

Abstract
The ring-shaped cohesin complex is thought to topologically hold sister chromatids together from their synthesis in S phase until chromosome segregation in mitosis. How cohesin stably binds to chromosomes for extended periods, without impeding other chromosomal processes that also require access to the DNA, is poorly understood. Budding yeast cohesin is loaded onto DNA by the Scc2–Scc4 cohesin loader at centromeres and promoters of active genes, from where cohesin translocates to more permanent places of residence at transcription termination sites. Here we show that, at the GAL2 and MET17 loci, pre-existing cohesin is pushed downstream along the DNA in response to transcriptional gene activation, apparently without need for intermittent dissociation or reloading. We observe translocation intermediates and find that the distribution of most chromosomal cohesin is shaped by transcription. Our observations support a model in which cohesin is able to slide laterally along chromosomes while maintaining topological contact with DNA. In this way, stable cohesin binding to DNA and enduring sister chromatid cohesion become compatible with simultaneous underlying chromosomal activities, including but maybe not limited to transcription.
Keywords: cohesin, genome stability, Saccharomyces cerevisiae, sister chromatid cohesion, transcription
1. Introduction
Cohesin is a ring-shaped multi-subunit protein complex, which is best known for its essential role in promoting sister chromatid cohesion [1–4]. The cohesin ring topologically encircles DNA and, in mitosis, cohesin entraps and thereby holds together pairs of sister chromatids. The cohesin complex consists of at least four essential subunits. At the core of the complex lies a dimer of structural maintenance of chromosomes (SMC) ATPases, Smc1 and Smc3. These form the ring via interaction interfaces at both ends of long flexible stretches of coiled-coil, an SMC hinge and the ATPase heads [5,6]. Two additional subunits of the complex are essential in all organisms studied, called Scc1 and Scc3 in budding yeast. Scc1 stabilizes the interaction between the SMC ATPase domains. Scc3 in turn associates with Scc1 and plays an essential role in loading of the cohesin complex onto DNA [7,8]. It does so by providing interactions with a separate cohesin loader complex that in budding yeast consists of the Scc2 and Scc4 subunits [9]. In addition to this cohesin core machinery, additional subunits Pds5, Wapl and sororin, the latter restricted to higher eukaryotes, associate with the cohesin complex to regulate the stability of cohesin binding to chromosomes.
A crucial moment for cohesin comes at the time of DNA replication. Two newly replicated sister chromatids emerge from the replication fork, which cohesin will hold together until mitosis. To ensure stable sister chromatid cohesion from S phase until mitosis, the cohesin ring is modified during DNA replication. The replication fork-associated acetyltransferase Eco1 acetylates cohesin rings on their Smc3 subunit, which stabilizes cohesin's grip on chromosomes [10–12]. This is achieved in at least two ways. First, acetylation renders cohesin resistant to a DNA unloading activity contained in the Wapl subunit. Dynamic cohesin unloading by Wapl is important to fine-tune the chromosome condensation status in both interphase and mitosis, but during DNA replication a subset of cohesin is rendered Wapl-resistant by Eco1 [13–16]. In addition, acetylation stabilizes cohesin on DNA by a second mechanism [17]. An explanation for both mechanisms has recently been suggested by the finding that the acetylation sites form a DNA sensor that triggers ATP hydrolysis, required for DNA entry into and exit out of the cohesin ring [18]. As the consequence, acetylated cohesin gains close to permanent association with chromosomes.
In budding yeast, acetylated cohesin remains chromosome-bound for longer than could be directly measured, at least for an hour [14]. In human cells, following cohesion establishment during DNA replication and assisted by the additional sororin subunit [19], about a third of cohesin complexes are similarly close to permanently stabilized and turn over on chromosomes with a rate that was again slower than could be measured, remaining chromosome-bound for at least 6 h [13]. An argument has been made that, in mammalian female oocytes, cohesin rings might persist on chromosomes for weeks (if not decades) to maintain sister chromatid cohesion in meiotic prophase arrested cells [20,21].
While cohesin is stably bound to chromosomes to provide sister chromatid cohesion, other forms of chromosomal metabolism must continue unabated underneath. Much of the budding yeast genome consists of genes that are frequently transcribed. Almost the entirety of the human genome is also transcribed, at least occasionally [22,23]. In addition, numerous DNA lesions continuously accrue and must be repaired in every cell, involving repair pathways that require access to the DNA. This includes for example nucleotide excision repair or DNA recombination events, both of which require DNA resynthesis to proceed over substantial distances. How cohesin can maintain stable sister chromatid cohesion over long periods of time, without restricting access to genomic regions for transcription, repair and other chromosomal requirements is not known.
We have previously observed that transcriptional activation of budding yeast genes can lead to a striking repositioning of cohesin along chromosomes, with cohesin apparently moving downstream of active genes to accumulate at convergent transcriptional termination sites [24]. Lateral sliding of cohesin, while retaining topological contact, has been suggested as a possible explanation for how the genome stays accessible while cohesin remains bound [25]. However, the actual mechanism of cohesin translocation is not known. In particular, it is not known whether relocation in response to transcriptional activation occurs while cohesin remains in contact with DNA or whether it requires cohesin dissociation from DNA and subsequent reloading at a new location. In the latter scenario, sister chromatid cohesion would gradually deteriorate during cohesin translocation, as under normal growth conditions cohesin can establish sister chromatid cohesion only during S phase, but not afterwards [12,26]. Indeed, a previous study that analysed cohesin's response to changes in the transcriptional programme concluded that cohesin is displaced from chromosomes in response to transcription [27]. How a transcription-mediated decay of sister chromatid cohesion over time is prevented in this scenario has not been explained.
Given the importance of stable sister chromatid cohesion, we have revisited the nature of cohesin translocation along budding yeast chromosome arms in response to transcriptional changes. We study two model loci where transcriptional induction leads to downstream translocation of cohesin. Cohesin behaviour is observed by chromatin immunoprecipitation (ChIP) followed by analysis on high-resolution oligonucleotide tiling arrays. We find that, following gene activation, cohesin can move downstream of genes without requirement for known dissociation or reloading factors and we observe a translocation intermediate, consistent with cohesin sliding along genes while retaining topological association. At another locus, close to a centromere, transcriptional activation leads to the apparent loss of cohesin from chromosomes. Together, our results support a model in which, at least along chromosome arms, cohesin retains topological contact with DNA and thereby sister chromatid cohesion, while sliding laterally to accommodate requirements of transcription and probably other forms of chromosomal metabolism.
2. Results
2.1. Cohesin translocation without the cohesin loader
As a model locus to study cohesin translocation, we chose the GAL2 locus on the right arm of budding yeast chromosome 12 [27,28]. There, as an exception to the typical location of cohesin peaks at convergent intergenic regions, three sequential genes in tandem orientation, SIC1, EMP46 and GAL2 are covered by cohesin. This is the case in cells grown in medium containing raffinose as the carbon source and arrested in G2/M by nocodazole treatment, when the three genes are barely transcribed (figure 1a) [29]. Following addition of galactose to the culture, which induces GAL2 expression, the GAL2 gene is cleared of cohesin. Instead, in an aliquot of the same culture taken 1 h after galactose addition, a marked cohesin peak appears downstream of GAL2 (figure 1a). The remainder of the genomic cohesin distribution remained largely unchanged following galactose addition, allowing the relative quantitative comparison of cohesin at the GAL2 locus with the rest of the genome. An upstream cohesin binding region that remained unchanged is therefore included in our graphs for comparison. In addition, we analysed cohesin chromatin immunoprecipitates by quantitative real-time PCR using primer pairs both within the GAL2 gene and downstream in the SRL2/EMP70 intergenic region, before and after galactose addition. This confirmed the quantitative downstream relocation of cohesin from the GAL2 locus (figure 1b).
