

Short guanine repeats cause a REV1-dependent 4- to 18-fold increase in the substitution rate of the surrounding DNA sequence.
Keywords: Mutation at a distance, mutator, repeat sequence, evolution, translesion synthesis, homologous recombination
Abstract
Mutation provides the raw material from which natural selection shapes adaptations. The rate at which new mutations arise is therefore a key factor that determines the tempo and mode of evolution. However, an accurate assessment of the mutation rate of a given organism is difficult because mutation rate varies on a fine scale within a genome. A central challenge of evolutionary genetics is to determine the underlying causes of this variation. In earlier work, we had shown that repeat sequences not only are prone to a high rate of expansion and contraction but also can cause an increase in mutation rate (on the order of kilobases) of the sequence surrounding the repeat. We perform experiments that show that simple guanine repeats 13 bp (base pairs) in length or longer (G13+) increase the substitution rate 4- to 18-fold in the downstream DNA sequence, and this correlates with DNA replication timing (R = 0.89). We show that G13+ mutagenicity results from the interplay of both error-prone translesion synthesis and homologous recombination repair pathways. The mutagenic repeats that we study have the potential to be exploited for the artificial elevation of mutation rate in systems biology and synthetic biology applications.
INTRODUCTION
Mutation is the source of both adaptive genetic variation and deleterious genetic load. As such, the mutation rates that we observe in nature represent a balance between opposing selective forces. Because extant organisms are relatively well adapted, most mutations that occur will be deleterious (1). It is this aversion to mutation that has driven the evolution of complex systems for high-fidelity replication and maintenance of genome integrity. However, mutation rates vary greatly; RNA viruses have an error rate of 10−4 substitutions per nucleotide per cell infection (2), whereas bacteria make only a single error for every billion nucleotides synthesized (3). Although it is perhaps not surprising that widely diverged species exhibit large differences in mutation rate, this observation raises questions about the degree of mutation rate variation within and across genomes.
Studies of clinical and experimental populations have revealed that mutation rates can evolve on time scales observable to experimentalists and provided insights into the dynamics of mutation rate evolution (4). In microbes, mutator strains arise via mutations that inactivate DNA repair enzymes, resulting in elevated mutation rates. These strains can become established in adapting populations because of the higher rate at which they produce beneficial mutations (4). However, after the population has become better adapted, the increased rate of deleterious mutation experienced by a mutator will be selected against, providing selective pressure to drive mutation rates back down (5, 6).
The effects of mutator alleles that diminish polymerase accuracy extend to the whole genome. However, mutation rates can also vary within a single genome. Highly transcribed genes have been found to have elevated mutation rates proportional to the rate of transcription (7, 8), probably because DNA that is highly transcribed is more often in a single-stranded state and thus more vulnerable to mutagens (9, 10). Another well-established correlate of mutation rate variation is DNA replication timing. Experimental (11) and bioinformatic (12, 13) studies have shown that later replicated DNA has a higher mutation rate than the earliest replicated regions. A proposed explanation for this is that DNA that is copied later during cell division will have less time for slower, high-fidelity repair mechanisms and will rely on error-prone DNA repair (14).
Primary DNA sequence can also influence mutation rates. Homopolymeric repeats of nucleotides are prone to increase and decrease in length at a high frequency (15, 16) and have been found to play an important role in genome evolution (17, 18), especially genetic switching mechanisms in pathogenic bacteria (19, 20). Microsatellites or other short repeats have been implicated in several human diseases (21), and the high degree of polymorphism that results from their instability has been exploited as genetic markers (22). In some cases, the causes of genetic instability have been directly linked to physical properties of DNA sequence. For instance, the twist angle between two adjacent bases in a DNA double helix is predetermined by their identity: some combinations of nucleotides have twist angles that are fragile and thus more prone to breakage (23). The repeated evolution of mutations in the promoter of pitX1 in multiple independent populations of sticklebacks has been hypothesized to be due to both the strong selection upon these mutations and the proclivity of this region to sustain double-strand breaks. The part of the pitX1 promoter in which these adaptive mutations occur is the most fragile site in the stickleback genome, as predicted by DNA twist angles (24).
The mutagenic effects of some DNA sequences have been found to extend to flanking regions of DNA, with stretches of DNA that have the capacity to form secondary structures often being the culprit. Tang and colleagues (25) found that runs of 230 Friedreich’s ataxia repeats (GAA•TTC) were able to induce large deletions [>50 bp (base pairs)] and point mutations in a reporter gene more than a kilobase downstream of the repeat itself. Others working with the same repeat unit found that this repeat sequence could induce mutagenesis in sequences up to 8 kb away (26). Directly inducing double-strand breaks using HO (27) or I-SCE1 (28) sharply increases not only the mutation rate in the surrounding 2 kb of sequence but also has a detectable but weak long-range mutator effect that decays exponentially across 60 kb of DNA sequence (28). In all of these cases, a combination of double-strand break and translesion repair was directly implicated (25–28).
In previous work, we found that short repeat sequences are positively correlated with the substitution rate in the surrounding DNA sequence (29), distinct from the well-known repeat length polymorphism associated with repetitive DNA sequences, and that the experimental insertion of repeat sequences in yeast could elevate mutation rates in the downstream sequence. We have proposed that repeat sequences are more likely than other sequences to recruit error-prone DNA repair polymerases, leading to an increased mutation rate in DNA sequence surrounding the repeat. Here, we test our hypothesis by investigating one particular type of repeat in more detail: homopolymeric runs of guanine nucleotides. We demonstrate which DNA replication repair pathways are necessary for mutagenesis and show that these sequences interact with other known causes of mutation rate variation.
RESULTS
Homopolymeric runs of guanines 13 bp or longer (G13+) cause an increase in mutation rate
Using Saccharomyces cerevisiae, we engineered runs of 11 to 14 guanine nucleotides four bases upstream of the URA3 coding region (Fig. 1A), and measured mutation rates (Fig. 2A). The mutation rate in the URA3 coding region, downstream of a G13 or G14 repeat sequence, increased by up to fourfold. We measured the mutation rate at another locus encoding the CAN1 gene (Fig. 2B), confirming that mutation rates obtained at a site that did not have a G14 sequence upstream did not increase relative to the wild type. Moving G14 from the coding strand to the template strand and from upstream of the URA3 translation start site to downstream of the transcription termination site abolished the mutagenic effect of G14 (Fig. 2C).
Fig. 1. Experimental approach to quantifying mutagenicity of G13+ DNA sequences.

