

Matrix-bound vesicles within ECM bioscaffolds provide mechanistic insight into inductive properties.
Keywords: Matrix Bound Nano Vesicles (MBV), Microvesicles (MV), Extracellular Matrix (ECM), Extracellular vesicles (EV), Exosomes
Abstract
Biologic scaffold materials composed of extracellular matrix (ECM) have been used in a variety of surgical and tissue engineering/regenerative medicine applications and are associated with favorable constructive remodeling properties including angiogenesis, stem cell recruitment, and modulation of macrophage phenotype toward an anti-inflammatory effector cell type. However, the mechanisms by which these events are mediated are largely unknown. Matrix-bound nanovesicles (MBVs) are identified as an integral and functional component of ECM bioscaffolds. Extracellular vesicles (EVs) are potent vehicles of intercellular communication due to their ability to transfer RNA, proteins, enzymes, and lipids, thereby affecting physiologic and pathologic processes. Formerly identified exclusively in biologic fluids, the presence of EVs within the ECM of connective tissue has not been reported. In both laboratory-produced and commercially available biologic scaffolds, MBVs can be separated from the matrix only after enzymatic digestion of the ECM scaffold material, a temporal sequence similar to the functional activity attributed to implanted bioscaffolds during and following their degradation when used in clinical applications. The present study shows that MBVs contain microRNA capable of exerting phenotypical and functional effects on macrophage activation and neuroblastoma cell differentiation. The identification of MBVs embedded within the ECM of biologic scaffolds provides mechanistic insights not only into the inductive properties of ECM bioscaffolds but also into the regulation of tissue homeostasis.
INTRODUCTION
Biologic scaffold materials composed of mammalian extracellular matrix (ECM) are commonly used for surgical repair or reconstruction of ventral hernias (1), skeletal muscle (2), esophagus (3), dura mater (4), tendon (5), breast tissue (6), and others (7). These ECM-based materials are either xenogeneic or allogeneic in origin and elicit a variety of favorable cellular responses that promote functional tissue reconstruction including angiogenesis, stem cell recruitment, antimicrobial activity, and modulation of innate immune response (8–10). However, the mechanisms by which these ECM bioscaffolds influence cell behavior are largely unknown.
Plausible and logical mechanisms by which ECM bioscaffolds interact with cells include cues generated by specific surface topography (11, 12), mechanobiology-related cell signaling (13, 14), integrin-mediated cell response to the ECM ligand landscape (12, 15), release of embedded growth factors/cytokines/chemokines (10, 16), and/or the creation or exposure of signaling cryptic peptide motifs (17). Extracellular vesicles (EVs) are membrane vesicles 30 to 1000 nm in diameter and are released from most cell types into the extracellular space (18, 19). To date, neither the presence nor the functional role of EVs as regulators of cell behavior in tissue ECM or in recipients of ECM bioscaffolds has been reported.
EVs are commonly found in cell culture supernatants and in most biologic fluids such as saliva, plasma, and cerebrospinal fluid (20). Different cell types release exosomes (that is, an EV subgroup that is 30 to 100 nm in diameter) containing microRNA (miRNA), cytokines, chemokines, and other types of proteins that can be internalized by, and function within, recipient cells (21). The contents of EVs have been shown to regulate diverse physiologic and pathologic processes, such as angiogenesis, immune cell phenotype, cell differentiation and fate, epithelial to mesenchymal transition, and apoptosis (22–24). Here, we identify EVs, which we have termed matrix-bound nanovesicles (MBVs), embedded within biologic scaffolds composed of ECM. The miRNA and protein cargo of MBVs and their ability to influence cell behavior were investigated.
RESULTS
Different enzymatic digestions affect the extractable amount of nucleic acids in ECM bioscaffold materials
Although the quantification of double-stranded DNA (dsDNA) is commonly used as a metric to assess thoroughness of tissue decellularization during the manufacturing of biologic scaffold materials (25, 26), the quantification of other forms of nucleic acid, such as RNA or single-stranded DNA (ssDNA), is neglected in these analyses. To determine whether alternative forms of nucleic acid were present after decellularization of the native tissue, nucleic acid was extracted from comminuted (acellular) ECM scaffold materials using the phenol/chloroform method. Quantification of dsDNA was performed using the PicoGreen assay, and quantification of total nucleic acid was performed by ultraviolet (UV) absorbance at 260 nm, which detects all forms of nucleic acid, including RNA. Results show that the amount of dsDNA present was only approximately 25 to 40% of the total nucleic acid present in ECM scaffold materials (Fig. 1 and table S1). When these ECM scaffolds were enzymatically degraded after treatment with various proteases before nucleic acid extraction, the amount of total extractable nucleic acid markedly increased compared to untreated samples. This pattern was observed for all forms of ECM scaffold materials tested, which included laboratory-produced and commercially available equivalents of urinary bladder matrix (UBM) and ACell MatriStem (Fig. 1A), small intestinal submucosa (SIS) and Cook Biotech Biodesign (Fig. 1B), and dermis and C.R. Bard XenMatrix (Fig. 1C). Results show that laboratory-produced scaffolds had similar nucleic acid concentrations to their respective commercially available counterparts, indicating that these results were not an artifact of laboratory manufacturing protocols. Furthermore, the nucleic acid content is much lower in dermis-derived ECM scaffolds compared to SIS- and UBM-derived ECM scaffolds, a result possibly due to the differences in anatomic location, physiologic function, and/or matrix density of the various tissues.
Fig. 1. Comparison of nucleic acid concentration from UBM, SIS, or dermis and their commercially available equivalents.
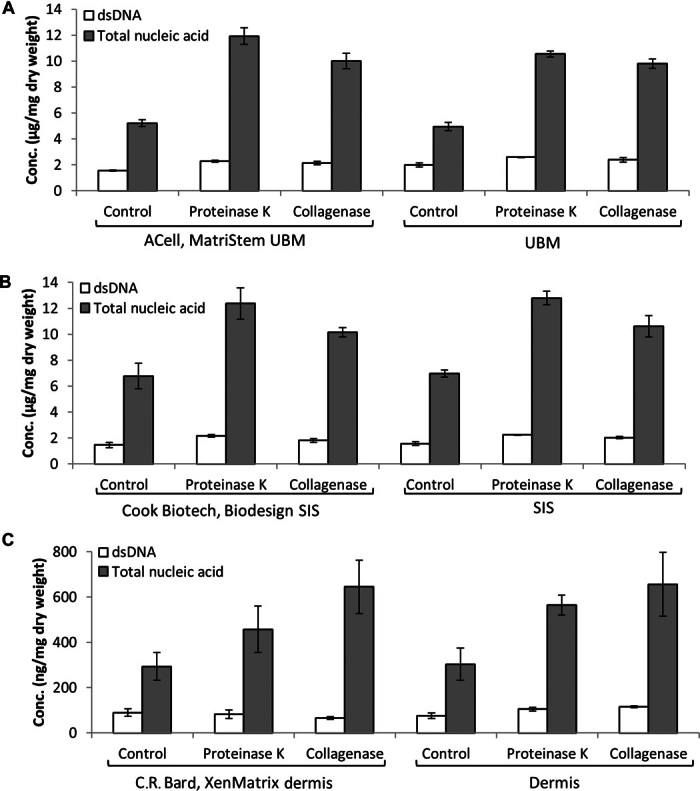
(A to C) Concentration of total nucleic acid and dsDNA per milligram dry weight of ECM scaffold from untreated (control) and proteinase K– or collagenase-treated samples of (A) UBM and ACell MatriStem (porcine UBM), (B) SIS and Cook Biotech Biodesign (porcine SIS), and (C) dermis and C.R. Bard XenMatrix (porcine dermis). Total nucleic acid concentration was assessed by UV absorbance at 260 nm. dsDNA concentration was assessed using PicoGreen dsDNA quantification reagent. Variability from isolation to isolation is depicted by SD. Data are means ± SD; n = 3 isolations per sample.
