

Researchers demonstrate that encapsulation is not necessary to achieve controlled, long-term protein release.
Keywords: controlled release, affinity release, drug delivery, PLGA, protein, central nervous system, hydrogel
Abstract
Encapsulation of therapeutic molecules within polymer particles is a well-established method for achieving controlled release, yet challenges such as low loading, poor encapsulation efficiency, and loss of protein activity limit clinical translation. Despite this, the paradigm for the use of polymer particles in drug delivery has remained essentially unchanged for several decades. By taking advantage of the adsorption of protein therapeutics to poly(lactic-co-glycolic acid) (PLGA) nanoparticles, we demonstrate controlled release without encapsulation. In fact, we obtain identical, burst-free, extended-release profiles for three different protein therapeutics with and without encapsulation in PLGA nanoparticles embedded within a hydrogel. Using both positively and negatively charged proteins, we show that short-range electrostatic interactions between the proteins and the PLGA nanoparticles are the underlying mechanism for controlled release. Moreover, we demonstrate tunable release by modifying nanoparticle concentration, nanoparticle size, or environmental pH. These new insights obviate the need for encapsulation and offer promising, translatable strategies for a more effective delivery of therapeutic biomolecules.
INTRODUCTION
Controlled biomolecule release is a key strategy for reducing both the systemic side effects associated with high drug concentrations and the frequency of drug administration. One of the most widely studied methods for controlling biomolecule release is encapsulation within a polymeric matrix, which slows diffusion. Poly(lactic-co-glycolic acid) (PLGA) is widely used for encapsulation because of its biocompatibility, biodegradability, wide range of degradation rates, and long clinical history (1, 2).
PLGA is typically formulated into injectable, protein-containing microparticles or nanoparticles by double-emulsion solvent evaporation, phase separation, or spray-drying techniques (3). All of these methods involve organic solvents, high shear forces, and/or high temperatures that result in protein denaturation and aggregation (4). In addition, PLGA protein formulations often suffer from low encapsulation efficiency and low protein loading (5, 6). The modification of process parameters—such as solvent type, volume (7, 8), or co-encapsulation with various excipients such as poly(ethylene glycol) (9), sugars (10), or bases (11)—can improve encapsulation efficiency and protein stability, yet, the requirement for encapsulation to achieve sustained release remains unchanged.
Embedding PLGA nanoparticles (PLGA np) within a hydrogel enables their localization at the site of hydrogel injection (12, 13), and is known to reduce the initial burst and to extend the release of encapsulated proteins (10, 14). While studying the release of three growth factors relevant to central nervous system (CNS) repair strategies [stromal cell–derived factor 1α (SDF1α; hereafter referred to as simply SDF), neurotrophin-3 (NT-3), and brain-derived neurotrophic factor (BDNF)], we found that their release profiles were independent of encapsulation. Sustained, burst-free release profiles were obtained whether the proteins were encapsulated in PLGA np that were then dispersed within a hydrogel (Fig. 1A) or simply mixed into the identical hydrogel with blank PLGA np (Fig. 1B). To the best of our knowledge, this is the first study of PLGA np being used for long-term controlled release without encapsulation. We hypothesize that adsorption due to short-range electrostatic interactions between the proteins and the PLGA np is the governing factor for release in this system, and we tested this hypothesis by controlling the release through nanoparticle concentration, nanoparticle size, and pH. To substantiate the data, we performed Monte Carlo simulations based on our release mechanism hypothesis and obtained similar release profiles.
Fig. 1. Two different PLGA np systems are compared for controlled protein release.
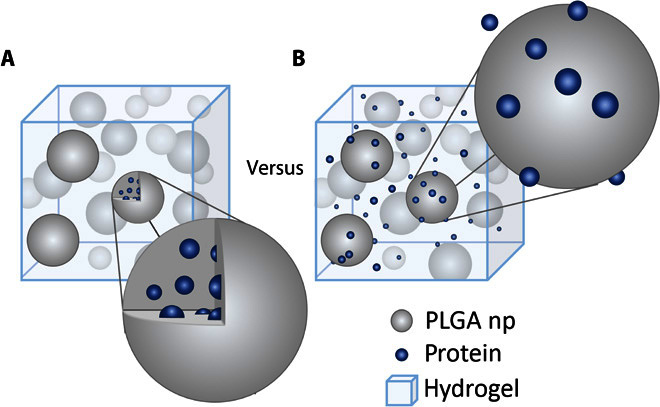
(A) Protein encapsulated in PLGA np dispersed in a hydrogel. (B) Protein and blank PLGA np dispersed in a hydrogel. For the latter, protein adsorbs to the PLGA np but is not encapsulated within them.
RESULTS
Sustained release without encapsulation
We tested the release of three proteins from two hydrogels with and without embedded PLGA np: SDF from a click cross-linked methylcellulose (XMC) hydrogel (15) (Fig. 2A), and NT-3 (Fig. 2B) and BDNF (Fig. 2C) from a physical blend of hyaluronan and methylcellulose (HAMC) (16). The diffusional release of SDF, NT-3, and BDNF from the hydrogel alone is rapid, generally reaching completion by day 2. By encapsulating the proteins in PLGA np before mixing them into the hydrogel (composite XMC and composite HAMC), we achieved sustained release for at least 28 days with no burst; however, we observed a 4- to 7-day delay in the release. Encapsulation efficiencies in PLGA np of 49 ± 4%, 47 ± 2%, and 47 ± 7% were achieved for SDF, NT-3, and BDNF, respectively.
Fig. 2. Controlled sustained release of positively charged proteins from PLGA np does not require encapsulation.
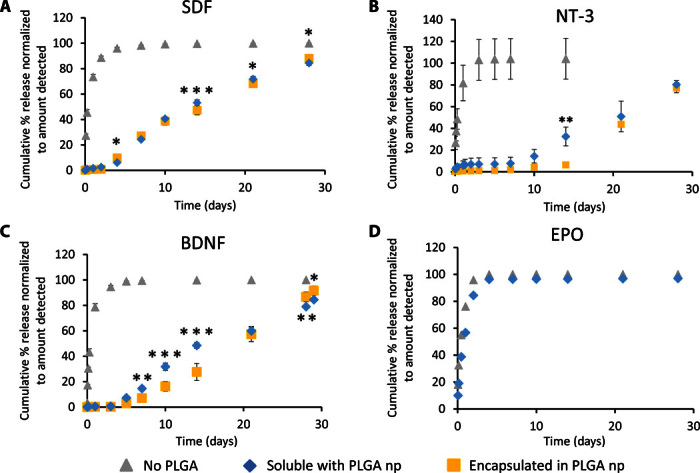
(A to C) Three different proteins show nearly identical release profiles whether encapsulated within PLGA np or simply mixed with blank PLGA np in a hydrogel: (A) SDF (pI 10.9, molecular mass of 8 kD, 200 ng/release), (B) NT-3 (pI 9.5, molecular mass of 27 kD, 1000 ng/release), and (C) BDNF (pI 10.9, molecular mass of 27 kD, 300 ng/release). The cumulative percentage released is significantly greater for soluble proteins with no PLGA np compared to soluble proteins with blank PLGA np or proteins encapsulated in PLGA np at all time points (P < 0.05, n = 3 for all releases, mean ± SD plotted). Asterisks indicate significant differences between release of soluble protein with PLGA np and release of protein encapsulated in PLGA np (*P < 0.05, **P < 0.01, ***P < 0.001). Curves for soluble SDF with no PLGA np and SDF encapsulated in PLGA np were taken with permission (15). (D) EPO (pI ~4, molecular mass of 30 kD, 84 ng/release) shows no attenuated release when mixed into HAMC with PLGA np versus just HAMC alone (without PLGA np), indicating that the phenomena observed with SDF, NT-3, and BDNF are based on short-range electrostatic interactions between positively charged proteins and negatively charged PLGA (n = 3, mean ± SD plotted).
