

Abstract
Although genetically compact, HIV-1 commandeers vast arrays of cellular machinery to sustain and protect it during cycles of viral outgrowth. Transposon-mediated saturation linker scanning mutagenesis was used to isolate fully replication-competent viruses harbouring a potent foreign epitope tag. Using these viral isolates, we performed differential isotopic labelling and affinity-capture mass spectrometric analyses on samples obtained from cultures of human lymphocytes to classify the vicinal interactomes of the viral Env and Vif proteins as they occur during natural infection. Importantly, interacting proteins were recovered without bias, regardless of their potential for positive, negative or neutral impact on viral replication. We identified specific host associations made with trimerized Env during its biosynthesis, at virological synapses, with innate immune effectors (such as HLA-E) and with certain cellular signalling pathways (for example, Notch1). We also defined Vif associations with host proteins involved in the control of nuclear transcription and nucleoside biosynthesis as well as those interacting stably or transiently with the cytoplasmic protein degradation apparatus. Our approach is broadly applicable to elucidating pathogen–host interactomes, providing high-certainty identification of interactors by their direct access during cycling infection. Understanding the pathophysiological consequences of these associations is likely to provide strategic targets for antiviral intervention.
Due to its small genome, many biological processes that occur during HIV-1 infection and propagation are probably achieved by a heavy reliance on its host. In turn, some of these cellular factors impart antiviral defences. Elucidating these virus–host protein interactions—the viral interactome—may therefore help fully reveal how HIV-1 gains control of the host and how the cell attempts to counteract infection.
RNA interference library screening has previously been used to identify cellular genes that negatively impact HIV-1 replication1–4. However, identity sets of host genes identified by this approach are discouragingly incongruent between different studies. Host protein affinity capture is a more direct approach for deciphering the HIV-1 interactome and, unlike RNA interference library screening, can identify factors with a positive, negative or neutral impact on viral replication5. Nevertheless, applying affinity capture screens to HIV-1 has been hampered by the lack of high-affinity antibodies against viral proteins and the lack of a systematic tagging methodology for the recovery of replication-competent viruses. As a result, a previous approach has involved the individual ectopic expression of tagged viral subunits6. Our new approach overcomes this bottleneck: by recovering fully replication-competent viral derivatives that harbour a potent epitope tag followed by probing virus–host interactions in their natural context, our approach identifies functionally relevant new interactors.
Generation of replication-competent, epitope-tagged HIV-1
We took a two-step approach to introduce tags into targeted HIV-1 proteins, without sacrificing replication competency (Fig. 1). First, to identify sites that can tolerate exogenous sequences, we made a large library in which a rare restriction endonuclease site (PmeI, Fig. 1a, red) was randomly and uniquely introduced by Tn7 transposition into either the env or vif gene segments of an otherwise replication-competent proviral plasmid DNA clone. These viral gene-specific mutant libraries were comprehensive, consisting of 100,000 to 300,000 independent clones per <1 kilobase of targeted gene segment (Fig. 1a), achieving a theoretical saturation of insertion sites within the region. As verification, a heterogeneous viral stock was prepared from the PmeI-saturated env C1-V3 proviral plasmid library (Fig. 1b, lib) and used to initiate infection of CEM cells. Replication-competent survivors emerged over extended periods of viral outgrowth, but were comparatively rare, as determined by diagnostic restriction site mapping (Fig. 1b). Four days post-infection, the previously diverse population quickly resolved into viruses harbouring an insertion into two positions, with one location (Fig. 1b, black arrows) being more prevalent than the other (grey arrows). Deep sequencing analyses confirmed mutational saturation (Fig. 1c) and allowed mapping of the replicative fitness landscape of these libraries in order to determine well-tolerated sites of insertion (Fig. 2a)7,8.
Figure 1. Selection of replication-competent, tagged derivatives of HIV-1.
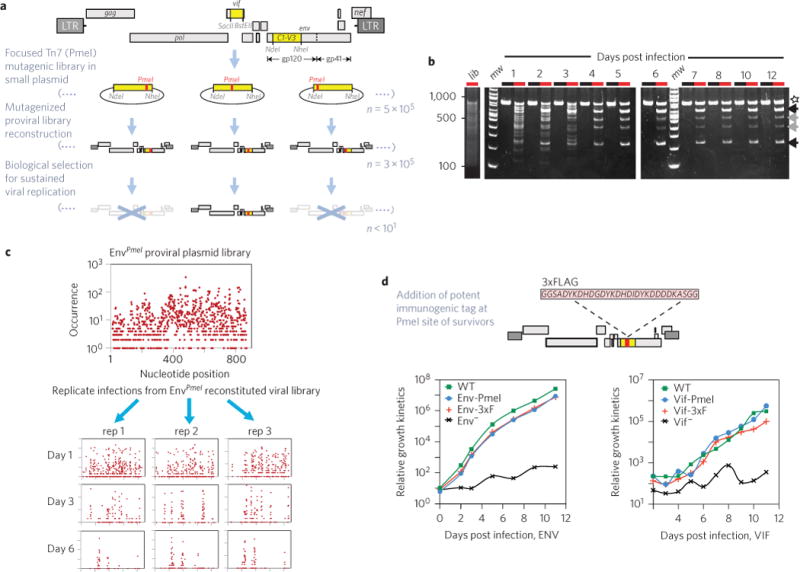
a, Viral gene segments (yellow) were independently subcloned to focus a step of Tn7-linker scanning-saturation mutagenesis. A proviral plasmid library was then reconstituted that contained ~3 × 105 to 5 × 105 independent clones, each harbouring a random PmeI insertion (red). DNA was pooled to generate an equally diverse library of PmeI-modified viruses by DNA transfection and used to infect a human T cell line. A stringent biological step selected for those viruses that can accommodate a five-amino-acid insertion yet remain fully replication-competent over several cycles of growth. Defective viruses (crossed out, X) are vastly overrepresented at early time points. Some mutant viruses capable of gaining entry into target cells do persist initially. However, the vast majority of these initially persisting viruses are non-competitive and are rapidly lost. Surviving PmeI-inserted HIV-1 derivative proviral clones exhibiting high replication competency at the end of selection are candidates for further recombinant manipulation and can be used as the recipient vectors for the insertion of foreign immunoaffinity tags (that is, 3xFLAG). b, Representative time course (n = 3; 1–12 d.p.i.) detailing the emergence of survivors from the Env-PmeI-modified clonal library (8% TBE PAGE). Lanes: black, uncut PCR product spanning the mutagenic targeted env gene segment; red, PmeI-digested PCR product. An uncleaved product is observed for any virus that has lost its PmeI insertion post day one (star). This species is (on sequence analysis) WT DNA, and indicates the occurrence of either intragenic retroviral recombination during reverse transcription or, less probably, an artefact of PCR recombination. c, Deep sequencing analysis details the mutational complexity of the plasmid DNA PmeI insertional library of the targeted env gene segment (top). The DNA sequences of viral survivors were determined 1, 3 and 5 d.p.i. Triplicate infection experiments were used to reveal the initially stochastic nature of successful selection pathways for viability. d, Amino acid sequence of the 3xFLAG (flanked by glycine/serine linkers) used to epitope tag a subset of replication-competent PmeI inserted viruses. Replication kinetics of Env- and Vif-modified viruses in comparison with the untagged and otherwise isogenic WT virus over several days of growth in their respective T cell line (n = 3; CEM cells for env recombinants; MT-2 cells for vif recombinants).
Figure 2. Permissible sites of insertion within Env and Vif and visualization of each tagged protein during infection.
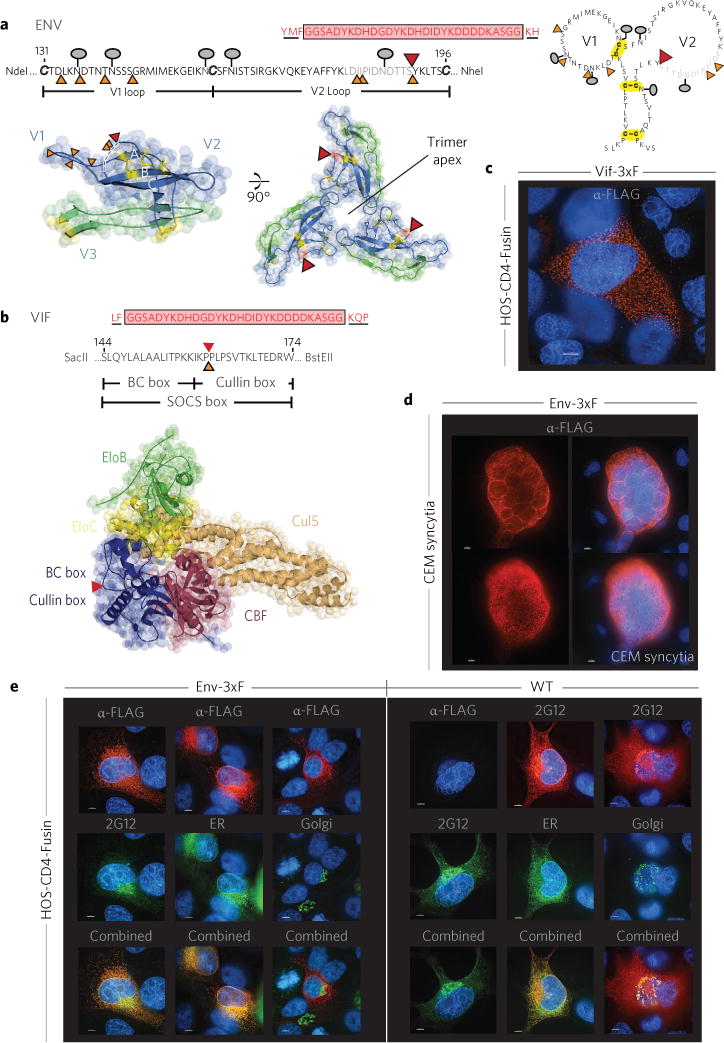
a,b,PmeI (orange) and 3xFLAG tag (red) insertion sites compatible with viral replication competency within the primary sequence and redrawn using MacPyMol for trimerized Env (PDB 4NCO)7 (a) and a pentameric Vif-containing complex (PDB 4N9F)8 (b). Five amino acids flanking the tag/linker and originating from transposition are underlined. The lollipop-like symbols denote N-linked Env glycosylation, and disulfide bonds are in yellow (a). Amino acids in the disordered/unresolved portion of the crystal structure are in light grey (a, top), and the dashed line (a, bottom left) shows an unresolved section in the crystal structure7; two post-selection Env PmeI insertional survivors map here. The 3xFLAG tag is immediately adjacent to this region, perhaps reflecting a flexible and/or alternative structural configuration(s). The epitope tag in Vif-3xF is between the Vif BC and Cullin boxes, shown within the primary sequence (b, top) and in the context of a pentameric complex where it is appointed away from all other protein–protein interfaces (b, bottom). c, The 3xFLAG-tagged Vif protein exhibits a cytoplasmic punctate staining pattern within Vif-3xF infected HOS-CD4-Fusin cells. d, 3xF-tagged Env protein (red) elaborates a highly reticulated distribution consistent with the endoplasmic reticulum (ER) in an infected CEM cell syncytium (two Z-slices from the same syncytium are shown: a medial and a peripheral slice). e, The 3xFLAG epitope is preferentially immunoreactive early during its maturation pathway within the ER. In contrast, the subcellular localization of 3xFLAG-tagged Env in HOS-CD4-Fusin cells is indistinguishable from WT Env using an antibody directed against a common gp120/gp160 epitope (2G12) as the Env is stained both within the ER and Golgi (the presentation is displayed vertically for all lanes of each panel, and also in Supplementary Fig. 2). Cells were first infected with baculovirus encoding an ER- or Golgi-specific fluorescent green probe and then infected with HIV Env-3xF or WT and incubated for an additional 48 h. Env was stained with mouse anti-FLAG mAb (red) or human anti-Env Ab (2G12; green or red). Counterstaining for nuclear DNA is in blue. For all fluorescent studies shown, the bar denotes 5 μm. All images are representative of three independent experiments with almost all stained cells in the population illustrating the respective phenotype displayed.
Interestingly, we found that after extensive selection for viability, different genetic solutions were obtained for each of three Env replicates, providing compelling evidence for the stochastic nature of selection. The position of the insertions in survivors was intriguing. Within the targeted N-terminal C1–V3 region of Env (amino acids 60–346), surviving viruses contained a linker insertion only within variable loops V1V2, but not V3 (Fig. 2a). Although the V1V2 loops, located at the Env apex, help to stabilize the trimeric form of gp120 crucial for receptor binding and entry, its variable sequence, inherent dynamic conformational flexibility as well as differential patterns of glycosylation combine to confound antibody-mediated neutralization. In surviving clones, N-linked glycosylation signals and disulfide bonds (C126:C196, C131:C157) in the V1V2 loop were preserved (Fig. 2a). V1 and V2 insertions did, however, cluster within a small, relatively unstructured region within the topmost, membrane-distal crown of the gp120 structure7,9 (Fig. 2a). In the CD4-unliganded trimer, although V2 amino acid residues 165–181 appear to be more protected from solvent exposure, residues 184–190 exist as the most flexible portion of V2 (refs 10,11). However, following CD4 binding, both of these regions undergo extensive restructuring and increased solvent exposure11. We chose a PmeI viral library survivor with insertion between residues 190 and 191 for further modification to create our prototype, 3xFLAG-tagged Env viral clone (Env-3xF). Although the variable loops of both HIV and SIV Env proteins have previously been targeted for foreign epitope tagging, the viability of these recombinant viruses was unpredictable, exhibiting a wide range of replication kinetics (in some cases, completely defective for viral passage)12–14. In contrast, our approach selects for tagged viruses in possession of robust replication competency, isolated without reference to prior structural information.