Enzymatic digestion of biologic scaffolds releases small RNA molecules
To determine whether RNA was present in ECM scaffolds, nucleic acid extractions from untreated or pepsin-, proteinase K–, or collagenase-treated UBM were exposed to deoxyribonuclease I (DNase I) or ribonuclease A (RNase A) nucleases and were analyzed by agarose gel electrophoresis (Fig. 2A). Results show that DNase I removed all nucleic acid material except for a smeared band that ran approximately between 25 and 200 base pairs (bp). Reciprocally, RNase A removed this small base pair nucleic acid fraction, leaving the larger base pair material, indicating that these short-length nucleic acid molecules were small RNA molecules. Furthermore, when compared to untreated (no digest) samples, these small RNA molecules could only be efficiently extracted after the ECM scaffolds were enzymatically degraded with pepsin, proteinase K, or collagenase (Fig. 2A). Nucleic acid preparations were further analyzed using the Agilent 2100 Bioanalyzer (Fig. 2B). Results show that, compared to samples not exposed to nuclease (Fig. 2B, top panel), DNase I removed all nucleic acid material except for the small RNA molecules (Fig. 2B, bottom panel). These small RNA molecules were identified in all biologic scaffold materials tested (Fig. 2C). The ability to remove DNA and RNA molecules from ECM scaffolds by exposure to nuclease before nucleic acid extraction was investigated. Results show that exposure of untreated or collagenase-treated UBM to DNase I or RNase A nucleases did not completely degrade the nucleic acid material (Fig. 2D), suggesting that there was a subset of nucleic acid incorporated within and protected by the ECM from nucleases other than the “free” nucleic acid (that is, genomic DNA or cellular RNA) that was present as a result of the decellularization process. We hypothesized that these nuclease-protected nucleic acid molecules may be packaged within vesicular bodies, such as microvesicles or exosomes, which have been previously shown to protect RNA/DNA cargo from nuclease activity (27).
Fig. 2. Enzymatic digestion of decellularized ECM scaffolds releases small RNA molecules.

(A) Nucleic acid extracted from untreated UBM (no digest) and pepsin-, proteinase K–, or collagenase-treated UBM was exposed to RNase A, DNase I, or no-nuclease treatment (control). (B) Electropherogram depicting the small RNA pattern of nucleic acid in fluorescence units (FU) before (top panel) and after (bottom panel) DNase I treatment. (C) Electropherogram depicting small RNA pattern from the indicated samples in FU. (D) A subset of nucleic molecules in biologic scaffolds is protected from nuclease degradation.
MBVs are present within the ECM
MBVs embedded within the ECM of biologic scaffolds were identified by transmission electron microscopy (TEM) on an osmium tetroxide–postfixed UBM. Rounded structures that stained positive for osmium were shown, indicating the presence of lipid membranes (Fig. 3A, left panel). Treatment with pepsin (Fig. 3A, middle panel), which only partially digests the ECM scaffold, showed that these MBVs were closely associated with the collagen network of the matrix. However, after more complete digestion of the ECM scaffold is attempted with proteinase K treatment (Fig. 3A, right panel), these MBVs could be separated from the fiber network. The structure (round vesicles) and size (50 to 400 nm) of these vesicles were similar to previously reported values for microvesicles and exosomes (21). Using a strategy of enzymatic digestion coupled with ultracentrifugation, MBVs could be purified from all tested forms of ECM scaffold materials, including commercially available products, after proteinase K treatment (Fig. 3B). Nanoparticle Tracking Analysis (NTA) (20) was used to identify the particle size and quantity of MBVs for each sample. All samples showed MBVs with the size range reported for EVs (21), ranging from 10 to 1000 nm (Fig. 3C). To determine whether MBVs contained surface antigen markers commonly attributed to exosomes, immunoblot analysis was performed for CD63, CD81, CD9, and Hsp70 (28). Results show that in contrast to porcine serum exosomes, human serum exosomes, or human mesenchymal stem cell (hMSC)–derived exosomes, no detectable CD63, CD81, CD9, and Hsp70 was observed in the bioscaffold-derived MBVs (Fig. 3D). MBVs were analyzed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 3E). The silver-stained gel revealed that MBVs contain protein cargo that appears to be distinctively different than exosomes isolated from hMSCs.
Fig. 3. Identification of ECM-embedded MBVs.
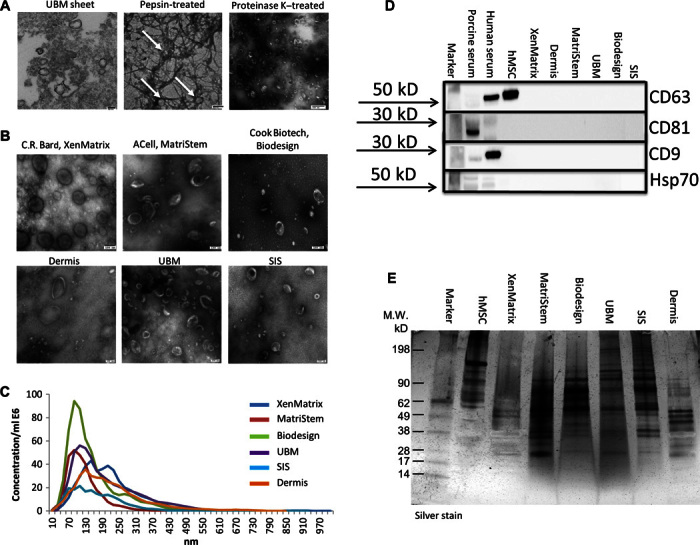
(A) TEM imaging of MBVs identified in a UBM sheet stained positive with osmium (left panel), pepsin-treated UBM (middle panel), or proteinase K–treated UBM (right panel). (B) TEM imaging of MBVs identified in proteinase K–treated ECM from three commercial and three laboratory-produced scaffolds. Scale bars, 100 nm. (C) Validation of MBV size was measured with NanoSight. (D) Western blot analysis was performed on four exosomal surface markers: CD63, CD81, CD9, and Hsp70. Expression levels were not detectable as compared to porcine serum, human serum, and human bone marrow–derived mesenchymal stem cell controls. (E) MBV protein cargo signature was different between MBVs and hMSCs as evaluated using SDS-PAGE and silver stain imaging.
Bioscaffold-derived MBVs contain miRNA
MBVs isolated from laboratory-produced and equivalent commercial products were treated with RNase A for 30 min before the RNA isolation step. RNase A treatment was performed to ensure that RNA sequencing data represent only RNA within the MBVs (29). Similar to exosomes and microvesicles (27), MBVs protect their nucleic acid cargo from nuclease degradation (fig. S1). After extraction of small RNA molecules, the RNAs that have less than 200 nucleotides (nt) were used to construct small RNA cDNA libraries. The libraries were read with the Ion Proton platform. Sequencing data were trimmed on the basis of size and Phred score before alignment to the human genome, which revealed that more than 34% of the small RNA reads were mapped. Posterior reads were annotated to miRBase (release 21). Between 33 and 240 miRNAs were identified per sample, and more than 50% were mutual between the commercial products and the in-house devices manufactured from the same source tissue (Fig. 4A). Twenty-two miRNAs were found to be expressed in all matrices regardless of the source tissue. Ingenuity Pathway Analysis (IPA) was used to identify pathways and cell and physiologic functions. All samples were found to contain miRNAs involved in cellular development, cellular growth and proliferation, cell death and survival, cellular movement, and cell cycle activity (Fig. 4B). Additionally, identified miRNAs were found to play a role in connective tissue development and function, organism development, and organ development (Fig. 4C).