(A) Poly-G sequences were engineered 4 bp upstream of the URA3 translation start site (the 5′UTR region). The G14-ORF (open reading frame) construct (G14-ORF) allowed detection of loss-of-function mutations in the URA3 reading frame by plating on medium containing 5-fluoroorotic acid (5-FOA), which selects for individual cells that contain mutations that inactivate URA3. The red asterisk indicates the mutation site. (B) Using a weak URA3 allele (URA3-w), this construct (G14-repeat) facilitated the detection of the polyguanine repeat expansion mutation, because the mutation, G14 to G15 or longer, results in the 5′-FOA–resistant phenotype. wt, wild type.
Fig. 2. Polyguanine sequences cause a localized, directional effect on mutation rate.
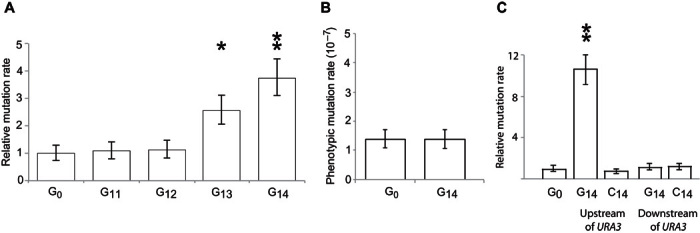
(A) Mutation rates of homopolymeric guanine repeat sequences of increasing length. The estimated phenotypic mutation rate of G0-ORF, G13-ORF, and G14-ORF is 5.4 × 10−7, 13.5 × 10−7, and 20.3 × 10−7, respectively. G11 and G12 had no detectable increase in mutation rate. (B) Mutation rate was measured using CAN1, at a site distal from the URA3 locus. (C) The G14 repeat does not cause an increase in mutation rate if engineered on the template strand (C14-URA3) or on either the coding strand (ORF-G14) or the template strand (ORF-C14) downstream of the URA3 terminator sequence. Significant differences were calculated using t tests. *P < 0.05; **P < 0.005. Error bars show 95% confidence intervals.
Sequence analysis of G0- and G14-ORF mutants reveals similar spectrum of mutations
We sequenced 113 independent G14-ORF ura3 mutants and 101 G0-ura3 mutants and analyzed the distribution and identity of the mutations (Fig. 3). Although both sets of mutations were significantly different from a uniform distribution (Kolmogorov-Smirnov test, G14, P = 0.023; G0, P = 0.002), they were not different from each other (Kolmogorov-Smirnov test, P = 0.99). The identities of the mutations were highly similar except for a slight enrichment of indels relative to substitutions (Fisher’s exact test, P = 0.048) and transitions relative to transversions (Fisher’s exact test, P = 0.035) in the set of G14-ORF ura3 mutants. Our sequence data incorporated the G14 repeat as well as the entire URA3 ORF, confirming that changes in guanine repeat length (repeat expansion or contraction) and large deletions were not responsible for any of the elevated mutation rate detected in this assay (table S1).
Fig. 3. Mutational spectrum of 183 5′-FOA–resistant mutants at the URA3 locus.

5′-FOA–resistant mutants were collected from G0-URA3 (brown) and G14-ORF (blue) strains. Each point mutation is shown directly above the wild-type sequence, and indels and complex mutations are shown directly below.
DNA replication timing correlates with the degree of G14 mutagenicity
To test whether genome position would influence G14 mutagenicity, we engineered G0-URA3 and G14-ORF genes into different positions on chromosomes XII and XV (Fig. 4 and table S2). We found that G14-URA3 inserts sustained a higher mutation rate than G0-URA3 inserts at the same site. The change in mutation rate associated with G14 ranged from a 4-fold to an 18-fold increase, supporting the fact that G14 mutagenicity occurs regardless of genome position.
Fig. 4. The effect of G14 mutagenicity is correlated with DNA replication timing.

(A and B) The regression of mutation rate and replication timing for G14-ORF alleles inserted at six different sites on chromosome XII and two sites on chromosome XV (Spearman’s R = 0.89, P = 0.007) (A) and for insertion of G0-URA3 at the same sites (Spearman’s R = 0.63, P = 0.086) (B). Replication timing is shown in minutes after the release of cells into synchronized S phase, as reported by Nieduszynski et al. (30). Error bars show 95% confidence intervals.
The direction of DNA replication for the chromosome V version of the G14-ORF construct indicates that the replication fork moves in the same direction as transcription for URA3. Because moving the G14 sequence to the template strand or downstream of the transcription termination site abolishes the mutagenic effect, this suggests a relationship between G14 mutagenicity and DNA replication timing. DNA replication timing correlates with mutation rate variation in organisms ranging from bacteria to humans (11, 12). We calculated correlation coefficients for mutation rates of the G14-ORF inserts and replication timing [from Nieduszynski et al. (30)] and found a significant positive correlation (Fig. 4A; Spearman’s R = 0.89, P = 0.007). The G0-URA3 inserts in equivalent positions showed a nonsignificant positive correlation (Fig. 4B; Spearman’s R = 0.638, P = 0.086) consistent with previous work (11).
G13 repeats and transcription promote the accumulation of replication fork intermediates
Repeat sequences are known to incur an increased risk of replication fork stalling (31). Upon fork stalling, replication reinitiates downstream, leaving a single-stranded gap that is filled in using either homologous recombination (HR) or translesion synthesis (TLS) (32–34), with a bias toward TLS for gaps requiring repair later in S phase (14, 34). TLS, mediated by the Rev1/Polζ complex, often introduces errors, even when synthesizing undamaged DNA in vivo (35, 36), and is responsible for approximately 50% of mutations in wild-type S. cerevisiae (37, 38). Replication fork intermediates can be visualized using the two-dimensional (2D) gel technique (39). We made the doxycycline-repressible constructs tet-G13-URA3 and tet-G0-URA3 for 2D analysis (we were unable to generate a tet-G14-URA3 construct). These were designed so that when running the 2D gel, replication fork intermediates that stall at the beginning of URA3 (Y molecules) should accumulate at the point indicated in Fig. 5B. We observed a 2.5-fold increase of Y molecules in G13 constructs compared to G0 constructs when cells were grown in the absence of doxycycline (Fig. 5, A to C).
Fig. 5. G13 repeats are implicated in the accumulation of replication fork intermediates.

(A) 2D gels for tet-G0-URA3 and tet-G13-URA3 under the control of a repressible tetR promoter (65) for time points taken at 15, 30, 45, and 60 min after synchronized cells were released from G1 cell cycle arrest. (B) The circled area show replication fork intermediates that are putatively stalled at the beginning of URA3 (Y molecules, blue circle). The reference region (red hexagon) is used to normalize between samples and determine the relative amounts of Y molecules in each treatment. (C) When transcription is enabled (−dox), the G13 construct has ~2.5 as much putative fork stalling as the G0 construct (calculated at the 30-min time point). When transcription is repressed, both G13 and G0 constructs appeared to have more Y molecules, although G13 has only ~1.13 as much replication fork intermediates as G0 (calculated at the 30-min time point). (D) Mutation rates for G0-URA3 and G13-ORF under the control of a repressible tetR promoter. −dox indicates the high-expression treatment, whereas +dox indicates repression of transcription. Significant differences were calculated using t tests; *P < 0.05. Error bars show 95% confidence intervals.