Figure 1.

Contribution of Scc2–Scc4 to cohesin translocation at the GAL2 locus. (a) Cohesin translocation following GAL2 induction. Cells were grown in raffinose-containing medium, synchronized in G1 and released into nocodazole-induced G2/M arrest. After 2 h, the culture was shifted to 35°C and after a further hour galactose was added to induce GAL2 expression. Scc1 association with chromatin was analysed before (− galactose) and one hour following induction (+ galactose). Enrichment in the immunoprecipitate relative to a whole genome DNA sample is shown along a region surrounding the GAL2 locus. Each bar represents the average of 25 oligonucleotide probes within adjacent 125 bp windows. Blue bars above and below the midline are genes transcribed, respectively, from left to right, and from right to left. The location of two primer pairs, P1 and P2, used for analysis of chromatin immunoprecipitates by quantitative real-time PCR in (b), are indicated. (b) Analysis of Scc1 chromatin immunoprecipitates using primer pairs P1 and P2 by quantitative real-time PCR, before and 1 h after galactose addition. The means and standard error of three repeats of the experiment are shown. (c) Schematic of cohesin translocation by either reloading or sliding. Indicated is the predicted requirement for the Scc2–Scc4 cohesin loader in the former, but not the latter, case. (d) Cohesin translocates at a reduced rate following inactivation of the Scc2–Scc4 cohesin loader. As (a), but cells carried temperature-sensitive alleles of the cohesin loader subunits. (e) Compromised GAL2 induction following cohesin loader inactivation. GAL2 mRNA induction following galactose addition, relative to actin, was assessed using quantitative real-time PCR in both a wild-type and scc2–4 strain. The means and standard deviation from two biological and four technical replicates are shown.
The following experiments were designed to explore two possible explanations for cohesin translocation. It could be that cohesin is removed from chromosomes following transcriptional activation of the GAL2 locus and a new cohesin binding site is established downstream, where cohesin is newly loaded. Alternatively, cohesin that topologically encircles DNA could be pushed downstream along chromosomes, without dissociation and therefore without need for reloading (figure 1c). To distinguish between these two possibilities, we first asked whether the Scc2–Scc4 cohesin loader is required for cohesin translocation. In the reloading model, this is likely to be the case. We therefore repeated the GAL2 induction experiment in strains in which the cohesin loader can be inactivated owing to a temperature-sensitive scc2–4 or scc4–4 mutation [9]. Once cells were arrested in G2/M, they were shifted to 35°C for 1 h, a restrictive temperature for the scc2–4 and scc4–4 alleles, before galactose addition. Following either Scc2 or Scc4 inactivation, GAL2 induction still caused cohesin translocation, but the extent of both cohesin loss from the GAL2 locus and its downstream accumulation were reduced (figure 1d). This suggests that, while not absolutely required, the cohesin loader facilitates cohesin repositioning at the GAL2 locus.
The cohesin translocation defect at the GAL2 locus, following Scc2–Scc4 inactivation, could be for two reasons. Scc2–Scc4-dependent cohesin loading might augment a less efficient, loading-independent translocation reaction. This scenario would explain the reduced downstream cohesin peak, but would not explain why clearance of the GAL2 locus is also less efficient in the absence of the cohesin loader. Alternatively, Scc2–Scc4 inactivation might hamper transcriptional activation of the GAL2 locus, which could explain both reduced cohesin loss from the GAL2 locus as well as reduced downstream accumulation. To investigate this possibility, we quantified GAL2 induction by real-time PCR analysis of transcript levels. This revealed that GAL2 induction was markedly reduced in scc2–4 mutant cells, compared with the wild-type control (figure 1e). While we do not know how the cohesin loader promotes GAL2 gene induction, we recently reported on its role in promoter nucleosome eviction, which might pertain to reduced gene activation [30]. Attenuated transcriptional activation, in turn, could explain the reduced efficiency of cohesin translocation. We conclude that cohesin translocation is possible without new cohesin loading. The mechanism by which Scc2–Scc4 augments the process at the GAL2 locus remains to be further explored.
2.2. Loading-deficient cohesin is translocation proficient
Given the complications with the interpretation of the above experiment, we used a complementary approach to test the contribution of new cohesin loading to transcription-induced translocation. Cohesin loading onto DNA requires hydrolysis of ATP, bound to the Smc1 and Smc3 head domains [8,31,32]. Mutation of an arginine finger in both ATPases (Smc1R58A/Smc3R58A) greatly reduces the rate of cohesin loading onto chromosomes. Starting from the G1/S transition, when the cohesin complex is assembled in budding yeast, it takes about 2 h longer for Smc1R58A/Smc3R58A cohesin to reach equal levels on chromosomes compared with wild-type cohesin [12] (figure 2a). If cohesin translocation involves ATP hydrolysis-dependent reloading, then downstream accumulation of Smc1R58A/Smc3R58A cohesin is expected to be reduced or delayed. To analyse whether this was the case, we performed the following experiment. Wild-type and Smc1R58A/Smc3R58A cells were synchronized in raffinose-containing medium by α-factor block and release. Cell cycle progression was halted by hydroxyurea (HU) treatment in early S phase for 2 h, to allow Smc1R58A/Smc3R58A cohesin to accumulate on chromosomes. Cultures were now released into nocodazole-containing medium, and the cohesin distribution in the G2/M-arrested cells was assessed at the GAL2 locus before and after induction by galactose addition. Expression of budding yeast galactose-inducible genes becomes detectable around 8–12 min following galactose addition to the growth medium [33]. To sensitively detect a cohesin translocation difference due to compromised loading, we therefore took our second sample 15 min following galactose addition. In wild-type cells, cohesin translocation at the GAL2 locus was largely complete by this time. Notably, translocation of Smc1R58A/Smc3R58A cohesin was similarly fast and efficient (figure 2b). Thus, while causing a marked delay in loading onto chromosomes, Smc1R58A/Smc3R58A cohesin was fully proficient in transcription-induced downstream relocation. This suggests that renewed ATP hydrolysis, and therefore renewed cohesin loading onto chromosomes, are unlikely to be part of the translocation mechanism.
Figure 2.

Altered cohesin dynamics do not impact on cohesin translocation. (a) Schematic of the respective consequences of the smc1R58A/smc3R58A and wpl1Δ mutations on dynamic cohesin association with chromosomes. (b) Cohesin translocation remains unaffected by the smc1R58A/smc3R58A mutations. Following release from α-factor arrest, cell cycle progression was halted for 2 h in HU-containing medium to allow cohesin accumulation on chromosomes, before cell cycle progression was allowed to resume and cells were arrested in G2/M by nocodazole treatment. Cohesin chromatin immunoprecipitation (ChIP) was then performed before and 15 min after GAL2 induction by galactose addition. (c) Cohesin translocation remains unaffected in the absence of Wapl. Cohesin ChIP was performed in G2/M arrested cells before and 1 h after GAL2 induction.
