

Abstract
A selection of heteroaryl fluorosulfates were readily synthesized using commercial SO2F2 gas. These substrates are highly efficient coupling partners in the Suzuki reaction. Through judicious selection of Pd-catalysts, the fluorosulfate functionality is differentiated from bromide and chloride; the order of reactivity being: -Br > -OSO2F > -Cl. Exploiting this trend allowed the stepwise chemoselective synthesis of a number of polysubstituted pyridines, including the drug, Etoricoxib.
Keywords: Fluorosulfates, Suzuki reaction, polysubstituted pyridine, chemoselectivity, Palladium
Graphical abstract
Introduction
Polysubstituted pyridines comprising aryl substituents are important functional moieties, finding use in applications as diverse as chemosensors,[1] molecular electronics[2] and pharmaceuticals.[3] Figure 1 illustrates a selection of functional polysubstituted pyridines including, the COX-2 selective inhibitor, Etoricoxib.[4,5] Over the years, numerous synthetic strategies have been developed to access these valuable structures.[8–10] Many involve classical multi-component condensation reactions (e.g. Knoevenagel and Hantzsh condensation using β-keto esters etc.), which often rely upon the formation of the heterocyclic ring in the last step. Conceptually, the controlled stepwise installation of substituents directly into an aromatic core is a more direct and modular strategy (Scheme 1). In discovery research, this is often accomplished by a sequence of one or more Suzuki cross coupling reactions.[11] By exploiting the distinct reactivity prolife of common electrophilic leaving groups, which follows the general order: -I > -Br ≥ -OTf ≫ -Cl, the chemoselective introduction of substituents directly into the aromatic core can sometimes be accomplished.[12,13]
Figure 1.

Examples of functional polysubstituted pyridines.
Scheme 1.

Synthetic approaches to polysubstituted pyridines.
Fluorosulfates have long been known to participate in transition-metal catalyzed reactions, serving as triflate surrogates in Negishi and Stille cross-couplings,[14] and in palladium-catalyzed alkoxy carbonylation reactions. Early published syntheses of aryl fluorosulfates include the pyrolysis of arenediazonium fluorosulfonate salts, and the reaction of sodium phenolates with ClSO2F or fluorosulfonic acid anhydride in the presence of pyridine or sodium hydride. These methods suffer from dangerous reaction conditions, expensive starting materials and/or low yields.[15]
In 2014, the Sharpless group reported a simple and reliable method for the synthesis of aryl fluorosulfates from phenols and sulfuryl fluoride (SO2F2), in the presence of triethylamine.[16] The efficient Suzuki cross couplings of aryl fluorosulfates with aryl boronic acids was subsequently disclosed in early 2015,[17] demonstrating the –OSO2F group as atom efficient and economically viable alternative to triflate.[18] More recently, Hanley[19] and coworkers published a study on how the fluorosulfate handle fits into the hierarchy of leaving group in Suzuki aryl couplings. This work confirmed the following: (i) almost no chemoselective discrimination was observed between the competing triflate and fluorosulfate substrates, (ii) there was reasonable selectivity for the bromide over the fluorosulfate, and (iii) there is absolute preference for the fluorosulfate vs. the chloride.
Here we report that heteroaryl fluorosulfates give outstanding results in the Suzuki coupling, demonstrating distinct reactivity from the corresponding halogens and triflates. Harnessing the unique reactivity of fluorosulfates, enabled the synthesis of a number of polysubstituted pyridines with exquisite chemoselective control.