Previous studies have shown that plasmid-based G20 and G32 repeats cause transcription-dependent replication fork stalling (40, 41). When we repressed transcription by supplementing the growth media with doxycycline, we found that the enrichment of Y molecules in G13 compared to G0 was reduced compared to the treatment without doxycycline (Fig. 5C). The ratio of G13 to G0 decreased, whereas the amount of actual detected replication fork intermediates increased; possible explanations for this will be discussed below. The change in replication fork intermediates caused by repression of transcription raises the possibility that the mutagenic effect of G13 is also changed. We measured the effect of transcriptional repression on mutation rates in tet-G13-URA3 and tet-G0-URA3. The mutation rates of tet-G13-URA3 and tet-G0-URA3 were reduced when URA3 transcription was repressed, although not significantly. Even after repression of transcription by doxycycline, the mutation rate of tet-G13-URA3 remained significantly higher than that of tet-G0-URA3 (Fig. 5D).
Mutagenesis downstream of G14 repeats is Rev1-dependent
Previously, we had proposed that repeat sequence–mediated increases in downstream mutation rate were caused by frequent recruitment of error-prone translesion DNA polymerases by sequences prone to stall the high-fidelity, housekeeping DNA polymerase (29). To test this, we deleted REV1, which is required for all TLS in yeast (42, 43). We found that ablation of REV1 significantly reduced the mutation rate of both G14-ORF (t test, P < 0.005) and G0-URA3 (t test, P < 0.005) (Fig. 6A). That deletion of REV1 decreases mutation rate suggests that the replication fork interruptions that are typically accommodated by Rev1-mediated TLS DNA synthesis are lethal in the rev1 mutant (44). If this is the case, G13+ sequences would cause an increased likelihood of mutation in the surrounding DNA sequence by increasing the rate of error-prone translesion DNA synthesis in that region.
Fig. 6. Expansion of polyguanine repeats occurs at a high rate and is RAD52-dependent.
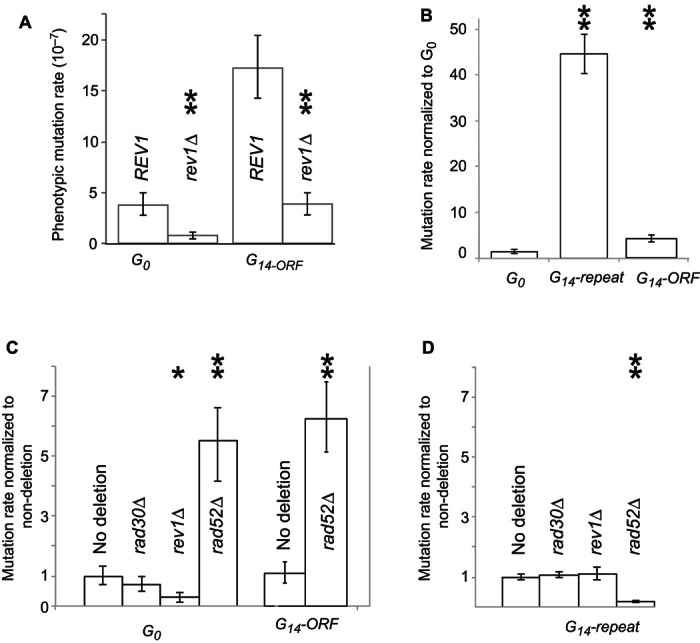
(A) Mutation rates of G0 and G14-ORF strains compared to their respective rev1 deletion mutants. (B) Mutation rates of G0, G14-repeat, and G14-ORF strains, shown relative to the G0 mutation rate. (C and D) In each panel, the mutation rates of deletion mutants are shown relative to their respective nondeletion progenitor, either G0 (C), G14-ORF (C), or G14-repeat (D). Significant differences calculated using t tests are indicated by asterisks: *P < 0.05 and **P < 0.005. Error bars represent 95% confidence intervals.
Expansion of homopolymeric repeats also occurs at the G14 repeat and is Rad52-dependent
It has long been established that homopolymeric repeat sequences are unstable, increasing and decreasing in repeat length at a high rate (19, 20). In the experiment described above, only mutations that occur in the ORF of URA3 can be recovered, even though mutations that change repeat length almost certainly occur in the G14 sequences of some of the individuals within the populations of yeast cells used to measure mutation rate. This is because mutations changing the length of the G14 repeat, which is in the 5′ untranslated region (5′UTR), do not cause the loss of URA3 function that is selected in the assay.
To facilitate the capture of mutations that change the number of G’s in the G14 repeat, we constructed a new strain containing the G14 sequence, engineered upstream of an alternative URA3 sequence (URA3-w), whose function is mildly compromised (G14-repeat, Fig. 1B). We had previously observed that a G15 repeat in the 5′UTR region of URA3-w could cause a reduction in protein translation (fig. S1). Although G14-ORF-w exhibits the Ura+ phenotype of the wild-type allele, a mutation from G14 to G15 (or longer) results in the assayable loss of URA3 function, probably due to a combined effect of impaired function and reduced translation (Fig. 1B). We used this construct (G14-repeat) to directly measure the mutation rate of repeat length increase from G14 to G15 or longer (contraction of the G repeat would not generate the phenotype). We found that repeat length–dependent increases in mutation rate were higher than the downstream mutation rate, with a 45-fold difference between G14-repeat and G0 cells (Fig. 6B). Sequencing of 105 independent ura3 mutant clones of G14-repeat confirmed that most of them carried an increased polyguanine repeat (G15) but there were no mutations in the coding region and no large deletions (table S1).
Repeat expansion occurs independently of REV1 and RAD30
Although deletion of REV1 reduced the mutation rate within the URA3 ORF (as detected by the G14-ORF construct), we found that deletion of REV1 had no effect on the mutation rate in the homopolymeric repeat as measured using the G14-repeat construct (Fig. 6D). To confirm that another translesion DNA synthesis pathway was not involved, a gene essential for another translesion pathway, RAD30, was deleted and also had no effect on mutation rate. We next turned to the alternative mechanism for rescuing the stalled replication fork, HR. Rad52 is essential for the annealing of DNA strands during HR (45), and its ablation causes an increase in mutation rate of approximately fivefold in G0 cells (Fig. 6D). The reason for this increase is that rad52 mutants depend on error-prone DNA polymerases to synthesize over the single-stranded gaps resulting from replication fork stalling (32, 33). Conversely, we found that deletion of RAD52 in the G14-repeat strain markedly reduced URA3 mutation rates (Fig. 6D), consistent with previous work examining recombination and frameshifts underlying “adaptive mutation” in Escherichia coli (46–48).
