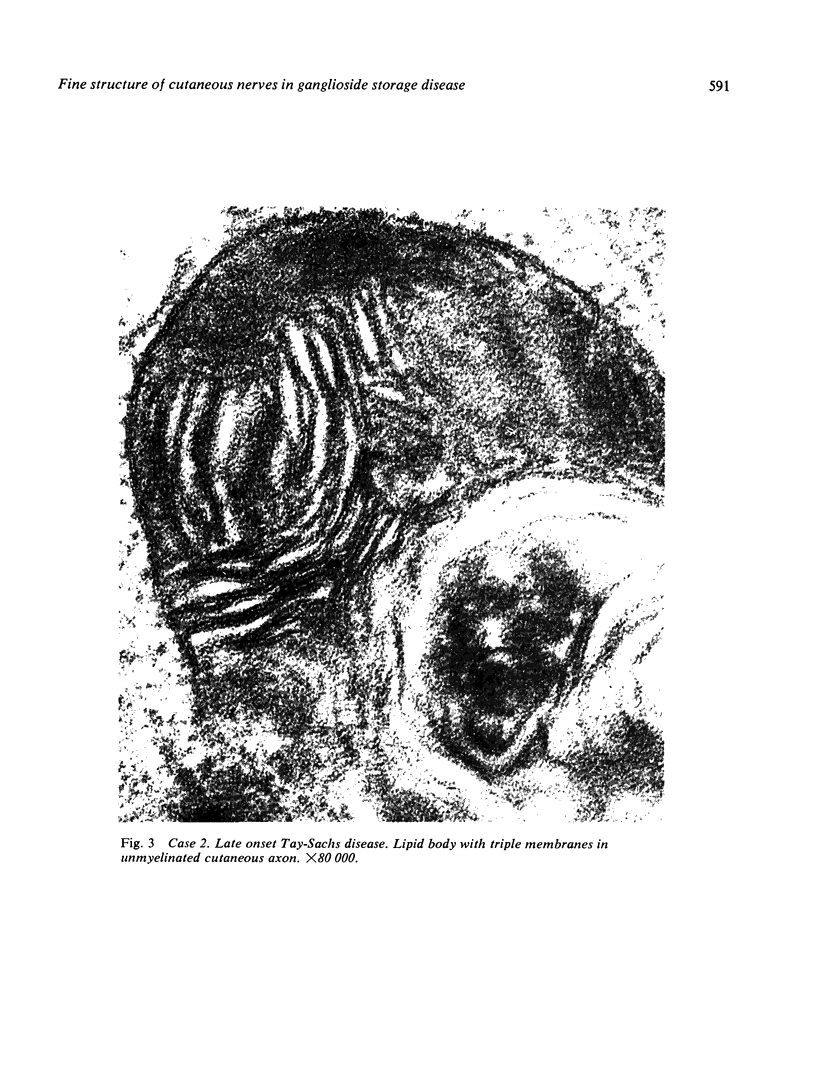
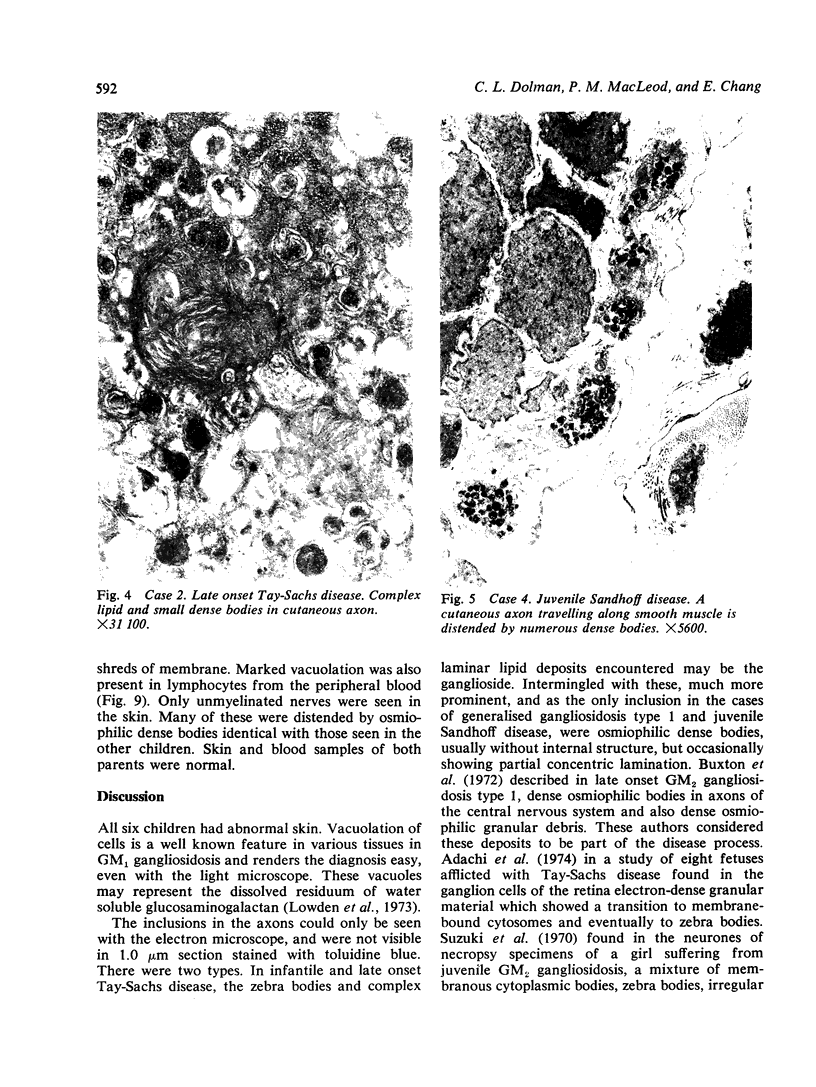
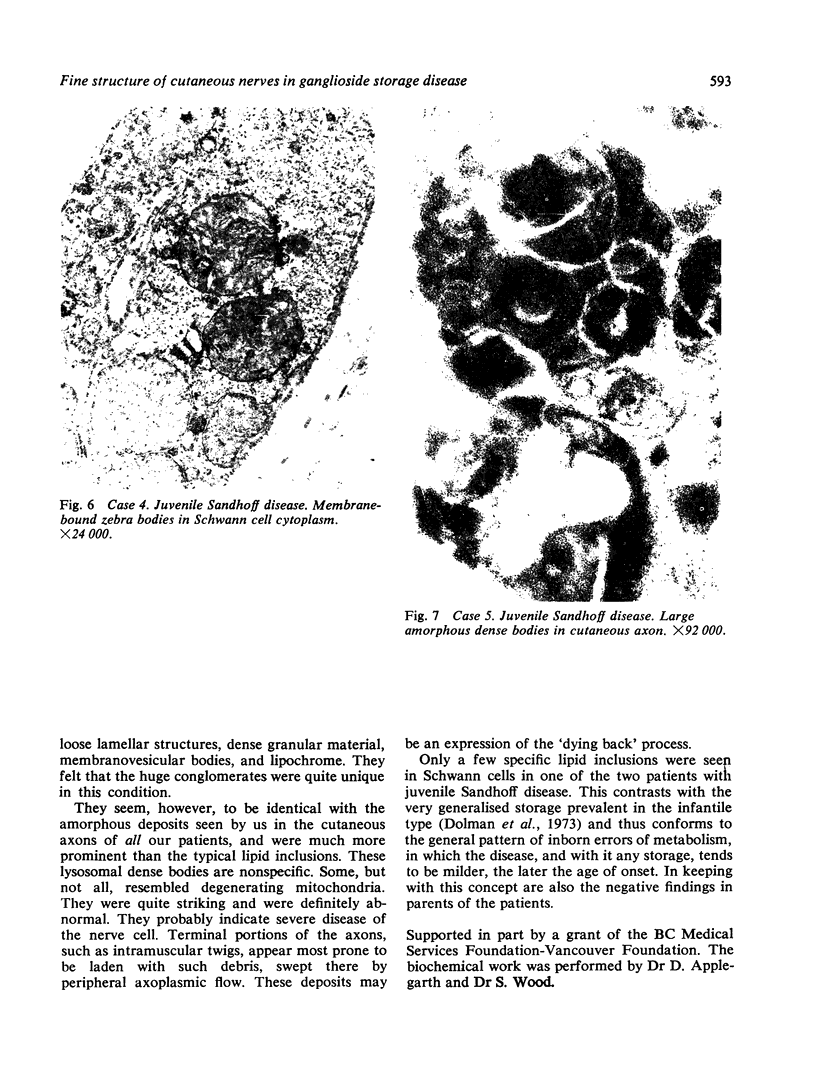
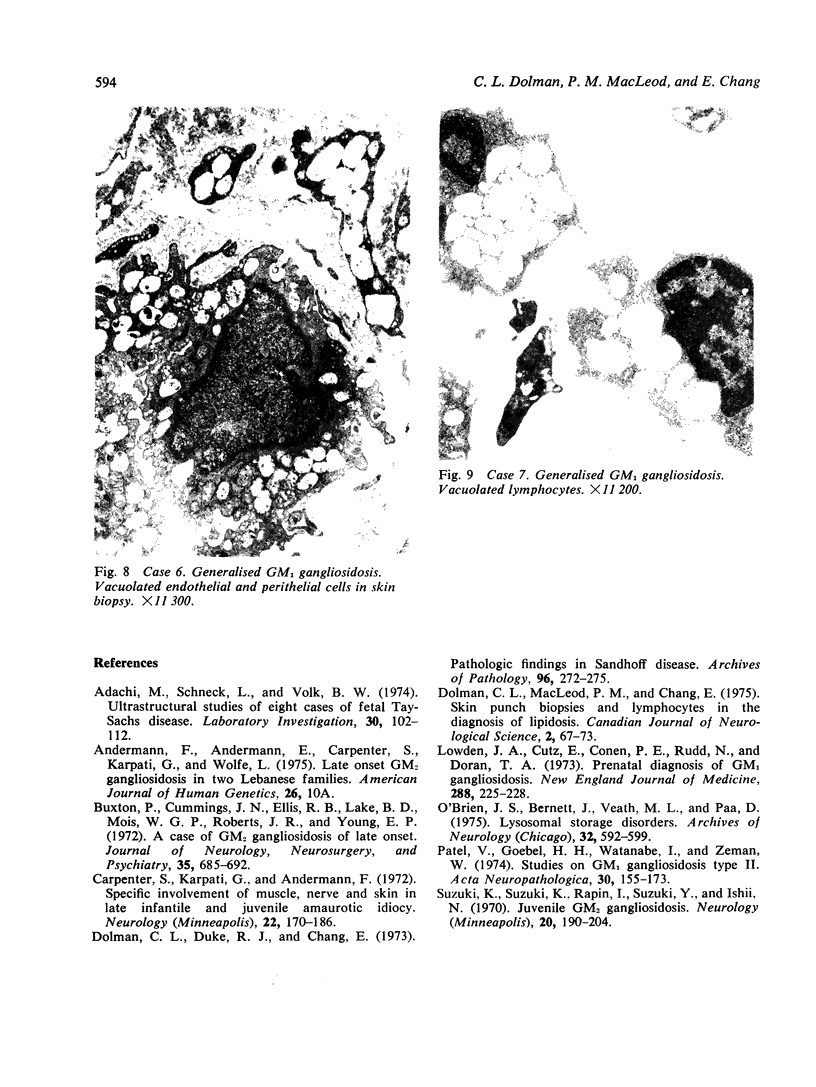
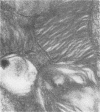
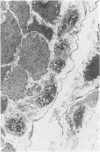
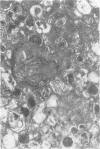
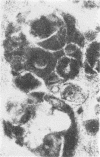
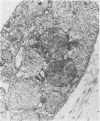
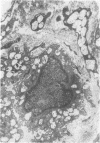
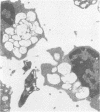
Skin punch biopsies of six children suffering from infantile or late onset Tay-Sachs disease, juvenile Sandhoff disease, or GM gangliosidosis type I, contained axons which, when viewed with the electron microscope, were distended by large amorphous black deposits. These are nonspecific residual bodies. Their large numbers indicate severe disturbance of the nerve cell and may be part of the dying back process. The three cases with Tay-Sachs disease had also axonal zebra or complex membranous bodies which appeared to be specific. Cytoplasmic vacuolation of other cells was a feature in the patient with GM1 gangliosidosis. Biopsies of three parents were negative.