In an effort to overcome this delay, we mixed additional proteins directly into the hydrogel with the protein-loaded PLGA np. We anticipated an initial burst release followed by a sustained release, reflecting fast diffusion from the hydrogel and slower diffusion from the nanoparticles. Instead, we observed an identical release profile, with only the total amount of released proteins increased (fig. S1) (17). We also observed a similar release profile when all of the proteins were simply mixed into the hydrogel with blank PLGA np (Fig. 2, A to C). All proteins remained bioactive when released using this encapsulation-free method (fig. S2), with all of the proteins incorporated into the system being available for release. This overcomes the <50% efficiency obtained with PLGA np encapsulation.
Adsorption governed by electrostatic interactions
To understand the release mechanism, we examined the physical characteristics of this encapsulation-free release system. Nanoparticle morphology and size were characterized by transmission electron microscopy (TEM) and dynamic light scattering (DLS). Nanoparticles were spherical, with some visible surface morphology, an average diameter of 293 ± 19 nm, and a polydispersity index of 0.21 ± 0.02 by DLS (fig. S3). ζ potential measurements showed that the surface of the PLGA np was negatively charged (−14.0 ± 0.7 mV), in agreement with previous reports (18, 19). The swelling ratio of HAMC was compared to that of composite HAMC (that is, PLGA np dispersed in HAMC). Composite HAMC had a significantly higher swelling ratio after 3 days compared to HAMC alone (fig. S4).
SDF, NT-3, and BDNF all have a net positive charge at physiological pH, with an isoelectric point (pI) of 10.9 for SDF, 9.5 for NT-3, and 10.1 for BDNF (20). We postulated that the delayed release that we observed was primarily caused by adsorption due to short-range electrostatic interactions between the protein and the PLGA np. To test this hypothesis, we examined the release of erythropoietin (EPO)—another protein that has shown benefits for CNS repair strategies (21) and has a pI of 3.3 to 4.3 (22), making it negatively charged at physiological pH. Soluble EPO was released just as rapidly from composite HAMC as from HAMC alone, with complete release observed after 2 days (Fig. 2D). This is consistent with our hypothesis that adsorption is mediated by electrostatic interactions.
The hydrogel itself seems to have a minimal role in controlling release because the release of SDF from XMC and that of NT-3 and BDNF from HAMC show similar profiles. We examined this further by replacing both XMC and HAMC with another common gel, agarose. Release of soluble BDNF from agarose alone was the same as that from HAMC alone, whereas release of soluble BDNF from agarose containing PLGA np exhibited the same delayed-release profile that we observed with HAMC containing PLGA np (Fig. 3A). Incubation of BDNF with free PLGA np in artificial cerebrospinal fluid (aCSF) at the same protein/PLGA ratio as in the hydrogels resulted in almost complete adsorption within minutes (Fig. 3B), suggesting that the mechanism for controlled release is mediated by the nanoparticles, not the hydrogel.
Fig. 3. Sustained release of BDNF from agarose containing PLGA np is disrupted by increased salt.
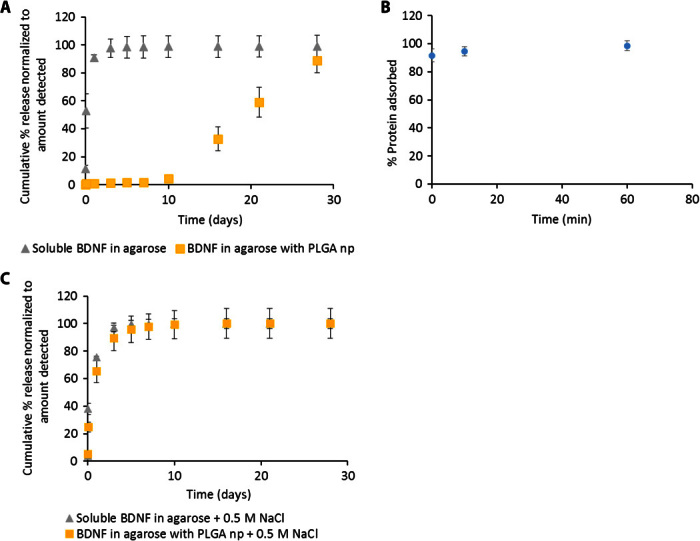
(A) BDNF shows the same delayed- and sustained-release profile from agarose containing PLGA np as from HAMC with PLGA np while diffusional release from agarose alone is still fast. (B) Almost all soluble BDNF is adsorbed to PLGA np after incubation, even at short times. (C) NaCl (0.5 M) completely disrupts the interaction of BDNF with PLGA np, resulting in purely diffusional release (P > 0.05 for all time points, n = 3 for all releases, mean ± SD plotted).
Adsorption mediated by electrostatic interactions can be disrupted by raising the level of competing ions in solution. When the salt concentration of the release media was raised from 149 mM (physiologically relevant) to 0.5 M, the release of BDNF from the agarose PLGA np composite was fast and almost identical to the release profile observed in the absence of the nanoparticles (Fig. 3C).
Tuning release by controlling the surface area of PLGA np
To further investigate the role of PLGA np in controlling release, we examined the role of PLGA np surface area because the amount of protein adsorbed is proportional to the available surface area. We investigated the release of BDNF from composite HAMC containing either 300-nm- or 1000-nm-diameter nanoparticles. Assuming similar densities and spherical nanoparticles, the 1000-nm nanoparticles would have three times less available surface area than the 300-nm nanoparticles for the same mass (see calculations in the Supplementary Materials). Consistent with our hypothesis that the interaction between PLGA np and positively charged proteins controls release, the larger nanoparticles (with the overall lower surface area) resulted in faster release (Fig. 4A).
Fig. 4. Release of positively charged proteins from composite HAMC can be tuned by changing the available nanoparticle surface area.
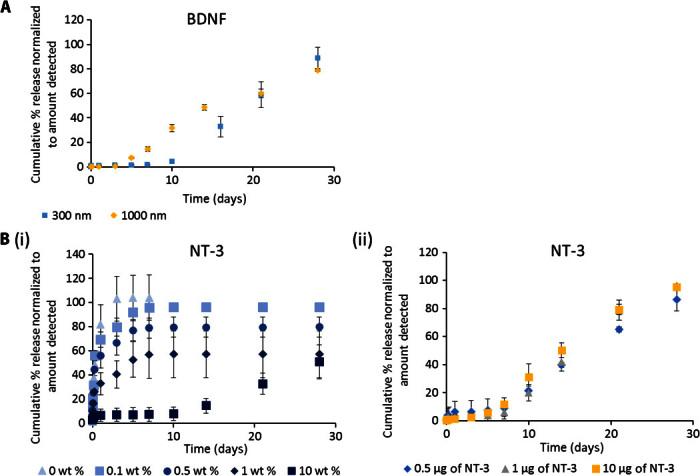
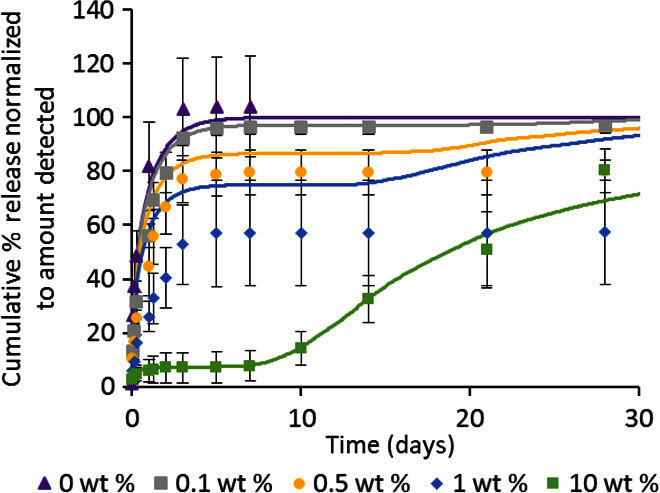
(A) Cumulative percentage release of BDNF from composite HAMC is higher with 1000-nm-diameter PLGA np than with 300-nm-diameter PLGA np while keeping PLGA np mass constant. (B) (i) Release of NT-3 from composite HAMC with 0, 0.1, 0.5, 1, and 10 wt % PLGA np. Cumulative percentage of NT-3 released is significantly lower for 10 wt % PLGA np than for all other curves at t > 1 day (P < 0.05). Cumulative percentage of NT-3 released is significantly higher with 0 wt % PLGA np than with 0.1 wt % (t = 1 day, P < 0.05), 0.5 wt % (1 day ≤ t ≤ 3 days, P < 0.05), and 1 wt % (t > 3 hours, P < 0.05). (ii) Release of 0.5, 1, or 10 μg of NT-3 from composite HAMC with 10 wt % PLGA np. The concentration of NT-3 incorporated can be increased up to 20 times with virtually no change in release profile. Cumulative percentage of NT-3 released is significantly higher for 10 μg than for 1 and 0.5 μg at 10 and 14 days (P < 0.05) and is significantly lower for 0.5 μg than for 1 and 10 μg at 21 and 28 days (P < 0.05) (n = 3 for all releases, mean ± SD plotted).