Insertion in Vif was similarly accommodated in a relatively unstructured region between the domains that associate with Elongin B/C (EloB/C) and Cullin15, a region reconstructed and ‘overprinted’ during adaptation of primate lentiviruses to humans16. The insertion splits the SOCS box, a region of Vif engaged with Elongin B/C that undergoes ‘induced-folding’ to restructure a Vif/EloB/C ternary complex capable of accommodating further assemblage with the Cul5-containing ubiquitin ligase15 (Fig. 2b). Remarkably, the original Vif PPXP sequence critical for multimerization and function17 is restored by target site DNA sequence duplication during transposition. In a pentameric complex8 consisting of Vif, Cul5, EloB/C and CBFβ, the site of foreign insertion faces away from all points of contact between pentamer components (Fig. 2b). Importantly, this virus emerged in two independent selections for sustained growth in MT-2 cells, in which viral replication requires functional Vif to counteract the host restriction factors (that is, APOBEC family members) that would normally act to inhibit viral propagation. Again, success in tag placement highlights the strength of our random insertion coupled with a powerful biological selection approach, without relying on preconceived notions of structure or function. Thus, our method establishes a validating strategy to pinpoint those rare positions within the viral proteome that are most amenable for tagging within essential viral gene products.
The second step of our strategy involved inserting the larger 3xFLAG tag (Fig. 1d, red triangle in Fig. 2a,b) into the PmeI sites that were previously introduced into the env (Env-3xF, Fig. 2a) and vif (Vif-3xF, Fig. 2b) viral genes. Each epitope-tagged recombinant virus exhibited robust replication kinetics comparable to that of the original wild-type (WT) clone (Fig. 1d). Remarkably, in both cases, the additions of a total of 35 amino acids at select locations within the viral proteome were stable and refractory to detectable deletion or further mutation or reversion to the WT sequence over at least a two-week period of viral outgrowth and expansion in human T cells (Supplementary Fig. 1). This allowed us to clonally amplify the tagged viruses, permitting quantitative affinity capture of endogenous viral–host macromolecular complexes from large cultures of infected cells.
Visualization of viral proteins during infection
The epitope tag provides an opportunity for the visualization of tagged viral proteins during infection (Fig 2c–e and Supplementary Figs 2–4). The Vif protein localized within mostly cytoplasmic foci, in agreement with its ectopic expression18. This provided, for the first time, the opportunity to directly visualize this viral protein during productive infection.
Env is expected to reside in the ER, the Golgi and the plasma membrane as it matures through the secretory pathway, as we observed using an antibody (2G12) that recognizes a common gp120 epitope. Importantly, 2G12-staining patterns for both Env-3xF and WT control viruses were indistinguishable (Fig. 2e and Supplementary Fig. 2), indicating that the cellular distribution of Env was unperturbed by the 3xFLAG insertion. Unexpectedly, however, the anti-FLAG antibody only stained a subset of these anticipated intracellular positions. Env-3xF was predominantly immunoreactive to anti-FLAG in the ER, elaborating a highly reticulated structure in adherent cells and within infected T cell syncytia (Fig. 2d), but not in the Golgi or at the cell surface (Fig. 2e and Supplementary Figs 2 and 3), reappearing only near sites of syncytial contact (Supplementary Fig. 3b). We further confirmed that the tag did not disrupt either the overall glycosylation profile of Env within productively infected T cells (Supplementary Fig. 5), or its incorporation or abundance in virions isolated from either transfected 293T or infected CEM cell cultures (Supplementary Fig. 6). An investigation of the temporal immunoreactivity of the 3xFLAG epitope revealed distinct staining patterns at early (2 h post infection, Supplementary Fig. 4a) versus late (22 h post infection, Supplementary Fig. 4b) stages of infection, when new Env is being synthesized in the producer cell after proviral DNA establishment. Our results are therefore consistent with the notion that the 3xFLAG epitope is not always accessible, being antibody-reactive only at specific points along the maturational and functional pathway of Env.
Direct identification of viral interactomes during infection
The retrieval of tagged Env or Vif from infected cells allows for quantitative recovery of their respective host interactomes. To differentiate the specifically interacting proteins from possible nonspecific associations, we used a quantitative mass spectrometric technique, isotopic differentiation of interactions as random or targeted (I-DIRT19; Figs 3 and 4 and Supplementary Fig. 7)20. As illustrated in Fig. 3a and Supplementary Fig. 12 (Env) and Fig. 4a and Supplementary Fig. 13 (Vif), specific interacting proteins are revealed as clusters exhibiting high ratios in both forward and reverse I-DIRT experiments, while nonspecific proteins are distributed throughout the lower-left quadrant. I-DIRT interaction results for Env were confirmed for selected interactors by reciprocal co-immunoprecipitation (Fig. 5a).
Figure 3. Reciprocal mass spectrometric isotopic differentiation of interactions as random or targeted (MS I-DIRT) (ref. 19) plot for Env (including inset of region from 0.97 to 1.0) and histogram of the number of proteins versus average I-DIRT ratios for Env.
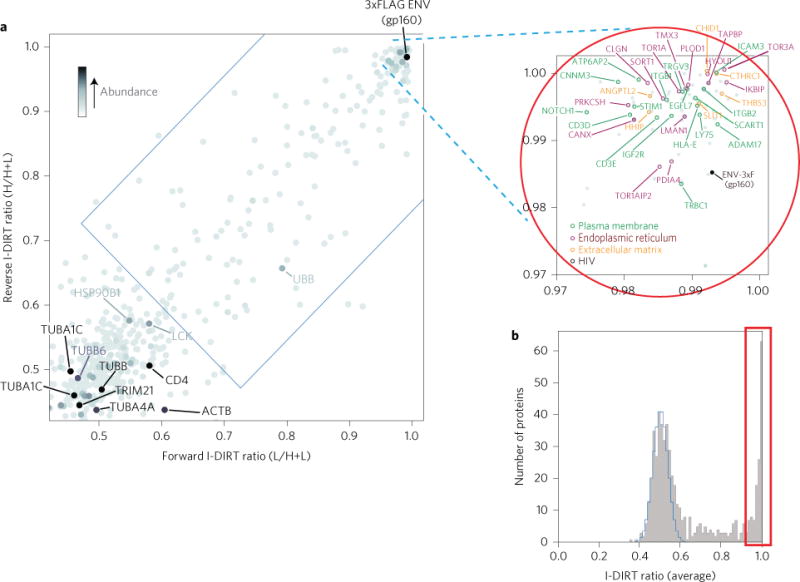
a, I-DIRT ratios for forward and reverse I-DIRT affinity isolations for the 3xFLAG-tagged HIV-1 Env protein. The blue rectangular region delimits the cutoff for statistical significance (<0.01% chance of being nonspecific). Selected proteins of interest are labelled. The average protein abundance within the two-affinity isolation is indicated by darkness of shading. Proteins registering greater than 0.97 on both I-DIRT and reverse I-DIRT are circled in red. b, Histogram showing the number of identified proteins in the Env affinity isolation plotted versus their average (forward and reverse) I-DIRT ratios. The calculated nonspecific distribution is indicated in blue, and putative specific interactors (>0.97) are boxed in red.
Figure 4. Vif interactors identified by reciprocal MS I-DIRT analysis and documentation of differential binding kinetics of its interactors.
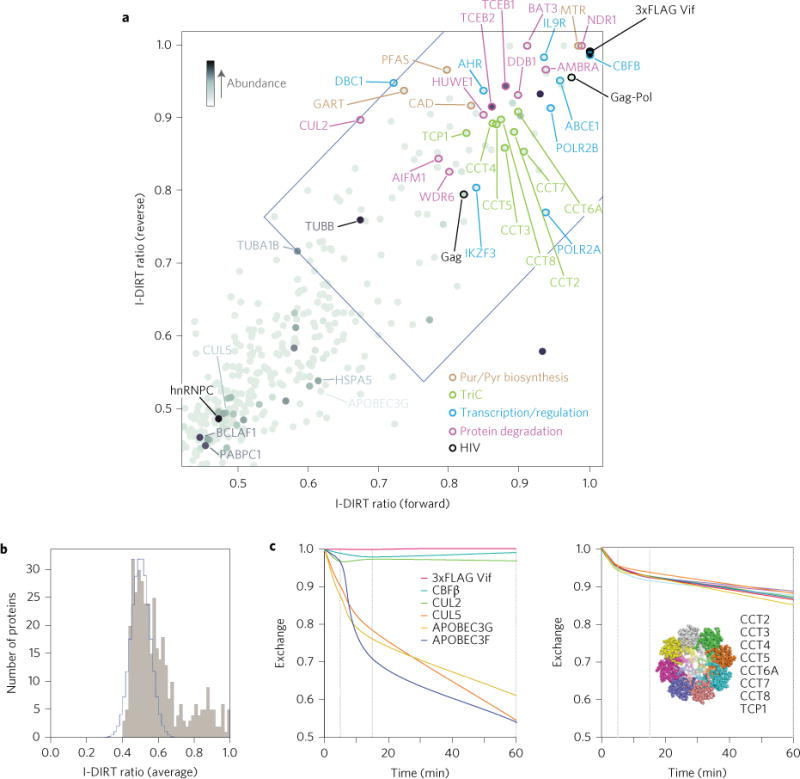
a, Plot of I-DIRT ratios for forward and reverse I-DIRT affinity isolations for the 3xFLAG-tagged HIV-1 Vif protein. The blue region delimits the cutoff for statistical significance (<0.01% chance of being nonspecific). Selected proteins of interest are labelled. Protein abundance is indicated by the darkness of shading. b, Histogram showing the number of identified proteins in the Vif affinity isolation plotted versus their average (forward and reverse) I-DIRT ratios. The calculated nonspecific distribution is indicated in blue. c, Left: Results of the isotopic exchange experiment in which immunoaffinity-isolated, heavy isotope-labelled tagged Vif and heavy isotope-labelled associated proteins were exposed to protein extract of normal (light) peptide isotopic content for the indicated times. Per cent exchange between the heavy labelled proteins on the beads and their light counterparts in the extract was plotted for tagged Vif itself and the known interactors CBFβ, Cul2, Cul5, APOBEC3F and APOBEC3G (left), showing that although CBFβ and Cul2 exhibit very little exchange, Cul5, APOBEC3F and APOBEC3G exchange rapidly. c, Right: Percentage exchange curves of Vif interactors TCP1, CCT2, CCT3, CCT3, CCT5, CCT6A, CCT7 and CCT8, which together form the T-complex (inset: End-on view of a cryogenic-electron microscopy (cryo-EM) density map Protein Data Bank (PDB) accession code 3IYG (ref. 20) as redrawn using MacPyMol), were almost superimposable, indicating that these proteins dissociate from the Vif affinity pull down as a complex, rather than as individual factors.
Figure 5. Experimental documentation for select interactors.
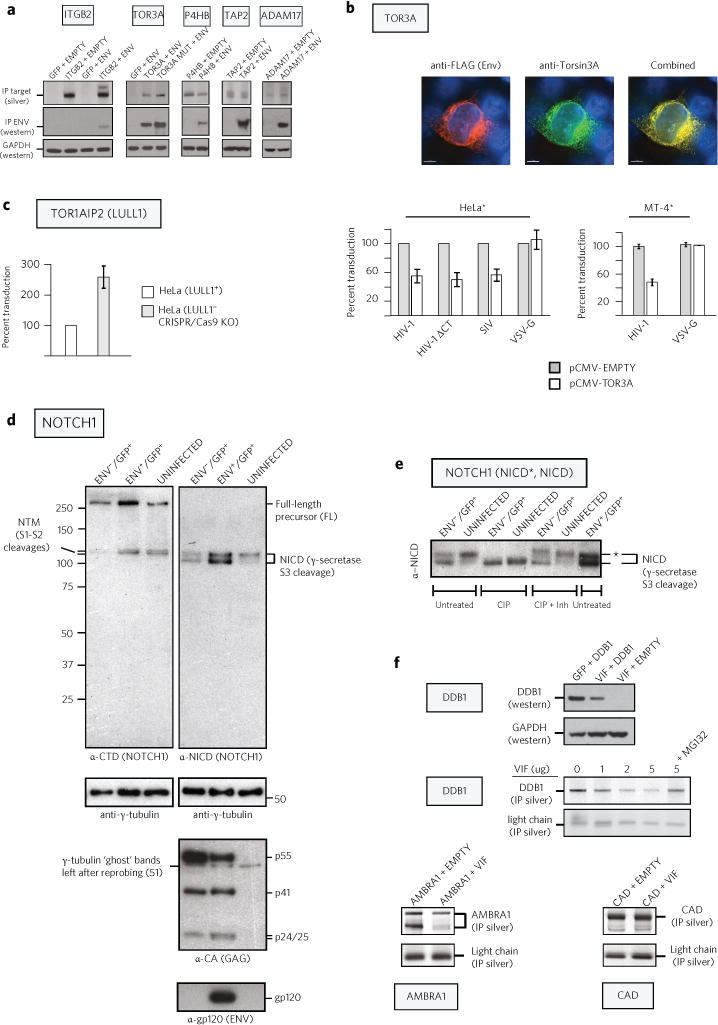
a, Co-immunoprecipitation of selected Env interactors confirms I-DIRT results. 293T cells were co-transfected with gp140 Env and the indicated plasmids. Extracts were immunoprecipitated, electrophoresed and silver stained (top) or western blotted against Env or GAPDH control. b, Tor3A occupies the same intracellular compartments as Env, and when expressed in producer cells, inhibits the next round of infection. Immunofluorescence: 293T cells co-transfected with Env-3xFLAG proviral DNA and an expression vector for human Tor3A were co-stained for FLAG (red) and Tor3A (green). Infection: 293T cells were co-transfected individually with WT env− proviral DNA, a pseudotype expression vector for the envelope proteins of HIV-1 (WT), HIV-1 ΔCT, SIV or VSV and with either pCMV-empty or pCMV-TOR3A. TZM-bl (HeLa*; n = 4) or MT4-GFP (MT4*; n = 2) indicator cells were infected with p24-normalized viral stocks. c, WT HIV-1 infectivity is increased when produced in TOR1AIP2 (LULL1) knockout cells. Virus produced in LULL1+ or LULL1− cells was normalized for reverse transcriptase activity and scored for infectivity on MT4* indicator cells (Supplementary Fig. 11a). d, HIV-1 infection alters Notch1 proteolytic processing while Env increases Notch1 levels. GFP-positive cells infected with either Env− or Env+ virus were purified by FACS (Supplementary Fig. 11b) and compared with uninfected cells. Western blots were probed against the Notch1 C-terminal domain (anti-CTD) or against a neo-epitope created upon γ-secretase cleavage (anti-NICD). Gamma (γ)-tubulin is a loading control, while Gag-and Env-specific antibodies confirm lane identities. e, HIV-1 infection leads to hypophosphorylation of the NICD independently of Env. Precalibrated lysates used in d were either untreated, treated with calf intestinal alkaline phosphatase (CIP) or treated with CIP plus phosphatase inhibitors and the lysates were electrophoresed, blotted and probed with anti-NICD. Upper bands (asterisk), hyperphosphorylated NICD; lower bands, hypophosphorylated NICD. f, Vif expression reduces DDB1 and AMBRA1 levels in 293T cells. For the DDB1 Vif titration (top right), indicated amounts of Vif construct were co-transfected with 5 μg of DDB1 plasmid (total DNA, 10 μg). For other transfections, Vif and the interactor or control plasmid were present at 5 μg each. Extracts were immunoprecipitated, electrophoresed and silver stained or probed by western blot (DDB1 and GAPDH). Residual antibody light chain was used as a loading control. n ≥ 2 for all experiments. Error bars represent standard deviation.