Fig. 4. Identification of miRNA packaged within MBVs.
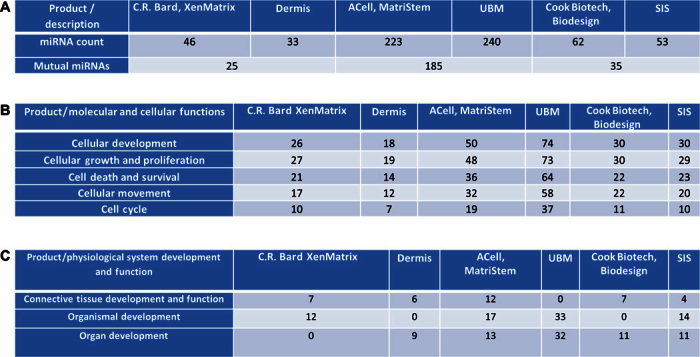
MBV small RNA sequencing analysis reveals specific miRNA signature between commercial products and comparable in-house products (n = 1). (A) Numbers in each box represents different miRNAs within each sample. (B and C) Molecular and cellular functions (B) and physiological system development and function pathways (C) associated with identified miRNAs were generated using IPA. Each box represents the numbers of different miRNAs involved in each pathway.
MBVs are biologically active
MBVs isolated from UBM were labeled with acridine orange. Successful labeling of MBVs was achieved, and these labeled vesicles were then detected within C2C12 cells following coculture, confirming cellular uptake (Fig. 5A). To determine whether the isolated MBVs could influence cell behavior, macrophages were exposed to MBVs (Fig. 5B). Macrophages were stimulated with interferon-γ (IFN-γ) and lipopolysaccharide (LPS) to induce an M1-like macrophage phenotype, interleukin-4 (IL-4) to induce an M2-like phenotype, a pepsin control, pepsin-solubilized UBM, collagenase control, or MBVs isolated from collagenase-treated UBM. Results show that macrophages expressed the Fizz-1 marker in response to UBM-derived MBVs, similar to the expression pattern of the IL-4–stimulated (M2) cells, an effect comparable to that induced by the parent ECM (pepsin-solubilized UBM) substrate. Neuroblastoma cells (N1E-115) have been shown to have neurite extension 5 days after treatment with pepsin-solubilized UBM (30). Using only MBVs isolated from collagenase-treated UBM, N1E-115 cells showed neurite extension within 3 days, whereas no change was observed in the control group (Fig. 5C).
Fig. 5. MBVs are biologically active.
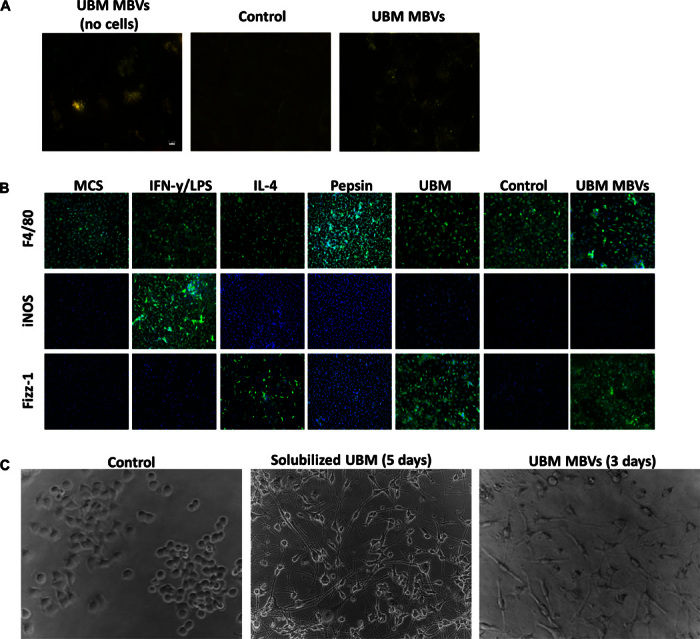
MBVs isolated from UBM were labeled with Exo-Glow. (A) C2C12 cells were exposed to labeled MBVs for 4 hours. The left panel shows a representative image of successful labeling of MBVs before exposure to cell culture. The right panel represents exposure of labeled MBVs in C2C12 compared to the middle panel image (control). Green fluorescence represents DNA, whereas red fluorescence represents RNA MBV cargo that is successfully integrated with target cells. (B) Bone marrow was isolated from C57bl/6 mice and cultured in medium supplemented with macrophage colony-stimulating factor (M-CSF) to derive macrophages. Macrophages were treated with IFN-γ (20 ng/ml) and LPS (100 ng/ml) to derive M1 macrophages, IL-4 (20 ng/ml) to derive M2 macrophages, and isolated MBVs (5 μg/ml) from a UBM source. Macrophages were fixed and immunolabeled for the pan-macrophage marker (F4/80) and markers associated with the M1 (iNOS) and M2 (Fizz-1) phenotype. MBV-treated macrophages are predominantly F4/80 + Fizz-1 + macrophages, indicating an M2-like phenotype. Experiment was conducted with n = 2 samples with four technical replicates. (C) N1E-115 neuroblastoma cells were exposed to pepsin-solubilized UBM and MBVs. Five days (solubilized UBM) and three days (MBVs) after exposure, neurite extensions were visible in treated cells compared to control.
DISCUSSION
The present study shows that 10- to 1000-nm MBVs are present in ECM bioscaffolds produced by the decellularization of source tissues, such as urinary bladder, SIS, and dermis. These MBVs survive chemical, enzymatic, and detergent-based decellularization protocols and can induce changes in cell behavior, as shown by the differential expression of macrophage surface markers that are associated with functional phenotypes, and the rapid formation of dendritic processes in a neural progenitor cell line. Of potential clinical significance, these MBVs are present in commercially manufactured ECM bioscaffolds.