Results and Discussion
The present study commenced with the gram-scale synthesis of the heteroaryl fluorosulfates 1-1 – 1–7,[16] accomplished by exposure of the corresponding pyridinol substrates to SO2F2[20] under our standard conditions [triethylamine (TEA, 1.5 equiv.), DCM, r.t. 2–24 h] (Table 1). The conversion of the more challenging 2-pyridones into the corresponding fluorosulfates required additional optimization of conditions. Despite a thermodynamic preference for the formation of the N-fluoro sulfamoyl fluoride,[21] the substituted pyridin-2-yl fluorosulfates were obtained by substituting TEA for N,N-diisopropylethylamine (DIPEA) in acetonitrile (ACN) (Table 1, 1–8 – 1–17). The N- and S- containing hydroxyheterocyles, including, quinolin-8-ol, 1H-indol-5-ol, methyl 3-hydroxythiophene-2-carboxylate and benzo[b]thiophen-4-ol, were also converted into their fluorosulfates in good yield (Table 1, 1–18 – 1–20), whereas N-fluorosulfonation of the 1H-indol-5-ol produced the fluorosulfate-fluorosulfamate 1–21.
Table 1.
Synthesis of heteroaryl fluorosulfates.

|
Reaction conditions: A) TEA (1.5 equiv), DCM, rt 2–24 h. B) DIPEA (1.5 ~ 3 equiv), CH3CN, rt, 24 h, C) 1–18, 1–19, the same as conditions A. 1–20, 1–21, the same as conditions B.
We next evaluated the practicality of the heteroaryl fluorosulfates as substrates in the Suzuki cross coupling reaction. An extensive survey of reaction parameters revealed optimal conditions (Supporting Information; Table S1, entry 7), that were subsequently employed in the coupling of fluorosulfate (1–8) with the boronic acids (2-1 – 2–18). Gratifyingly, the fluorosulfate substrates performed well, yielding the corresponding biaryl products 3-1 – 3–18 in excellent yield (Table 2). Likewise, the Suzuki coupling of the heteroaryl fluorosulfates (1-1, 1–2, 1–9 – 1–13, 1–18 – 1–20) and the phenyl boronic acid 2-1, proceeded smoothly to give the biaryl products 4-1 – 4–11 in good yields (Table 3).
Table 2.
Suzuki reaction of pyridin-2-yl fluorosulfates with a range of aryl- and heteroaryl boronic acids (2).[a]

|
Reaction conditions: 1–8 (1.0 mmol), (hetero)aryl boronic acid 2 (1.5 mmol), Pd-PEPPSI-IPr (1 mol%), K2CO3 (3.0 mmol), EtOH/H2O (10 mL, 3/1), in nitrogen at 35 °C.
The reaction was run in EtOH.
The reaction was run in 10 mL 1,4-dioxane/H2O (3/1) at refluxing temperature.
Table 3.
Suzuki reactions of heteroaryl fluorosulfates (1) with phenylboronic acid 2-1.[a]

|
Reaction conditions: 1 (1.0 mmol), phenylboronic acid 2-1 (1.5 mmol), Pd-PEPPSI-IPr (1 mol%), K2CO3 (3.0 mmol), EtOH/H2O (10 mL, 3/1), in nitrogen at 35 °C.
The reaction was run with 2 mol% of Pd-PEPPSI-IPr.
Transesterification to the ethyl ester occurred under the reaction conditions.
The relative reactivity of the pyridyl fluorosulfates and other common electrophiles were then calibrated by performing competition experiments. For example, when a 1:1 mixture of the 2-fluorosulfate pyridine (1–8) and the 2-bromopyridine (5-1) was subjected to the phenylboronic acid 2-1 (1.5 equiv) and Pd(PPh3)4/KF in refluxing toluene, the 2-bromopyridine (5-1) was completely consumed within 3 hours; the unreacted 2-fluorosulfate pyridine (1–8) was fully recovered (Figure 2A).
Figure 2.