We checked whether rad52 deletion was able to reduce the mutation rate in G14-ORF cells. However, similar to G0 cells, the mutation rate was increased approximately fivefold (Fig. 6C), showing that Rad52-mediated HR affects only the change of G14 length, not the downstream mutagenic effect of G14.
G14 mutagenesis is not caused by formation of a G-quadruplex structure
G-quadruplex structures, which can induce double-strand breaks and replication fork pausing in Yeast pif1 (G-quadruplex resolvase) deletion mutants, are another potential cause of DNA replication stress (49). To test whether our polyguanine sequences could form G-quadruplex structures, we compared a known G-quadruplex–forming sequence from Tetrahymena (50) to G11, G12, G13, and G14 sequences. We designed five oligomers (given in Materials and Methods) that included either the G-quadruplex control sequence or 11 to 14 guanines in a row, each integrated into the same sequence context as the URA3 constructs used for fluctuation tests in this study. We first performed circular dichroism analysis of the oligos in ionic solutions that promote G-quadruplex formation. Circular dichroism analysis showed that the control was able to form a G-quadruplex, whereas the G11 to G14 repeat sequences could not (fig. S2, A to E). We then used DNA polymerase stop assays to test whether DNA polymerase could synthesize the complementary DNA across the single-stranded template, based on the principle that a stable secondary structure should inhibit DNA synthesis. The results show that the G-quadruplex control blocked the polymerase, whereas G11 to G14 sequences did not have the same effect (fig. S2F).
DISCUSSION
“Mutation at a distance” has been shown to occur in a number of different sequence contexts. For instance, DNA sequences that are prone to double-strand breaks have been indirectly linked to kataegis, a catastrophic mutational event that causes large clusters of mutations dispersed over tens of kilobases in cancer genomes (51, 52). Break-induced replication (BIR) is often initiated in response to double-strand breaks (53) and produces long regions of DNA in single-stranded form, which are more susceptible to mutation than double-stranded DNA. Experiments in yeast have shown that the widely distributed clusters of point mutations characteristic of kataegis result from the attack by mutagenic agents upon extended tracts of single-stranded DNA (53), whereas studies in yeast (54, 55) and mammalian cells (52) have implicated the deregulation of AID/APOBEC deaminases as an alternative cause of kataegis-like mutational events. The elevated mutation rate that we observed in this study is unlikely to be caused by a similar mechanism because G14-ORF mutagenesis requires REV1 whereas BIR functions independently of any translesion polymerase (56).
Double-strand breaks have also been implicated as the cause of the mutagenicity of long repeats known to form hairpin secondary structures (25, 26). The importance of double-strand breaks as a trigger for induced mutagenesis has been further reinforced by studies that directly induce double-strand breaks using HO (27) or I-SCE1 (28). In all of these systems, increases in mutation rate are detectable in the surrounding 1 to 2 kb of sequence and dependent on TLS (25–28, 57). Conversely, the repeat-induced mutagenesis system studied by Shishkin et al. (58) and Shah et al. (59) occurs independently of TLS and can induce mutations both upstream and downstream of the repeat sequence. Mutagenesis in this system only occurs when repeat tracts exceed the length of the Okazaki fragment.
Here, we provide a tentative mechanism for how G14 repeats can lead to elevated mutation rates in the surrounding DNA sequence. The 2D gel results suggest that the replication fork is more prone to stall at the G13-URA3 sequence than at the G0-URA3 sequence. Mutation rates of the C14-ORF, ORF-C14, and ORF-G14 shown in Fig. 2C suggest that the mutagenic effect of G14 was dependent on the direction of replication. In this experiment, the guanines are seemingly required to be in the leading strand, and the mutagenic effect is only conferred on sequences that are replicated downstream of the guanine repeat sequence. However, this result is also consistent with the direction of transcription playing a role. G14 mutagenesis only occurs when transcription proceeds through G14 with the URA3 downstream.
The question of the relative importance of replication or transcription is addressed by the insertion of G14-ORF constructs in various positions in chromosomes XII and XV (Fig. 4 and table S2). Although all of these constructs are transcribed through G14 to URA3, the direction of replication and whether the genes are on the Watson or Crick strand vary by chromosome position (table S2). All constructs had a 4- to 18-fold increase in mutation rate, regardless of the direction of replication or the position of G14 on the leading or lagging strand. These results concur with previous work that has shown that G20 to G32 repeats on plasmids caused high rates of replication fork stalling that depend on transcription (40, 41). In agreement with our results, the equivalent cytosine repeats (C20 to C32) did not have an effect on replication fork stalling (40, 41). Mutation rates were not measured in these previous experiments, but in our study, even with repressed transcription, the mutation rate remained elevated (Fig. 5D). The tet repressor is known to be extremely effective at repressing expression; thus, it is unlikely that leaky expression can account for the remaining mutagenic effect in the noninduced tet-G13-URA3. However, it has been shown that RNA polymerase II (RNAPII) (sufficient for hindering the DNA replication fork) can bind to the tet promoter even in the absence of induction (60). Figure 5 shows that tet repression of transcription causes the G0 strain to accumulate more Y replication fork intermediates. This is consistent with RNAPII binding the repressed tet promoter. However, these accumulated Y fork intermediates do not translate into an increased mutation rate for G0. That G0 does not have an elevated mutation rate despite an increase in replication fork intermediates suggests that the G13+ sequence does more than stall the replication fork; it may also interfere with homologous repair, biasing toward TLS repair. The precise role of transcription in G13+ mutagenesis requires further work to be fully resolved.
One model proposed by Lopes and co-workers (32), and later by Huang et al. (33), postulates that after replication fork stalling, the replication complex decouples from the fork and reinitiates replication downstream. This leaves a patch of single-stranded DNA to be filled using either HR or TLS. We propose that G14 repeats confer a higher probability of replication fork stalling than other DNA sequences (Fig. 7). This leads to more TLS activity in the region and, therefore, a higher mutation rate. There is mounting evidence that in early S phase, high-fidelity HR is the synthesis mechanism of choice, whereas in late S phase and early G2 phase, TLS is preferred (11, 14, 33). Our finding of a strong correlation between G14-induced mutation and DNA replication timing adds support to this hypothesis by suggesting that G14 repeats in later-replicating DNA are more likely to depend on TLS for repair, leading to a higher average mutation rate in this region.