2.3. Wapl-dependent unloading is dispensable for cohesin translocation
We took an additional approach to test whether a dissociation and reloading cycle forms part of cohesin translocation, by asking whether the dissociation activity of cohesin's Wapl subunit is involved in the process. Before DNA replication, cohesin associates with chromosomes in a dynamic fashion, undergoing cycles of loading and Wapl-mediated dissociation (figure 2a). Following DNA replication, acetylation turns cohesin resistant to Wapl, though Wapl retains an influence over at least a fraction of cohesin [15,16,18,30]. To test whether cohesin unloading from chromosomes by Wapl is involved in cohesin translocation, we compared a wild-type and a wpl1Δ strain, deleted for the gene encoding Wapl. Following synchronization, cells were again arrested in G2/M by nocodazole treatment and then GAL2 gene expression was induced by galactose addition. Cohesin translocation occurred in an indistinguishable fashion in both wild-type and wpl1Δ cells (figure 2c). This observation again suggests that cohesin does not dissociate and reload during the translocation process.
2.4. Pre-existing cohesin relocates downstream
Another way to assess whether cohesin translocates by lateral movement, or by dissociation and reloading, is to determine the composition of the newly formed downstream cohesin pool. This pool could consist of previously chromosome-bound cohesin, as expected following lateral movement. Alternatively, it could include cohesin that was not previously chromosome-bound. To distinguish between these possibilities, we devised the following experiment. As before, cells containing the temperature-sensitive scc2–4 allele were synchronized by α-factor block and release and arrested in G2/M by nocodazole treatment under permissive conditions. Endogenous cohesin, containing a Pk epitope-tagged Scc1 subunit for detection, is bound to chromosomes under these conditions. The cohesin loader was now inactivated by temperature shift. After this, we induced synthesis of an HA epitope-tagged Scc1 subunit, under control of the galactose-inducible GAL1 promoter, for 1 h. The differentially epitope-tagged Scc1 assembles into functional new cohesin complexes [12], so that cells now contained two cohesin populations: endogenous Pk epitope-tagged cohesin that is bound to chromosomes, as well as HA epitope-tagged cohesin in the nucleoplasm (figure 3a). Now, we induced cohesin translocation and analysed the composition of the downstream cohesin pool.
Figure 3.

Pre-existing cohesin translocates at the MET17 locus. (a) Schematic of the experiment to compare the behaviour of endogenous cohesin (containing Pk epitope-tagged Scc1) and ectopic cohesin (containing HA epitope-tagged Scc1) that was expressed after cohesin loader inactivation, but before cohesin translocation was induced. (b) Cohesin translocation at the MET17 locus. Cohesin chromatin immunoprecipitation (ChIP) was performed in G2/M arrested cells, before (YP) and 30 min after transfer to medium lacking amino acids (YNB—aa). (c) Only pre-existing cohesin translocates. After 1.5 h following release from α-factor arrest into G2/M, cells were shifted to a restrictive temperature for the scc2–4 allele for 1 h before Scc1-HA expression was induced by galactose addition for one further hour. Then cells were transferred to medium lacking amino acids. Cohesin ChIP was performed against both endogenous (Scc1-Pk) and ectopic (Scc1-HA) cohesin just before and 30 min following amino acids starvation. (d) Ectopic Scc1-HA levels at the indicated times were compared by immunoblotting to those in a control strain in which endogenous Scc1 was fused to the same HA epitope. Tubulin served as a loading control. (e) The efficiency of Scc1 chromatin immunoprecipitation using an HA and Pk epitope, respectively, was quantitatively compared at five cohesin binding sites and three negative control regions.
As we used galactose to induce Scc1-HA synthesis, we used another translocation model locus, MET17, whose transcription is activated by amino acid starvation [27,28]. In rich medium, containing raffinose and galactose, endogenous Scc1-Pk containing cohesin covers the MET17 gene, while the downstream, convergently transcribed ACO1 gene is cohesin free (figure 3b). Thirty minutes after transfer to synthetic medium lacking amino acids, endogenous cohesin translocated downstream. MET17 was now free of cohesin, whereas the ACO1 locus was partly covered. This confirms our earlier results that cohesin translocation can occur independently of the Scc2–Scc4 cohesin loader. We next quantitatively analysed the composition of the new cohesin peak by ChIP followed by quantitative real-time PCR, using seven primer pairs across the MET17–ACO1 locus. This analysis confirmed downstream translocation of endogenous Scc1-Pk-containing cohesin, whereas we did not detect HA epitope-tagged Scc1 from soluble cohesin above background levels at any of the locations, either before or after MET17 induction (figure 3c). This suggests that the ‘new’ cohesin peak consists largely, if not exclusively, of previously chromosome-bound cohesin. The closest possible origin of cohesin in the new peak is those cohesin molecules that previously occupied the upstream location and appear to have moved downstream along the chromosome following transcriptional activation.
As a control for the above experiment, we confirmed that ectopic Scc1-HA, following 1 h of induction, reached levels comparable to those in a strain in which the endogenous SCC1 locus is tagged with the same epitope (figure 3d). We furthermore confirmed that, under these conditions, both HA and Pk epitope-tagged cohesin is retrieved with similar efficiency by ChIP (figure 3e). This validates the quantitative comparison between ectopic and endogenous cohesin following MET17 induction and supports the conclusion that little if any ectopic cohesin is incorporated into a newly established cohesin peak following transcriptional induction.
2.5. A translocation intermediate suggestive of cohesin sliding
Our above results suggest that pre-existing chromosomal cohesin is repositioned during cohesin translocation. To observe the process at a greater time resolution, we returned to the GAL2 locus (figure 4a). We again used synchronized cells that were arrested in G2/M by nocodazole treatment. To enhance the time resolution of our observations, we lowered the temperature of the culture to 16°C before GAL2 induction by galactose addition. Samples were now collected every 3 min and processed for ChIP analysis. Cohesin along the GAL2 locus decreased sequentially, starting 6 min after galactose addition at the 5′ end of the gene, over time progressing towards the 3′ end (figure 4b). At 9–12 min after galactose addition, a transient intermediate peak formed that continued to move further downstream to reach its final position 15 min after induction. These observations are consistent with a sliding movement of cohesin along chromosomes, being pushed downstream while retaining topological contact with DNA.
Figure 4.