First identified by electron microscopy in 1967 as a product of platelets (31, 32), EVs have attracted considerable attention over the past decade as potent vehicles of intercellular communication because of their ability to transfer RNA, proteins, enzymes, and lipids, thereby affecting various physiologic and pathologic processes. The production and release of EVs is evolutionarily conserved in both prokaryotic and eukaryotic organisms, thus underscoring the importance of vesicle-mediated processes in mammalian cell physiology (33). EVs are nanosized, membrane-bound vesicles with diameters ranging from 30 to 1000 nm and are categorized into three main groups (exosomes, microvesicles, and apoptotic bodies) on the basis of their size, origin, markers, and mode of release (19). EVs are secreted by a variety of cell types and have been isolated from biologic fluids or in cell culture supernatants (20). Several reports have shown that EVs are capable of anchoring to ECM constituents through the presence of adhesion molecules, such as intercellular adhesion molecule–1 (ICAM-1) and members of the integrin family such as αM and β2 integrins (34–36). For example, B cell–derived exosomes have been found to express β1 and β2 integrins capable of mediating anchorage to collagen-1, fibronectin, and tumor necrosis factor–α (TNF-α)–activated fibroblasts (37). In addition, EVs that have been termed “matrix vesicles” or “calcification vesicles” were shown to anchor selectively to the matrix of bone, cartilage, and predentin (38, 39), which are the product of chondrocytes, osteoblasts, and odontoblasts, and have been shown to serve as the initial site of calcification in skeletal tissues (40). However, it is still uncertain whether calcification vesicles participate in intercellular signaling similar to exosomes and microvesicles (41). Although MBVs share features similar to exosomes and microvesicles, including nanometer size and the presence of miRNA cargo, their presence within the interstitial matrix of soft tissue and the lack of identifiable markers (for example, CD63, CD81, CD9, and Hsp70) suggest that MBVs represent a different population of signaling vesicles. It is also possible that treatment with proteinase K may affect the presence of MBV surface markers as part of its enzymatic activity. Furthermore, the protein signature of hMSC exosomes was distinctively different from MBVs as shown by the silver stain. Notably, the signature of MBVs varies between the different source tissues from which the bioscaffolds were prepared. UBM and SIS MBVs have a similar protein cargo signature compared to dermis MBVs. Further studies are needed to identify the specific protein cargo of MBVs and how quickly this cargo can change in response to microenvironmental stimuli, such as hypoxia, mechanical loading, and others.
The present study shows that MBVs are an integral and functional component of ECM bioscaffolds. In both laboratory-produced and commercially available products, MBVs can only be isolated from ECM scaffolds after disruption of the matrix by enzymatic digestion. It is possible that the close association of MBVs with collagen fibers provides protection from the decellularization agents used in the manufacturing of bioscaffolds. Other plausible explanations may be attributed to their nanometer size and the inherent stability of EVs, including resistance to RNase degradation and capacity to withstand lyophilization and extreme changes in temperature and pH (42–45).
Results of the present study show that MBVs isolated from anatomically distinct source tissue have differential miRNA signatures, a finding that may be exploited to develop a more defined and effective use of ECM scaffold materials for clinical applications. We identified 22 miRNAs that are expressed in all samples regardless of their tissue origin. Although further studies are required, this result suggests the potential use of specific miRNAs as an MBV marker. Note that sequencing analysis was done with n = 1 sample, and therefore, definitive conclusions cannot be made regarding the consistency of the tissue-specific miRNA profiles. The biologic effects of the profile of MBV miRNA found in the present study are consistent with the recognized inductive properties of biologic scaffolds in both preclinical and clinical applications, including cell growth and proliferation (46), cell survival (47), and cell migration and differentiation (48, 49). Although it is unlikely that MBVs mediate all biologic effects attributed to ECM scaffolds, the present data suggest that MBVs are sufficient to recapitulate some of the biologic effects of the parent ECM. miRNAs have been shown to be highly conserved in both vertebrate and invertebrate animal species (50–53). Because of this high degree of homology, recent studies have used computational methods to identify miRNA orthologs in pigs by performing a homology search with known miRNA sequences from human and mouse genomes (54). Therefore, it is not surprising that MBV miRNA isolated from porcine-derived bioscaffolds are capable of exerting biologic effects on murine-derived macrophages and likely with human cells when used in clinical applications. However, future studies are required to fully characterize the MBV miRNA signature and to more finely delineate their functional roles during constructive tissue remodeling.
Numerous studies have investigated the role of EVs in pathologic processes, such as cancer and autoimmune disorders; however, the role of EVs in cellular and tissue homeostasis, response to injury, and regulation of physiologic and regenerative processes are unknown. The biologic effects of EV signaling, which include inhibition of cell senescence and apoptosis (55, 56), transfer of proangiogenic proteins and miRNAs (57, 58), ECM production and remodeling (59, 60), promotion of anti-inflammatory cytokine secretion, and modulation of the M1/M2 macrophage phenotype (61, 62), all have functional relevance in wound healing and regenerative processes. Our results show that MBVs are capable of influencing macrophage polarization toward anti-inflammatory M2-like phenotype and of stem cell differentiation, which are phenomena previously identified as hallmarks of ECM-mediated constructive remodeling (2, 63). These results also suggest possible physiologic roles for MBVs during tissue development, homeostasis, and wound healing. Studies that use EVs isolated from biologic fluids have shown a differential miRNA signature that is dependent on cell origin and is reflective of the disease state of the tissue from which they were secreted (20). Similarly, we speculate that MBV cargo may also be altered in disease states where dynamic remodeling of the ECM occurs, such as fibrosis, chronic inflammation, and neoplastic progression, raising the possibility that MBVs could be used as prognostic biomarkers for health and disease. A significant body of work has shown that the age and species of the native source tissue used for preparation of bioscaffolds are critical factors that determine the outcome in clinical applications (64, 65), raising the possibility that MBVs isolated from bioscaffolds might also be used as a quality control metric for clinical devices manufactured from these source tissues. Results reported here represent the first identification of MBVs in biologic scaffold materials and offer insights into the mechanisms by which ECM scaffolds modulate cell behavior.
MATERIALS AND METHODS
Chemicals and reagents
Pepsin from porcine stomach mucosa was obtained from MP Biomedicals. Collagenase from Clostridium histolyticum was obtained from Sigma-Aldrich. The proteinase K solution, Quant-iT PicoGreen dsDNA Assay Kit, and RNase A were obtained from Thermo Fisher Scientific. RNase-free DNase was obtained from Qiagen. All reagents were assessed by TEM to ensure that they were free of contaminating EVs.
ECM bioscaffold production
Dermal ECM
Dermal ECM was prepared as previously described (16). Briefly, full-thickness skin was harvested from market-weight (~110 kg) pigs (Tissue Source Inc.), and the subcutaneous fat and epidermis were removed by mechanical delamination. This tissue was then treated with 0.25% trypsin (Thermo Fisher Scientific) for 6 hours, 70% ethanol for 10 hours, 3% H2O2 for 15 min, 1% Triton X-100 (Sigma-Aldrich) in 0.26% EDTA/0.69% tris for 6 hours with a solution change for an additional 16 hours, and 0.1% peracetic acid/4% ethanol (Rochester Midland) for 2 hours. Water washes were performed between each chemical change with alternating water and phosphate-buffered saline (PBS) washes following the final step. All chemical exposures were conducted under agitation on an orbital shaker at 300 rpm. Dermal ECM was then lyophilized and milled into particulate form using a Wiley Mill with a #40 mesh screen.
Urinary bladder matrix
UBM was prepared as previously described (66). Porcine urinary bladders from market-weight animals were acquired from Tissue Source, LLC. Briefly, the tunica serosa, tunica muscularis externa, tunica submucosa, and tunica muscularis mucosa were mechanically removed. The luminal urothelial cells of the tunica mucosa were dissociated from the basement membrane by washing with deionized water. The remaining tissue consisted of basement membrane and subjacent lamina propria of the tunica mucosa and was decellularized by agitation in 0.1% peracetic acid with 4% ethanol for 2 hours at 300 rpm. The tissue was then extensively rinsed with PBS and sterile water. The UBM was then lyophilized and milled into particulate form using a Wiley Mill with a #60 mesh screen.