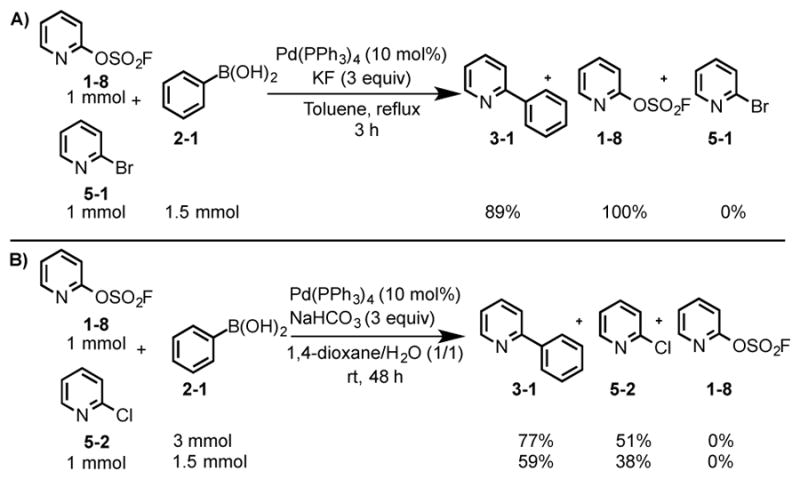
Competition experiments based on the Suzuki cross coupling of 2-fluorosulfate pyridine, 2-chloropyridine, and 2-bromopyridine with phenyl boronic acid.
A similar experiment was performed with a 1:1 mixture of the pyrid-2-yl fluorosulfate (1–8) and the 2-chloropyridine (5-2). When subjected to the phenylboronic acid (2-1) (3 equiv) [Pd(PPh3)4/NaHCO3 (dioxane/water (1/1) at room temperature], the pyrid-2-yl fluorosulfate (1–8) was consumed within 48 hours, whereas 51% of the unreacted 2-chloropyridine was recovered (Figure 2B). Based upon these observations, we conclude that the relative reactivity of the electrophiles examined in these coupling reaction follows: -Br > -OSO2F > -Cl.
Experiments were then performed on pyridines with both fluorosulfate and halide substituents juxtaposed on the same ring. In concert with Hanley’s related work on aryl fluorosulfates, the coupling of the bis-substituted chloropyridin fluorosulfates 1–14, 1–17, 1–3 with the phenylboronic acid (2-1), realized the chlorophenylpyridines 6-1, 6-2 and 6-3, respectively (Figure 3A). More importantly, when the bromopyridin fluorosulfates (1–15, 1–16, 1–4 – 1–6) were reacted with the boronic acid 2-1, exclusive formation of the mono-substituted fluorosulfate products 7-1 – 7-5 was observed (Figure 3B).
Figure 3.

Suzuki reactions of pyridinyl fluorosulfates and pyridinyl triflates bearing -Cl or -Br. [a]1 mmol of pyridinyl fluorosulfates were used in all of the reactions. [b] The ratio of Br- and -OSO2F substituted product in 7-2 is 9:1.
Unlike its fluorosulfate analogue 1–4, the triflate 8, when treated under the same cross coupling conditions generated a [1.4: 1.0] mixture of the coupling products 9 and 10, respectively (Figure 3C). This unprecedented level of chemoselectivity demonstrated by the fluorosulfate group presents a window of reactivity distinct from the triflate. From these particular experiments, we rank the relative order of leaving group potential for substituted pyridines as: Br ≥ -OTf > -OSO2F > -Cl.
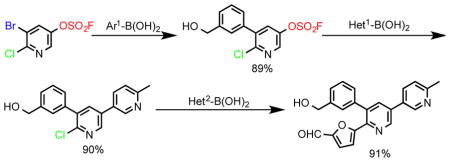
We next explored the possibility of exploiting the fluorosulfate group in the synthesis of polysubstituted heteroarenes. The 5-bromo-6-chloropyridin-3-yl fluorosulfate (1–7) building block was selected as a model substrate. Treatment of 1–7 with the phenylboronic acid (2-1) and Pd(PPh3)4/KF/toluene, gave the 6-chloro-5-phenylpyridin-3-yl fluorosulfate 11 as the sole product (Figure 4A). This impressive chemoselectivity was mirrored in the subsequent Suzuki coupling of 11 with the methylphenylboronic acid (2-2), giving the 2-chloro-3-phenyl-5-(p-tolyl)pyridine (12) in 91% yield. Finally, Suzuki coupling of the heteroaryl chloride 12 with 4-trifluoromethylpheylboronic acid (2–5) [Pd-PEPPSI-IPr, aqueous ethanol], afforded the tri-substituted pyridine 13 in 98% yield. This concise synthesis of the tri-substituted pyridine 13 was achieved in just three steps with an overall yield of 73%. Moreover, each step in the reaction sequence could be scaled up to gram-quantities without compromising the yield.