Fig. 7. Model for the outcome of G13+-induced replication fork stalling.
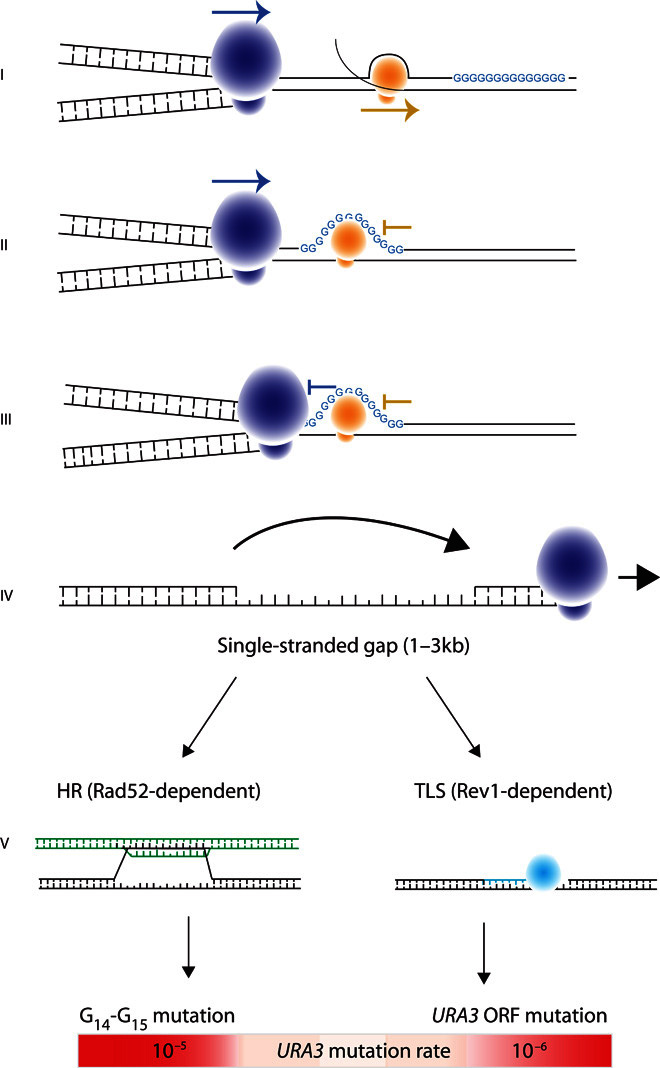
(I) The replication fork can proceed in either direction, but the transcription complex must encounter the G13+ repeat before the transcribed gene. (II) Transcription stalls at the G13+ sequence. (III) The replication fork stalls at the G13+ sequence; stalling is more likely if there is a stalled transcription complex already present. (IV) The replication fork detaches from the template and reinitiates replication downstream, leaving a patch of singlestranded DNA that is 800 to 3000 bp in length. (V) The DNA complementary to the single-stranded gap is synthesized using either Rad52-dependent HR (detected using the G14-repeat construct) or Rev1-dependent TLS (detected using the G14-ORF construct) to bypass the difficult-to-replicate region.
In one recent study, mismatch repair efficiency was found to vary with time of replication (15). We show that most of the difference in mutation rates between G14-URA3 and G0-URA3 is dependent on having a functional REV1. We also show that G14 inserts have a stronger correlation between replication timing and mutation rate than G0 inserts. It follows that this stronger correlation is also REV1-dependent, and in support of this, a previous comprehensive analysis of G0-URA3 inserts at 49 sites on chromosome 6 found that the deletion of REV1 abolished the correlation between replication timing and mutation rate (11). These results suggest that mismatch repair plays a background role in the correlation between mutation rate and replication timing that we observed in this study.
The G14 repeats that we study do not require an artificial increase in mutation rate or polymerase deletion for the effect to be detectable. Moreover, G14 repeats are short, simple, and present in most genomes, suggesting that G14 repeats could play an important role in genome evolution. The tight correlation between repeat-induced mutation and replication timing (R = 0.89, P = 0.007) suggests that the degree to which a repeat sequence will alter mutation rate can be directly combined with knowledge of replication timing. Because such repeats are common throughout all genomes, it is not implausible that these repeats could have implications as locally acting mutator sequences. In addition, short repeat sequences are easy to engineer and could be applied to synthetic biology applications for speeding up evolution of a target gene.
MATERIALS AND METHODS
Strain construction
All strains were constructed in a strain isogenic with W303 (MATa his3-11,15 leu2-3,112 trp1-1 ura3 ade2-1). Homopolymeric nucleotide strains were constructed by amplifying URA3, with primers containing a homopolymeric nucleotide tract at the position between −4 and −5 of URA3, and the resultant polymerase chain reaction (PCR) product was transformed into ura− yeast cells using the LiAc transformation method. The URA3 genes of transformants were amplified using PCR, and the sequences were confirmed by Sanger sequencing. Different mutant strains were constructed by amplifying the G418 insertion mutant for each gene of interest from the whole-genome deletion collection. Strains were transformed with PCR products and deletion mutants selected based on their resistance to G418. G14-repeat was constructed using an alternative URA3 sequence that carries a point mutation at position 736 (val247ile), which has slightly reduced function compared to the wild-type URA3 gene. Change in repeat length from G14 to G15 in the G14-repeat construct reduced protein translation such that cells containing this mutation were 5-FOA–resistant and detectable using the mutation rate assay.
Fluctuation assays
Strains to be assayed were grown overnight in 3-ml complete supplement mixture (CSM)–uracil medium, diluted 10−4, and then inoculated into 100-μl cultures so that there were approximately 1000 cells per culture. At least 24 independent cultures were used per assay, and each assay was repeated at least three times. Cultures were left overnight at 30°C until the cultures were assessed to have reached a suitable density, and then the entire culture, except for 5 μl, was plated onto predried 5-FOA plates to detect ura3 mutants that were 5-FOA–resistant. The remaining culture was pooled and diluted, and then the cell count was assayed using a Scepter cell counter. Mutation rates were calculated using the maximum likelihood method (61). To measure the background mutation rate at a site distal from the URA3 locus, the wild-type CAN1 locus was restored in G0-URA3 and G14-ORF strains. Mutation rate assays were carried out the same as above except that mutations in the CAN1 gene were detected by plating on CSM-arginine plates supplemented with canavanine (60 μg/ml).