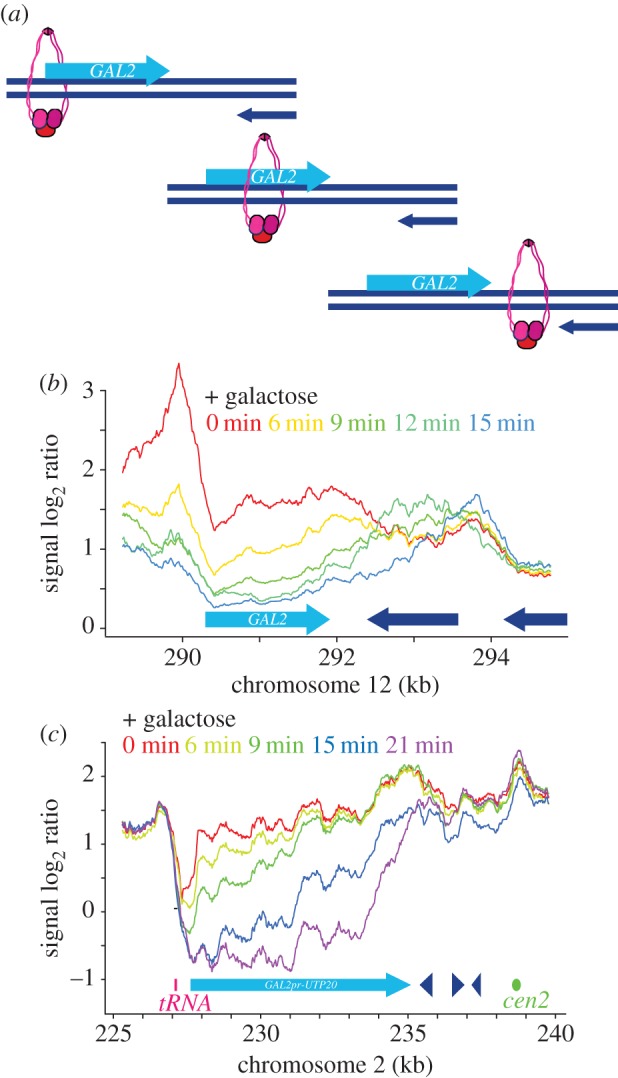
A translocation intermediate suggestive of cohesin sliding. (a) A model depicting a cohesin sliding intermediate. (b) Time course analysis of cohesin translocation at the GAL2 locus in cells arrested in G2/M and shifted to 16°C for 1 h before galactose addition. (c) Cohesin behaviour at the synthetic GAL2 promoter-UTP20 locus, following galactose induction as in (b).
2.6. Apparent cohesin loss following transcriptional activation close to a centromere
With an aim to improve the spatial resolution when visualizing cohesin translocation, we engineered a synthetic inducible locus, making use of the 7.5 kb long UTP20 gene, the 13th longest gene in the budding yeast genome. UTP20 lies close to the centromere on chromosome 2, within the cohesin-enriched region that surrounds budding yeast centromeres [34]. We replaced the UTP20 promoter with the GAL2 promoter, so that we could switch UTP20 expression off and on at will. The UTP20 gene product is essential, with a role in 18S rRNA maturation [35]. Following GAL2-UTP20 repression, cells continued to proliferate for approximately three divisions without notable adverse effect, before proliferation slowed and eventually terminated. Therefore, cultures were grown in galactose-containing medium to maintain UTP20 expression, then shifted to medium containing raffinose just before cell synchronization by α-factor block and release. After release, cells were arrested in G2/M by nocodazole treatment, and the temperature reduced to 16°C. Galactose was added, and the cohesin distribution observed in 3 min intervals. Starting at 6 min after galactose addition, cohesin levels at the 5′ end of UTP20 declined, after which cohesin loss spread in the 5′ to 3′ direction over time (figure 4c). Unlike at the GAL2 locus, abundant cohesin was present downstream of UTP20 already at the start of the experiment, and we did not observe further cohesin accumulation there. Instead, it appeared as if cohesin was sequentially cleared from the UTP20 locus in the direction of transcription. Cohesin might be dislodged from chromosomes as the consequence of transcription at this locus. Alternatively, we cannot exclude that cohesin was in fact pushed downstream, but entered a more dynamic cohesin pool in the vicinity of the centromere that might have obscured the arrival of new cohesin. In either case, our findings confirm that cohesin is cleared from genes by transcription. It also reconciles our results with previous observations that, at certain loci, cohesin is apparently lost from chromosomes following transcriptional activation [27].
2.7. Cohesin translocation along unreplicated DNA
So far, all our cohesin translocation experiments were performed in G2/M arrested cells in which cohesin is engaged in holding sister chromatids together. We wondered whether the ability to slide along chromosomes is a feature that cohesin gains during the establishment of sister chromatid cohesion, or whether chromosome-bound cohesin can do so already before DNA replication. To address this, we arrested cells in early S phase using HU treatment. We used a strain engineered to take up bromodeoxyuridine (BrdU) and performed ChIP against BrdU to assess which parts of the genome were replicated in this arrest. This analysis revealed BrdU incorporation at regions surrounding the early origins ars1211 and ars1213, 60 kb upstream and 80 kb downstream of the GAL2 locus, respectively (figure 5a). The GAL2 locus itself remained unreplicated in the arrest. When we induced GAL2 transcription by galactose addition in HU-arrested cells, we observed cohesin translocation in a similar manner to what is seen in G2/M arrested cells (figure 5b). Thus, cohesin is able to move along chromosomes both on unreplicated and replicated DNA. This suggests that the mode of cohesin binding to DNA that endows its ability to translocate, most likely topological DNA embrace, is gained already before S phase and remains unaltered during the establishment of sister chromatid cohesion.
Figure 5.

Cohesin translocation along unreplicated DNA. (a) Cells were synchronized in G1 and released into medium containing BrdU and 200 mM HU for 1 h. BrdU incorporation around early replicating origins was visualized by ChIP against BrdU [14]. (b) Cohesin translocation following GAL2 induction in similarly synchronized and arrested cells. Scc1 association with chromatin was analysed before (− galactose) and one hour following induction (+ galactose).
2.8. The cohesin translocation pattern is shaped by transcription termination
Scc2–Scc4 cohesin loader binding sites are found in the promoters of strongly expressed genes. Cohesin can be transiently detected at these loading sites in G1 before the cohesin distribution becomes dominated by its more permanent places of residence at convergent transcriptional termination sites [7,24,30]. This pattern could be explained if most cohesin moves along chromosomes after loading, in response to transcription, until reaching places where transcription converges. To test whether the chromosomal cohesin distribution is defined by transcriptional termination, we used the temperature-sensitive rat1-1 mutation in the Rat1 exonuclease, required for efficient transcriptional termination in budding yeast. At a non-permissive temperature for the rat1-1 allele, RNA polymerase II progresses past normal termination points at an increased frequency [36,37] (figure 6a). When we compared the cohesin association pattern between wild-type and rat1-1 cells in G2/M-arrested cells, we noted that cohesin peaks were broader and less well defined in rat1-1 mutant cells already at the permissive temperature (figure 6b). Following shift to the restrictive temperature, numerous cohesin peaks disappeared, exemplified at the strongly expressed RPL40A gene, where the downstream cohesin peak was lost as the consequence of compromised transcriptional termination (figure 6b). Genome-wide, of a total of 981 prominent cohesin peaks that we counted, 167 disappeared following rat1-1 inactivation. Additional examples from different chromosomes are shown in the electronic supplementary material, figure S1.
Figure 6.

The cohesin distribution is determined by transcription termination. (a) A schematic of how the Rat1 nuclease aids transcription termination and of how its absence might affect cohesin positioning. (b) Cohesin distribution surrounding the RPL40A gene in G2/M arrested cells in a rat1-1 strain and a wild-type control under the indicated conditions. (c) As (b), but the region surrounding the GAL2 gene is shown, before the temperature shift and before and after galactose addition for 1 h.
At the GAL2 locus, the cohesin distribution was also broader in rat1-1 cells at permissive temperature, compared with the wild-type control. Following shift to the restrictive temperature, the cohesin peak downstream of RPL10 and FMP25 disappeared. Furthermore, in response to GAL2 induction, cohesin translocation reached further downstream with an increased proportion of cohesin accumulating as far downstream as the RAX2 gene (figure 6c). These observations are consistent with the possibility that cohesin moves downstream along genes, until reaching places where transcription terminates. This further suggests that much of chromosomal cohesin reaches its final distribution by moving from promoter loading sites towards places of convergent transcriptional termination.