Small intestinal submucosa
Preparation of SIS bioscaffold has been previously described (67). Briefly, jejunum was harvested from 6-month-old market-weight (~110 to ~120 kg) pigs and split longitudinally. The superficial layers of the tunica mucosa were mechanically removed. Likewise, the tunica serosa and tunica muscularis externa were mechanically removed, leaving the tunica submucosa and basilar portions of the tunica mucosa. Decellularization and disinfection of the tissue were completed by agitation in 0.1% peracetic acid with 4% ethanol for 2 hours at 300 rpm. The tissue was then extensively rinsed with PBS and sterile water. The SIS was then lyophilized and milled into particulate form using a Wiley Mill with a #60 mesh screen.
Three commercially available biologic scaffolds, each composed of one of the three tissue ECMs described above, were also evaluated in this study: MatriStem (ACell Inc.), XenMatrix (C.R. Bard–Davol Inc.), and Biodesign (Cook Biotech Inc.).
Enzymatic digestion of ECM samples
ECM samples were lyophilized and ground into a powder using a Wiley Mill with #40 or #60 mesh screen. Enzymatic digestion was performed by treating each sample (5 mg dry weight) with either proteinase K (0.1 mg/ml) in buffer [50 mM tris-HCl (pH 8) and 200 mM NaCl] for 24 hours at room temperature, collagenase (0.1 mg/ml) in buffer [50 mM tris (pH 8), 5 mM CaCl2, and 200 mM NaCl] for 24 hours at room temperature, or pepsin (1 mg/ml) in 0.01 M HCl for 24 hours at room temperature. Before nucleic acid extraction, pepsin-solubilized samples were neutralized to pH 7.4 with NaOH. Untreated samples (control) were prepared by resuspension of each sample (5 mg/ml dry weight) in 50 mM tris-HCl (pH 7.4) and 200 mM NaCl. Before ECM addition, all enzymatic solutions were passed through a 0.22-μm filter (Millipore).
Nucleic acid extraction and profiling
Nucleic acid was extracted from ECM samples by the addition of an equal volume of phenol/chloroform (pH 7.4). Samples were briefly vortexed and centrifuged at 12,000g for 10 min, and the aqueous phase was then transferred to a new tube. Nucleic acid was precipitated by the addition of 1/10 volume of 3 M sodium acetate and 3 volumes of 100% ethanol, mixed by inversion, and centrifuged for 20 min at 20,000g and 4°C. Nucleic acid pellets were washed once with 75% ethanol and resuspended in nuclease-free water. Nucleic acid was analyzed using the Agilent 2100 Bioanalyzer (Agilent Technologies) or by electrophoresis in 2% (w/v) agarose gels and ethidium bromide staining. Quantification of total nucleic acid was performed by UV absorbance at 260 nm using the Thermo Fisher Scientific NanoDrop 1000 Spectrophotometer. Quantification of dsDNA was performed using the Quant-iT PicoGreen dsDNA Assay Kit, according to the manufacturer’s recommended protocol.
MBV isolation
Enzymatically digested ECM was subjected to successive centrifugations at 500g (10 min), 2500g (20 min), and 10,000g (30 min) to remove collagen fibril remnants. Each of the above centrifugation steps was performed three times. The fiber-free supernatant was then centrifuged at 100,000g (Beckman Coulter Optima L-90K ultracentrifuge) at 4°C for 70 min. The 100,000g pellets were washed and suspended in 500 μl of PBS and passed through a 0.22-μm filter (Millipore). All solutions used for enzymatic digestion of the samples were subjected to the same procedure to ensure the absence of EVs in the preparations.
MBV imaging
TEM imaging was conducted on ECM vesicles loaded on carbon-coated grids and fixed in 4% paraformaldehyde. Grids were imaged at 80 kV with a JEOL 1210 TEM with a high-resolution Advanced Microscopy Techniques digital camera. Size of MVs was determined from representative images using JEOL TEM software.
MBV size determination
MBVs were diluted in particle-free PBS, and their size was determined using NTA (20), as described (68). Briefly, NTA measurements were performed using a NanoSight NS500 instrument (NanoSight NTA 2.3 Nanoparticle Tracking and Analysis Release Version Build 0025). Size distribution of MBVs was determined by measuring the rate of Brownian motion with a NanoSight LM10 system (NanoSight) equipped with fast video capture and particle-tracking software. MBVs were diluted in particle-free PBS and injected into a NanoSight sample cubicle. The mean ± SD size distribution was determined.
Gel electrophoresis, Western blotting, and silver stain
MBV protein concentration was determined using bicinchoninic acid assay quantification kit (Pierce Chemical). Equal concentrations of MBVs were resuspended in Laemmli buffer (R&D Systems) containing 5% β-mercaptoethanol (Sigma-Aldrich) and fractionated on a 4 to 20% gradient SDS-PAGE (Bio-Rad). The gels were run using Mini-PROTEAN electrophoresis module assembly (Bio-Rad) at 150 mV in running buffer (25 mM tris base, 192 mM glycine, and 0.1% SDS), followed by semidry transfer to polyvinylidene difluoride membranes (Millipore) for 45 min at constant voltage in transfer buffer [25 mM tris (pH 7.5), 192 mM glycine, 20% methanol, and 0.025% SDS]. The membranes were then blocked for 45 min with Pierce protein-free blocking buffer (Pierce Chemical) and incubated overnight with the following primary antibodies: rabbit anti-CD63, rabbit anti-CD81, rabbit anti-CD9, and rabbit anti-Hsp70, at 1:1000 dilution (System Biosciences). Membranes were washed three times for 15 min each before and after they were incubated with goat anti-rabbit secondary antibody, at 1:10,000 dilution (System Biosciences). The washed membranes were exposed to chemiluminescent substrate (Bio-Rad) and then visualized using a ChemiDoc Touch instrument (Bio-Rad). Silver staining of gels was performed using the Silver Stain Plus Kit (Bio-Rad) according to the manufacturer’s instruction and visualized using a ChemiDoc Touch instrument (Bio-Rad).
RNA isolation
MBV RNA was isolated using the SeraMir Kit (System Biosciences) according to the manufacturer’s instructions. Before RNA isolation, MBV samples were treated with RNase A (10 μg/ml) (Applied Biosystems) at 37°C for 30 min to degrade any contaminating “free RNA” that remained from the decellularization process. The reaction was terminated by the addition of RNase inhibitor (Applied Biosystems). RNA quantity was determined using NanoDrop spectrophotometer (NanoDrop), and its quality was determined by Agilent Bioanalyzer 2100 (Agilent Technologies).
RNA sequencing and data analysis
Small RNA libraries were prepared using Ion Total RNA-Seq Kit version 2, according to the manufacturer’s instructions. Following bead-based size selection of RNA in the 10- to 20-nt range, cDNA was created by hybridization and ligation of indexed sequencing adapters followed by reverse transcription and polymerase chain reaction (PCR). Amplified library was again size-selected using a bead-based method and was run on a bioanalyzer to verify library size distribution. The Ion One Touch 2 System was used to perform automated emulsion PCR of the prepared libraries and templated Ion Sphere Particle enrichment. Sequencing was performed on the Ion Proton platform using a single P1 sequencing chip. Obtained data were imported into CLC Genomics Workbench 8 (Qiagen). All of the following steps were performed on CLC, unless mentioned otherwise. The adaptors were trimmed, and all reads that had two ambiguous nucleotides, had a Phred score <30, or were lower than 15 nt or above 100 nt were removed. Conserved reads were then aligned to the human genome (hg38) to verify valid reads. Reads were extracted, counted, and then annotated on miRBase 21 (human reference); a 2-nt mismatch was allowed per read. Only sequences that matched a mature miRNA were used for downstream analysis.