The available PLGA np surface area can similarly be controlled by the concentration of nanoparticles within the gel. We observed a direct correlation between the number of PLGA np dispersed in the hydrogel and the release rate of NT-3 from composite HAMC. With the lowest concentration of nanoparticles (0.1 wt %; Fig. 4B, i), there was a faster initial release rate that approached the diffusion-controlled release profile of NT-3 from HAMC alone (0 wt %; Fig. 4B, i). As the concentration of nanoparticles increased, the release rate slowed. We were able to increase the concentration of NT-3 by 20-fold (from 0.5 to 10 μg) in the 10 wt % PLGA np composite while maintaining a virtually identical release profile (Fig. 4B, ii). Thus, we have a facile method to control both the release rate and the amount of protein released.
Tuning release by modifying adsorption affinity
Because adsorption can be controlled by modifying the interaction strength between the proteins and the PLGA np, we next examined the role of supernatant pH in protein release from our drug delivery system (DDS), as supernatant pH would affect both the charge of the protein and that of the PLGA np surface. SDF was released from composite XMC into media at pH 3, 5, and 7. The release rate of SDF from composite XMC (that is, in the presence of nanoparticles) significantly increased with decreasing pH (Fig. 5A); yet, in the absence of nanoparticles, the release rate of SDF was unaffected by decreasing pH (fig. S5). Moreover, the difference in release cannot be explained by destabilization of the gel at lower pH, because there was no significant mass loss of PLGA from the hydrogel over the first 7 days of release at either pH 3 or pH 7 (fig. S6). Thus, these data indicate that pH can be used to control release and that, at lower pH (where the carboxylate anions of PLGA are protonated), the electrostatic interaction between PLGA anions and protein cations is diminished.
Fig. 5. Release of positively charged proteins can be tuned by changing the environmental pH.
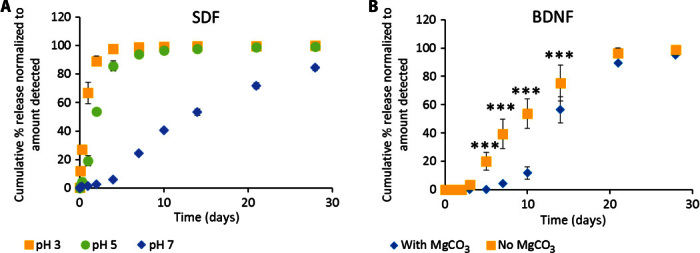
(A) Release of SDF from composite XMC into media at pH 3, pH 5, or pH 7. Cumulative percentage release of SDF is significantly greater at pH 3 than at pH 5 (t < 10 days, P < 0.01) and pH 7 (t > 0 days, P < 0.0001), likely because PLGA carboxylate anions are protonated to carboxylic acids, thereby reducing electrostatic interactions with positively charged proteins. (B) Release of BDNF from HAMC with PLGA np with or without encapsulated magnesium carbonate (MgCO3). Release is delayed with encapsulated MgCO3 (a basic salt), which neutralizes acidic degradation products and thereby maintains a higher/neutral local pH (n = 3 for all releases, mean ± SD plotted).
To investigate the effects of local pH on protein release, we compared the release of BDNF from composite HAMC containing PLGA np with and without encapsulated MgCO3. Because PLGA is known to degrade into acidic products by bulk degradation, the encapsulation of basic salts has been used to delay pH changes within PLGA np (11). Release of soluble BDNF from composite HAMC had a delay of approximately 7 to 10 days with encapsulated MgCO3 (Fig. 5B). In contrast, in the absence of MgCO3, the delay was shortened to just 4 days. This suggests that the mechanism for protein release is related to a change in PLGA charge from negative carboxylate anions to neutral carboxylic acids, which is slowed in the presence of MgCO3.
Monte Carlo simulations
As a further test of our hypothesis for release mechanism, we performed three-dimensional on-lattice Monte Carlo simulations of protein release from a hydrogel with a stochastic logistic-population model for the degradation of the PLGA np. The nanoparticles were modeled as cubic obstacles having short-range interactions with the proteins. We included both a deep attractive potential well to model electrostatic adsorption and a low repulsive barrier to adsorption, with both potentials approaching zero as the nanoparticles degrade. This scheme is observable qualitatively in heuristic two-dimensional simulations (see movie S1). We used this model to reproduce the release curves obtained in Fig. 4B(i) by fitting the simulation parameters to best match the case of 10 wt % PLGA np, then using those same parameters for all other simulations without any additional fitting (Fig. 6). The combined reduced χ2 value for all simulations was 3.9.
Fig. 6. Monte Carlo simulations agree well with experimental results.
Release of NT-3 from HAMC with 10 wt % PLGA np (Fig. 4B, i) was fitted using three-dimensional on-lattice Monte Carlo simulations. Simulations were then run for all other nanoparticle concentrations in Fig. 4B(i) using the same parameters. The combined reduced χ2 value for all simulations was 3.9. Symbols represent data, whereas solid lines represent the simulation results.
Although the simulation parameters were chosen to best reproduce the 10 wt % curve with its clear plateau region and secondary rise, the 0, 0.1, and 0.5 wt % curves are also well described. Intermediate concentrations, such as 1 wt %, show the least agreement, demonstrating the implicit effects of a finite gel size on the relative significance of protein-nanoparticle interactions and diffusion in Monte Carlo simulations.
DISCUSSION
Models of drug release from PLGA np usually consider polymer degradation, erosion, and diffusion of the drugs through the resulting water-filled pores as rate-determining (1, 23). However, our results show very similar release profiles for three different proteins whether they are encapsulated in PLGA np or simply mixed with them, indicating that another mechanism must be involved.
We postulate that initially the protein is fully adsorbed to the negatively charged nanoparticle surface (Fig. 7, i). As the nanoparticle begins to degrade, acidic components build up and decrease the local pH (Fig. 7, ii). At a certain threshold, the original negative carboxylate anion on the nanoparticle surface becomes protonated to carboxylic acid, thereby weakening the electrostatic interactions with the positively charged proteins and initiating release (Fig. 7, iii). Release can then be governed by nanoparticle degradation and protein diffusion and/or adsorption/desorption, depending on the system.
Fig. 7. Adsorption may be rate-limiting for the release of positively charged proteins from PLGA np.