The Env interactome
ER proteins that associate with gp160 Env
The components of the Env interactome are summarized in Fig. 6 and Supplementary Figs 12 and 15 (and arranged with respect to averaged I-DIRT scores in Supplementary Table 1). The majority of the interactors were recovered from the ER, consistent with the availability of the 3xFLAG epitope (Fig. 2 and Supplementary Figs 2–6). Interactors include members of the glycosylation, thio/redox and chaperone-mediated folding pathways, which generally associate with proteins targeted for secretion or plasma membrane incorporation. Consistent with the immunocytochemical accessibility of the Env 3xF tag (Fig. 2e and Supplementary Figs 2, 3) in the ER, we observed that when Env was competitively eluted with 3xFLAG peptide from the antibody support under native conditions, it was predominantly recovered as gp160, the Env precursor being in its uncleaved and trimerized form (Supplementary Fig. 8a). The isolated gp160 largely bears N-linked high-mannose glycans that are not processed further by terminal glycosylation, as occurs within the Golgi apparatus (Supplementary Fig. 8b). Furthermore, in agreement with the trimer structure and function, this tagged Env was recognized by its receptor, soluble CD4 (sCD4), and by a subset of broadly neutralizing monoclonal antibodies (bNAbs), but was not recognized by bNAbs that have epitopes near the site of 3xFLAG-tag insertion in the V1V2 loop (PG9, PG16 and to a lesser extent, PGT145), or by bNAbs (PGT121) that require post-ER sugar modification for binding (Supplementary Fig. 8c). Taken together, these results confirm our fluorescence visualization observations that 3xFLAG epitope exposure is temporally and spatially conditional, reactive in the ER but not in the Golgi apparatus, such that the tagged protein is only recoverable from certain intracellular sites during its extensive carbohydrate elaboration and transportation.
Figure 6. Summary of biological subclasses of cellular interactors engaged with Vif and Env during active viral growth.
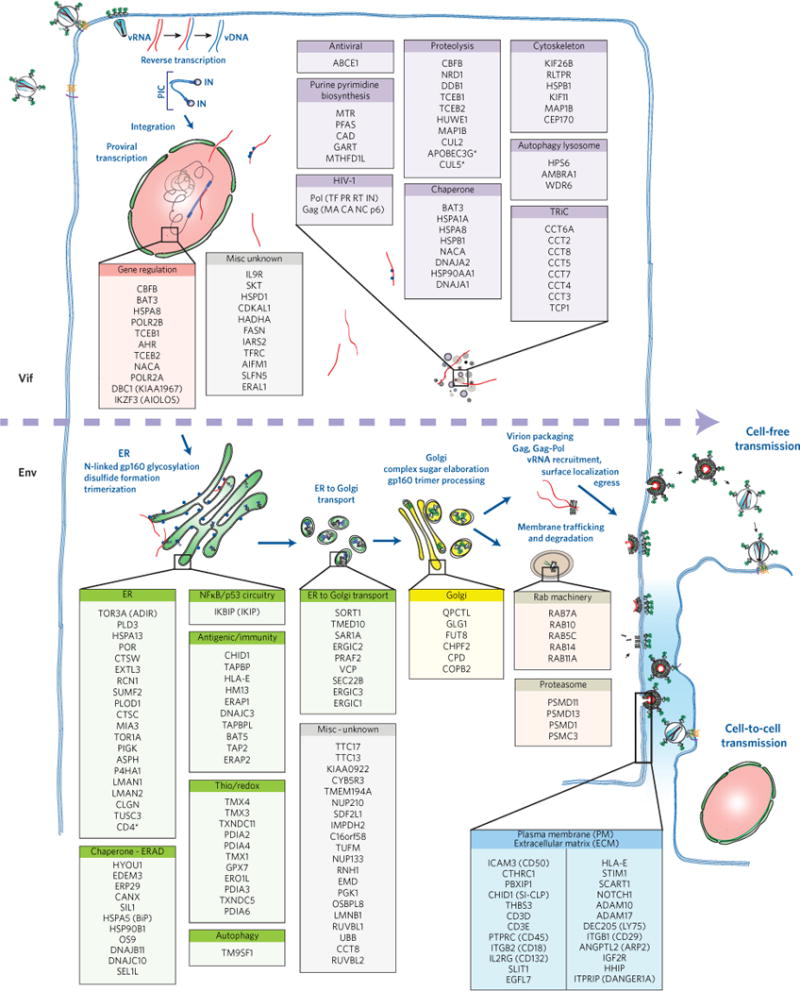
An abbreviated schematic of the HIV life cycle starting at the upper left with infection of a susceptible target cell and at the bottom right showing perpetuation of the infectious cycle by viral egress and transmission via cell-free infection of both nearby and distant cells or by cell-to-cell transmission of the virus to an immediately adjacent cell. The members of each class of cellular interactor are colour-coded based on their predicted intracellular location and/or function. The dashed horizontal arrow intersecting the figure indicates the progression of the viral life cycle and separates the members of the Vif interactome from those interacting with Env during the series of steps that occur during the Env biosynthetic and maturation pathways.
Because most proteins were recovered along with gp160 from the ER, we sought to provide evidence for impact on the viral life cycle for a set of identified Env interactors. Thus, we selected the highest scoring I-DIRT candidate (TOR3A) and its heterodimeric catalytic partner (TOR1AIP2/LULL1) for further study. Tor3A (ADIR) is a member of the torsin subfamily of the AAA+ ATPase chaperones21 that are thought to manage protein folding as well as intracellular trafficking. Mutations in Tor1A result in early onset torsion dystonia, a poorly characterized human disease. Intriguingly, Tor3A and TOR1AIP2/LULL1 are both interferon-induced proteins, suggesting a possible association with host cell antiviral defence. We find that Tor3A occupies the same intracellular compartment(s) as Env, the inherent infectivity of virus produced from cells co-transfected with WT proviral DNA and a vector overexpressing human Tor3A being compromised in either a fibroblast or lymphoid indicator cell line (Fig. 5b). This effect is specific for the HIV-1 and SIV Envs, but without effect on HIV-1 Env− virus pseudotyped with VSV-G, suggesting that Tor3A acts against HIV-1 during a late phase of the viral life cycle, perhaps modulating the folding/oligomerization of this large, highly glycosylated protein. In contrast, compared with its progenitor cell line, virus produced in CRISPR/Cas9 TOR1AIP2/LULL1 knockout cells22 increased the infectivity of WT HIV-1 (Fig. 5c and Supplementary Fig. 11a), suggesting that HIV-1 Env maturation and therefore function may be constrained by Tor3A and LULL1. Interestingly, TOR1AIP2 was recently shown to be required for efficient growth of HSV-1 as deletion of TOR1AIP2 affected a late step of the viral life cycle preceding viral egress but not earlier steps of viral entry, transcription or protein production22.
Innate immune factors
Host defence components implicated in the evasion of host antiviral response and specifically targeted for inactivation by many types of virus appear within the Env interactome. We found three components of the transporter associated with antigen processing (TAP) complex—TAPBP (tapasin), TAPBPL and TAP2—that are essential for transport of viral peptides from the cytoplasm onto major histocompatibility class I (MHC-I) molecules within the ER, the earliest step in the process of exposure of virus-derived epitopes on the surface of infected cells. Viruses often interfere with the host cell’s MHC-I antigen processing/presentation pathways in an attempt to evade cytotoxic lymphocytes23; indeed, HIV-1 Nef down-modulates MHC-I molecules from the cell surface24. Whether the interactions of these TAP complex members with HIV-1 Env diminish TAP antiviral activity, is, however, unknown. HLA-E, another factor in innate immune response, exhibits potent inhibition of natural killer cell cytotoxic activity, and HM13, a peptidase required for the genesis of nonomeric peptides subsequently loaded into the HLA-E cleft, was also found to be a member of the HIV-1 Env interactome. Interestingly, HM13 processes the hepatitis C virus (HCV) viral core protein (p18), liberating a short peptide derived from the core, a ligand for HLA-E that stabilizes its expression and favours the inhibition of natural killer cell cytotoxicity against HCV infection25.
The putative virological synapse and components of the Notch1 signalling pathway
Unexpectedly, using green fluorescent protein (GFP) or sNLuc reporter viruses (Supplementary Fig. 9), we found that compared to the WT, Env-3xF virus prepared from either 293T transfection or by collecting virions from ongoing CEM cell infection is preferentially transmitted via cell-to-cell contact, being profoundly defective for cell-free transmission (Supplementary Fig. 10). In a single-step infection, Env-3xF exhibits nearly 100-fold defect compared to the WT virus cell-free infection (Supplementary Fig. 10a). However, when Env-mediated viral entry is bypassed and an unrelated viral envelope transfected pseudotype (that is, VSV-G ‘super-pseudotyping’) is used as an alternative to initiate the first round of infection (all subsequent viral passages relying on provirally encoded HIV-1 env), the kinetics of viral growth are nearly identical for the WT and tagged virus (Fig. 1d and Supplementary Fig. 10b). This result, combined with results obtained using methodologies26,27 designed to distinguish between the two modes of transmission (Supplementary Fig. 10c,d) is consistent with the idea that the V1V2-tagged28 Env-3xF virus preferentially utilizes cell-to-cell transmission. Intriguingly, we found that it is only during cell-to-cell transmission—and not cell-free infection—that the Env-3xF virus can be effectively neutralized by the addition of anti-FLAG antibody (Supplementary Fig. 10e). This result may reveal intrinsic structural differences between the two forms of Env during distinct modes of viral transmission.
Taken together, the above results suggest that, in addition to the ER compartment, the 3xFLAG epitope is conditionally available during cell–cell receptor and/or co-receptor engagement and entry. This probably explains why a significant number of host proteins from the extracellular matrix (ECM) and the plasma membrane (PM) are found within the Env interactome (Fig. 6 and Supplementary Figs 12 and 15 and Supplementary Table 1). Some of these may be putative components of the still vaguely characterized HIV-1 virological synapse (VS) required for cell-to-cell virus transmission29. In vivo, the VS may provide a relatively protective environment for the virus against antibody-mediated neutralization26,30,31, host restriction factor inhibition32,33 and/or antiretroviral therapy34 and therefore presents a challenging barrier to viral eradication.
The identity of PM proteins associated with HIV-1 Env during ongoing viral growth is provocative. As expected, we recovered cell adhesion molecules, some of which mediate lymphocytic uropod elongation and migration, that is, integrin subunits along with the integrin receptor, ICAM3. In addition, we recovered receptors and modulators that affect various signalling pathways, namely, T cell receptor subunits, Notch components and Hedgehog (HHIP) as well as factors involved in calcium signalling35,36. HIV-1 Env also associated with the mannose-6-phosphate/insulin-like growth factor receptor 2 (ILGF2), which is thought to facilitate HIV-1 replication in microglial cells37. The mannose-6-phosphate modification of gp120 being host-cell-line-specific, perhaps preferentially added to the viral envelopes of transmitted founder viruses38. In summary, the associations of Env with host factors residing in proximate locations near the cellular plasma membrane and extracellular space are noteworthy and may further help to resolve some of the salient interactomic features of the HIV-1 VS.
The Notch signalling pathway possesses unique features that make it particularly amenable for interdependent yet non-reciprocal, direct cell-to-cell communication between adjacent cells. Historically known for its role in embryonic differentiation and development39, Notch has also been linked to memory T-cell homeostasis and a proposed model of HIV-1 latency40. CSL (CBF-1, Su(H), Lag-1), a key effector of the Notch pathway, has been shown to be a potent and specific inhibitor of the HIV-1 long terminal repeat (LTR) promoter41. Co-isolation of Notch signalling components with Env prompted us to examine the steady-state levels and phosphorylation status of the Notch protein in the context of viral infection. To this end, VSV-G super-pseudotyped Env+ or Env− GFP viral stocks (Supplementary Fig. 11b) were used to infect CEM cells in single-cycle format, the GFP+ cells from each type of infected cell isolated by flow cytometric purification and cell extracts probed by western blot analysis with antibodies specific for Notch1 (Fig. 5d,e). We observed the following: (1) the steady-state levels of both the Notch1 full-length (FL) protein as well as its intracellular proteolytic derivative (NICD) are increased in the context of Env+ infection when compared to the otherwise isogenic infection with the Env− virus or when contrasted to Notch1 levels from uninfected CEM cells (Fig. 5d); (2) although several domains of the NICD are targets of phosphorylation modification (a key determinant in the regulation of its transactivation activity, stability and nuclear localization42), infection with either the Env+ or Env− viral derivative results in the hypophosphorylation of the NICD relative to uninfected CEM cells (Fig. 5e). The latter result suggests that although Env expression in infected cells increases Notch1 protein levels, possibly by stabilizing the protein, NICD phosphorylation is not dependent on Env expression per se but rather via an unidentified consequence of productive viral infection itself.
Further connection with the Notch1 pathway may be provided by the Vif interactor, IKZF3 (Aiolos), a strong transcriptional repressor of the Ikaros family, in part shaping the repertoire of genes targeted by the Notch pathway in T cells43 and adhesion-related genes, disrupting cell–cell and cell–matrix interactions.