2.9. Cohesin translocation independent of gene looping
Gene loops have been proposed to bring gene promoters and terminators into spatial proximity in budding yeast [38]. An alternative possibility for cohesin translocation from promoters to terminators could therefore be envisioned, in which cohesin hops across loops from promoter to terminator, instead of sliding along the length of the gene (figure 7a). While loop hopping should not lead to translocation intermediates of the sort that we observed along the GAL2 locus (figure 4b), we nevertheless wanted to investigate whether gene looping affects the chromosomal cohesin pattern. We took advantage of the TFIIB mutant sua7-1, which is defective in gene looping [39]. The cohesin localization pattern remained virtually unchanged between wild-type and sua7-1 cells, with most of the cohesin concentrated at convergent transcriptional termination sites (figure 7b). This demonstrates that gene looping is not required for cohesin to reach terminators. Despite a largely identical cohesin pattern, we observed small differences in sua7-1 cells, e.g. a minor new cohesin peak at ‘58 kb’ along chromosome 6 (figure 7b). We do not currently know whether this difference is due to the absence of gene looping, or whether it is caused by another difference in transcriptional dynamics in sua7-1 cells [39].
Figure 7.

Cohesin translocation independent of gene looping. (a) Schematic of cohesin translocation either by sliding, or by hopping between promoter and terminator, brought together by gene looping. (b) Scc1 association with chromatin was compared between sua7-1 and wild-type cells, grown at 30°C and synchronized in G2/M using nocodazole treatment. The enrichment of DNA in the Scc1 chromatin immunoprecipitate relative to a whole genome DNA sample is shown along a section of chromosome 6. See figure 1 for further details.
2.10. Cohesin translocation in response to T7 RNA polymerase transcription
We finally wanted to address what aspect of transcription makes cohesin slide along chromosomes. It could be that a particular component of the RNA polymerase II machinery contacts cohesin, or simply that the sheer overall size of the greater than 2 MDa polymerase holocomplex pushes cohesin [40]. Alternatively, the RNA transcript that emerges from and moves along the chromosome with the advancing polymerase might drag cohesin along. To address this question, we engineered the endogenous GAL2 locus such that it can be transcribed by the bacteriophage T7 RNA polymerase [41,42] (figure 8a). The T7 RNA polymerase is a 99 kDa monomeric protein, much smaller than the eukaryotic RNA polymerase II complex. For this purpose, we replaced the endogenous GAL2 promoter by a T7 promoter consensus sequence and expressed the T7 RNA polymerase, fused to a nuclear localization signal, under control of the yeast housekeeping ADH1 promoter. The cohesin distribution in cells harbouring the T7 promoter-GAL2 locus, but lacking T7 RNA polymerase, was indistinguishable from what is observed in wild-type cells (figure 8b). Addition of T7 RNA polymerase resulted in striking clearance of the GAL2 locus and downstream sequences, with hardly any cohesin detectable up to the SMC4-CSF1 convergence site 15 kb further along the chromosome. This suggests that transcription by the T7 RNA polymerase efficiently removes cohesin.
Figure 8.

Cohesin sliding in response to T7 RNA polymerase transcription. (a) Schematic of how the GAL2 gene was turned into a bacteriophage T7 RNA polymerase-transcribed locus. (b) Cohesin distribution at the T7 promoter-GAL2 locus in strains that either do not (− T7 polymerase) or do (+ T7 polymerase) express a T7 RNA polymerase-NLS fusion protein. (c) Timecourse analysis of the cohesin distribution along the T7 promoter-GAL2 locus, following 20 mM IPTG addition in a strain that expresses the lacI repressor together with the T7 RNA polymerase. (d) Cohesin distribution as in (c), following overnight culture in the absence or presence of IPTG. (e) Comparison of GAL2 transcript levels in wild-type cells, grown overnight under conditions in which GAL2 expression was repressed or induced, and T7 promoter-GAL2 cells that were grown under the indicated conditions.
We next wanted to establish whether T7 RNA polymerase pushes cohesin along chromosomes, similar to yeast RNA polymerase II, or displaces cohesin from chromosomes. Therefore, we rendered T7 transcription inducible by co-expressing the LacI transcriptional repressor that binds to a lac operator sequence that we included as part of the T7 promoter. Cohesin distribution in the presence of both T7 RNA polymerase and LacI was again comparable to that in the absence of both proteins, indicating that LacI efficiently repressed GAL2 transcription (figure 8c–e). We now added isopropyl β-d-1-thiogalactopyranoside (IPTG) to the culture to relieve LacI repression and induce T7 RNA polymerase transcription. Over the course of 2 h, this led to successive clearance of the GAL2 locus with concomitant accumulation of cohesin at the terminator of the opposing EMP70 gene (figure 8c). After overnight growth in medium containing IPTG, cohesin was also detected further downstream at the RAX2 terminator (figure 8d). The transcriptional strength of the induced T7 promoter in the presence of LacI and IPTG was lower than if LacI was altogether absent (figure 8e), which could be the reasons for the less complete clearance of cohesin downstream of GAL2. Together, these results suggest that transcription even by the relatively small T7 RNA polymerase leads to downstream translocation of cohesin along chromosomes towards places where transcription converges. While we cannot exclude direct interactions between cohesin and cellular RNA polymerases, a driving force that is sufficient for cohesin movement is provided by a feature in common with T7 RNA polymerase, possibly the RNA transcript that emerges from the polymerase.
3. Discussion
In this study, we analyse the nature of the striking cohesin pattern changes along chromosomes that result from transcriptional alterations. We find that factors with known roles in dynamic cohesin loading and turnover on DNA, such as the Scc2–Scc4 cohesin loader, the cohesin ATPase or Wapl, have little or no impact on cohesin movement in response to transcriptional changes. While we cannot exclude that an as-yet-unidentified mechanism unloads and re-loads cohesin during transcription, we did not find evidence that cohesin repositioning involves recruitment of new cohesin. Rather, pre-existing cohesin appears to slide along the chromosome to reach new places of residence, defined by transcription termination. These observations support a model in which cohesin that is topologically bound to DNA is able to slide laterally to adapt to the needs of other aspects of chromosome biology including, but probably not limited to, transcription.
A previous study concluded that cohesin is displaced from chromosomes by transcription [27]. While at first sight this contradicts our conclusions, on closer inspection the previous results differ only slightly from our current observations. At most of the investigated loci, both studies, as well as our previous study [24], have seen cohesin translocation in response to transcriptional activation. We do not know why at a subset of loci, e.g. the MET17 locus, translocation is more obvious in this study than previously seen. The greater resolution of our oligonucleotide tiling microarrays, compared with a relatively sparse probe distribution [27], could be a contributing factor. At the same time, we imagine that transcription could lead to loss of cohesin at certain loci. This could be the case at places where an equilibrium between bound and free cohesin is more actively maintained, e.g. close to the centromere [34]. For the purpose of this study, we focused on analysing cohesin behaviour at places where cohesin translocation is clearly observed. Given that most cohesin loading sites in the budding yeast genome are at promoters of active genes, while most lasting places of cohesin residence correspond to areas of convergent transcriptional termination [24,28,30], we consider it likely that most of the cohesin reaches its final destination by sliding along chromosomes.