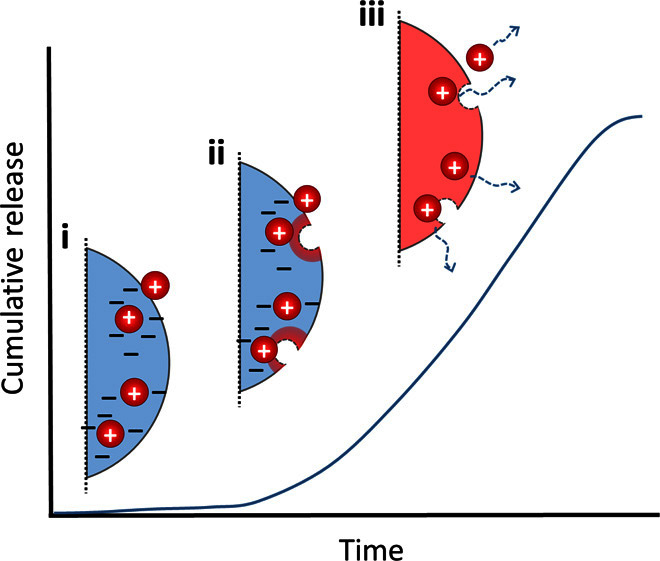
(i) Initially, the protein is fully adsorbed to the negatively charged nanoparticle surface. (ii) As the nanoparticle begins to degrade, acidic components build up and decrease the local pH. (iii) At a certain threshold, the nanoparticle surface becomes neutral, weakening the electrostatic interactions with the positively charged proteins and initiating release.
It is possible that the presence of the PLGA np affects the release of proteins simply by changing the gel mechanics. Methylcellulose forms a gel via hydrophobic interactions, and its shear modulus increases upon incorporation of PLGA np, suggesting additional hydrophobic interactions between methylcellulose and PLGA np (12). These hydrophobic interactions have been postulated to affect the release rate of proteins from PLGA np (10). However, our results show that the addition of PLGA np actually increases the hydrogel swelling ratio, likely as a result of the presence of carboxylate anions on the PLGA np surface. Increased swelling of poly(acrylic acid) gels with the addition of polystyrene nanoparticles has been attributed to the sulfonic acid groups on the nanoparticle surface (24), and carboxylate anions have previously been found to affect the swelling of hydrogels. Increased swelling should lead to an increased mesh size and subsequently increased release rate of a protein (25); however, our data show the opposite effect, indicating an interaction between PLGA np and the protein. In addition, changes in mesh size would be expected to affect all proteins, depending on their size; however, EPO, which has a molar mass larger than that of SDF and similar to those of NT-3 and BDNF, does not show sustained release in the presence of PLGA np. Thus, gel mechanics are unlikely the mechanism controlling the release of proteins from composite PLGA np hydrogels.
Adsorption is recognized as one of the factors affecting the release of biomolecules encapsulated in PLGA particles (26) and is a major cause of incomplete release (27, 28). Adsorption of proteins at the surface of PLGA particles is also often cited as the cause of the initial burst observed in the release from PLGA np formulations (29) and is attributed to a combination of electrostatic and hydrophobic interactions (18, 30). The release of proteins adsorbed to the surface of PLGA particles is often fast (within the first 1 to 8 hours of release) and has not been fully studied (18, 31).
To examine the possibility of a hydrogel effect, we tested release from agarose, an uncharged polymer with a mechanism of gelation (32) different from that of either XMC (15) or HAMC (16, 33). The release profiles of BDNF were the same whether from composite HAMC or composite agarose, suggesting a specific interaction between the protein and the PLGA np, not the hydrogel. When we incubated BDNF with PLGA np alone at the same ratio used in the release studies, almost all the BDNF was removed from the supernatant within minutes, demonstrating a strong interaction with the PLGA np.
Adsorption is governed by noncovalent interactions, including electrostatic, hydrophobic, and van der Waals forces. Given that SDF, NT-3, and BDNF are all positively charged at neutral pH, and a negatively charged protein (EPO) did not show the same release behavior, we hypothesized that electrostatic interactions were governing protein adsorption. The range of electrostatic interactions is governed by the Debye length. For our release system, the Debye length is very small (<1 nm), so long-range electrostatic interactions are not important; however, short-range interactions likely still exist. Electrostatic interactions such as these can be disrupted with an excess of ions. In fact, BDNF is often purified using ion exchange chromatography, and a salt concentration of 0.5 M is within the range used for elution (34, 35). When we examined the release of BDNF from composite agarose in the presence of 0.5 M NaCl, the release rate approached diffusional values. This supports a hypothesis of predominantly electrostatically controlled adsorption to the PLGA np, with little contribution from hydrophobic and van der Waals forces (36).
Control over the release rate is essential for the success of sustained delivery formulations. We explored different parameters of our DDS and their effects on the release rate of soluble proteins. Models of drug release controlled by adsorption/desorption demonstrate that release is governed by the number of available binding sites and the strength of the interaction (18, 37).
Nanoparticle size can be readily varied by modifying the formulation conditions (4) and is therefore a facile method to control release. Assuming a similar density, smaller nanoparticles provide an overall larger surface area and would therefore increase the extent of adsorption. We show that BDNF released from HAMC with 300-nm nanoparticles showed a longer delay and slower release than that from HAMC with 1000-nm nanoparticles.
Because protein loading and nanoparticle concentration are decoupled in our DDS, changing the nanoparticle concentration provides a simple method to tune release. In general, release of NT-3 is faster with lower nanoparticle concentrations. When the nanoparticle concentration is decreased from 10 to 1 wt % with a constant amount of protein, a significant burst release is seen. On the other hand, increasing the protein concentration 10-fold while maintaining the nanoparticle concentration at 10 wt % does not change the release profile of NT-3 despite an identical nanoparticle/protein mass ratio as the 1 wt % case above. A similar effect is seen in affinity-controlled release systems: as long as the available binding sites are in excess, the release rate depends on the concentration of binding ligand (here the PLGA np), not on the binding ligand/protein ratio (38).
Theoretically, complete surface coverage of our PLGA np occurs at 1.41 wt % protein loading (see calculations in the Supplementary Materials). When increasing protein concentration, we never exceeded 0.1 wt % protein loading by mass because of the significant costs of these proteins; however, on the basis of our calculations, we expect that a 1–wt % loading could be achieved without significantly altering the release profile. This suggests that our DDS could deliver substantially higher loadings than previously reported for PLGA encapsulation of therapeutic proteins such as NT-3 (39), BDNF (40–42), or nerve growth factor (43, 44).
PLGA np lose their negative charge at or below pH 5 (19), thus decreasing their charge attraction to positively charged proteins. We observed increased release rates of SDF at pH 3 and 5 compared to pH 7. This is not attributable to gel destabilization at low pH because there is no significant mass loss of PLGA np over 7 days at pH 3 or pH 7. The ability to increase the release rate at acidic pH may have interesting implications for in vivo delivery at acidic sites, such as the tumor microenvironment (45).
In all of the release profiles, a constant, slow release phase was observed after the initial delay or burst. PLGA degrades by bulk degradation into acidic components, which are often trapped within the nanoparticles before their dissolution, decreasing the pH inside the nanoparticles (11, 28). We postulate that this reduction in local pH reduces the magnitude of the protein-polymer interaction (as shown in our pH study) and initiates protein release as a certain pH threshold is reached. Thus, by controlling the pH change within the PLGA np, it should be possible to eliminate or extend this delay. Indeed, encapsulation of a basic salt, MgCO3, slowed the release of BDNF from PLGA np dispersed in HAMC, likely by decreasing the rate of PLGA degradation and pH change within the nanoparticles. Similarly, PLGA polymers with different properties (molecular weight, end group, and lactide/glycolide ratio) could also be used to formulate nanoparticles with various degradation rates to achieve different release profiles.