The Vif interactome
The Vif interactome reveals diverse and dynamic associations with host factors
The Vif Interactome as determined from our I-DIRT analyses is composed of 53 host proteins in addition to proteins derived from viral Pr55 Gag and Pr185 Gag/Pol polyproteins (Figs 4 and 6, Supplementary Figs 13 and 15 and Supplementary Table 2). Among these were several previously documented interactors, including EloB/C (TCEB1/2) and CBFβ. Novel factors identified include members of the cytoplasmic purine/pyrimidine biosynthetic pathways (also shown to be altered in activity during human cytomegalovirus infection44), as well as members of the nuclear transcription and alternative RNA splicing/elongation machinery, hinting at possible unexpected roles of Vif in modulating nucleoside biosynthesis and host gene expression. It is interesting to note that Vif, via its cytoplasmic sequestration of transcription factor CBFβ (Fig. 4c), has recently been shown to affect the expression of several of CBFβ’s cofactors, RUNX1-regulated genes involved in immune cell function, including IKZF3 (ref. 45).
Although the I-DIRT approach can greatly improve the selectivity in determining specific interactions, proteins exhibiting rapid exchange within Vif complexes during affinity capture are detected as nonspecific, resulting in false negatives and confounding their authentication as true interactors. A cogent demonstration of this effect is provided by a subset of the identified host proteins that are known to associate with Vif during viral replication. These proteins (APOBEC3G, APOBEC3F and CUL5) were selectively recovered during Vif immunoisolation, but, paradoxically, initially scored as nonspecific because of their very low I-DIRT ratios. We reasoned, however, that certain proteins might exhibit fast exchange within Vif complexes during the hour-long affinity capture procedure, thus lowering their inherent I-DIRT ratio. To test this possibility, we performed an isotopic differentiated, in vitro exchange experiment (Fig. 4c). Indeed, APOBEC3G, APOBEC3F and CUL5 are rapidly exchanged within the first 5 min of incubation (Fig. 4c left, lower traces). In contrast, CBFβ, a known interactor46,47 identified by its high I-DIRT ratios, was detected as a stable interactor across all time points in the isotope exchange assay (Fig. 4c left, upper traces) consistent with its suspected roles in folding, stability and solubility of primate Vif. It is also noteworthy that all eight members of the TCP1 ring complex (TRiC, T-complex), an ATP-dependent chaperone, show both congruent high I-DIRT ratios and overlapping exchange rates with Vif (Fig. 4c right). These results suggest that TCP1 associates with Vif as a single integrated complex, rather than as individually dispersed subunits. Overall, these results show that I-DIRT and in vitro exchange studies have the potential to dissect which factors associate and exchange as subcomplexes. Finally, in contrast to previous reports48,49, we and others6 find that CUL2 can associate with Vif. Although CUL5 exchanges rapidly, CUL2 does not appreciably dissociate from Vif during in vitro experimentation (Fig. 4c, upper). Vif is known to use CUL5 for degradation of APOBEC3G50, consistent with the observation that APOBEC3G exhibits rapid exchange kinetics similar to CUL5. The more stable profile of CUL2 with Vif may therefore predict a different functional association.
Vif interaction and loss of steady-state levels of specific host proteins
Given that Vif is known to mediate the degradation of host factors in the APOBEC family through the ubiquitin/proteasome pathway, we examined Vif’s ability to modulate the expression of a subset of the putative host interactors we had identified. Steady-states levels of the given host proteins were determined after ectopic expression with or without Vif in the absence of viral replication. We found that DDB1 and AMBRA1 levels were detectably reduced to diminished levels when ectopically co-expressed with Vif, but not with a control construct, while CAD remained unaffected by the presence of Vif (Fig. 5f). Moreover, downregulation of the DDB1 protein was blocked by the addition of proteasome inhibitor MG-132 (Fig. 5f), suggesting that the downregulation occurs via the proteasome pathway, as is the case with the APOBEC family of Vif substrates. It is possible that DDB1 and AMBRA1, which are known to co-associate and stimulate the autophagy pathway, might be targeted for Vif-mediated degradation to prevent autophagy of HIV-1 viral subcomplexes within the cell. However, we acknowledge an obvious and cautious caveat—these observations were revealed during ectopic over-expression of Vif in isolation—so the impact on natural viral replication remains to be determined.
The current work is, in part, corroborated by previous work from the Krogan laboratory in which viral genes were individually expressed in tissue culture cells6. In these studies, ~40% of the cellular identifications made with Env and Vif by Krogan and colleagues6 were also present in our list of specific interactors (Supplementary Fig. 14). However, the reverse was not true (compare Supplementary Figs 14–16). Indeed, apart from the proteins shared between the two studies, between 72 and 82% of those host proteins associated with HIV were unique to our study, as directly recovered from all stages of ensuing HIV-1 replication.
Conclusion
We have established and applied an genetic–proteomic–interactomic approach to systematically identify and validate host components of the HIV–host interactome, those host protein arrays in vicinal association with the viral machinery directly during productive, cycling infection. The candidates identified in this study represent cellular factors impacting viral propagation or immune escape pathways, contributing to our understanding of the strategies employed by HIV-1 to gain control of the host cell and those employed by the host cell in response to viral infection. Finally, our methodologies can be adapted to other viruses or pathogens, thus benefiting the systematic elucidation and validation of host–pathogen interactions. Information from these types of study will promote the comprehensive understanding of the consequences of these interactions as well as provide new therapeutic targets for intervention.
Methods
Selection strategy for the recovery of epitope-tagged, replication-competent viruses
We used saturation linker scanning mutagenesis to introduce a unique endonuclease restriction site at all possible nucleotide positions within a given targeted segment of the HIV-1 genome. This new site then served as an acceptor for the insertion of DNA sequences that encode an exogenous epitope tag. In this way, we recovered replication-competent viral derivatives that harbour a well-characterized peptide tag (3xFLAG)51 that is efficiently detected by a cognate antibody. This allowed for large-scale affinity capture of any virus–host macromolecular complexes formed during infection with these tagged viruses, which were then analysed by mass spectrometry.
We chose two key distinct HIV-1 proteins for analysis: the abundant, extensively glycosylated envelope protein Env, which is the target of anti-HIV-1 vaccine initiatives, and the relatively poorly expressed, cytosolic viral infectivity ‘accessory’ factor, Vif, which usurps an otherwise nuclear transcriptional regulatory cofactor, CBFβ, along with components of the host proteasomal degradation machinery to counteract intrinsic host-specified defence factors that seek to destroy the genomic integrity of viral DNA during the next cycle of replication. A differential isotopic labelling strategy19 was used and confirmed in reciprocal, replicate experiments to unambiguously determine specific interactions that occur during ensuing viral replication. Taken together, our work promises to open a new chapter in the study of HIV and viral infection generally—by using an orthogonal approach, compared with previous HIV interactor screens—to provide high-quality identification of in vivo interactions that occur during viral replication.
For cloning purposes, the LAI-derived HIV-1 proviral clone R7/3 (refs 52–55) was engineered (overlap-PCR site-directed mutagenesis) to contain a pair of unique restriction endonuclease sites immediately flanking each of the targeted gene segments. This allows for the isolated mutagenesis and manipulation of the targeted viral gene segment without disruption of the remainder of the proviral genome or plasmid backbone. None of the mutations altered the identity of the corresponding amino acid when decoded. The new restriction sites and the primers used for vif mutagenesis were SacII (5′-CCTCATCCgcGgATAAGTTCAGAAG-3′) and BstEII (5′-GACCAAGGGtCACcGAGGGAGC-3′), the lower case letters indicating the nucleotides altered by mutagenesis. The engineered HIV-1 vif and env (constant region C1 through variable loop V3) regions were then subcloned into pBluescript II (pBS) KS(−) (Stratagene), to generate pBS-KS-vif and pBS-KS-env (C1–V3). These small pBS-KS(−) subclones served as substrates for a focused in vitro transposition reaction using the Tn7-derived, GPS-LS linker scanning system (New England Biolabs). More than 106 recombinants were selected based on an ampicillin/kanamycin double-resistant phenotype (the transposed element specifying kanamycin resistance, and ampicillin resistance conferred by the proviral plasmid DNA clone). Viral gene segments that received a transposon were gel-purified, pooled and cloned back into the modified R7/3 proviral vectors using each of the respective unique restriction enzyme pairs, again selecting the ampicillin/kanamycin double-resistant phenotype. After digestion with PmeI, the linearized DNA was acrylamide-gel-purified, self-ligated and used to generate plasmid DNA libraries of the vif or env viral coding region, each clone uniquely exceptional with respect to the location of 15 base pairs containing a unique PmeI restriction endonuclease site within the targeted viral gene segment.
Diverse proviral DNA libraries were used to prepare a heterogeneous population of viral particles by efficient transfection of either 293T (env) or 293-APOBEC3G-HA (vif) producer cells. Clones were selected for sustained viral replication in a suitable immortalized human T cell line (CEM cells for env recombinants and MT-2 cells for vif recombinants, respectively). Clonal survival indicates that a five-amino-acid insertion can be accommodated within the viral proteome, mediating all of its necessary functions without impinging on viral replication-competency. Viral DNA was recovered from survivor cultures, and the individual sites of insertion were identified by DNA sequencing analysis.
Random, high-density, transposon-mediated insertion paired with next-generation sequencing, followed by selective screens for viability, is a powerful genetic footprinting tool to construct maps of the replicative fitness landscape of a given viral protein in its natural functional context. This technique has been applied to interrogate the genetic plasticity of a given viral genome in a single step of mutagenic/viability selection, with the results interpreted in the context of previously solved three-dimensional structures (for example, HCV56). Probing of the genetic landscape for the HCV genome was made possible by the simple mechanics of its replication of single-stranded, genomic RNA. However, unlike HCV, retroviral genomes are diploid (comprising two intravirion strands of RNA) and extensively recombine during reverse transcription and conversion to a single DNA molecule, which is a mosaic of its viral RNA progenitors57. In this context, an intravirion crossover event between two distinct, PmeI-marked viral genomes could result in the restoration of the WT virus from a completely mutant viral population. Indeed, when reconstituted, the WT virus can quickly overtake the mutant viruses during extended periods of selection. This is true even when a PmeI-marked virus is only slightly replication-deficient. Because recombination occurs less frequently between adjacent sequences, small regions of the proviral genome (that is, the 857-base-pair N-terminal gp120 C1–V3 portion of env) encoding single viral genes, or their subdomains (Fig. 1a, yellow), were individually targeted for Tn7-mediated PmeI-mutagenesis. Although this limits insertional genetic footprinting to much smaller targeted regions of the HIV genome, as we show here, this type of analysis is quite feasible and can provide informational landscapes even within the confines of much smaller regions of the viral genome.
Following infection (as described below), replication-competent PmeI-tagged HIV-1 clones produced from sustained infection were recovered by Hirt extraction58 or by purification of total DNA from the infected cell culture using a QIAamp DNA Blood Mini Kit (Qiagen). The viral coding regions (vif or env) harbouring unique PmeI insertions were amplified by PCR using the following primer pairs: vif (5′-TATAGACATCACTATGAAAGC-3′ and 5′-CATAAGTTTCATAGATATGTTG-3′), env (C1-V3) (5′-CATCAGATGCTAAAGCATATG-3′ and 5′-GTTCTCTTAATTTGCTAGC-3′). PCR-amplified PmeI-containing fragments were cloned back into proviral R7/3, sequenced to determine the location of the PmeI insertion, and the resulting proviruses tested for replication competency. Replication-competent clones represent mutant HIV-1 that can tolerate a 15-base-pair insertion yet remain viable over several cycles of viral outgrowth. The PmeI insert clones served as universal substrates for further addition of epitope tags at the unique PmeI site within the env and vif proteins. Oligonucleotide complementary pairs encoding the appropriate 1xFLAG (NH2-GGSADYDDDDKASGG) or 3xFLAG epitope (NH2-GGSADYKDHDGDYKDHDIDYKDDDDKASGG-COOH) reading frame were 5′-phosphorylated, annealed and cloned into the PmeI site. A restriction endonuclease site (BlpI, GC/TNAGC) absent from the proviral plasmid DNA vector was also included within the DNA sequence of each oligonucleotide pair as complete BlpI digestion after oligonucleotide pair ligation prevents multiple insertions of the foreign epitope tag sequence. The resulting epitope-tagged proviral clones were then confirmed for tag DNA sequence and orientation, the correct plasmid clones were used to generate viral stocks, the 3xFLAG-tagged viruses were retested for sustained viral growth and replication competent clones were selected for further experimentation.
The plasmid pCAGGS was used to express JR-FL Env on virus-like particles (VLPs)59. JR-FL Env was truncated at amino acid 709 to produce a mutant termed gp160ΔCT, which was shown to increase native trimer expression and produces a pseudotyped virus that exhibits a similar neutralization sensitivity profile as the full-length JR-FL gp160 (ref. 60). ‘SOS’ mutations introduce an intermolecular disulfide bond between gp120 and gp41 (ref. 61). This VLP (ref. 59) is known as JR-FLSOS.
Antibodies
Monoclonal antibodies (mAbs) were obtained from their producers, the NIH AIDS Reagent Repository, the Center for AIDS Reagents or commercial suppliers. The mAbs included the following: 2G12 (H. Katinger), directed to a glycan-dependent epitope of gp120 (refs 62,63); CA13 or ARP3119 (C. Arnold, gift of J. Moore), a murine mAb directed to gp120 C1 (ref. 64); CO11, 39F (J. Robinson) and 902 (B. Chesebro), directed to the gp120 V3 loop60; PGT121 (D. Burton), directed to epitope clusters involving the base of the V3 loop of gp120 and dependent on N332 glycan65; VRC01 and VRC07 (J. Mascola), directed to epitopes overlapping the CD4 binding site66,67; PG9, PG16 and PGT145 (D. Burton), a quaternary-specific, glycan-dependent mAb that binds to the V1V2 region68,69; 2F5 and 4E10 (H. Katinger) and 10E8 (J. Mascola and M. Connors), directed to the gp41 membrane-proximal ectodomain region (MPER)70,71; Chessie 8 (G. Lewis), a murine mAb that binds to the gp41 cytoplasmic domain72; 246-D (S. Zolla-Pasner), a non-neutralizing human mAb to gp41 (refs 73–75); a goat polyclonal raised against HIV-1 gp120 (Ab21179; Abcam). Other antibodies used in this study include the following: MAb319 (M. Malim), a murine mAb that binds to HIV-1 Vif protein76–78; 183-12H-5C (M. Malim), a murine mAb that is specific for the HIV-1 capsid protein p24 (CA)78; M2-FLAG (F3165; Sigma-Aldrich), a murine mAb that interacts with internal/terminal FLAG-derived sequences79; a rabbit polyclonal anti-GFP antibody80; a murine mAb specific for GAPDH (6C5, SC-32233; Santa Cruz); rabbit polyclonal antibody against Torsin 3A protein (AV33117; Sigma-Aldrich); a sheep polyclonal antibody to the Notch1 C-terminal domain (aa 2428–2556) (AF3647; R&D Systems); a rabbit mAb specifc for the Notch1 NICD (V1744) (D3B8; Cell Signalling). The latter antibody detects endogenous levels of the NICD only when released by cleavage between Gly1753 and Val1754, the antibody not recognizing full-length Notch1 or Notch1 cleaved at other positions. Anti-γ-tubulin was obtained from Sigma-Aldrich (T5326).