Cohesin is topologically loaded onto DNA by the cohesin loader and holds together sister chromatids by topological embrace [8,43]. If cohesin retains contact with DNA during its translocation, as suggested by our results, it implies that cohesin rings indeed slide along chromosomes. The inner diameter of cohesin of close to 35 nm [5] should enable cohesin to move along a duplex DNA, even if it is packed into 10 nm nucleosome particles and even if cohesin encircles two sister chromatids. Electrostatic interactions of the cohesin complex with DNA [8,18] are likely to add a certain friction to the movement. Thus, an active driving force is undoubtedly required to achieve translocation. This force probably stems from the movement of RNA polymerases along the DNA. The features of the transcription apparatus that lead to translocation are surprisingly generic. They are shared by the yeast RNA polymerase II holocomplex that is responsible for transcription of much of the budding yeast genome, with the evolutionarily distant T7 bacteriophage RNA polymerase. In common between these distant polymerases is the RNA transcript that emerges from them. It could be the sheer size and steric properties of the RNA transcript that drags cohesin along. We cannot exclude that endogeneous RNA polymerase machineries employ more specific ways to move cohesin. An open question of outstanding importance is why cohesin readily moves along chromosomes in response to transcription, but remains stationary during DNA replication [3,12]. Given the prevalence of cohesin translocation in response to transcription, it is tempting to speculate that a specific mechanism is in place to prevent cohesin from sliding away during replication.
In most organisms that have been studied, cohesin loader binding sites are distinct from where much of the cohesin accumulates. This includes fission yeast and mammals [44–46]. In mammalian cells, akin to what is seen in budding yeast, the cohesin loader subunit Nipped B is found in promoter regions of active genes. In budding yeast, the promoter specificity is achieved by the nucleosome landscape and the RSC chromatin remodelling complex [30]. The identifying features in mammalian cells remain to be fully understood. The mediator complex and the AP1 and other transcription factors have been implicated, but causal relationships remain to be ascertained [45,46]. After being loaded, mammalian cohesin accumulates at CTCF binding sites [47,48]. How it arrives there from its promoter loading sites is not known. If transcription moves cohesin along chromosomes in human cells, then we might expect to see a broad cohesin distribution along genes. Human genes are far longer than those found in yeasts. At a transcription elongation speed of a few kb per minute and considering that most cohesin resides on chromosomes for less than 25 min [13,49], cohesin would seldom reach a gene's 3′ end. A reported, albeit weak, cohesin enrichment upstream and downstream of genes is consistent with this scenario [48]. Notably, when acetylated and therefore more stably chromosome-bound cohesin is analysed, a marked shift to the 3′ end of short genes is observed, reminiscent of what is found in yeast [50]. In Drosophila, cohesin and its loader colocalize with each other, both at promoters of expressed genes and along their gene bodies [51]. Such a distribution could be explained if Drosophila cohesin and its loader engage in a somewhat tighter complex, compared with what is observed in other organisms. In this way, cohesin and its loader could stay associated during and after the loading reaction while cohesin moves along the gene body.
Do SMC complexes other than the cohesin complex also slide along chromosomes? In analogy to cohesin, the budding yeast condensin complex has been demonstrated to bind chromosomes by topological embrace [52]. While most of budding yeast condensin is found close to its chromosomal loading sites, that coincide with those of the cohesin complex, short-range movement of this dynamically chromosome-bound complex in response to both RNA polymerase I and II transcription has been observed [53,54]. Fission yeast condensin in turn is found in a bimodal distribution along expressed genes, enriched at both promoters and terminators [55,56], consistent with sliding along genes following loading at promoters. A third SMC complex in all eukaryotes, the Smc5/6 complex, shares many of its localization features with cohesin, including prominent enrichment at centromeres and sites of convergent transcription termination [57,58]. We therefore suggest that sliding along chromosomes is a universal feature of SMC complexes, by which these ring-shaped protein complexes stably hold onto DNA while not obstructing other chromosomal activities that have to occur at the same time.
4. Material and methods
4.1. Yeast strains and culture
All yeast strains were of the Saccharomyces cerevisiae W303 background, apart from the rat1-1 mutant strain and its control, which were of FY23 background. The strain genotype details can be found in the electronic supplementary material, table S1. Cells were cultured in rich YP medium containing 2% glucose, 2% raffinose or 2% raffinose + 2% galactose as the carbon source at 25°C, or at the indicated respective restrictive temperatures for the various temperature-sensitive alleles. For amino acid starvation, cells were filtered, washed and transferred to synthetic complete SC medium lacking amino acids [59]. Cell synchronization was in all cases achieved by first arresting cells in G1 by pheromone α-factor treatment. Following release from the α-factor arrest, cells passed through a synchronous cell cycle and were arrested in early S phase or G2/M by inclusion of 200 mM HU or 5 µg ml−1 nocodazole in the growth medium, respectively. Cell cycle progression and cell synchrony were in all cases confirmed by FACS analysis of cellular DNA content.
4.2. Chromatin immunoprecipitation
At the indicated times, aliquots of the cultures were harvested and processed for ChIP of the cohesin complex as described [24]. Antibodies used for ChIP were α-Pk (clone SV5-Pk1, AbD Serotec), α-myc (clone 9E10) and α-HA (clone 12CA5). Processed chromatin immunoprecipitates and input DNA samples were hybridized to Affymetrix GeneChip S. cerevisiae Tiling 1.0R arrays. Presented is the genome-normalized ratio of the hybridization signals of the chromatin immunoprecipitate over the input DNA. Each bar in the bar graphs represents the average of 25 oligonucleotide probes within neighbouring 125 bp windows. For the line graphs, a smoothed moving average of 200–500 bp is shown. The microarray data are available from the GEO database under the accession number GSE80464. The quantitative analysis of cohesin binding to individual loci was performed as described, using previously described qPCR primer pairs [30], as well as primer pairs flanking the GAL2 locus, listed in the electronic supplementary material, table S2.
4.3. Gene expression analysis
Cells were collected by centrifugation and total RNA was extracted using RNeasy reagents (Qiagen), following the manufacturer's instructions. Double-stranded cDNA (dscDNA) was synthesized from 10 µg of RNA using SuperScript II reverse transcriptase (Invitrogen), using random primers. Quantitative analysis of cDNA levels was then performed using a SYBR Green real-time PCR master mix (Life Technologies) and a ViiA 7 real-time PCR system (Thermo Fisher). The sequences of the primer pairs used are listed in electronic supplementary material, table S3.
Supplementary Material
Acknowledgements
We thank Y. Kakui, Y. Katou, G. Kelly and K. Shirahige for their help with microarray analyses, C. Cole and M. Rosbash for reagents, A. Lengronne and L. Lopez-Serra for sharing expertise and results and all our laboratory members for discussions and comments on the manuscript.
Authors' contributions
M.O.-H. and F.U. conceived the study, M.O.-H. performed most experiments, S.M. and C.P.S. performed the experiments during the revision process, M.O.-H. and F.U. wrote the manuscript.
Competing interests
The authors declare that no conflict of interest exists.
Funding
This study was supported by an EMBO Long Term Fellowship to S.M., by The Francis Crick Institute and by the European Research Council.