Agreement between the minimal Monte Carlo simulations and the experimental data further suggests that the release curves result from (i) an initial diffusive regime during which proteins either escape the hydrogel or adsorb to a nanoparticle, (ii) a plateau region, and, finally, (iii) a secondary rise attributable to the release of the proteins following PLGA np degradation. The model overpredicts the initial burst for lower nanoparticle concentrations, especially the 1 wt % case, which is likely a result of the small finite size of the simulated system. The model also predicts an eventual rise (phase 3) for lower nanoparticle concentrations that we expected but did not observe experimentally. It is possible that the releases were simply not taken to long-enough time points to observe this phase, or perhaps a portion of the protein was adsorbing irreversibly at longer time points when the pH changes were not as rapid. However, reproducing the general shape of the release curves and obtaining adequate agreement for five different release curves using a single set of parameters show that the release hypothesis is robust.
Although we cannot rule out the possibility that the polymer degradation, irrespective of the pH change, decreases the affinity of the protein for the nanoparticles, previous studies have shown that the available surface area for adsorption actually increases as the nanoparticles degrade, which would likely result in increased adsorption and slower release (27). Therefore, it seems more likely that polymer degradation indirectly affects protein release through pH change.
Using PLGA for the release of bioactive proteins without encapsulation is a paradigm shift in controlled delivery, and the use of the well-established PLGA facilitates clinical translation. Furthermore, controlled release is obtained without any modifications to the protein that may affect activity. Tunable release is achieved by varying simple formulation parameters, and stimuli-responsive release is demonstrated by modifying environmental pH. The next step is the extension of these concepts to additional proteins. Although we show similar release profiles for three different proteins, not all proteins can be expected to interact in the same way. A simple adsorption experiment could quickly ascertain whether this method is suitable for a given protein. Polymer composition (for example, end group and lactide/glycolide ratio) might also be tailored to suit a particular protein. Although not explored herein, for controlled release of negatively charged proteins, PLGA np could be coated with positively charged polymers, such as chitosan (46), or positively charged polymeric nanoparticles that degrade into basic components could be used instead. The results presented here provide the basis for a fundamental change in the way we use PLGA for protein delivery.
MATERIALS AND METHODS
PLGA was purchased from Sigma-Aldrich (50:50 lactide/glycolide, carboxy-terminated, molecular mass of 7 to 17 kD), and LACTEL Absorbable Polymers were purchased from Durect Corp. (50:50 lactide/glycolide, carboxy-terminated, 0.15 to 0.25 dl/g in hexafluoroisopropanol). Proteins were sourced as follows: SDF (R&D Systems), NT-3 (PeproTech), BDNF (PeproTech), and EPO (Janssen). Enzyme-linked immunosorbent assay (ELISA) kits for SDF and NT-3 were purchased from R&D Systems, and those for BDNF were purchased from Promega. All other materials were obtained as indicated.
PLGA np formation
PLGA np were formed by water/oil/water double-emulsion solvent evaporation, as previously described by Baumann et al. (47). Briefly, 120 mg of PLGA and 0.05% Pluronic NF-127 (BASF) were dissolved in 900 μl of dichloromethane (DCM; Caledon Laboratories) and vortexed with 100 μl of bovine serum albumin (BSA; 120 mg/ml; Sigma Aldrich) in aCSF [149 mM NaCl, 3 mM KCl, 0.8 mM MgCl2, 1.4 mM CaCl2, 1.5 mM Na2HPO4, and 0.2 mM NaH2PO4 (pH 7.4)] to achieve 10% w/w BSA/PLGA in the final formulation. The mixture was then sonicated for 2 min on ice. For encapsulation of SDF, NT-3, BDNF, or EPO, these proteins were dissolved in the aqueous phase before sonication. The primary emulsion was then added to 3 ml of a 2.5% (w/v) **solution of poly(vinyl alcohol) (30 to 70 kg/mol; Sigma Aldrich), vortexed, and sonicated for 2 min on ice. This secondary emulsion was added to a hardening bath of poly(vinyl alcohol) (PVA) and stirred for a minimum of 4 hours to allow the DCM to evaporate. The resultant nanoparticles were washed four times by ultracentrifugation, lyophilized, and stored at −20°C. Nanoparticles were sized by DLS (Malvern Instruments). For 1000-nm nanoparticles, the volume of PVA added in the secondary emulsion was increased to 5 ml.
Measurement of encapsulation efficiency
Stromal cell–derived factor 1α
Encapsulation efficiency was measured by dissolving 10 mg of dry protein-loaded PLGA np in 1 ml of dimethyl sulfoxide for 1 hour at 37°C. Ten milliliters of 0.05 M NaOH containing 0.5% (w/v) sodium dodecyl sulfate was then added, and the sample was left for another hour at room temperature on a rocker. SDF concentration was then measured by ELISA at a 1:10 dilution as directed.
BDNF/NT-3
Encapsulation efficiency was measured by dissolving 5 mg of dry protein-loaded PLGA np in 5 ml of 0.05 M NaOH on a shaker at room temperature for 30 min. The protein concentration was determined by ELISA.
Preparation of PLGA np/hydrogel DDS
Composite HAMC
To form the HAMC hydrogel, we dissolved methylcellulose (300 kg/mol; Shin-Etsu) and sodium hyaluronate (HA; 1.4 to 1.8 × 106 g/mol; NovaMatrix) in aCSF, using a dual asymmetric centrifugal mixer (FlackTek), to a final concentration of 2.8% (w/v) HA and 6% (w/v) methylcellulose. Nanoparticles were dispersed in aCSF at 20% (w/w) by 5 min of bath sonication, and protein (NT-3, BDNF, or EPO) was then added. Composite HAMC was formed by blending the nanoparticle dispersion and HAMC at a 1:1 ratio using a dual asymmetric centrifugal mixer.
Composite XMC
XMC was prepared as previously described (15), except that empty PLGA np dispersed in aCSF and SDF were added before mixing in the cross-linker.
Composite agarose
Ultralow gelling temperature SeaPrep Agarose (Lonza) was dissolved in aCSF to a concentration of 3% (w/v) by microwaving on high power in 4-s bursts and vortexing until clear. The solution was allowed to cool to room temperature before further handling. Nanoparticles were dispersed in aCSF at 20% (w/w) by 5 min of bath sonication, and BDNF was then added. Room temperature agarose was added to the nanoparticle dispersion at a 1:1 volume ratio, and a dual asymmetric centrifugal mixer was used to create the composite agarose. One hundred microliters of composite agarose was pipetted into a 2-ml centrifuge tube and allowed to gel at 4°C for 1 hour.
In vitro release assays
Composite HAMC/XMC
One hundred microliters of either HAMC, XMC, composite HAMC, or composite XMC, with or without dispersed protein, was injected into a 2-ml centrifuge tube. After 10 min of gelation at 37°C, 900 μl (for release of BDNF and NT-3) or 400 μl (for release of SDF) of prewarmed aCSF was added. For BDNF releases, the aCSF also contained 0.1% (w/v) BSA to ensure sustained bioactivity. The supernatant was completely replaced at designated time points for the duration of the release study, and the protein concentration was assessed by the relevant ELISA. At the conclusion of the study, the remaining gel was processed as described above for encapsulation efficiency, and the remaining protein was quantified by ELISA. Releases were performed in triplicate. For release of SDF at different pH values, the pH of the aCSF was adjusted to pH 5 or pH 3 and used in both the gel formation and the supernatant.
Agarose/agarose with high salt
Agarose or agarose composite with or without dispersed BDNF, previously prepared in a 2-ml centrifuge tube, was placed in an incubator at 37°C to warm for 10 min. Nine hundred microliters of prewarmed aCSF (0.1% w/v BSA) was added to the composite, and this supernatant was completely replaced at designated time points for the duration of the release study. For the high-salt agarose composite, aCSF with 0.5 M NaCl was used to create the gel and the release supernatant. At the end of the study, 0.5 ml of 0.5 M NaCl was added to the gel to release any remaining BDNF. ELISA was used to determine BDNF concentration at each time point and the BDNF remaining in the gel. Releases were performed in triplicate.