2D gel analysis
Strains for the 2D gel analysis were constructed by amplifying tet-GN-URA3 fragments from a plasmid for insertion by HR into the intergenic region between YCL052C and YCL054W at position 34,028 on chromosome III. After we introduced the tet-GN-URA3 fragments into S. cerevisiae, we plated the cells on selection plates lacking uracil. Colonies were picked and transformation was confirmed by PCR and Sanger sequencing. For the 2D gel experiments, log-phase cells [OD600 (optical density at 600 nm) ≈ 0.5] were synchronized at G1 by α factor (10 μg/ml) [with or without doxycycline (10 μg/ml)] for 2.5 hours and then released in YPD (yeast extract, peptone, and dextrose) medium (with or without doxycycline) at 25°C. Cells were harvested at 15, 30, 45, and 60 min and terminated immediately using sodium azide, and then in vivo psoralen cross-linking was undertaken as previously described (62, 63). Genomic DNA was digested using Hind III. First-dimension gels were made of 0.35% agarose and second-dimension gels were made of 1% agarose. A DNA fragment correlated to the yeast genome positions 34,028 to 34,999 was used to make the probe. Replication intermediate signals were quantified as previously described (39).
DNA synthesis stop assay
To determine whether G11 to G14 sequences could form G-quadruplex structures, we conducted experiments comparing a known G-quadruplex–forming sequence from Tetrahymena (GGGTTGGGTTGGGTTGGGTT) (50) to G11, G12, G13, and G14 sequences. We designed oligonucleotides composed of either homopolymeric runs of 11 to 14 G’s in a row or the G-quadruplex sequence, integrated into the same sequence context as the genetic constructs used to measure mutation rate in this study. Following Han and co-workers (64), a radiolabeled primer (Γ-32P), shown below, was annealed with template DNA (10 nM) in buffer containing 5 mM KCl. To initiate the sequencing reactions, MgCl (3 μM), Taq polymerase (2.5 U per reaction), and deoxynucleotide triphosphates (final concentration of 100 μM) were added and the mix was incubated at either 37° or 55°C. The reactions were stopped and then run on 12% polyacrylamide gel. If the template forms a G-quadruplex, then DNA synthesis will not be completed, and no band can be visualized on the polyacrylamide gel.
Primer: [CTGCACAGAACAAAAACCTGCAGGAAACG]. Templates: G-quadruplex control, GCTTTCGACATGATT(GGGTTGGGTTGGGTTGGGTT)TATCTTCGTTTCCTGCAGGTTTTTGTTCTGTGCAG; G11, d[GCTTTCGACATGATTGGGGGGGGGGGTATCTTCGTTTCCTGCAGGTTTTTGTTCTGTGCAG; G12, GCTTTCGACATGATTGGGGGGGGGGGGTATCTTCGTTTCCTGCAGGTTTTTGTTCTGTGCAG; G13, GCTTTCGACATGATTGGGGGGGGGGGGGTATCTTCGTTTCCTGCAGGTTTTTGTTCTGTGCAG; and G14, GCTTTCGACATGATTGGGGGGGGGGGGGGTATCTTCGTTTCCTGCAGGTTTTTGTTCTGTGCAG.
Circular dichroism
Following the work by Dexheimer et al. (50), we incubated cuvettes containing 5 μM of oligomeric DNA dissolved in tris-HCl (50 mM, pH 7.6) containing either 100 mM KCl or 100 mM NaCl for 5 min at 90°C and then let them slowly cool to 25°C. Circular dichroism spectra were measured on a spectropolarimeter (J-815, JASCO) using a 1-cm path length quartz cuvette, over a range of 200 to 320 nm, with a response time of 1 s and a scanning speed of 100 nm min−1. Three replicate measurements were taken and measured at 25°C.
Supplementary Material
Acknowledgments
We thank A. Murray, P. Rainey, M. Fumasoni, and three anonymous reviewers for their comments on the manuscript. We thank members of the Leu laboratory for helpful discussions. Funding: M.J.M. was funded by an Academia Sinica distinguished postdoctoral fellowship. J.-Y.L. was supported by Academia Sinica of Taiwan (100-CDA-L04) and the Taiwan Ministry of Science and Technology (NSC103-2321-B-001-001). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Author contributions: M.J.M. and J.-Y.L. designed the study. M.J.M., Y.-H.Y., S.Y.C., C.-F.K., and J.-F.G. performed experiments. M.J.M. and J.-Y.L. analyzed the data and wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/5/e1501033/DC1
fig. S1. Expansion of the polyguanine repeat (G14 to G15) reduces the Ura3 protein abundance but not the mRNA level.
fig. S2. G11 to G14 sequences do not stop DNA polymerase from synthesizing DNA, whereas G-quadruplex does.
table S1. Summary table of 318 sequenced ura3 mutants from G0, G14-ORF, and G14-repeat strains.
table S2. Chromosome insertion position and replication timing for engineered G14-URA inserts.
REFERENCES AND NOTES
- 1.Keightley P. D., Lynch M., Toward a realistic model of mutations affecting fitness. Evolution 57, 683–685 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Sanjuán R., Nebot M. R., Chirico N., Mansky L. M., Belshaw R., Viral mutation rates. J. Virol. 84, 9733–9748 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drake J. W., The distribution of rates of spontaneous mutation over viruses, prokaryotes, and eukaryotes. Ann. N. Y. Acad. Sci. 870, 100–107 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Giraud A., Radman M., Matic I., Taddei F., The rise and fall of mutator bacteria. Curr. Opin. Microbiol. 4, 582–585 (2001). [DOI] [PubMed] [Google Scholar]
- 5.McDonald M. J., Hsieh Y.-Y., Yu Y.-H., Chang S.-L., Leu J.-Y., The evolution of low mutation rates in experimental mutator populations of Saccharomyces cerevisiae. Curr. Biol. 22, 1235–1240 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Wielgoss S., Barrick J. E., Tenaillon O., Wiser M. J., Dittmar W. J., Cruveiller S., Chane-Woon-Ming B., Médigue C., Lenski R. E., Schneider D., Mutation rate dynamics in a bacterial population reflect tension between adaptation and genetic load. Proc. Natl. Acad. Sci. U.S.A. 110, 222–227 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Datta A., Jinks-Robertson S., Association of increased spontaneous mutation rates with high levels of transcription in yeast. Science 268, 1616–1619 (1995). [DOI] [PubMed] [Google Scholar]