Full text
PDF

Images in this article
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Adachi M., Schneck L., Volk B. W. Ultrastructural studies of eight cases of fetal Tay-Sachs disease. Lab Invest. 1974 Jan;30(1):102–112. [PubMed] [Google Scholar]
- Buxton P., Cumings J. N., Ellis R. B., Lake B. D., Mair W. G., Roberts J. R., Young E. P. A case of G M2 gangliosidosis of late onset. J Neurol Neurosurg Psychiatry. 1972 Oct;35(5):685–692. doi: 10.1136/jnnp.35.5.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter S., Karpati G., Andermann F. Specific involvement of muscle, nerve, and skin in late infantile and juvenile amaurotic idiocy. Neurology. 1972 Feb;22(2):170–186. doi: 10.1212/wnl.22.2.170. [DOI] [PubMed] [Google Scholar]
- Dolman C. L., Chang E., Duke R. J. Pathologic findings in Sandhoff disease. Arch Pathol. 1973 Oct;96(4):272–275. [PubMed] [Google Scholar]
- Dolman C. L., MacLeod P. M., Chang E. Skin punch biopsies and lymphocytes in the diagnosis of lipidoses. Can J Neurol Sci. 1975 Feb;2(1):67–73. doi: 10.1017/s0317167100019995. [DOI] [PubMed] [Google Scholar]
- Lowden J. A., Cutz E., Conen P. E., Rudd N., Doran T. A. Prenatal diagnosis of G M1 -gangliosidosis. N Engl J Med. 1973 Feb 1;288(5):225–228. doi: 10.1056/NEJM197302012880502. [DOI] [PubMed] [Google Scholar]
- O'Brien J. S., Bernett J., Veath M. L., Paa D. Lysosomal storage disorders. Diagnosis by ultrastructural examination of skin biopsy specimens. Arch Neurol. 1975 Sep;32(9):592–599. doi: 10.1001/archneur.1975.00490510048002. [DOI] [PubMed] [Google Scholar]
- Patel V., Goebel H. H., Watanabe I., Zeman W. Studies on GM1-gangliosidosis, type II. Acta Neuropathol. 1974;30(2):155–173. doi: 10.1007/BF00685440. [DOI] [PubMed] [Google Scholar]
- Suzuki K., Rapin I., Suzuki Y., Ishii N. Juvenile GM2-gangliosidosis. Clinical variant of Tay-Sachs disease or a new disease. Neurology. 1970 Feb;20(2):190–204. doi: 10.1212/wnl.20.2.190. [DOI] [PubMed] [Google Scholar]