Adsorption study
Two micrograms of BDNF was mixed with 20 mg of PLGA np in 200 μl of aCSF and incubated for 0 min (as fast as possible), 10 min, and 60 min at 37°C. At each designated time point, the mixture was centrifuged at 12,000 rpm for 2 min to pellet the nanoparticles. The supernatant was carefully removed and analyzed by ELISA. Controls of soluble BDNF in aCSF with no nanoparticles were processed in the same way at each time point to take into account any losses resulting from centrifugation or adsorption to the tube. Samples of PLGA np in aCSF with no BDNF were used as blanks for the ELISA. The amount of BDNF adsorbed to the nanoparticles was calculated as 2 μg minus the amount detected in the supernatant. Each time point was performed in triplicate.
Bioactivity assays
All animal procedures were performed in accordance with the Guide to the Care and Use of Experimental Animals (Canadian Council on Animal Care), and protocols were approved by the Animal Care Committee at the University of Toronto.
NT-3: Dorsal root ganglia explant assay
The bioactivity of released NT-3 was quantified as previously described by Stanwick et al. (10). Sprague-Dawley rat embryo dorsal root ganglia (DRG; E17) were harvested and pooled in neural basal media containing 1 volume % fetal bovine serum, 2 volume % B-27 serum-free supplement, 1 volume % penicillin-streptomycin, and 1 volume % l-glutamine. The DRG were placed on glass coverslips (12 mm in diameter) coated with poly-d-lysine (50 μg/ml in sterile water) and laminin (5 μg/ml in phosphate-buffered saline) in a 24-well plate. Media with aCSF alone (control media), media with soluble (standard) NT-3, or media with released NT-3 were added to each well. The DRG were fixed after a 48-hour incubation in 4% paraformaldehyde and stained with NF200 and 4′,6-diamidino-2-phenylindole (DAPI; Sigma Aldrich). DRG were imaged on an Olympus FV1000 confocal microscope. Image analysis was performed with ImageJ (National Institutes of Health). For each DRG, the DAPI-positive area of the multicellular DRG body was subtracted from the total NF200-positive area (center + extended neurites) to yield the area of neurite outgrowth. A minimum of three DRGs were assayed per group.
SDF: Neurosphere migration assay
In vitro activity of SDF was quantified as previously described (15). Briefly, 125 μl of SDF release samples or controls was diluted to 2000 ng/ml (or as high as possible) in serum-free media and adjusted to pH 7. Each was then added to 125 μl of adult rat spinal cord neurosphere suspension in a fibronectin-coated 48-well tissue culture plate. Neurospheres were allowed to adhere for 1 hour in a tissue culture incubator at 37°C and 5% CO2. Images of four neurospheres per well were collected using an inverted Olympus laser scanning confocal microscope with automated stage at ×10 magnification. The coordinates of each image were saved. The plate was then returned to the incubator. Using the saved coordinates, we again imaged the same neurospheres 24 hours later. Images were analyzed using ImageJ (National Institutes of Health). The initial area of the neurosphere (A1) and the final area of cells migrated out from the neurosphere (A2) were measured and subtracted to yield the total area of migration (A2 − A1).
BDNF: DRG neurite outgrowth assay
In vitro activity of BDNF was determined using a DRG bioassay. Rat embryo DRG (E17 female Sprague-Dawley rats, n = 3) were removed and pooled in media composed of neural basal media supplemented with 2% B-27 serum-free supplement, 1% penicillin-streptomycin, and 1% l-glutamine (Life Technologies). The DRG were then placed on 12-mm-diameter glass coverslips coated with poly-d-lysine (50 μg/ml) and laminin (5 μg/ml) in a 24-well plate. Each release sample replicate was tested on a separate plate. All wells contained three DRG and were treated with 0.5 ml of media and 0.5 ml of the BDNF release study supernatant (7, 28, 35, and 42 days). For the controls, 0.5 ml of media and 0.5 ml of aCSF (without BDNF) were added to the wells. The DRG were grown for 48 hours at 37°C and 5% CO2, fixed with 4% paraformaldehyde, and processed for immunocytochemistry. The DRG were imaged using an inverted confocal laser scanning microscope (Olympus FV1000). Neurite outgrowth area was calculated by subtracting the cell body area from the total area of the DRG neurite outgrowth. To account for any differences in cell body size, we standardized the neurite outgrowth area to the cell body area for statistical analysis. Neurite outgrowth of DRG treated with release samples was compared to controls to assess bioactivity.
Transmission electron microscopy
PLGA np were suspended in water at 2 mg/ml with sonication for 5 min. A sample (5 μl) was deposited onto a freshly glow-discharged 400-mesh carbon-coated copper TEM grid (Ted Pella Inc.) and allowed to adhere for 4 min. Excess liquid was removed with filter paper, and nanoparticles were then stained with 2% uranyl acetate (UA, w/v, 5 μL, pH 4.3, 15 seconds). Stain was wicked away with filter paper, and samples were immediately imaged using a Hitachi H-7000 microscope operating at 75 kV. Images were captured using an Advanced Microscopy Techniques XR-60 CCD camera with typical magnifications between ×30,000 and ×75,000.
ζ potential measurements
PLGA np were suspended at 0.1 mg/ml in distilled water with 1 mM KCl. ζ potential was measured in a disposable folded capillary cell using a Zetasizer Nano ZS (Malvern Instruments).
Swelling study
An in vitro swelling study was performed as previously described (15). Briefly, 180 μl of HAMC or composite HAMC was injected into a 2-ml centrifuge tube. After incubation at 37°C for 10 min, 1620 μl of prewarmed aCSF was added. At each time point, the tube was weighed after complete removal of the aCSF. The normalized swelling ratio, Q, was determined by the equation
where mt is the total mass at each time point, m0 is the total mass at time 0, and mtube is the mass of the centrifuge tube.
Monte Carlo modeling
The on-lattice Monte Carlo simulations model proteins as noninteracting point particles stochastically diffusing on a three-dimensional cubic lattice of size a, representing the hydrogel, with an effective diffusion coefficient (48). Proteins that diffuse to the edge of the cubic simulation box permanently escape the hydrogel. For determination of the Monte Carlo time step, τMC (in hours), for a set of simulations of a hydrogel of size L, a simulation is run in the absence of PLGA np, and the rapid diffusional release rate is matched to the experimental cumulative release. For L = 100a, the conversion (0.0148 ± 0.0001) hours/τMC is found. To simulate embedded PLGA np, we introduce a number of impenetrable obstacle lattice sites that exclude proteins. Each randomly placed, nonoverlapping nanoparticle (labeled i) is cubic with a hard-core size 2R + a. In addition to the hard core, each nanoparticle interacts with the proteins through a double step-function potential. Immediately adjacent to the core is a deep attractive potential well of depth ui(t) per thermal energy and range a. Large negative values of ui(t) cause proteins to be tightly bound to nanoparticles. In addition, there is a repulsive step-potential height εi(t) per thermal energy and of range a, which represents the barrier to adhesion. Adsorption is modeled via a simple Metropolis algorithm. For a simulated decrease in attraction due to PLGA hydrolysis, the potentials approach zero after long times as and , where c is the number density of nanoparticles, K is the nanoparticle-hydrogel degradation-product carrying capacity per hydrogel volume, and Ni(t) is the stochastically increasing number of degradation products. The number of degradation products grows randomly via an algorithm that reproduces logistic-population statistics , where the summation occurs over all nanoparticles in the hydrogel and r0 represents the first-order PLGA degradation rate constant. This captures the increase in the degradation rate due to the increase in acidic degradation products over time (self-catalyzed PLGA degradation), while limiting the degradation rate by the degradation-product carrying capacity. The free simulation parameters are r0, K, u0, and ε0. We presume from the flatness of the plateaus in the cumulative percentage released (Figs. 2 to 5) that |u0| ≫ kBT and thus chose the large value u0 = −10kBT. This leaves the degradation rate, the near-unity carrying capacity, and the barrier height. These are determined by fitting the highest concentration (10 wt %) release curve, which has the most pronounced effects due to degradation. The parameters found are K = (0.350 ± 0.025)a−3, ε0 = (1.025 ± 0.025)kBT, and r0 = (0.37 ± 0.02)d−1. All other simulations were run using the same parameters with a combined reduced χ2 value of 3.9.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software. For all tests, *P < 0.05, **P < 0.01, and ***P < 0.001. Releases were analyzed using two-way analysis of variance (ANOVA), followed by the Sidak test for multiple comparisons if there were two groups or Tukey’s test for multiple comparisons if there were more than two groups. Activities of NT-3, SDF, and BDNF were analyzed using one-way ANOVA followed by Dunnett’s post hoc test comparing them to controls.