Ingenuity Pathway Analysis
miRNAs identified by RNA sequencing were analyzed by IPA to determine an miRNA signaling pathway signature.
MBV fluorescence labeling
MBV nucleic acid cargo was labeled using Exo-Glow (System Biosciences), according to the manufacturer’s instructions. Briefly, 500 μl of resuspended MBVs was labeled with Exo-Glow and incubated at 37°C for 10 min. ExoQuick-TC (100 μl) was added to stop the reaction, and samples were placed on ice for 30 min. Samples were then centrifuged for 10 min at 14,000g. The supernatant was removed, and the pellet was resuspended with 500 μl of 1× PBS; 50 μl of this MBV suspension was added to C2C12 in culture. The cells were cultured for 4 hours, and the transfer of the MBV cargo to the cells was determined by imaging using a 100× objective and Axio Observer Z1 microscope.
Cell culture
C2C12 mouse muscle myoblast cells were obtained from the American Type Culture Collection (ATCC) and cultured following the ATCC guidelines in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Sigma-Aldrich) at 37°C in 5% CO2. N1E-115 mouse neuroblastoma cells were obtained from ATCC and maintained in DMEM (Invitrogen) supplemented with 5% FBS, 1% l-glutamine (2 mM), and 1% penicillin/streptomycin (Sigma-Aldrich) at 37°C in 5% CO2. One million cells were plated in a six-well plate before addition of MBVs. Murine bone marrow–derived macrophages (BMDMs) were isolated and characterized as previously described (69). Briefly, bone marrow was harvested from 6- to 8-week-old C57bl/6 mice. Harvested cells from the bone marrow were washed and plated at 1 × 106 cells/ml and were allowed to differentiate into macrophages for 7 days with complete medium changes every 48 hours.
Macrophage immunolabeling
Primary mouse BMDMs were isolated, and the presence of two markers (iNOS and Fizz-1) that are associated with the proinflammatory “M1-like” and anti-inflammatory “M2-like” (respectively) phenotypes was determined (69). The primary antibodies used for immunofluorescence staining were (i) rat monoclonal anti-F4/80 (Abcam) at 1:200 dilution for a pan-macrophage marker, (ii) rabbit polyclonal anti-iNOS (Abcam) at 1:100 dilution for an M1 marker, and (iii) rabbit polyclonal anti–Fizz-1 (PeproTech) for an M2 marker. Cells were incubated in blocking solution consisting of PBS, 0.1% Triton X, 0.1% Tween 20, 4% goat serum, and 2% bovine serum albumin to prevent nonspecific binding for 1 hour at room temperature. Blocking solution was removed, and cells were incubated in primary antibodies for 16 hours at 4°C. After washing in PBS, cells were incubated in fluorophore-conjugated secondary antibodies (Alexa Fluor donkey anti-rat 488 or donkey anti-rabbit 488; Invitrogen) for 1 hour at room temperature. After washing again with PBS, nuclei were counterstained with 4′,6-diamidino-2-phenylindole before imaging. Images of three 20× fields were taken for each well using a live-cell microscope. Light exposure times for ECM- and MBV-treated macrophages were standardized on the basis of those set for cytokine-treated macrophages to determine the presence of the markers on the macrophages.
Supplementary Material
Acknowledgments
This project used the University of Pittsburgh HSCRF Genomics Research Core. We thank H. Deborah for conducting the small RNA sequencing assay and Y. Sadovsky for his contribution in acquiring the NTA data. Funding: No extramural funding was used to support this study. Author contributions: L.H., G.S.H., J.D.N., and S.F.B. conceptualized, designed, and funded the research; L.H., G.S.H., J.D.N., L.Z., and J.L.D. performed experiments; L.H., G.S.H., J.D.N., L.Z., J.L.D., and N.J.T. analyzed the data; L.H., G.S.H., J.D.N., L.Z., J.L.D., N.J.T., D.B.S., and S.F.B. interpreted the results of experiments; L.H., G.S.H., J.D.N., L.Z., and J.L.D. prepared the figures; L.H., G.S.H., J.D.N., N.J.T., and S.F.B. drafted the manuscript; L.H., G.S.H., N.J.T., and S.F.B. edited and revised the manuscript; L.H., G.S.H., J.D.N., and S.F.B. approved the final version of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/6/e1600502/DC1
table S1. Comparison of nucleic acid concentration from UBM, SIS, or dermis and their commercially available equivalents.
fig. S1. Nuclease protection assay.
REFERENCES AND NOTES
- 1.Alicuben E. T., DeMeester S. R., Onlay ventral hernia repairs using porcine non-cross-linked dermal biologic mesh. Hernia 18, 705–712 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sicari B. M., Rubin J. P., Dearth C. L., Wolf M. T., Ambrosio F., Boninger M., Turner N. J., Weber D. J., Simpson T. W., Wyse A., Brown E. H., Dziki J. L., Fisher L. E., Brown S., Badylak S. F., An acellular biologic scaffold promotes skeletal muscle formation in mice and humans with volumetric muscle loss. Sci. Transl. Med. 6, 234ra258 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Badylak S. F., Hoppo T., Nieponice A., Gilbert T. W., Davison J. M., Jobe B. A., Esophageal preservation in five male patients after endoscopic inner-layer circumferential resection in the setting of superficial cancer: A regenerative medicine approach with a biologic scaffold. Tissue Eng. Part A 17, 1643–1650 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bejjani G. K., Zabramski J. Durasis Study Group , Safety and efficacy of the porcine small intestinal submucosa dural substitute: Results of a prospective multicenter study and literature review. J. Neurosurg. 106, 1028–1033 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Longo U. G., Lamberti A., Petrillo S., Maffulli N., Denaro V., Scaffolds in tendon tissue engineering. Stem Cells Int. 2012, 517165 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salzberg C. A., Nonexpansive immediate breast reconstruction using human acellular tissue matrix graft (AlloDerm). Ann. Plast. Surg. 57, 1–5 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Badylak S. F., Freytes D. O., Gilbert T. W., Reprint of: Extracellular matrix as a biological scaffold material: Structure and function. Acta Biomater. 23, S17–S26 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Brown B. N., Valentin J. E., Stewart-Akers A. M., McCabe G. P., Badylak S. F., Macrophage phenotype and remodeling outcomes in response to biologic scaffolds with and without a cellular component. Biomaterials 30, 1482–1491 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maquart F. X., Bellon G., Pasco S., Monboisse J. C., Matrikines in the regulation of extracellular matrix degradation. Biochimie 87, 353–360 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Agrawal V., Tottey S., Johnson S. A., Freund J. M., Siu B. F., Badylak S. F., Recruitment of progenitor cells by an extracellular matrix cryptic peptide in a mouse model of digit amputation. Tissue Eng. Part A 17, 2435–2443 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ning L.-J., Zhang Y.-J., Zhang Y., Qing Q., Jiang Y.-L., Yang J.-L., Luo J.-C., Qin T.-W., The utilization of decellularized tendon slices to provide an inductive microenvironment for the proliferation and tenogenic differentiation of stem cells. Biomaterials 52, 539–550 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Mammoto T., Jiang E., Jiang A., Mammoto A., Extracellular matrix structure and tissue stiffness control postnatal lung development through the lipoprotein receptor–related protein 5/Tie2 signaling system. Am. J. Respir. Cell Mol. Biol. 49, 1009–1018 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Bissell M. J., Aggeler J., Dynamic reciprocity: How do extracellular matrix and hormones direct gene expression? Prog. Clin. Biol. Res. 249, 251–262 (1987). [PubMed] [Google Scholar]