Plasmid clones
Addgene: ITGB2 (Integrin-β2 mYFP; 8638), DDB1 (pcDNA3-FLAG-DDB1; 19918), P4HB (hPDI1_18-508_WT_pFLAG-CMV1; 31382), CAD (pcDNA3.1-HisFlag-CAD; 31615), ADAM17 (pRK5F-TACE; 31713), VIF (pGEX-4T-1-p19-T7-Vif; 46313). OriGene: TAP2 (pCMV6-Entry-TAP2-Myc-DDK; RC239305), AMBRA1 (pCMV6-Entry-AMBRA1-Myc-DDK; RC206255), TOR3A (pCMV6-AC-TOR3A. untagged; SC320221, pCMV6-AC-tGFP-TOR3A; RG200896). Clontech: pEGFP-N1 (6085-1). Other: A tagged expression construct for a gp140 trimeric derivative of HIV-1 Env was generously supplied by the Michel Nussenzweig laboratory81,82 (CCR5-tropic YU-2 Env; amino acids 1–683 fused to the T4 phage trimerization domain)83.
Immunoblotting analyses
Protein samples were resolved using 4–12% or 10% NuPAGE Bis-Tris gels with NuPAGE MOPS SDS running buffer (Novex, Life Technologies), transferred to polyvinylidene difluoride (PVDF) membranes and blocked in 5% milk. Membranes were incubated with primary antibody diluted in phosphate buffered saline (PBS) with 5% milk and 0.2% Tween 20 at 4 °C overnight or 1 h at room temperature. Primary antibodies were as follows: 1 μg ml−1 mouse anti-FLAG mAb M2 (Sigma) for 1xFLAG and 3xFLAG tagged viral protein expression; 1:1,000 monoclonal anti-Vif (#316)78. Proteins were detected with the ECL Plus Chemiluminescent System (Amersham Biosciences), Immobilon Western HRP Chemiluminescent Substrate (Millipore) or SuperSignal West Pico Chemiluminescence System (Thermo Scientific).
Endoglycosidase analysis
The 3xFLAG-tagged Env protein was immunopurified from CEM cells infected under continuous culture conditions and eluted by the addition of 3xFLAG peptide. The eluate was digested at 37 °C for 1 h with recombinant N-glycosidase F (PNGase F) or endoglycosidase H (endo H) (New England Biolabs) or left untreated. Western blot analysis was carried out using antibodies specific for gp41, gp120 or the FLAG epitope.
Native polyacrylamide gel electrophoresis (PAGE) immunoblotting
A modified BN-PAGE protocol59 was used to determine the oligomeric status of the immunoisolated Env protein recovered under native conditions (elution with 3xFLAG peptide) from infected CEM cells. Briefly, the eluate, various Env-VLPs and soluble JR-FL gp120 (Progenics) were incubated in an equal volume of solubilization buffer (0.12% Triton X-100, 1 mM EDTA/1.5 M aminocaproic acid) containing protease inhibitor cocktail (Sigma). An equal volume of 2× sample buffer containing 150 mM MOPS, 100 mM Tris-HCl (pH 7.7), 40% glycerol and 0.1% Coomassie blue was then added and samples were loaded onto a 4–12% Bis-Tris native-PAGE gel (Invitrogen). Samples were electrophoresed at 4 °C for 3 h at 100 V with 50 mM MOPS/50 mM Tris (pH 7.7) containing 0.002% Coomassie blue as cathode buffer and the same buffer without Coomassie blue as the anode buffer. The gel was then transferred onto PVDF membrane. Excess Coomassie blue dye was removed after blotting by washing with 30% methanol/10% acetic acid then 100% methanol. The blot was probed using 1 μg ml−1 each of human monoclonal antibodies: 2G12, b12 and 447-52D (anti-gp120 cocktail) and 20 μg ml−1 each of 4E10 and 2F5 (anti-gp41 cocktail). Goat anti-human alkaline phosphatase conjugates were used to detect the primary antibodies at 1:3,000 (Jackson).
Immunofluorescent microscopy
The adherent cell lines HeLa-CD4+-CXCR4+, HOS-pBABE-puro, HOS-CD4-Fusin, 293T and 293T-APOBEC3G-HA cells were plated on glass coverslips treated with either poly-L-lysine (0.01% solution, Sigma) or 200 g ml−1 type I collagen (BD Biosciences) or plated onto pre-treated collagen glass bottom tissue culture plates (MetTek) one day before infection. Infected suspension cultures of CEM cells were concentrated and spun onto slides containing mounting media (Vector Laboratories) and fixed before staining. For continuous infection, 10 ng p24 of 3xFLAG-tagged or the untagged control virus was incubated with target cells for two days. For time course immunofluorescence, target cells were spinoculated with 1 μg p24 of 3xFLAG-tagged or the untagged control parental virus at 700g for 1.5 h at 16 °C. After 1 h incubation at 37 °C, cells were washed once and fresh media were added to remove input viruses, and cells were incubated at 37 °C for an additional 2 and 22 h infection, respectively. For single-step infections, viruses were VSV-G envelope pseudotyped and used to infect HOS-pBABE-puro, 293T or 293T-APOBEC3G-HA cells and examined 72 h.p.i. for the distribution of signal. Cells were fixed with freshly made 3% formaldehyde/PBS for 30 min, permeabilized with 0.2% Triton in 3% formaldehyde/PBS for 10 min and blocked with 1% goat serum/PBS for 30 min. The cells were then incubated with mouse anti-FLAG M2 mAb (Sigma) and then stained with Alexa Fluor 594-coupled goat anti-mouse antibody (Molecular Probes). Nuclei were stained with bis-benzimide (Sigma, Hoechst no. 33342) in PBS. F-actin was labelled using Alexa Fluor 488 Phalloidin (Invitrogen), and ER and Golgi were labelled using BACMAM 2.0 CellLight ER-GFP and Golgi-GFP (Invitrogen). Images were collected with an inverted DeltaVision optical sectioning microscope (Applied Precision). The images were processed and deconvolved through 15 iterations using the DeltaVision deconvolution software (Applied Precision).
Cell culture, viral production and infection
TOR1AIP2 (LULL1) CRISPR/Cas9 knockout HeLa cell line was provided by C. Zhao and C. Schlieker (Yale University). Other cell lines were obtained from the NIH AIDS Reagent Repository, the Center for AIDS Reagents. Adherent cell lines, HeLa-CD4+-CXCR4/CCR5-β-gal-Luc (TZM-bl) (J.C. Kappes, X. Wu and Tranzyme), HOS-pBABE-puro (N. Landau), HOS-CD4-Fusin (N. Landau), 293T and 293-APOBEC3G-HA (X.F. Yu) and cells were maintained in DMEM supplemented with 10% FBS plus pen/strep. Suspension T cell lines, CEM (CEM-SS; P. Nara), MT-2 and MT-4 (D. Richman) were cultured in RPMI supplemented with 10% FBS and Pen/Strep. To produce virus, subconfluent monolayers of 293T cells in T-25 culture flasks were transfected with 3 μg proviral DNA using 25 μg polyethylenimine (PEI) (Polysciences). For the R7/3-PmeI vif library and R7/3 clones with 1x or 3xFLAG insertion into vif, 293-APOBEC3G-HA cells were used to produce the viral stock. This cell line is derived from 293T cells stably transfected with an expression vector encoding HA-tagged, human APOBEC3G49. Viral supernatants were collected 42 h post-transfection (h.p.tx.) by centrifugation at 1,000 r.p.m. for 5 min and filtered (0.45 μm, Millipore). Viral production was determined by p24 enzyme-linked immunosorbent assay (ELISA) performed using the HIV-1 p24 Antigen EIA kit (Beckman Coulter) or by a modified product-enhanced reverse transcriptase (PERT) assay84. For selection of the subset of replication-competent viruses containing PmeI inserts, viral infections were initiated with the equivalent of 10 ng of p24 capsid antigen, the target cells were 1 × 106 CEM (Env tagged) or MT-2 (Vif-tagged) cells. Cell density was maintained at ~0.5 × 106 cells per ml with a split ratio of 1:3. VLPs were produced by transiently transfecting 293T cells with a pCAGGS Env-expressing plasmid and a sub-genomic plasmid pNL4-3.Luc.R-E- using PEI. Two dptx, supernatants were collected and pre-cleared by low-speed centrifugation, before pelleting at 50,000g in a Sorvall SS34 rotor. To remove residual medium, VLP pellets were diluted with 1 ml PBS, then re-centrifuged in a microfuge at 15,000 r.p.m. and resuspended in PBS at 1,000× the original concentration. Standard cell lines were employed, each with the expected phenotype (that is, infectivity, reporter activity and morphology) although none have been directly tested for mycoplasma.
Profiling of the site of PmeI insertion over time during biological selection for viral survivors within the Env (C1–V3) library
Libraries of viral supernatants with insertions in env produced by transfection were incubated with 40 U ml−1 Turbo DNase (Ambion) at 37 °C for 1 h prior to infections. Three biological replicates of the infection were performed. Viral DNA was recovered using QIAamp DNA Blood Mini Kit (Qiagen) over 1–12 d.p.i. The recovered DNA was digested with DpnI to degrade any possible input, GA−CH3 TC-methylated, transfected plasmid DNA originating from growth in Escherichia coli. PCR (primers: 5′-CATCAGATGCTAAAGCATATG-3′, 5′-GTTCTCTTAATTTGCTAGC-3′) was used to amplify (Expand High Fidelity polymerase, Roche) tagged, viral DNAs from cell culture. The resulting PCR fragment was TA-cloned into pGEM-T Easy (Promega). The ligation products were transformed into DH10B and more than 105 colonies were pooled then purified using QIAprep Spin Miniprep Kit (Qiagen), restricted with NdeI/NheI±PmeI and analysed by 8% TBE PAGE (Invitrogen).
Deep sequencing
Purified DNA was treated with the restriction enzyme DpnI to digest any residual proviral plasmid constructs. DNA amplification was conducted via a nested PCR strategy with the following primers:
HIV forward 1 5′-GGCGTTACTCGACAGAGGAG-3′
HIV reverse 1 5′-CCAAGAACCCAAGGAACAAA-3′
HIV forward 2 5′-GCAGAAGACAGTGGCAATGA-3′
HIV reverse 2 5′-GGGAGGGTGATTGTGTCACT-3′
The PCR product was then gel-purified through a 0.8% agarose gel using a silica column (Qiagen QiaQuik). For Ion Torrent sequencing, 500 ng of PCR product DNA from each of the samples was submitted for sequencing on two 316 chips (Ion Torrent). Each sample was fragmented individually and ligated to a MID-coded Ion Torrent primer/adaptor before sequencing.
Assessment of foreign insertion stability within the HIV-1 genome over time
To investigate the individual stabilities of the PmeI insert and the 3xFLAG tag over an extended period of 12 days, two procedures were undertaken: (1) western blot analysis of infections initiated with viruses encoding a PmeI or 3xFLAG insertion; (2) PCR amplification and restriction endonuclease scoring of the targeted viral region. The total DNA was purified from infected MT-2 (Vif-PmeI, Vif-3xF) or CEM (Env-PmeI, Env-3xF) cells, the vif or env regions PCR amplified as described above and compared to the untagged virus grown in the same cell lines. After purification, the PCR fragments were digested with PmeI restriction enzyme to assess the integrity of this restriction site for viruses with this type of insertion or BlpI, a restriction site contained within the 3xFLAG tag. The sizes of the products of these digestions were displayed by agarose gel electrophoresis. Multiple biological replicates were performed for each procedure.