Supplementary Material
Acknowledgments
We would like to thank S. Doyle and B. Calvieri from the University of Toronto Microscopy Imaging Laboratory for assistance with TEM imaging. We are grateful to the members of the Shoichet Laboratory for helpful discussions. Funding: We received funding from the Canadian Institutes of Health Research [foundation grant to M.S.S. (FDN-143276) and Training program in regenerative medicine scholarship to A.T.], the Natural Sciences and Engineering Research Council of Canada (NSERC) [Discovery (RGPIN-2014-04679) to M.S.S., Vanier to M.M.P., post-doctoral fellowship to C.K.M., Canada graduate scholarship - doctoral program to J.M.O., and post-graduate scholarship - doctoral program and NSERC Collaborative Research and Training Experience Program in M3 Materials, Mimetics, and Manufacturing (CREAT 432258-13) to I.E.D.], Ontario Graduate Scholarship (to M.M.P.), the Canadian Partnership in Stroke Recovery (A.T. and M.S.S.), the Heart and Stroke Foundation of Canada (000170 to M.S.S.), the European Molecular Biology Organization (ALTF181-2013 to T.N.S.), and the European Research Council (Advanced Grant MiCE 291234 to T.N.S.). Author contributions: M.M.P., I.E.D., J.M.O., and M.S.S designed the experiments. M.M.P., I.E.D., J.M.O., A.T., and C.K.M. performed the experiments. T.N.S. performed the modeling and stochastic simulations. All authors analyzed the data and wrote the paper. Competing interests: M.S.S., M.M.P., I.E.D., and J.M.O. have submitted a patent application that covers encapsulation-free protein release related to the content of this paper. A.T., C.K.M., and T.N.S. declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/5/e1600519/DC1
fig. S1. Release of NT-3 from a PLGA np/hydrogel DDS containing encapsulated NT-3 and soluble NT-3.
fig. S2. Bioactivity of proteins released from hydrogels with embedded PLGA np.
fig. S3. Characteristics of PLGA np used in this study.
fig. S4. Swelling of HAMC hydrogel with and without PLGA np.
fig. S5. Release of soluble SDF from XMC alone into aCSF at pH 3, pH 5, or pH 7.
fig. S6. Mass loss of PLGA from the release system at pH 3 and pH 7.
movie S1. Two-dimensional Monte Carlo simulations of protein release from a hydrogel with embedded cubic degrading nanoparticles.
Calculation of the relative surface area for PLGA np of different sizes
Calculation for maximum surface coverage
REFERENCES AND NOTES
- 1.Hines D. J., Kaplan D. L., Poly (lactic-co-glycolic) acid–controlled-release systems: Experimental and modeling insights. Crit. Rev. Ther. Drug Carrier Syst. 30, 257–276 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi J.-S., Seo K., Yoo J.-W., Recent advances in PLGA particulate systems for drug delivery. J. Pharm. Invest. 42, 155–163 (2012). [Google Scholar]
- 3.Makadia H. K., Siegel S. J., Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 3, 1377–1397 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ye M., Kim S., Park K., Issues in long-term protein delivery using biodegradable microparticles. J. Control. Release 146, 241–260 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Bao W., Zhou J., Luo J., Wu D., PLGA microspheres with high drug loading and high encapsulation efficiency prepared by a novel solvent evaporation technique. J. Microencapsul. 23, 471–479 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Govender T., Stolnik S., Garnett M. C., Illum L., Davis S. S., PLGA nanoparticles prepared by nanoprecipitation: Drug loading and release studies of a water soluble drug. J. Control. Release 57, 171–185 (1999). [DOI] [PubMed] [Google Scholar]
- 7.van de Weert M., Hennink W. E., Jiskoot W., Protein instability in poly(lactic-co-glycolic acid) microparticles. Pharm. Res. 17, 1159–1167 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Coleman J., Lowman A., Biodegradable nanoparticles for protein delivery: Analysis of preparation conditions on particle morphology and protein loading, activity and sustained release properties. J. Biomater. Sci. Polym. Ed. 23, 1129–1151 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Pérez-Rodriguez C., Montano N., Gonzalez K., Griebenow K., Stabilization of α-chymotrypsin at the CH2Cl2/water interface and upon water-in-oil-in-water encapsulation in PLGA microspheres. J. Control. Release 89, 71–85 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Stanwick J. C., Baumann M. D., Shoichet M. S., Enhanced neurotrophin-3 bioactivity and release from a nanoparticle-loaded composite hydrogel. J. Control. Release 160, 666–675 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Liu Y., Schwendeman S. P., Mapping microclimate pH distribution inside protein-encapsulated PLGA microspheres using confocal laser scanning microscopy. Mol. Pharm. 9, 1342–1350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baumann M. D., Kang C. E., Tator C. H., Shoichet M. S., Intrathecal delivery of a polymeric nanocomposite hydrogel after spinal cord injury. Biomaterials 31, 7631–7639 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Wood M. D., Gordon T., Kim H., Szynkaruk M., Phua P., Lafontaine C., Kemp S. W. P., Shoichet M. S., Borschel G. H., Fibrin gels containing GDNF microspheres increase axonal regeneration after delayed peripheral nerve repair. Regen. Med. 8, 27–37 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Jeon O., Kang S.-W., Lim H.-W., Hyung Chung J., Kim B.-S., Long-term and zero-order release of basic fibroblast growth factor from heparin-conjugated poly(l-lactide-co-glycolide) nanospheres and fibrin gel. Biomaterials 27, 1598–1607 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Pakulska M. M., Vulic K., Tam R. Y., Shoichet M. S., Hybrid crosslinked methylcellulose hydrogel: A predictable and tunable platform for local drug delivery. Adv. Mater. 27, 5002–5008 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Gupta D., Tator C. H., Shoichet M. S., Fast-gelling injectable blend of hyaluronan and methylcellulose for intrathecal, localized delivery to the injured spinal cord. Biomaterials 27, 2370–2379 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Elliott Donaghue I., Tator C. H., Shoichet M. S., Sustained delivery of bioactive neurotrophin-3 to the injured spinal cord. Biomater. Sci. 3, 65–72 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Singh M., Kazzaz J., Chesko J., Soenawan E., Ugozzoli M., Giuliani M., Pizza M., Rappouli R., O’Hagan D. T., Anionic microparticles are a potent delivery system for recombinant antigens from Neisseria meningitidis serotype B. J. Pharm. Sci. 93, 273–282 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Sahoo S. K., Panyam J., Prabha S., Labhasetwar V., Residual polyvinyl alcohol associated with poly (d,l-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J. Control. Release 82, 105–114 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Bleul C. C., Fuhlbrigge R. C., Casasnovas J. M., Aiuti A., Springer T. A., A highly efficacious lymphocyte chemoattractant, stromal cell–derived factor 1 (SDF-1). J. Exp. Med. 184, 1101-1109 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y., Cooke M. J., Sachewsky N., Morshead C. M., Shoichet M. S., Bioengineered sequential growth factor delivery stimulates brain tissue regeneration after stroke. J. Control. Release 172, 1–11 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Lai P. H., Everett R., Wang F. F., Structural characterization of human erythropoietin. J. Biol. Chem. 261, 3116–3121 (1986). [PubMed] [Google Scholar]