- 14.Gilbert T. W., Stolz D. B., Biancaniello F., Simmons-Byrd A., Badylak S. F., Production and characterization of ECM powder: Implications for tissue engineering applications. Biomaterials 26, 1431–1435 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Ruoslahti E., RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 12, 697–715 (1996). [DOI] [PubMed] [Google Scholar]
- 16.Reing J. E., Brown B. N., Daly K. A., Freund J. M., Gilbert T. W., Hsiong S. X., Huber A., Kullas K. E., Tottey S., Wolf M. T., Badylak S. F., The effects of processing methods upon mechanical and biologic properties of porcine dermal extracellular matrix scaffolds. Biomaterials 31, 8626–8633 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis G. E., Bayless K. J., Davis M. J., Meininger G. A., Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am. J. Pathol. 156, 1489–1498 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nawaz M., Camussi G., Valadi H., Nazarenko I., Ekström K., Wang X., Principe S., Shah N., Ashraf N. M., Fatima F., Neder L., Kislinger T., The emerging role of extracellular vesicles as biomarkers for urogenital cancers. Nat. Rev. Urol. 11, 688–701 (2014). [DOI] [PubMed] [Google Scholar]
- 19.van der Pol E., Böing A. N., Harrison P., Sturk A., Nieuwland R., Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 64, 676–705 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Yáñez-Mó M., Siljander P. R.-M., Andreu Z., Zavec A. B., Borràs F. E., Buzas E. I., Buzas K., Casal E., Cappello F., Carvalho J., Colás E., Cordeiro-da Silva A., Fais S., Falcon-Perez J. M., Ghobrial I. M., Giebel B., Gimona M., Graner M., Gursel I., Gursel M., Heegaard N. H. H., Hendrix A., Kierulf P., Kokubun K., Kosanovic M., Kralj-Iglic V., Krämer-Albers E.-M., Laitinen S., Lässer C., Lener T., Ligeti E., Linē A., Lipps G., Llorente A., Lötvall J., Manček-Keber M., Marcilla A., Mittelbrunn M., Nazarenko I., Nolte-’t Hoen E. N. M., Nyman T. A., O’Driscoll L., Olivan M., Oliveira C., Pallinger E., Del Portillo H. A., Reventós J., Rigau M., Rohde E., Sammar M., Sánchez-Madrid F., Santarém N., Schallmoser K., Ostenfeld M. S., Stoorvogel W., Stukelj R., Van der Grein S. G., Vasconcelos M. H., Wauben M. H. M., De Wever O., Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 4, 27066 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Dommelen S. M., Vader P., Lakhal S., Kooijmans S. A. A., van Solinge W. W., Wood M. J. A., Schiffelers R. M., Microvesicles and exosomes: Opportunities for cell-derived membrane vesicles in drug delivery. J. Control. Release 161, 635–644 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Greening D. W., Gopal S. K., Mathias R. A., Liu L., Sheng J., Zhu H.-J., Simpson R. J., Emerging roles of exosomes during epithelial–mesenchymal transition and cancer progression. Semin. Cell Dev. Biol. 40, 60–71 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Quesenberry P. J., Dooner M. S., Goldberg L. R., Aliotta J. M., Pereira M., Amaral A., Del Tatto M. M., Hixson D. C., Ramratnam B., A new stem cell biology: The continuum and microvesicles. Trans. Am. Clin. Climatol. Assoc. 123, 152–166 (2012). [PMC free article] [PubMed] [Google Scholar]
- 24.Robbins P. D., Morelli A. E., Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 14, 195–208 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keane T. J., Londono R., Turner N. J., Badylak S. F., Consequences of ineffective decellularization of biologic scaffolds on the host response. Biomaterials 33, 1771–1781 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Gilbert T. W., Freund J. M., Badylak S. F., Quantification of DNA in biologic scaffold materials. J. Surg. Res. 152, 135–139 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jenjaroenpun P., Kremenska Y., Nair V. M., Kremenskoy M., Joseph B., Kurochkin I. V., Characterization of RNA in exosomes secreted by human breast cancer cell lines using next-generation sequencing. PeerJ 1, e201 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stickney Z., Losacco J., McDevitt S., Zhang Z., Lu B., Development of exosome surface display technology in living human cells. Biochem. Biophys. Res. Commun. 472, 53–59 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Lunavat T. R., Cheng L., Kim D.-K., Bhadury J., Jang S. C., Lässer C., Sharples R. A., López M. D., Nilsson J., Gho Y. S., Hill A. F., Lötvall J., Small RNA deep sequencing discriminates subsets of extracellular vesicles released by melanoma cells—Evidence of unique microRNA cargos. RNA Biol. 12, 810–823 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Medberry C. J., Crapo P. M., Siu B. F., Carruthers C. A., Wolf M. T., Nagarkar S. P., Agrawal V., Jones K. E., Kelly J., Johnson S. A., Velankar S. S., Watkins S. C., Modo M., Badylak S. F., Hydrogels derived from central nervous system extracellular matrix. Biomaterials 34, 1033–1040 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolf P., The nature and significance of platelet products in human plasma. Br. J. Haematol. 13, 269–288 (1967). [DOI] [PubMed] [Google Scholar]
- 32.Hargett L. A., Bauer N. N., On the origin of microparticles: From “platelet dust” to mediators of intercellular communication. Pulm. Circ. 3, 329–340 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deatherage B. L., Cookson B. T., Membrane vesicle release in bacteria, eukaryotes, and archaea: A conserved yet underappreciated aspect of microbial life. Infect. Immun. 80, 1948–1957 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Escola J.-M., Kleijmeer M. J., Stoorvogel W., Griffith J. M., Yoshie O., Geuze H. J., Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J. Biol. Chem. 273, 20121–20127 (1998). [DOI] [PubMed] [Google Scholar]
- 35.Théry C., Regnault A., Garin J., Wolfers J., Zitvogel L., Ricciardi-Castagnoli P., Raposo G., Amigorena S., Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J. Cell Biol. 147, 599–610 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Théry C., Boussac M., Véron P., Ricciardi-Castagnoli P., Raposo G., Garin J., Amigorena S., Proteomic analysis of dendritic cell-derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 166, 7309–7318 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Clayton A., Turkes A., Dewitt S., Steadman R., Mason M. D., Hallett M. B., Adhesion and signaling by B cell-derived exosomes: The role of integrins. FASEB J. 18, 977–979 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Anderson H. C., Vesicles associated with calcification in the matrix of epiphyseal cartilage. J. Cell Biol. 41, 59–72 (1969). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson H. C., Matrix vesicles and calcification. Curr. Rheumatol. Rep. 5, 222–226 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Anderson H. C., Molecular biology of matrix vesicles. Clin. Orthop. Relat. Res. 314, 266–280 (1995). [PubMed] [Google Scholar]