Figure 4.
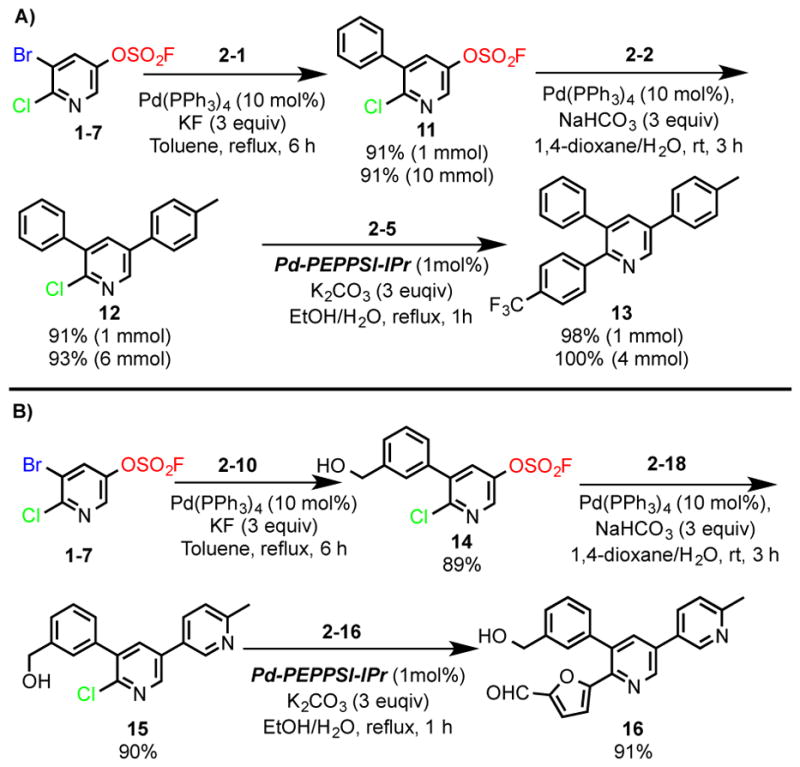
Preparation of tri-substituted pyridines through successive and selective Suzuki reactions.
In another example, the tri-substituted pyridine (16) was assembled in excellent yield through sequential cross couplings of the fluorosulfate 1–7 with the functionalized aryl- (2–10) and heteroaryl boronic acids (2–18 & 2–16), respectively (Figure 4B).
We next turned our attention to the synthesis of Etoricoxib,[4,5] which has previously been synthesized via a six-step sequence from the 3-bromo-5-chloropyridin-2-ol (17) building block (Scheme 2A).[5] We anticipated that our fluorosulfate chemistry would enable a more direct access to Etoricoxib, by circumventing protecting group chemistry. Beginning with the same 5-bromo-6-chloropyridin-3-yl fluorosulfate (17) starting material, the formation of the pyridin-2-yl fluorosulfate (18) was readily achieved by reaction with sulfuryl fluoride. The synthesis of Etoricoxib was successfully completed by the sequential installation of the 4-(methylsulfonyl)phenyl and 6-methylpyridin-3-yl substituents into the pyridine core respectively, in an overall 40.3% yield (Scheme 2B). To the best of our knowledge, the synthetic approach to Etoricoxib described here is the most concise to date.[22]
Scheme 2.