References
- 1.Onn I, Heidinger-Pauli JM, Guacci V, Ünal E, Koshland DE. 2008. Sister chromatid cohesion: a simple concept with a complex reality. Annu. Rev. Cell Dev. Biol. 24, 105–129. (doi:10.1146/annurev.cellbio.24.110707.175350) [DOI] [PubMed] [Google Scholar]
- 2.Nasmyth K, Haering CH. 2009. Cohesin: its roles and mechanisms. Annu. Rev. Genet. 43, 525–558. (doi:10.1146/annurev-genet-102108-134233) [DOI] [PubMed] [Google Scholar]
- 3.Uhlmann F. 2009. A matter of choice: the establishment of sister chromatid cohesion. EMBO Rep. 10, 1095–1102. (doi:10.1038/embor.2009.207) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peters J-M, Nishiyama T. 2012. Sister chromatid cohesion. Cold Spring Harb. Perspect. Biol. 4, a011130 (doi:10.1101/cshperspect.a011130) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson DE, Losada A, Erickson HP, Hirano T. 2002. Condensin and cohesin display different arm conformations with characteristic hinge angles. J. Cell Biol. 156, 419–424. (doi:10.1083/jcb.200111002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haering CH, Löwe J, Hochwagen A, Nasmyth K. 2002. Molecular architecture of SMC proteins and the yeast cohesin complex. Mol. Cell 9, 773–788. (doi:10.1016/S1097-2765(02)00515-4) [DOI] [PubMed] [Google Scholar]
- 7.Hu B, et al. 2011. ATP hydrolysis is required for relocating cohesin from sites occupied by its Scc2/4 loading complex. Curr. Biol. 21, 12–24. (doi:10.1016/j.cub.2010.12.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murayama Y, Uhlmann F. 2014. Biochemical reconstitution of topological DNA binding by the cohesin ring. Nature 505, 367–371. (doi:10.1038/nature12867) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ciosk R, Shirayama M, Shevchenko A, Tanaka T, Toth A, Shevchenko A, Nasmyth K. 2000. Cohesin's binding to chromosomes depends on a separate complex consisting of Scc2 and Scc4 proteins. Mol. Cell 5, 1–20. (doi:10.1016/S1097-2765(00)80420-7) [DOI] [PubMed] [Google Scholar]
- 10.Ben-Shahar TR, Heeger S, Lehane C, East P, Flynn H, Skehel M, Uhlmann F. 2008. Eco1-dependent cohesin acetylation during establishment of sister chromatid cohesion. Science 321, 563–566. (doi:10.1126/science.1157774) [DOI] [PubMed] [Google Scholar]
- 11.Ünal E, Heidinger-Pauli JM, Kim W, Guacci V, Onn I, Gygi SP, Koshland DE. 2008. A molecular determinant for the establishment of sister chromatid cohesion. Science 321, 566–569. (doi:10.1126/science.1157880) [DOI] [PubMed] [Google Scholar]
- 12.Lengronne A, McIntyre J, Katou Y, Kanoh Y, Hopfner K-P, Shirahige K, Uhlmann F. 2006. Establishment of sister chromatid cohesion at the S. cerevisiae replication fork. Mol. Cell 23, 787–799. (doi:10.1016/j.molcel.2006.08.018) [DOI] [PubMed] [Google Scholar]
- 13.Gerlich D, Koch B, Dupeux F, Peters J-M, Ellenberg J. 2006. Live-cell imaging reveals a stable cohesin–chromatin interaction after but not before DNA replication. Curr. Biol. 16, 1571–1578. (doi:10.1016/j.cub.2006.06.068) [DOI] [PubMed] [Google Scholar]
- 14.Lopez-Serra L, Lengronne A, Borges V, Kelly G, Uhlmann F. 2013. Budding yeast Wapl controls sister chromatid cohesion maintenance and chromosome condensation. Curr. Biol. 23, 64–69. (doi:10.1016/j.cub.2012.11.030) [DOI] [PubMed] [Google Scholar]
- 15.Kueng S, Hegemann B, Peters BH, Lipp JJ, Schleiffer A, Mechtler K, Peters J-M. 2006. Wapl controls the dynamic association of cohesin with chromatin. Cell 127, 955–967. (doi:10.1016/j.cell.2006.09.040) [DOI] [PubMed] [Google Scholar]
- 16.Tedeschi A, et al. 2013. Wapl is an essential regulator of chromatin structure and chromosome segregation. Nature 501, 564–568. (doi:10.1038/nature12471) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guacci V, Stricklin J, Bloom MS, Guo X, Bhatter M, Koshland D. 2015. A novel mechanism for the establishment of sister chromatid cohesion by the ECO1 acetyltransferase. Mol. Biol. Cell 26, 117–133. (doi:10.1091/mbc.E14-08-1268) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murayama Y, Uhlmann F. 2015. DNA entry into and exit out of the cohesin ring by an interlocking gate mechanism. Cell 163, 1628–1640. (doi:10.1016/j.cell.2015.11.030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmitz J, Watrin E, Lénárt P, Mechtler K, Peters J-M. 2007. Sororin is required for stable binding of cohesin to chromatin and for sister chromatid cohesion in interphase. Curr. Biol. 17, 630–636. (doi:10.1016/j.cub.2007.02.029) [DOI] [PubMed] [Google Scholar]
- 20.Tachibana-Konwalski K, Godwin J, van der Weyden L, Champion L, Kudo NR, Adams DJ, Nasmyth K. 2010. Rec8-containing cohesin maintains bivalents without turnover during the growing phase of mouse oocytes. Genes. Dev. 24, 2505–2516. (doi:10.1101/gad.605910) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jessberger R. 2012. Age-related aneuploidy through cohesion exhaustion. EMBO Rep. 13, 539–546. (doi:10.1038/embor.2012.54) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goffeau A, et al. 1997. The yeast genome directory. Nature 387, 5– 6. [PubMed] [Google Scholar]
- 23.Consortium TEP. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. (doi:10.1038/nature11247) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lengronne A, Katou Y, Mori S, Yokobayashi S, Kelly GP, Itoh T, Watanabe Y, Shirahige K, Uhlmann F. 2004. Cohesin relocation from sites of chromosomal loading to places of convergent transcription. Nature 430, 573–578. (doi:10.1038/nature02742) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ocampo-Hafalla MT, Uhlmann F. 2011. Cohesin loading and sliding. J. Cell Sci. 124, 685–691. (doi:10.1242/jcs.073866) [DOI] [PubMed] [Google Scholar]
- 26.Ström L, Lindroos HB, Shirahige K, Sjögren C. 2004. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol. Cell 16, 1003–1015. (doi:10.1016/j.molcel.2004.11.026) [DOI] [PubMed] [Google Scholar]
- 27.Bausch C, Noone S, Henry JM, Gaudenz K, Sanderson B, Seidel C, Gerton JL. 2007. Transcription alters chromosomal locations of cohesin in Saccharomyces cerevisiae. Mol. Cell. Biol. 27, 8522–8532. (doi:10.1128/MCB.01007-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glynn EF, Megee PC, Yu HG, Mistrot C, Unal E, Koshland DE, DeRisi JL, Gerton JL. 2004. Genome-wide mapping of the cohesin complex in the yeast Saccharomyces cerevisiae. PLoS Biol. 2, 1325–1339. (doi:10.1371/journal.pbio.0020259) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santos A, Wernersson R, Jensen LJ. 2014. Cyclebase 3.0: a multi-organism database on cell-cycle regulation and phenotypes. Nucl. Acids Res. 43, D1140–D1144. (doi:10.1093/nar/gku1092) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez-Serra L, Kelly G, Patel H, Stewart A, Uhlmann F. 2014. The Scc2–Scc4 complex acts in sister chromatid cohesion and transcriptional regulation by maintaining nucleosome-free regions. Nat. Genet. 46, 1147–1151. (doi:10.1038/ng.3080) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arumugam P, Gruber S, Tanaka K, Haering CH, Mechtler K, Nasmyth K. 2003. ATP hydrolysis is required for cohesin's association with chromosomes. Curr. Biol. 13, 1941–1953. (doi:10.1016/j.cub.2003.10.036) [DOI] [PubMed] [Google Scholar]