- 8.Schuster-Böckler B., Lehner B., Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 488, 504–507 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Yang Y., Sterling J., Storici F., Resnick M. A., Gordenin M. A., Hypermutability of damaged single-strand DNA formed at double-strand breaks and uncapped telomeres in yeast Saccharomyces cerevisiae. PLOS Genet. 4, e1000264 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan K., Sterling J. F., Roberts S. A., Bhagwat A. S., Resnick M. A., Gordenin D. A., Base damage within single-strand DNA underlies in vivo hypermutability induced by a ubiquitous environmental agent. PLOS Genet. 8, e1003149 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lang G. I., Murray A. W., Mutation rates across budding yeast chromosome VI are correlated with replication timing. Genome Biol. Evol. 3, 799–811 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stamatoyannopoulos J. A., Adzhubei I., Thurman R. E., Kryukov G. V., Mirkin S. M., Sunyaev S. R., Human mutation rate associated with DNA replication timing. Nat. Genet. 41, 393–395 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lawrence M. S., Stojanov P., Polak P., Kryukov G. V., Cibulskis K., Sivachenko A., Carter S. L., Stewart C., Mermel C. H., Roberts S. A., Kiezun A., Hammerman P. S., McKenna A., Drier Y., Zou L., Ramos A. H., Pugh T. J., Stransky N., Helman E., Kim J., Sougnez C., Ambrogio L., Nickerson E., Shefler E., Cortés M. L., Auclair D., Saksena G., Voet D., Noble M., DiCara D., Lin P., Lichtenstein L., Heiman D. I., Fennell T., Imielinski M., Hernandez B., Hodis E., Baca S., Dulak A. M., Lohr J., Landau D.-A., Wu C. J., Melendez-Zajgla J., Hidalgo-Miranda A., Koren A., McCarroll S. A., Mora J., Lee R. S., Crompton B., Onofrio R., Parkin M., Winckler W., Ardlie K., Gabriel S. B., Roberts C. W. M., Biegel J. A., Stegmaier K., Bass A. J., Garraway L. A., Meyerson M., Golub T. R., Gordenin D. A., Sunyaev S., Lander E. S., Getz G., Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waters L. S., Walker G. C., The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G2/M phase rather than S phase. Proc. Natl. Acad. Sci. U.S.A. 103, 8971–8976 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lujan S. A., Clausen A. R., Clark A. B., MacAlpine H. K., MacAlpine D. M., Malc E. P., Mieczkowski P. A., Burkholder A. B., Fargo D. C., Gordenin D. A., Kunkel T. A., Heterogeneous polymerase fidelity and mismatch repair bias genome variation and composition. Genome Res. 24, 1751–1764 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lang G. I., Parsons L., Gammie A. E., Mutation rates, spectra, and genome-wide distribution of spontaneous mutations in mismatch repair deficient yeast. G3 3, 1453–1465 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levinson G., Gutman G. A., Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 4, 203–221 (1987). [DOI] [PubMed] [Google Scholar]
- 18.Hammerschmidt K., Rose C. J., Kerr B., Rainey P. B., Life cycles, fitness decoupling and the evolution of multicellularity. Nature 515, 75–79 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Mirkin S. M., Expandable DNA repeats and human disease. Nature 447, 932–940 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Moxon R., Bayliss C., Hood D., Bacterial contingency loci: The role of simple sequence DNA repeats in bacterial adaptation. Annu. Rev. Genet. 40, 307–333 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Usdin K., Grabczyk E., DNA repeat expansions and human disease. Cell. Mol. Life Sci. 57, 914–931 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tautz D., Hypervariability of simple sequences as a general source for polymorphic DNA markers. Nucleic Acids Res. 17, 6463–6471 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zlotorynski E., Rahat A., Skaug J., Ben-Porat N., Ozeri E., Hershberg R., Levi A., Scherer S. W., Margalit H., Kerem B., Molecular basis for expression of common and rare fragile sites. Mol. Cell Biol. 23, 7143–7151 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan Y. F., Marks M. E., Jones F. C., Villarreal G. Jr, Shapiro M. D., Brady S. D., Southwick A. M., Absher D. M., Grimwood J., Schmutz J., Myers R. M., Petrov D., Jónsson B., Schluter D., Bell M. A., Kingsley D. M., Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327, 302–305 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang W., Dominska M., Gawel M., Greenwell P. W., Petes T. D., Genomic deletions and point mutations induced in Saccharomyces cerevisiae by the trinucleotide repeats (GAA.TTC) associated with Friedreich’s ataxia. DNA Repair 12, 10–17 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saini N., Zhang Y., Nishida Y., Sheng Z., Choudhury S., Mieczkowski P., Lobachev K. S., Fragile DNA motifs trigger mutagenesis at distant chromosomal loci in saccharomyces cerevisiae. PLOS Genet. 9, e1003551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rattray A. J., Shafer B. K., McGill C. B., Strathern J. N., The roles of REV3 and RAD57 in double-strand-break-repair-induced mutagenesis of Saccharomyces cerevisiae. Genetics 162, 1063–1077 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shee C., Gibson J. L., Rosenberg S. M., Two mechanisms produce mutation hotspots at DNA breaks in Escherichia coli. Cell Rep. 2, 714–721 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonald M. J., Wang W. C., Huang H.-D., Leu J.-Y., Clusters of nucleotide substitutions and insertion/deletion mutations are associated with repeat sequences. PLOS Biol. 9, e1000622 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nieduszynski C. A., Knox Y., Donaldson A. D., Genome-wide identification of replication origins in yeast by comparative genomics. Genes Dev. 20, 1874–1879 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mirkin E. V., Mirkin S. M., Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 71, 13–35 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lopes M., Foiani M., Sogo J. M., Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell 21, 15–27 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Huang D., Piening B. D., Paulovich A. G., The preference for error-free or error-prone postreplication repair in Saccharomyces cerevisiae exposed to low-dose methyl methanesulfonate is cell cycle dependent. Mol. Cell Biol. 33, 1515–1527 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu C., Gan H., Han J., Zhou Z.-X., Jia S., Chabes A., Farrugia G., Ordog T., Zhang Z., Strand-specific analysis shows protein binding at replication forks and PCNA unloading from lagging strands when forks stall. Mol. Cell 56, 551–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shcherbakova P. V., Fijalkowska I. J., Translesion synthesis DNA polymerases and control of genome stability. Front. Biosci. 11, 2496–2517 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Zhong X., Garg P., Stith C. M., Nick McElhinny S. A., Kissling G. E., Burgers P. M. J., Kunkel T. A., The fidelity of DNA synthesis by yeast DNA polymerase zeta alone and with accessory proteins. Nucleic Acids Res. 34, 4731–4742 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quah S.-K., von Borstel R. C., Hastings P. J., The origin of spontaneous mutation in Saccharomyces cerevisiae. Genetics 96, 819–839 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cassier C., Chanet R., Henriques J. A. P., Moustacchi E., The effects of three PSO genes on induced mutagenesis: A novel class of mutationally defective yeast. Genetics 96, 841–857 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fumasoni M., Zwicky K., Vanoli F., Lopes M., Branzei D., Error-free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polα/Primase/Ctf4 complex. Mol. Cell 57, 812–823 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krasilnikova M. M., Samadashwily G. M., Krasilnikov A. S., Mirkin S. M., Transcription through a simple DNA repeat blocks replication elongation. EMBO J. 17, 5095–5102 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Belotserkovskii B. P., Liu R., Tornaletti S., Krasilnikova M. M., Mirkin S. M., Hanawalt P. C., Mechanisms and implications of transcription blockage by guanine-rich DNA sequences. Proc. Natl. Acad. Sci. U.S.A. 107, 12816–12821 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prakash S., Johnson R. E., Prakash L., Eukaryotic translesion synthesis DNA polymerases: Specificity of structure and function. Annu. Rev. Biochem. 