- 23.Mollica F., Biondi M., Ungaro F., Quaglia F., Immacolata M., Rotonda L., Netti P. A., Mathematical modelling of the evolution of protein distribution within single PLGA microspheres: Prediction of local concentration profiles and release kinetics. J. Mater. Sci. Mater. Med. 19, 1587–1593 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Kim B., Hong D., Chang W. V., Swelling and mechanical properties of pH-sensitive hydrogel filled with polystyrene nanoparticles. J. Appl. Polym. Sci. 130, 3574–3587 (2013). [Google Scholar]
- 25.Ende M. T. A., Peppas N. A., Transport of ionizable drugs and proteins in crosslinked poly(acrylic acid) and poly(acrylic acid-co-2-hydroxyethyl methacrylate) hydrogels. I. Polymer characterization. J. Appl. Polym. Sci. 59, 673–685 (1996). [Google Scholar]
- 26.Fredenberg S., Wahlgren M., Reslow M., Axelsson A., The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—A review. Int. J. Pharm. 415, 34–52 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Crotts G., Saha H., Park T. G., Adsorption determines in-vitro protein release rate from biodegradable microspheres: Quantitative analysis of surface area during degradation. J.Control. Release 47, 101–111 (1997). [Google Scholar]
- 28.Wei Y., Wang Y. X., Wang W., Ho S. V., Qi F., Ma G. H., Su Z. G., Microcosmic mechanisms for protein incomplete release and stability of various amphiphilic mPEG-PLA microspheres. Langmuir 28, 13984–13992 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Rafati A., Boussahel A., Shakesheff K. M., Shard A. G., Roberts C. J., Chen X., Scurr D. J., Rigby-Singleton S., Whiteside P., Alexander M. R., Davies M. C., Chemical and spatial analysis of protein loaded PLGA microspheres for drug delivery applications. J. Control. Release 162, 321–329 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Chesko J., Kazzaz J., Ugozzoli M., O’Hagan D. T., Singh M., An investigation of the factors controlling the adsorption of protein antigens to anionic PLG microparticles. J. Pharm. Sci. 94, 2510–2519 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Cai C., Bakowsky U., Rytting E., Schaper A. K., Kissel T., Charged nanoparticles as protein delivery systems: A feasibility study using lysozyme as model protein. Eur. J. Pharm. Biopharm. 69, 31–42 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Xiong J.-Y., Naranayan J., Liu X.-Y., Chong T. K., Chen S. B., Chung T.-S., Topology evolution and gelation mechanism of agarose gel. J. Phys. Chem. B 109, 5638–5643 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Li L., Thangamathesvaran P. M., Yue C. Y., Tam K. C., Hu X., Lam Y. C., Gel network structure of methylcellulose in water. Langmuir 17, 8062–8068 (2001). [Google Scholar]
- 34.Rosenfeld R. D., Zeni L., Haniu N., Talvenheimo J., Radka S. F., Bennett L., Miller J. A., Welcher A. A., Purification and identification of brain-derived neurotrophic factor from human serum. Protein Expr. Purif. 6, 465–471 (1995). [DOI] [PubMed] [Google Scholar]
- 35.Fukuzono S., Fujimori K., Shimizu N., Production of biologically active mature brain-derived neurotrophic factor in Escherichia coli. Biosci. Biotechnol. Biochem. 59, 1727–1731 (1995). [DOI] [PubMed] [Google Scholar]
- 36.Park T. G., Lee H. Y., Nam Y. S., A new preparation method for protein loaded poly(d, l-lactic-co-glycolic acid) microspheres and protein release mechanism study. J. Control. Release 55, 181–191 (1998). [DOI] [PubMed] [Google Scholar]
- 37.Barrow N. J., Madrid L., Posner A. M., A partial model for the rate of adsorption and desorption of phosphate by goethite. J. Soil Sci. 32, 399–408 (1981). [Google Scholar]
- 38.Vulic K., Pakulska M. M., Sonthalia R., Ramachandran A., Shoichet M. S., Mathematical model accurately predicts protein release from an affinity-based delivery system. J. Control. Release 197, 69–77 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Piantino J., Burdick J. A., Goldberg D., Langer R., Benowitz L. I., An injectable, biodegradable hydrogel for trophic factor delivery enhances axonal rewiring and improves performance after spinal cord injury. Exp. Neurol. 201, 359–367 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Bertram J. P., Rauch M. F., Chang K., Lavik E. B., Using polymer chemistry to modulate the delivery of neurotrophic factors from degradable microspheres: Delivery of BDNF. Pharm. Res. 27, 82–91 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Lampe K. J., Kern D. S., Mahoney M. J., Bjugstad K. B., The administration of BDNF and GDNF to the brain via PLGA microparticles patterned within a degradable PEG-based hydrogel: Protein distribution and the glial response. J. Biomed. Mater. Res. A 96A, 595–607 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Wang Y., Teng Y., Zu Z. H., Ju R. K., Guo M. Y., Wang X. M., Xu Q. Y., Cui F. Z., Combination of hyaluronic acid hydrogel scaffold and PLGA microspheres for supporting survival of neural stem cells. Pharm. Res. 28, 1406–1414 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Péan J.-M., Venier-Julienne M.-C., Boury F., Menei P., Denizot B., Benoit J.-P., NGF release from poly(d,l-lactide-co-glycolide) microspheres. Effect of some formulation parameters on encapsulated NGF stability. J. Control. Release 56, 175–187 (1998). [DOI] [PubMed] [Google Scholar]
- 44.de Boer R., Knight A. M., Spinner R. J., Malessy M. J. A., Yaszemski M. J., Windebak A. J., In vitro and in vivo release of nerve growth factor from biodegradable poly-lactic-co-glycolic-acid microspheres. J. Biomed. Mater. Res. A 95A, 1067–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fleige E., Quadir M. A., Haag R., Stimuli-responsive polymeric nanocarriers for the controlled transport of active compounds: Concepts and applications. Adv. Drug Deliv. Rev. 64, 866–884 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Ravi Kumar M. N. V., Bakowsky U., Lehr C. M., Preparation and characterization of cationic PLGA nanospheres as DNA carriers. Biomaterials 25, 1771–1777 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Baumann M. D., Kang C. E., Stanwick J. C., Wang Y., Kim H., Lapitsky Y., Shoichet M. S., An injectable drug delivery platform for sustained combination therapy. J. Control. Release 138, 205–213 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Hickey O. A., Mercier J.-F., Gauthier M. G., Tessier F., Bekhechi S., Slater G. W., Effective molecular diffusion coefficient in a two-phase gel medium. J. Chem. Phys. 124, 204903 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/5/e1600519/DC1
fig. S1. Release of NT-3 from a PLGA np/hydrogel DDS containing encapsulated NT-3 and soluble NT-3.
fig. S2. Bioactivity of proteins released from hydrogels with embedded PLGA np.
fig. S3. Characteristics of PLGA np used in this study.
fig. S4. Swelling of HAMC hydrogel with and without PLGA np.
fig. S5. Release of soluble SDF from XMC alone into aCSF at pH 3, pH 5, or pH 7.
fig. S6. Mass loss of PLGA from the release system at pH 3 and pH 7.
movie S1. Two-dimensional Monte Carlo simulations of protein release from a hydrogel with embedded cubic degrading nanoparticles.
Calculation of the relative surface area for PLGA np of different sizes
Calculation for maximum surface coverage