Characteristics of tagged viral infections
For standard growth curves, 1 × 106 CEM cells in T-25 flasks were infected with either Env-3xF or the untagged, parental control virus (R7/3), each infection normalized for the amount of p24 antigen (10 ng). Input virus was removed by washing twice with PBS following an overnight period of incubation to establish infection. Culture supernatants (200 μl) were collected daily through 12 d.p.i. The rate of viral growth was quantified by measuring the concentration of p24 in the culture supernatants, as determined by an HIV-1 p24 antigen EIA kit (Beckman Coulter), by a modified product-enhanced reverse transcriptase (PERT) assay84 or by measuring the amount of secreted deep sea shrimp nanoluciferase85 (sNLuc, Promega, Supplementary Fig. 9a,c). Infection was performed independently, three times, as biological replicates. PERT and p24 assays were performed with duplicate technical repeats. Infection in MT-2 cells with Vif-3xF or WT R7/3 produced from 293-APOBEC3G-HA were similarly performed. For single-step infections, lymphoid suspension cell lines, CEM and MT-4 were infected with eGFP indicator viral derivatives and scored for percent infectivity by flow cytometry. The epithelial indicator cell line HeLa (TZM-bl) was also used for these analyses, with case measurement of chemiluminescent β-galactosidase activity (Galacto-Star, Life Technologies) assays for the extent of successful infection. To test for the equivalency of infection using a super-pseudotyped protocol, eGFP derivatives of the WT, Env-PmeI or Env-3xF viruses were prepared by co-transfection of 293T cells with each of the respective replication-competent proviral DNA clones (1.5 μg) with that of a VSV-G Env expression vector (0.5 μg), the cells were washed and fed 24 h later, and the viral stocks were collected at 48 h and used to infect a 0.5 ml culture of 5 × 105 CEM cells in a 12-well plate format using duplicate 1:2 serial dilutions of virus starting from a viral inoculum of 10 ng p24. Multiple cycles of infection were prevented by the addition of an RT inhibitor 18 h.p.i. Cells were fixed two days later at 4 °C overnight in a final paraformaldehyde (PFA) concentration of 2%. The infectivity of proviral-encoded env gene VSV-G Env super-pseudotyped eGFP stocks across several MOI (multiplicity of infection) during single-step infection were measured by analytical cell sorting for the percentage of cells in the population transduced for green fluorescence. Evidence for highly efficient cell-to-cell transmission of the Env-3xF virus across several cycles of viral outgrowth was obtained as follows. Super-pseudotyped viral derivatives were prepared and equilibrated for RT activity and p24 content as described above using derivatives that encode sNLuc. Thus, instead of the eGFP, sNLuc is capable of the detection of viral expansion over time regardless of cell death or syncytia formation. CEM cells were inoculated with a set VSV-G super-pseudotyped sNLuc viruses such that approximately 1% of the cells were infected. After the first round of infection, any transmission to other cells is entirely dependent on the proviral-encoded HIV-1 Env. When graphed (Fig. 1d and Supplementary Fig. 10b), the growth kinetics of the two viruses nearly overlap, indicating efficient transmission of the 3xFLAG-tagged virus compared to the WT virus. Multiple biological replicates were performed. For viral neutralization studies and mode of viral transmission and cell-to-cell infections, CEM cells (3 × 105) were plated in 1.5 ml media in a 12-well format. Enough VSV-G super-pseudotyped eGFP virus of each stock was added to infect 1% of the cells of the population. The following day (2 d.p.i.), the cells were washed in PBS, resuspended in 1 ml medium, and 50 μl of the CEM cell suspension was plated in a flat-bottomed, 96-well format. Anti-FLAG M2 mAb, 2F5 or 246-D was adjusted to 40 μg ml−1 in medium, and 50 μl was added in as a biological duplicate to each infected well. The other rows of cells were treated with a 1:5 serial dilution of each different antibody and incubated at 37 °C for an additional 45 min; 2 × 104 uninfected CEM cells were added and mixed in each well to maintain a final volume of 200 μl per well. After an additional day of incubation (3 d.p.i.), an additional 2 × 104 uninfected CEM cells were again added. At 5 d.p.i. an equivalent amount of antibody was added as before and at 6 d.p.i. infection, the cells were fixed and quantified by flow cytometry scoring for green fluorescence.
Immunoaffinity isolation from infection
Human T cell lines were infected with 5 μg p24 virus (CEM, Env-3xF; MT-2, Vif-3xF). Cultures were infected by spinoculation at 700g at 16 °C for 1 h and then incubated at 37 °C to initiate infection. Cultures were expanded 1:3 when required in T-175 flasks. Additional uninfected CEM or MT-2 cells were added to each flask when they were ‘blossoming’ with microscopic syncytia formation. Infected cells were collected at 6–8 d.p.i. by centrifugation at 200g for 10 min to yield ~1 g of cell mass. At this point, the protocol and the lysis buffer extraction composition, individually tailored and optimized based on the specific biochemical characteristics of the tagged viral protein, diverged for the two 3xFLAG-tagged viral proteins. For Env-3xF infections, after washing the cells three times with PBS, the cells were resuspended in 300 μl per gram of cells in freezing buffer: 20 mM HEPES (pH 7.5) containing 1.2% polyvinylpyrrolidone (wt/vol), complete protease inhibitor cocktail (Roche) and 2 μM integrase inhibitor L-870812 (Merck)86. The cell mixture (~1 g) was then snap frozen in liquid nitrogen and combined with 2 ml of previously prepared and similarly formed pellets of lysis buffer (20 mM K-HEPES (pH 7.4), 110 mM KOAc, 2 mM MgCl2, 0.1% Tween 20, 1% Triton, 0.5% deoxycholate, 300 mM NaCl, 50 units/ml DNase I (Roche) supplemented with protease inhibitor (Roche) and 1 μM HIV-1 integrase inhibitor, L-870812 (Merck)). The combined frozen pellets were maintained at −196 °C and loaded into a precooled, 25 ml stainless steel jar containing a single 20 mm stainless steel grinding ball. A liquid nitrogen-adapted CryoMill (Retch) was used for efficient cellular fragmentation by cryogenic grinding80. Grinding was performed with 12 cycles at 25 Hz for 4 min each. The ground cellular material in freezing/lysis buffer was thawed and immediately mixed with additional lysis buffer (10 ml g−1 of original cells). For Vif-3xF infections, ~1 g of infected MT-2 cells, which require Vif expression for productive HIV-1 infection, was collected as described above. Lysis buffer (50 mM Tris-HCl (pH 7.4), 300 mM NaCl, 0.5% Triton, 50 units/ml DNase I (Roche) supplemented with protease inhibitor (Roche), 1 μM L-870812 (Merck) and 50 μM of the proteasome inhibitor, MG132 (Sigma)) was added at room temperature to frozen cell pellets at 10 ml g−1 of cells. For both extraction types described above, the cell lysates were slowly rotated at 4 °C for 10 min, vortexed vigorously three times for 10 s before centrifugation (10 min at 3,600g at 4 °C). A total of 8 mg M-270 Epoxy Dynabeads (Invitrogen) pre-conjugated with M2 anti-FLAG mAb (Sigma) at a ratio of 10 μg of antibody per mg of beads (as previously described80) was used for immunoaffinity isolations. Supernatants were incubated with beads at 4 °C for 1 h, and after six washes with the respective lysis buffer, one of two procedures was used for elution of the tagged viral protein and its associated complexes: (1) basic elution of the protein complexes from the beads with the addition of 700 μl of a freshly made 0.5 N NH4OH, 0.5 mM EDTA solution shaken for 20 min at room temperature, with the eluate frozen in liquid nitrogen and dried using vacuum centrifugation and the lyophilized samples resuspended in 30 μl 1x LDS– or SDS–PAGE sample buffer with reducing agent (Invitrogen); (2) native elution of 3xFLAG-tagged Env or -tagged Vif protein recovered by incubation of the reacted Dynabeads with 30 μl of a 5 mg ml−1 solution of the 3xFLAG peptide (Sigma) in TBS for 20 min at room temperature. All samples for mass spectrometry were then heated with agitation at 70 °C for 10 min, alkylated with 100 mM iodoacetamide at room temperature in the dark for 30 min, resolved by 1D gel electrophoresis on a 4–12% NuPAGE Novex Bis-Tris gel (Invitrogen) and stained with SimplyBlue SafeStain (Invitrogen).
Scoring for stable interaction between purified Env-3xF protein and anti-Env mAbs or soluble CD4
Magnetic beads (0.6 mg) were conjugated with VRC07, PGT121, PGT145, 10E8, VRC01, 4E10, PG9, PG16, 2G12 or sCD4 as previously described80. After blocking with blocking buffer (5% milk in PBS/0.05% Tween) for 30 min, each batch of conjugated beads was incubated in blocking buffer for 1 h with 50 μl of a 10 μg ml−1 solution of Env3xF protein eluted from fulminant infection as described above. After six PBS/0.05% Tween washes, samples were eluted in 1x LDS buffer by incubation at 95 °C for 10 min, and the eluted material was subjected to western blot analysis as probed with a 1:10,000 dilution of anti-FLAG mouse mAb (M2).
Cell culture for I-DIRT and reverse I-DIRT analyses
Sample preparation for I-DIRT17 analyses used cells infected with the untagged, parental control virus80, R7/3, grown in Lys/Arg deficient RPMI-1640, 10% dialysed FBS (Gibco) and Pen/Strep (Pierce) supplemented with heavy isotope 13C6 labelled Lys and Arg (Cambridge Isotopes). Cells were cultured in the heavy labelled media for more than six cell doublings before infection to reduce the level of the natural isotope (incorporation of the 13C6 label >99%). Cells infected with the 3xFLAG-tagged viruses were cultured in media supplemented with Lys and Arg with natural isotopic abundances (i.e., predominantly 12C6). Infections were collected as described above. Equal amounts of light and heavy frozen cell pellets (0.8–1.0 g each) were mixed before grinding, lysis and immunoaffinity isolation. For reverse I-DIRT, the isotopic characteristics of the RPMI media were switched—cells infected with the control virus were grown in the natural carbon isotope while cells infected with a 3xFLAG-tagged virus were cultured in media supplemented with 13C6 labelled Arg and Lys.
Mass spectrometry (MS)-based proteomic analysis
Following SDS–PAGE separation, in-gel digestion with trypsin was performed as previously described87. Briefly, each lane was divided into ten fractions and gel slices were cut into ~2 mm3 cubes. Gel cubes were destained in 50% acetonitrile (ACN) in 50 mM ammonium bicarbonate (ABC), dehydrated in 100% acetonitrile and incubated overnight at 37 °C with 12.5 ng μl−1 trypsin in 50 mM ABC. The reaction was quenched and peptides were extracted with 0.5% formic acid (FA) for 4 h, followed by a second extraction for 2 h with 50% ACN in 0.5% FA. Extracted peptides were desalted by C18-StageTips, concentrated by vacuum centrifugation to ~1 μl and suspended in 10 μl of 1% FA. Desalted peptides (4 μl) were analysed by direct injection onto a C18 analytical column (Acclaim PepMap RSLC, 1.8 μm; 75 μm × 25 cm) using a Dionex Ultimate 3000 nanoLC coupled online to an LTQ Orbitrap Velos mass spectrometer (Thermofisher Scientific). Peptide separation and MS detection were performed over 90 min at 250 nl min−1 using a discontinuous ACN gradient from a 4 to 16% mobile phase B over 60 min, followed by a 16 to 40% mobile phase B over 30 min (mobile phase A, 0.1% FA; mobile phase B, 97% ACN in 0.1% FA). The mass spectrometer was operated in data-dependent acquisition mode with dynamic exclusion enabled and preview scan mode disabled. One acquisition cycle comprised a single full-scan mass spectrum (m/z = 350–1700) in the orbitrap (resolution = 60,000 at m/z = 400), followed by collision-induced dissociation (CID) fragmentation in the linear ion trap of the top 15 most intense precursor ions. The orbitrap full scan target value was 1 × 106 (maximum ion time = 300 ms), and the ion trap target value was 1 × 104 (maximum ion time = 125 ms). CID fragmentation was performed with an isolation width of 2.0 Da, a normalized collision energy of 30 and an activation time of 10 ms. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE88 partner repository with data set identifier PXD003762.
MS data processing and precursor ion quantitation
MS/MS spectra were extracted from Thermo instrument (raw) files derived from a single gel lane and searched by Proteome Discoverer/SEQUEST (version 1.3, Thermo Fisher Scientific) against the human subset of the UniProt SwissProt protein sequence database (release 2010–11) appended with common contaminants and HIV-1 polyprotein and mature protein sequences (20,349 sequences). SEQUEST search parameters were as follows: full trypsin specificity with two missed cleavages, precursor and fragment tolerances of 10 ppm and 0.5 Da, fixed modification of carbamidomethylation of cysteine and variable modifications for methionine oxidation, 13C6-lysine and arginine, deamidation of asparagine, and phosphorylation of serine, threonine and tyrosine. Putative SEQUEST peptide-spectrum matches (PSMs) were scored by the Percolator algorithm89 to calculate individual (PEP) and global error probabilities (q-values) by a semi-supervised approach using reverse database searches. PSMs were filtered to achieve an estimated 1% peptide false discovery rate (q-value < 0.01) and then PSMs were assembled into protein groups requiring a minimum of two unique peptides and using the ‘strict maximum parsimony principle’ option. Isotope-based relative quantification was performed using the Proteome Discoverer precursor ions quantification workflow. Briefly, the ‘event detector’ module detected and filtered precursor (MS1) isotopic envelopes (mass precision = 2 ppm, signal-to-noise ratio = 5, RT tolerance = 0.2 min and single-peak/missing channels = 1). Each profile was m/z and RT-matched with corresponding PSMs and the cognate light and heavy isotope pairs were subjected to intensity-based summation. The following precursor ion parameters were used: Replace missing quan values with minimum intensity, True; Use single-peak quan channels, True; Reject all quan values if not all quan channels are present, False; Maximum allowed fold change, 50; Use ratios above maximum allowed fold change, True. After automated calculation of light and heavy peptide abundances, an additional quantitative peptide-level filter was applied. Quantified peptides with light (forward) or heavy (reverse) quantitative values that were less than ½ (Vif I-DIRT) or ¼ (Env I-DIRT) of the median value (across all fractions) were excluded. Isotope ratios were calculated as light/total for forward I-DIRT experiments or heavy/total for reverse I-DIRT, Vif isotope exchange and Env SILAC experiments. Peptides that corresponded to unique peptide sequences, that is, were not shared in other protein groups, were used to calculate the median protein isotope ratio (minimum of three quantified PSMs) and coefficient of variation. Protein groups, accessions numbers and quantitative values were exported to Excel for downstream statistical and functional analysis.
Identification of viral–host interactomes via differential isotopic labeling
CEM cells infected with tagged Env virus (performed identically for tagged Vif virus in the context of the restrictive MT-2 cell line) or uninfected cells were each passaged for one week in standard media (light, L) or media pre-equilibrated with isotopic label (heavy, H) such that, at steady state, >99% of the host proteome’s 12C6 lysine (K) and arginine (R) residues had been substituted for their 13C6 substituted, 6-Da-distinguishing counterparts. Light CEM (or MT-2) cells were then infected with the Env− or Vif− 3xFLAG-tagged virus, expanded in suspension culture (500 ml), and the terminal infected cell pellets were flash frozen in liquid nitrogen. At the same time, the companion heavy CEM (or MT-2) culture was infected with the untagged WT virus and expanded and processed as above. The frozen cell pellets from the two types of infection were mixed (1:1) before cellular disruption via mechanical cryogenic lysis.