74, 317–353 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Lemontt J. F., Mutants of yeast defective in mutation induced by ultraviolet light. Genetics 68, 21–33 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schürer K. A., Rudolph C., Ulrich H. D., Kramer W., Yeast MPH1 gene functions in an error-free DNA damage bypass pathway that requires genes from Homologous recombination, but not from postreplicative repair. Genetics 166, 1673–1686 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mortensen U. H., Bendixen C., Sunjevaric I., Rothstein R., DNA strand annealing is promoted by the yeast Rad52 protein. Proc. Natl. Acad. Sci. U.S.A. 93, 10729–10734 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Foster P. L., Trimarchi J. M., Adaptive reversion of a frameshift mutation in Escherichia coli by simple base deletions in homopolymeric runs. Science 265, 407–409 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harris R. S., Longerich S., Rosenberg S. M., Recombination in adaptive mutation. Science 264, 258–260 (1994). [DOI] [PubMed] [Google Scholar]
- 48.Rosenberg S. M., Longerich S., Gee P., Harris R. S., Adaptive mutation by deletions in small mononucleotide repeats. Science 265, 405–407 (1994). [DOI] [PubMed] [Google Scholar]
- 49.Lopes J., Piazza A., Bermejo R., Kriegsman B., Colosio A., Teulade-Fichou M.-P., Foiani M., Nicolas A., G-quadruplex-induced instability during leading-strand replication. EMBO J. 30, 4033–4046 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dexheimer T. S., Sun D., Hurley L. H., Deconvoluting the structural and drug-recognition complexity of the G-quadruplex-forming region upstream of the bcl-2 P1 promoter. J. Am. Chem. Soc. 128, 5404–5415 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nik-Zainal S., Alexandrov L. B., Wedge D. C., Van Loo P., Greenman C. D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L. A., Menzies A., Martin S, Leung K., Chen L., Leroy C., Ramakrishna M., Rance R., Lau K. W., Mudie L. J., Varela I., McBride D. J., Bignell G. R., Cooke S. L., Shlien A., Gamble J., Whitmore I., Maddison M., Tarpey P. S., Davies H. R., Papaemmanuil E., Stephens P. J., McLaren S., Butler A. P., Teague J. W., Jönsson G., Garber J. E., Silver D., Miron P., Fatima A., Boyault S., Langerød A., Tutt A., Martens J. W. M., Aparicio S. A. J. R., Borg Å., Salomon A. V., Thomas G., Børresen-Dale A.-L., Richardson A. L., Neuberger M. S., Futreal P. A., Campbell P. J., Stratton M. R., Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harris R. S., Molecular mechanism and clinical impact of APOBEC3B-catalyzed mutagenesis in breast cancer. Breast Cancer Res. 17, 8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakofsky C. J., Roberts S. A., Malc E., Mieczkowski P. A., Resnick M. A., Gordenin D. A., Malkova A., Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep. 7, 1640–1648 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lada A. G., Stepchenkova E. I., Waisertreiger I. S., Noskov V. N., Dhar A., Eudy J. D., Boissy R. J., Hirano M., Rogozin I. B., Pavlov Y. I., Genome-wide mutation avalanches induced in diploid yeast cells by a base analog or an APOBEC deaminase. PLOS Genet. 9, e1003736 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taylor B. J., Nik-Zainal S., Wu Y. L., Stebbings L. A., Raine K., Campbell P. J., Rada C., Stratton M. R., Neuberger M. S., DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. eLife 2, e00534 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lydeard J. R., Lipkin-Moore Z., Sheu Y. J., Stillman B., Burgers P. M., Haber J. E., Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes Dev. 24, 1133–1144 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holbeck S. L., Strathern J. N., A role for REV3 in mutagenesis during double-strand break repair in Saccharomyces cerevisiae. Genetics 147, 1017–1024 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shishkin A. A., Voineagu I., Matera R., Cherng N., Chernet B. T., Krasilnikova M. M., Narayanan V., Lobachev K. S., Mirkin S. M., Large-scale expansions of Friedreich’s ataxia GAA repeats in yeast. Mol. Cell 35, 82–92 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shah K. A., Shishkin A. A., Voineagu I., Pavlov Y. I., Shcherbakova P. V., Mirkin S. M., Role of DNA polymerases in repeat-mediated genome instability. Cell Rep. 2, 1088–1095 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Uhlmann T., Boeing S., Lehmbacher M., Meisterernst M., The VP16 activation domain establishes an active mediator lacking CDK8 in vivo. J. Biol. Chem. 282, 2163–2173 (2007). [DOI] [PubMed] [Google Scholar]
- 61.Hall B. M., Ma C. X., Liang P., Singh K. K., Fluctuation AnaLysis CalculatOR: A web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics 25, 1564–1565 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Friedman K. L., Brewer B. J., Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Methods Enzymol. 262, 613–627 (1995). [DOI] [PubMed] [Google Scholar]
- 63.Liberi G., Cotta-Ramusino C., Lopes M., Sogo J., Conti C., Bensimon A., Foiani M., Methods to study replication fork collapse in budding yeast. Methods Enzymol. 409, 442–462 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Han H., Hurley L. H., Salazar M., A DNA polymerase stop assay for G-quadruplex-interactive compounds. Nucleic Acids Res. 27, 537–542 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gossen M., Bujard H., Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. U.S.A. 89, 5547–5551 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Z.-F., Li M.-H., Hsu S.-T., Chang T.-C., Structural basis of sodium-potassium exchange of a human telomeric DNA quadruplex without topological conversion. Nucleic Acids Res. 42, 4723–4733 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sen D., Gilbert W., A sodium-potassium switch in the formation of four-stranded G4-DNA. Nature 344, 410–414 (1990). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/5/e1501033/DC1
fig. S1. Expansion of the polyguanine repeat (G14 to G15) reduces the Ura3 protein abundance but not the mRNA level.
fig. S2. G11 to G14 sequences do not stop DNA polymerase from synthesizing DNA, whereas G-quadruplex does.
table S1. Summary table of 318 sequenced ura3 mutants from G0, G14-ORF, and G14-repeat strains.
table S2. Chromosome insertion position and replication timing for engineered G14-URA inserts.