- 32.Weitzer S, Lehane C, Uhlmann F. 2003. A model for ATP hydrolysis-dependent binding of cohesin to DNA. Curr. Biol. 13, 1930–1940. (doi:10.1016/j.cub.2003.10.030) [DOI] [PubMed] [Google Scholar]
- 33.Baker SM, Okkema PG, Jaehning JA. 1984. Expression of the Saccharomyces cerevisiae GAL7 gene on autonomous plasmids. Mol. Cell. Biol. 4, 2062–2071. (doi:10.1128/MCB.4.10.2062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ocampo-Hafalla MT, Katou Y, Shirahige K, Uhlmann F. 2007. Displacement and re-accumulation of centromeric cohesin during transient pre-anaphase centromere splitting. Chromosoma 116, 531–544. (doi:10.1007/s00412-007-0118-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dez C, Dlakic M, Tollervey D. 2007. Roles of the HEAT repeat proteins Utp10 and Utp20 in 40S ribosome maturation. RNA 13, 1516–1527. (doi:10.1261/rna.609807) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amberg DC, Goldstein AL, Cole NC. 1992. Isolation and characterization of RAT1: an essential gene of Saccharomyces cerevisiae required for the efficient nucleocytoplasmic trafficking of mRNA. Genes Dev. 6, 1173–1189. (doi:10.1101/gad.6.7.1173) [DOI] [PubMed] [Google Scholar]
- 37.Kim M, Krogan NJ, Vasiljeva L, Rando OL, Nedea E, Greenblatt JF, Buratowski S. 2004. The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature 432, 517–522. (doi:10.1038/nature03041) [DOI] [PubMed] [Google Scholar]
- 38.Sullivan JM, Tan-Wong SM, Morillon A, Lee B, Coles J, Mellor J, Proudfoot NJ. 2004. Gene loops juxtapose promoters and terminators in yeast. Nat. Genet. 36, 1014–1018. (doi:10.1038/ng1411) [DOI] [PubMed] [Google Scholar]
- 39.Singh BN, Hampsey M. 2007. A transcription-independent role for TFIIB in gene looping. Mol. Cell 27, 806–816. (doi:10.1016/j.molcel.2007.07.013) [DOI] [PubMed] [Google Scholar]
- 40.Wild T, Cramer P. 2012. Biogenesis of multisubunit RNA polymerases. Trends Biochem. Sci. 37, 99–105. (doi:10.1016/j.tibs.2011.12.001) [DOI] [PubMed] [Google Scholar]
- 41.Tabor S, Richardson CC. 1985. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl Acad. Sci. USA 82, 1074–1078. (doi:10.1073/pnas.82.4.1074) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dower K, Rosbash M. 2002. T7 RNA polymerase-directed transcripts are processed in yeast and link 3' end formation to mRNA nuclear export. RNA 8, 686–697. (doi:10.1017/S1355838202024068) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haering CH, Farcas AM, Arumugam P, Metson J, Nasmyth K. 2008. The cohesin ring concatenates sister DNA molecules. Nature 454, 297–301. (doi:10.1038/nature07098) [DOI] [PubMed] [Google Scholar]
- 44.Schmidt CK, Brookes N, Uhlmann F. 2009. Conserved features of cohesin binding along fission yeast chromosomes. Genome Biol. 10, R52 (doi:10.1186/gb-2009-10-5-r52) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kagey MH, et al. 2010. Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435. (doi:10.1038/nature09380) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zuin J, Franke V, van Ijcken WFJ, van der Sloot A, Krantz ID, van der Reijden MIJA, Nakato R, Lenhard B, Wendt KS. 2014. A cohesin-independent role for NIPBL at promoters provides insight in CdLS. PLoS Genet. 10, e1004153 (doi:10.1371/journal.pgen.1004153) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parelho V, et al. 2008. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 132, 422–433. (doi:10.1016/j.cell.2008.01.011) [DOI] [PubMed] [Google Scholar]
- 48.Wendt KS, et al. 2008. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451, 796–801. (doi:10.1038/nature06634) [DOI] [PubMed] [Google Scholar]
- 49.Cannon D, Chubb JR. 2011. PolII caught speeding by single gene imaging. EMBO Rep. 12, 1208–1210. (doi:10.1038/embor.2011.217) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deardorff MA, et al. 2012. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 489, 313–317. (doi:10.1038/nature11316) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Misulovin Z, et al. 2008. Association of cohesin and Nipped-B with transcriptionally active regions of the Drosophila melanogaster genome. Chromosoma 117, 89–102. (doi:10.1007/s00412-007-0129-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cuylen S, Metz J, Haering CH. 2011. Condensin structures chromosomal DNA through topological links. Nat. Struct. Mol. Biol. 18, 894–901. (doi:10.1038/nsmb.2087) [DOI] [PubMed] [Google Scholar]
- 53.Johzuka K, Horiuchi T. 2007. RNA polymerase I transcription obstructs condensin association with 35S rRNA coding regions and can cause contraction of long repeat in Saccharomyces cerevisiae. Genes Cells 12, 759–771. (doi:10.1111/j.1365-2443.2007.01085.x) [DOI] [PubMed] [Google Scholar]
- 54.D'Ambrosio C, Schmidt CK, Katou Y, Kelly G, Itoh T, Shirahige K, Uhlmann F. 2008. Identification of cis-acting sites for condensin loading onto budding yeast chromosomes. Genes Dev. 22, 2215–2227. (doi:10.1101/gad.1675708) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakazawa N, Sajiki K, Xu X, Villar-Briones A, Arakawa O, Yanagida M. 2015. RNA pol II transcript abundance controls condensin accumulation at mitotically up-regulated and heat-shock-inducible genes in fission yeast. Genes Cells 20, 481–499. (doi:10.1111/gtc.12239) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sutani T, et al. 2015. Condensin targets and reduces unwound DNA structures associated with transcription in mitotic chromosome condensation. Nat. Commun. 6, 7815 (doi:10.1038/ncomms8815) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Betts Lindroos H, Ström L, Itoh T, Katou Y, Shirahige K, Sjögren C. 2006. Chromosomal association of the Smc5/6 complex reveals that it functions in differently regulated pathways. Mol. Cell 22, 755–767. (doi:10.1016/j.molcel.2006.05.014) [DOI] [PubMed] [Google Scholar]
- 58.Jeppsson K, et al. 2014. The chromosomal association of the Smc5/6 complex depends on cohesion and predicts the level of sister chromatid entanglement. PLoS Genet. 10, e1004680 (doi:10.1371/journal.pgen.1004680) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rose MD, Winston F, Hieter P. 1990. Laboratory course manual for methods in yeast genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.