For additional biological and technical replicates, we replicated the experiments but switched the light and heavy labels (‘reverse’ I-DIRT), increasing the robustness of this analysis and discriminating against possible exogenous contaminants. Thus, proteins from Env and Vif forward and reverse I-DIRT pull downs were run on an acrylamide gel. Gel lanes containing the proteins were excised and digested with trypsin, and the resultant peptides were subjected to liquid chromatography-tandem mass spectrometry analysis. Forward [L/(L+H)] versus reverse [H/(H+L)] I-DIRT ratios were calculated from the averages of the peptide ratios. We developed a statistical test to identify proteins not likely (<0.01% chance) to fall within the nonspecific distribution. The I-DIRT data were analysed based on the following: theoretically, IT/(IT+IUT) (where IT and IUT are the mass spectrometric intensities of peptides from the tagged and untagged strains, respectively) is 1 for specific interactors and 0.5 for nonspecific interactors. To apply the theory to experimental data, we performed simulations based on a simple model: (1) both IT and IUT are affected by normally distributed noise of the same magnitude; (2) the concentration of proteins is not precisely equal in the tagged and untagged samples. These simulated distributions can be used to estimate the probability that a measured protein is specific or nonspecific by taking the uncertainty of the measurement into account. This model explains the observed experimental distributions of specific and nonspecific proteins. Proteins with intermediate values (typically between 0.7 and 0.9) are probably specific interactors that are fast exchangers. Specific interactors are indicated in I-DIRT forward versus reverse plots in the upper right corner, while nonspecific proteins map to the lower left corner of the plot. We also plotted histograms of the average (forward and reverse) I-DIRT ratios versus the binned number of proteins exhibiting those averages.
Competitive isotopic exchange assay for detection of Vif–host dynamic interaction
The methodologies for the differential metabolic labelling of MT-2 cells infected with either Vif-3xF (heavy) or the untagged comparator (light) virus were the same as that used in the reverse I-DIRT experiments outlined above. In this assay, ~2.5 g of cells from each infection were collected and separately lysed in Vif lysis buffer. Anti-FLAG M2 mAb conjugated magnetic Dynabeads (12 mg) were then used to affinity purify Vif complexes from the heavy labelled Vif-3xF infected cells. After removing the heavy lysate from the beads, an equal volume of the light labelled lysate prepared from the untagged control infection was added to the beads at 4 °C. The mixture was then divided into three equivalent aliquots, which were then incubated at 4 °C for 5, 15 or 60 min. At each time point, the beads were washed six times with 1 ml Vif lysis buffer and then eluted with 25 ml of a 5 mg ml−1 3xFLAG peptide solution in TBS.
Evaluating the effects of TOR1AIP2 (LULL1) gene expression on viral infectivity
Super-pseudotyped HIV-1 stocks were generated by co-transfecting 1.5 μg of WT proviral reporter plasmid (R11-eGFP) with 0.5 μg of the pCI-VSV-G envelope expression vector into 293T cells and collecting cell free stocks 2 days post-transfection (d.p.tx.). This viral stock was used to infect ~50% confluent HeLa cell lines (parental derivative HeLa and LULL1 CRISPR/Cas9 KO HeLa)22 in a six-well plate format, with each cell line infected in duplicate as biological repeats. On the following day, viral input stock was removed by extensive washes with PBS, and the cultures were replenished with fresh media and viral supernatants from each cell type collected two days later. Stocks were normalized for reverse transcriptase activity and virus infectivity was gauged by infection of a T cell indicator cell line (MT-4-GFP (MT-4*) cells)90. Infectivity was restricted to one-step dynamics by the addition of a reverse transcriptase inhibitor ~18 h.p.i. Fluorescence activated cell sorting (FACS) 48 h.p.i. was used to quantify cells that were productively infected and expressing the transduced GFP fluorescent marker.
Potential effects of Env and viral infection on steady-state levels and modification of the Notch1 protein
Mid-log-phase CEM suspension cells were infected with HIV-1-GFP reporter viruses that were intact or defective for viral Env function. The infectivity of both viruses was greatly enhanced by pseudotyping virions with the VSV-G envelope glycoprotein during generation of the stocks in 293T cells. CEM cells were infected in parallel with equivalent titres of each stock, and reverse transcriptase inhibitor was added to both cultures 24 h.p.i. in order to prevent spread of the WT virus. GFP-positive and -negative cells were separated by FACS, and the isolated cells were lysed in radioimmunoprecipitation assay buffer supplemented with complete protease inhibitor cocktail (Roche), and sonicated and resolved by 4–12% NuPAGE Bis-Tris protein gels (ThermoFisher). Western blotting was performed, probing with antibodies as indicated.
Small-scale immunoprecipitation and construction of pEGFP-VIF, p3xF-VIF and pEGFP-TOR3A and pEGFP-TOR3A (E236Q)
For Env interactor immunoprecipitations (IPs), 150 mm dishes of 293T cells were transfected (Lipofectamine 2000) with the indicated plasmids for 24 h. For Vif IPs, transfected cells were incubated for 20 h, vehicle (DMSO) or MG132 was added (at 10 μM), and cells were incubated for an additional 10 h. Cells were collected and pelleted. Cell pellets stored at −80 °C were thawed, resuspended in 1 ml lysis buffer (20 mM HEPES (pH 7.4), 110 mM KOAc, 300 mM NaCl, 1% Triton) supplemented with protease inhibitor (Roche), sequentially disrupted with 23 and 26 gauge needles, incubated on ice for 10 min and centrifuged for 20 min at 3,600g at 4 °C. Supernatants were incubated with 0.2 mg Dynabeads conjugated to M2 anti-FLAG mAb (DDB1, P4HB, CAD, ADAM17, TAP2, AMBRA1) or rabbit polyclonal anti-GFP80 (EGFP, ITGB2, TOR3A, TOR3A (E236Q)) antibodies for 1 h at 4 °C with rotation, washed three times with lysis buffer and eluted with 750 μl 1 N NaOH 5 mM EDTA for 7–15 h. Eluates were lyophilized and resuspended in 30 µl SDS–PAGE loading buffer with reducing agent. Samples were electrophoresed on 4–12% NuPAGE Bis-Tris gels (Novex) and silver stained (Thermo-Scientific-Pierce) or transferred to PVDF membranes and blocked in 5% milk. Membranes were incubated with primary antibody diluted in Tris-buffered saline (TBS-T) with 5% milk and 0.1% Tween 20 at 4 °C overnight or 1 h at room temperature. Primary antibodies were as follows: 1:500 mouse anti-FLAG mAb M2 (to detect DDB1-FLAG), 1:1,000 monoclonal anti-GAPDH, 1:250 goat anti-gp120 (Env). Proteins were detected with the SuperSignal West Pico Chemiluminescence System.
To obtain an expression vector for GFP-tagged Vif (used for the co-IP studies with FLAG-tagged interactor constructs), the Vif coding region from a pGEX Vif vector (Addgene #46313) was amplified by PCR and cloned into pEGFP-N1, yielding pEGFP–VIF. To obtain a FLAG-tagged Vif expression construct (used for the co-IP studies with the GFP-tagged Vif interactor constructs), the EGFP coding region of pEGFP-VIF was excised and replaced with 3xFLAG to create p3xF-VIF. The TOR3A coding region was amplified by PCR from pCMV6-AC-tGFP-TOR3A (Origene #RG200896) and cloned into pEGFP-N1. A mutant expression clone specifying an E > Q point mutation at position 236 (LFIFDE > LFIFDQ) in the Walker B box of the TOR3A ATPase domain91–93 was also constructed using the QuikChange mutagenesis kit (Agilent). The ‘empty’ vector is pcDNA3.1(+). Clones were sequenced to ensure correctness. Further cloning details are available from the authors on request.
Status of Notch1 (NICD) phosphorylation
Whole cell extracts (15 μg) from each of the test conditions were incubated with ten units of calf intestinal alkaline phosphatase (CIP, NEB) for 1 h at 37 °C in the presence or absence of general phosphatase inhibitors (20 μM NaF and 1 μM sodium orthovanadate) in 1x CutSmart buffer (NEB). Proteins were examined by western immunoblot with antibodies specific for the γ-secretase-processed Notch1 NICD domain (V1744).
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health: R01AI047054, R21AI065321, R01AI081615 and R21AI097233 (to M.A.M.), P41GM103314 (to B.T.C. and D.F.), U54GM103511 (to M.P.R., B.T.C., M.A.M. and D.F.), P41GM109824 (to M.P.R. and B.T.C.), R01AI093278 and R33AI084714 (to J.M.B.) and DP1DA026192 and R21AI102187 (to I.M.C.). The authors thank V. Sahi (Aaron Diamond AIDS Research Center) for all flow cytometry employed in this study, C. Zhao and C. Schlieker (Yale University) for the TOR1AIP2 (LULL1) CRISPR/Cas9 knockout HeLa cell line, P. Nahirney (Rockefeller EM Resource Center) for imaging, M. Nussenzweig (Rockefeller University) for a trimerized derivative of YU-2 gp140 Env, K. Jacobs, J. Boland and D. Roberson of the NCI Core Genotyping Facility for ion torrent sequencing, and contributors to the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, for specified reagents described in the Supplementary Methods.
Footnotes
Author contributions
Y.L. and M.A.M. designed and developed the genetic strategy used to select epitope-tagged, replication-competent viruses. I.M.C., E.Y.J., M.P.R. and B.T.C. designed and developed the proteomic and MS approaches described. Y.L. generated all libraries and performed the genetic, virological, immunofluorescent, electron microscopic studies and immunoisolations. T.M.G. performed the mass spectrometric analyses. D.F., T.M.G., E.Y.J. and Y.L. compiled and evaluated the MS data. S.K., D.F., E.Y.J. and Y.L. analysed the deep sequencing results of the mutagenic landscape. Y.L. performed molecular characterization of tagged viral clones. T.T. and J.M.B. determined the oligomeric status of the HIV-1 tagged Env pullouts. Y.L. and K.D.M. performed the neutralization and super-pseudotyping experiments, and assayed Notch1 processing and phosphorylation, while E.Y.J. conducted Env reciprocal immunoprecipitations and Vif co-transfection experiments. Y.L., E.Y.J., T.M.G., T.T., J.M.B., M.P.R., B.T.C. and M.A.M. wrote the manuscript, with assistance from all authors.
Additional information
Supplementary information is available online.
Competing interests
The authors declare no competing financial interests.
References
- 1.Brass AL, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 2.Bushman FD, et al. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathogens. 2009;5:e1000437. doi: 10.1371/journal.ppat.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konig R, et al. Global analysis of host–pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135:49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou H, et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008;4:495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Luo Y, Muesing MA. Mass spectrometry-based proteomic approaches for discovery of HIV–host interactions. Future Virol. 2014;9:979–992. doi: 10.2217/fvl.14.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jager S, et al. Global landscape of HIV–human protein complexes. Nature. 2012;481:365–370. doi: 10.1038/nature10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Julien JP, et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science. 2013;342:1477–1483. doi: 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo Y, et al. Structural basis for hijacking CBF-β and CUL5 E3 ligase complex by HIV-1 Vif. Nature. 2014;505:229–233. doi: 10.1038/nature12884. [DOI] [PubMed] [Google Scholar]
- 9.Pancera M, et al. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature. 2014;514:455–461. doi: 10.1038/nature13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Taeye SW, et al. Immunogenicity of stabilized HIV-1 envelope trimers with reduced exposure of non-neutralizing epitopes. Cell. 2015;163:1702–1715. doi: 10.1016/j.cell.2015.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guttman M, et al. CD4-induced activation in a soluble HIV-1 Env trimer. Structure. 2014;22:974–984. doi: 10.1016/j.str.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laird ME, Desrosiers RC. Infectivity and neutralization of simian immunodeficiency virus with FLAG epitope insertion in gp120 variable loops. J Virol. 2007;81:10838–10848. doi: 10.1128/JVI.00831-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pantophlet R, Wang M, Aguilar-Sino RO, Burton DR. The human immunodeficiency virus type 1 envelope spike of primary viruses can suppress antibody access to variable regions. J Virol. 2009;83:1649–1659. doi: 10.1128/JVI.02046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang X, Lipchina I, Cocklin S, Chaiken I, Sodroski J. Antibody binding is a dominant determinant of the efficiency of human immunodeficiency virus type 1 neutralization. J Virol. 2006;80:11404–11408. doi: 10.1128/JVI.01102-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergeron JR, et al. The SOCS-box of HIV-1 Vif interacts with ElonginBC by induced-folding to recruit its Cul5-containing ubiquitin ligase complex. PLoS Pathogens. 2010;6:e1000925. doi: 10.1371/journal.ppat.1000925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Etienne L, Hahn BH, Sharp PM, Matsen FA, Emerman M. Gene loss and adaptation to hominids underlie the ancient origin of HIV-1. Cell Host Microbe. 2013;14:85–92. doi: 10.1016/j.chom.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernacchi S, Mercenne G, Tournaire C, Marquet R, Paillart JC. Importance of the proline-rich multimerization domain on the oligomerization and nucleic acid binding properties of HIV-1 Vif. Nucleic Acids Res. 2011;39:2404–2415. doi: 10.1093/nar/gkq979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wichroski MJ, Robb GB, Rana TM. Human retroviral host restriction factors APOBEC3G and APOBEC3F localize to mRNA processing bodies. PLoS Pathogens. 2006;2:e41. doi: 10.1371/journal.ppat.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tackett AJ, et al. I-DIRT, a general method for distinguishing between specific and nonspecific protein interactions. J Proteome Res. 2005;4:1752–1756. doi: 10.1021/pr050225e. [DOI] [PubMed] [Google Scholar]
- 20.Cong Y, et al. 4.0-Å resolution cryo-EM structure of the mammalian chaperonin TRiC/CCT reveals its unique subunit arrangement. Proc Natl Acad Sci USA. 2010;107:4967–4972. doi: 10.1073/pnas.0913774107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dron M, et al. Molecular cloning of ADIR, a novel interferon responsive gene encoding a protein related to the torsins. Genomics. 2002;79:315–325. doi: 10.1006/geno.2002.6709. [DOI] [PubMed] [Google Scholar]
- 22.Turner E, Brown RS, Laudermilch E, Tsai P, Schlieker C. The torsin activator LULL1 is required for efficient growth of HSV-1. J Virol. 2015;89:8444–8452. doi: 10.1128/JVI.01143-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hansen TH, Bouvier M. MHC class I antigen presentation: learning from viral evasion strategies. Nature Rev Immunol. 2009;9:503–513. doi: 10.1038/nri2575. [DOI] [PubMed] [Google Scholar]