- 41.Shapiro I. M., Landis W. J., Risbud M. V., Matrix vesicles: Are they anchored exosomes? Bone 79, 29–36 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalra H., Adda C. G., Liem M., Ang C.-S., Mechler A., Simpson R. J., Hulett M. D., Mathivanan S., Comparative proteomics evaluation of plasma exosome isolation techniques and assessment of the stability of exosomes in normal human blood plasma. Proteomics 13, 3354–3364 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Lv L.-L., Cao Y., Liu D., Xu M., Liu H., Tang R.-N., Ma K.-L., Liu B.-C., Isolation and quantification of microRNAs from urinary exosomes/microvesicles for biomarker discovery. Int. J. Biol. Sci. 9, 1021–1031 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zarovni N., Corrado A., Guazzi P., Zocco D., Lari E., Radano G., Muhhina J., Fondelli C., Gavrilova J., Chiesi A., Integrated isolation and quantitative analysis of exosome shuttled proteins and nucleic acids using immunocapture approaches. Methods 87, 46–58 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Ban J.-J., Lee M., Im W., Kim M., Low pH increases the yield of exosome isolation. Biochem. Biophys. Res. Commun. 461, 76–79 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Reing J. E., Zhang L., Myers-Irvin J., Cordero K. E., Freytes D. O., Heber-Katz E., Bedelbaeva K., McIntosh D., Dewilde A., Braunhut S. J., Badylak S. F., Degradation products of extracellular matrix affect cell migration and proliferation. Tissue Eng. Part A 15, 605–614 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Teodori L., Costa A., Marzio R., Perniconi B., Coletti D., Adamo S., Gupta B., Tarnok A., Native extracellular matrix: A new scaffolding platform for repair of damaged muscle. Front. Physiol. 5, 218 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crapo P. M., Gilbert T. W., Badylak S. F., An overview of tissue and whole organ decellularization processes. Biomaterials 32, 3233–3243 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vorotnikova E., McIntosh D., Dewilde A., Zhang J., Reing J. E., Zhang L., Cordero K., Bedelbaeva K., Gourevitch D., Heber-Katz E., Badylak S. F., Braunhut S. J., Extracellular matrix-derived products modulate endothelial and progenitor cell migration and proliferation in vitro and stimulate regenerative healing in vivo. Matrix Biol. 29, 690–700 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Pasquinelli A. E., Reinhart B. J., Slack F., Martindale M. Q., Kuroda M. I., Maller B., Hayward D. C., Ball E. E., Degnan B., Müller P., Spring J., Srinivasan A., Fishman M., Finnerty J., Corbo J., Levine M., Leahy P., Davidson E., Ruvkun G., Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 408, 86–89 (2000) [DOI] [PubMed] [Google Scholar]
- 51.Weber M. J., New human and mouse microRNA genes found by homology search. FEBS J. 272, 59–73 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Lagos-Quintana M., Rauhut R., Yalcin A., Meyer J., Lendeckel W., Tuschl T., Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12, 735–739 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Lagos-Quintana M., Rauhut R., Meyer J., Borkhardt A., Tuschl T., New microRNAs from mouse and human. RNA 9, 175–179 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim H.-J., Cui X.-S., Kim E.-J., Kim W.-J., Kim N.-H., New porcine microRNA genes found by homology search. Genome 49, 1283–1286 (2006). [DOI] [PubMed] [Google Scholar]
- 55.van Balkom B. W. M., de Jong O. G., Smits M., Brummelman J., den Ouden K., de Bree P. M., van Eijndhoven M. A. J., Pegtel D. M., Stoorvogel W., Würdinger T., Verhaar M. C., Endothelial cells require miR-214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood 121, 3997–4006 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Barile L., Lionetti V., Cervio E., Matteucci M., Gherghiceanu M., Popescu L. M., Torre T., Siclari F., Moccetti T., Vassalli G., Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc. Res. 103, 530–541 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Cantaluppi V., Biancone L., Figliolini F., Beltramo S., Medica D., Deregibus M. C., Galimi F., Romagnoli R., Salizzoni M., Tetta C., Segoloni G. P., Camussi G., Microvesicles derived from endothelial progenitor cells enhance neoangiogenesis of human pancreatic islets. Cell Transplant. 21, 1305–1320 (2012). [DOI] [PubMed] [Google Scholar]
- 58.Ekström E. J., Bergenfelz C., von Bülow V., Serifler F., Carlemalm E., Jönsson G., Andersson T., Leandersson K., WNT5A induces release of exosomes containing pro-angiogenic and immunosuppressive factors from malignant melanoma cells. Mol. Cancer 13, 88 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Distler J. H. W., Jüngel A., Huber L. C., Seemayer C. A., Reich C. F. III, Gay R. E., Michel B. A., Fontana A., Gay S., Pisetsky D. S., Distler O., The induction of matrix metalloproteinase and cytokine expression in synovial fibroblasts stimulated with immune cell microparticles. Proc. Natl. Acad. Sci. U.S.A. 102, 2892–2897 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Borges F. T., Melo S. A., Özdemir B. C., Kato N., Revuelta I., Miller C. A., Gattone V. H. II, LeBleu V. S., Kalluri R., TGF-β1-containing exosomes from injured epithelial cells activate fibroblasts to initiate tissue regenerative responses and fibrosis. J. Am. Soc. Nephrol. 24, 385–392 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang B., Yin Y., Lai R. C., Tan S. S., Choo A. B., Lim S. K., Mesenchymal stem cells secrete immunologically active exosomes. Stem Cells Dev. 23, 1233–1244 (2014). [DOI] [PubMed] [Google Scholar]
- 62.Mokarizadeh A., Delirezh N., Morshedi A., Mosayebi G., Farshid A.-A., Mardani K., Microvesicles derived from mesenchymal stem cells: Potent organelles for induction of tolerogenic signaling. Immunol. Lett. 147, 47–54 (2012). [DOI] [PubMed] [Google Scholar]
- 63.Brown B. N., Londono R., Tottey S., Zhang L., Kukla K. A., Wolf M. T., Daly K. A., Reing J. E., Badylak S. F., Macrophage phenotype as a predictor of constructive remodeling following the implantation of biologically derived surgical mesh materials. Acta Biomater. 8, 978–987 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Badylak S. F., Decellularized allogeneic and xenogeneic tissue as a bioscaffold for regenerative medicine: Factors that influence the host response. Ann. Biomed. Eng. 42, 1517–1527 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Sicari B. M., Johnson S. A., Siu B. F., Crapo P. M., Daly K. A., Jiang H., Medberry C. J., Tottey S., Turner N. J., Badylak S. F., The effect of source animal age upon the in vivo remodeling characteristics of an extracellular matrix scaffold. Biomaterials 33, 5524–5533 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mase V. J. Jr, Hsu J. R., Wolf S. E., Wenke J. C., Baer D. G., Owens J., Badylak S. F., Walters T. J., Clinical application of an acellular biologic scaffold for surgical repair of a large, traumatic quadriceps femoris muscle defect. Orthopedics 33, 511 (2010). [DOI] [PubMed] [Google Scholar]
- 67.Badylak S. F., Lantz G. C., Coffey A., Geddes L. A., Small intestinal submucosa as a large diameter vascular graft in the dog. J. Surg. Res. 47, 74–80 (1989). [DOI] [PubMed] [Google Scholar]
- 68.Webber J., Clayton A., How pure are your vesicles? J. Extracell. Vesicles 2, 24009896 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sicari B. M., Dziki J. L., Siu B. F., Medberry C. J., Dearth C. L., Badylak S. F., The promotion of a constructive macrophage phenotype by solubilized extracellular matrix. Biomaterials 35, 8605–8612 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/6/e1600502/DC1
table S1. Comparison of nucleic acid concentration from UBM, SIS, or dermis and their commercially available equivalents.
fig. S1. Nuclease protection assay.