- 24.Collins KL, Baltimore D. HIV’s evasion of the cellular immune response. Immunol Rev. 1999;168:65–74. doi: 10.1111/j.1600-065x.1999.tb01283.x. [DOI] [PubMed] [Google Scholar]
- 25.Schulte D, et al. The HLA-ER/HLA-ER genotype affects the natural course of hepatitis C virus (HCV) infection and is associated with HLA-E-restricted recognition of an HCV-derived peptide by interferon-γ-secreting human CD8+ T cells. J Infect Dis. 2009;200:1397–1401. doi: 10.1086/605889. [DOI] [PubMed] [Google Scholar]
- 26.Abela IA, et al. Cell–cell transmission enables HIV-1 to evade inhibition by potent CD4bs directed antibodies. PLoS Pathogens. 2012;8:e1002634. doi: 10.1371/journal.ppat.1002634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sourisseau M, Sol-Foulon N, Porrot F, Blanchet F, Schwartz O. Inefficient human immunodeficiency virus replication in mobile lymphocytes. J Virol. 2007;81:1000–1012. doi: 10.1128/JVI.01629-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brandenberg OF, et al. Partial rescue of V1V2 mutant infectivity by HIV-1 cell–cell transmission supports the domain inverted question marks exceptional capacity for sequence variation. Retrovirology. 2014;11:75. doi: 10.1186/s12977-014-0075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dale BM, Alvarez RA, Chen BK. Mechanisms of enhanced HIV spread through T-cell virological synapses. Immunol Rev. 2013;251:113–124. doi: 10.1111/imr.12022. [DOI] [PubMed] [Google Scholar]
- 30.Duncan CJ, et al. High-multiplicity HIV-1 infection and neutralizing antibody evasion mediated by the macrophage–T cell virological synapse. J Virol. 2014;88:2025–2034. doi: 10.1128/JVI.03245-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malbec M, et al. Broadly neutralizing antibodies that inhibit HIV-1 cell to cell transmission. J Exp Med. 2013;210:2813–2821. doi: 10.1084/jem.20131244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jolly C, Booth NJ, Neil SJ. Cell–cell spread of human immunodeficiency virus type 1 overcomes tetherin/BST-2-mediated restriction in T cells. J Virol. 2010;84:12185–12199. doi: 10.1128/JVI.01447-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong P, et al. Cell-to-cell transmission can overcome multiple donor and target cell barriers imposed on cell-free HIV. PLoS ONE. 2013;8:e53138. doi: 10.1371/journal.pone.0053138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sigal A, et al. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature. 2011;477:95–98. doi: 10.1038/nature10347. [DOI] [PubMed] [Google Scholar]
- 35.Shim AH, Tirado-Lee L, Prakriya M. Structural and functional mechanisms of CRAC channel regulation. J Mol Biol. 2014;427:77–93. doi: 10.1016/j.jmb.2014.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Rossum DB, et al. DANGER, a novel regulatory protein of inositol 1,4,5-trisphosphate-receptor activity. J Biol Chem. 2006;281:37111–37116. doi: 10.1074/jbc.M608760200. [DOI] [PubMed] [Google Scholar]
- 37.Suh HS, et al. Insulin-like growth factor 2 receptor is an IFNγ-inducible microglial protein that facilitates intracellular HIV replication: implications for HIV-induced neurocognitive disorders. Am J Pathol. 2010;177:2446–2458. doi: 10.2353/ajpath.2010.100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Go EP, et al. Characterization of host-cell line specific glycosylation profiles of early transmitted/founder HIV-1 gp120 envelope proteins. J Proteome Res. 2013;12:1223–1234. doi: 10.1021/pr300870t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nature Rev Genet. 2012;13:654–666. doi: 10.1038/nrg3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kulpa DA, et al. The immunological synapse: the gateway to the HIV reservoir. Immunol Rev. 2013;254:305–325. doi: 10.1111/imr.12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tyagi M, Karn J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007;26:4985–4995. doi: 10.1038/sj.emboj.7601928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee HJ, Kim MY, Park HS. Phosphorylation-dependent regulation of Notch1 signaling: the fulcrum of Notch1 signaling. BMB Rep. 2015;48:431–437. doi: 10.5483/BMBRep.2015.48.8.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geimer Le, Lay AS, et al. The tumor suppressor Ikaros shapes the repertoire of Notch target genes in T cells. Sci Signal. 2014;7:ra28. doi: 10.1126/scisignal.2004545. [DOI] [PubMed] [Google Scholar]
- 44.Munger J, Bajad SU, Coller HA, Shenk T, Rabinowitz JD. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathogens. 2006;2:e132. doi: 10.1371/journal.ppat.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim DY, et al. CBFβ stabilizes HIV Vif to counteract APOBEC3 at the expense of RUNX1 target gene expression. Mol Cell. 2013;49:632–644. doi: 10.1016/j.molcel.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jager S, et al. Vif hijacks CBF-β to degrade APOBEC3G and promote HIV-1 infection. Nature. 2012;481:371–375. doi: 10.1038/nature10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang W, Du J, Evans SL, Yu Y, Yu XF. T-cell differentiation factor CBF-β regulates HIV-1 Vif-mediated evasion of host restriction. Nature. 2012;481:376–379. doi: 10.1038/nature10718. [DOI] [PubMed] [Google Scholar]
- 48.Xiao Z, et al. Assembly of HIV-1 Vif-Cul5 E3 ubiquitin ligase through a novel zinc-binding domain-stabilized hydrophobic interface in Vif. Virology. 2006;349:290–299. doi: 10.1016/j.virol.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 49.Yu Y, Xiao Z, Ehrlich ES, Yu X, Yu XF. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 2004;18:2867–2872. doi: 10.1101/gad.1250204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu X, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 51.Hernan R, Heuermann K, Brizzard B. Multiple epitope tagging of expressed proteins for enhanced detection. Biotechniques. 2000;28:789–793. doi: 10.2144/00284pf01. [DOI] [PubMed] [Google Scholar]
- 52.Low A, et al. Natural polymorphisms of human immunodeficiency virus type 1 integrase and inherent susceptibilities to a panel of integrase inhibitors. Antimicrob Agents Chemother. 2009;53:4275–4282. doi: 10.1128/AAC.00397-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mohammed KD, Topper MB, Muesing MA. Sequential deletion of the integrase (Gag-Pol) carboxyl terminus reveals distinct phenotypic classes of defective HIV-1. J Virol. 2011;85:4654–4666. doi: 10.1128/JVI.02374-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Topper M, et al. Posttranslational acetylation of the human immunodeficiency virus type 1 integrase carboxyl-terminal domain is dispensable for viral replication. J Virol. 2007;81:3012–3017. doi: 10.1128/JVI.02257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wiskerchen M, Muesing MA. Human immunodeficiency virus type 1 integrase: effects of mutations on viral ability to integrate, direct viral gene expression from unintegrated viral DNA templates, and sustain viral propagation in primary cells. J Virol. 1995;69:376–386. doi: 10.1128/jvi.69.1.376-386.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Remenyi R, et al. A comprehensive functional map of the hepatitis C virus genome provides a resource for probing viral proteins. mBio. 2014;5:e01469. doi: 10.1128/mBio.01469-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levy DN, Aldrovandi GM, Kutsch O, Shaw GM. Dynamics of HIV-1 recombination in its natural target cells. Proc Natl Acad Sci USA. 2004;101:4204–4209. doi: 10.1073/pnas.0306764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 59.Moore PL, et al. Nature of nonfunctional envelope proteins on the surface of human immunodeficiency virus type 1. J Virol. 2006;80:2515–2528. doi: 10.1128/JVI.80.5.2515-2528.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crooks ET, et al. Characterizing anti-HIV monoclonal antibodies and immune sera by defining the mechanism of neutralization. Hum Antibodies. 2005;14:101–113. [PMC free article] [PubMed] [Google Scholar]
- 61.Binley JM, et al. A recombinant human immunodeficiency virus type 1 envelope glycoprotein complex stabilized by an intermolecular disulfide bond between the gp120 and gp41 subunits is an antigenic mimic of the trimeric virion-associated structure. J Virol. 2000;74:627–643. doi: 10.1128/jvi.74.2.627-643.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanders RW, et al. The mannose-dependent epitope for neutralizing antibody 2G12 on human immunodeficiency virus type 1 glycoprotein gp120. J Virol. 2002;76:7293–7305. doi: 10.1128/JVI.76.14.7293-7305.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scanlan CN, et al. The broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2G12 recognizes a cluster of alpha1→2 mannose residues on the outer face of gp120. J Virol. 2002;76:7306–7321. doi: 10.1128/JVI.76.14.7306-7321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Billington J, et al. Stability of a receptor-binding active human immunodeficiency virus type 1 recombinant gp140 trimer conferred by intermonomer disulfide bonding of the V3 loop: differential effects of protein disulfide isomerase on CD4 and coreceptor binding. J Virol. 2007;81:4604–4614. doi: 10.1128/JVI.02138-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walker LM, et al. Rapid development of glycan-specific, broad, and potent anti-HIV-1 gp120 neutralizing antibodies in an R5 SIV/HIV chimeric virus infected macaque. Proc Natl Acad Sci USA. 2011;108:20125–20129. doi: 10.1073/pnas.1117531108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li Y, et al. Mechanism of neutralization by the broadly neutralizing HIV-1 monoclonal antibody VRC01. J Virol. 2011;85:8954–8967. doi: 10.1128/JVI.00754-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu X, et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science. 2010;329:856–861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Walker LM, et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011;477:466–470. doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Walker LM, et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang J, et al. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature. 2012;491:406–412. doi: 10.1038/nature11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zwick MB, Saphire EO, Burton DR. gp41: HIV’s shy protein. Nature Med. 2004;10:133–134. doi: 10.1038/nm0204-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Abacioglu YH, et al. Epitope mapping and topology of baculovirus-expressed HIV-1 gp160 determined with a panel of murine monoclonal antibodies. AIDS Res Hum Retroviruses. 1994;10:371–381. doi: 10.1089/aid.1994.10.371. [DOI] [PubMed] [Google Scholar]
- 73.Gorny MK, Gianakakos V, Sharpe S, Zolla-Pazner S. Generation of human monoclonal antibodies to human immunodeficiency virus. Proc Natl Acad Sci USA. 1989;86:1624–1628. doi: 10.1073/pnas.86.5.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu JY, Gorny MK, Palker T, Karwowska S, Zolla-Pazner S. Epitope mapping of two immunodominant domains of gp41, the transmembrane protein of human immunodeficiency virus type 1, using ten human monoclonal antibodies. J Virol. 1991;65:4832–4838. doi: 10.1128/jvi.65.9.4832-4838.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dennison SM, et al. Nonneutralizing HIV-1 gp41 envelope cluster II human monoclonal antibodies show polyreactivity for binding to phospholipids and protein autoantigens. J Virol. 2011;85:1340–1347. doi: 10.1128/JVI.01680-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Simon JH, et al. The Vif and Gag proteins of human immunodeficiency virus type 1 colocalize in infected human T cells. J Virol. 1997;71:5259–5267. doi: 10.1128/jvi.71.7.5259-5267.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fouchier RA, Simon JH, Jaffe AB, Malim MH. Human immunodeficiency virus type 1 Vif does not influence expression or virion incorporation of gag-, pol-, and env-encoded proteins. J Virol. 1996;70:8263–8269. doi: 10.1128/jvi.70.12.8263-8269.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Simon JH, Southerling TE, Peterson JC, Meyer BE, Malim MH. Complementation of vif-defective human immunodeficiency virus type 1 by primate, but not nonprimate, lentivirus vif genes. J Virol. 1995;69:4166–4172. doi: 10.1128/jvi.69.7.4166-4172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brizzard BL, Chubet RG, Vizard DL. Immunoaffinity purification of FLAG epitope-tagged bacterial alkaline phosphatase using a novel monoclonal antibody and peptide elution. Biotechniques. 1994;16:730–735. [PubMed] [Google Scholar]
- 80.Cristea IM, Williams R, Chait BT, Rout MP. Fluorescent proteins as proteomic probes. Mol Cell Proteom. 2005;4:1933–1941. doi: 10.1074/mcp.M500227-MCP200. [DOI] [PubMed] [Google Scholar]
- 81.Scheid JF, et al. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature. 2009;458:636–640. doi: 10.1038/nature07930. [DOI] [PubMed] [Google Scholar]
- 82.Scheid JF, et al. A method for identification of HIV gp140 binding memory B cells in human blood. J Immunol Methods. 2009;343:65–67. doi: 10.1016/j.jim.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang X, Farzan M, Wyatt R, Sodroski J. Characterization of stable, soluble trimers containing complete ectodomains of human immunodeficiency virus type 1 envelope glycoproteins. J Virol. 2000;74:5716–5725. doi: 10.1128/jvi.74.12.5716-5725.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vermeire J, et al. Quantification of reverse transcriptase activity by real-time PCR as a fast and accurate method for titration of HIV, lenti- and retroviral vectors. PLoS ONE. 2012;7:e50859. doi: 10.1371/journal.pone.0050859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hall MP, et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol. 2012;7:1848–1857. doi: 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hazuda DJ, et al. Integrase inhibitors and cellular immunity suppress retroviral replication in rhesus macaques. Science. 2004;305:528–532. doi: 10.1126/science.1098632. [DOI] [PubMed] [Google Scholar]
- 87.Guise AJ, Greco TM, Zhang IY, Yu F, Cristea IM. Aurora B-dependent regulation of class IIa histone deacetylases by mitotic nuclear localization signal phosphorylation. Mol Cell Proteom. 2012;11:1220–1229. doi: 10.1074/mcp.M112.021030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vizcaino JA, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:D447–D456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Spivak M, Weston J, Bottou L, Kall L, Noble WS. Improvements to the percolator algorithm for peptide identification from shotgun proteomics data sets. J Proteome Res. 2009;8:3737–3745. doi: 10.1021/pr801109k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kutluay SB, et al. Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell. 2014;159:1096–1109. doi: 10.1016/j.cell.2014.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Goodchild RE, Dauer WT. Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc Natl Acad Sci USA. 2004;101:847–852. doi: 10.1073/pnas.0304375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nature Rev Mol Cell Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 93.Naismith TV, Heuser JE, Breakefield XO, Hanson PI. TorsinA in the nuclear envelope. Proc Natl Acad Sci USA. 2004;101:7612–7617. doi: 10.1073/pnas.0308760101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.