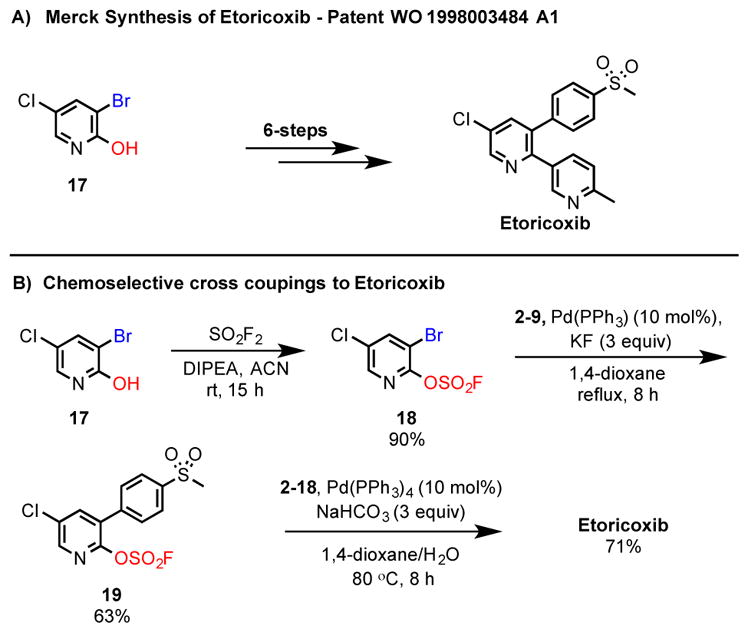
A) Merck synthesis of Etoricoxib; B) chemoselective sequential Suzuki cross coupling/SuFEx approach to Etoricoxib.
Conclusions
In summary, we have developed an efficient protocol for the synthesis of functional heteroaryl fluorosulfates. This was made possible by harnessing the incredible reactivity of the sulfur connector, SO2F2. The heteroaryl fluorosulfates were demonstrated as versatile electrophilic cross coupling partners in the Pd-catalyzed Suzuki reaction, exhibiting distinct reactivity from the corresponding halogens and triflates. Harnessing the unique reactivity of fluorosulfates, enabled the synthesis of a number of polysubstituted pyridines with exquisite chemoselective control.
The privileged nature of the fluorosulfate leaving group was further demonstrated with the concise synthesis of the drug, Etoricoxib, in just 3 steps. The scalability of heteroaryl fluorosulfates to kilogram quantities, coupled with the plethora of available Pd catalysts, render fluorosulfates ideal substrates for both process and discovery applications.
Experimental Section
General procedure for Preparation of heteroaryl fluorosulfates 1 with TEA in DCM
A three-neck round-bottom flask equipped with a thermometer was charged with the corresponding hydroxyheterocycle, DCM (10 mL/g) and TEA (1.5 equiv). The mixture was stirred at room temperature for 10 minutes. The reaction flask was then sealed with a septum. The atmosphere above the solution was removed under a gentle vacuum, and the SO2F2 gas (sulfuryl fluoride, Vikane) was introduced from a balloon through a three-way valve joint. For large scale reactions, the depletion of the sulfuryl fluoride from the balloon was observed, and additional SO2F2 gas introduced via a fresh balloon as required. The reaction mixture was stirred vigorously at room temperature between 2–24 hours, and the reaction progress monitored by TLC. The reaction mixture was then washed with saturated sodium bicarbonate and brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude residue was purified by flash column chromatography over silica gel to give the heteroaryl fluorosulfates 1.
General procedure for Preparation of heteroaryl fluorosulfates 1 with DIPEA in ACN
A three-neck round-bottom flask equipped with a thermometer was charged with the corresponding hydroxyheterocycle, ACN (10 mL/g) and DIPEA (1.5 – 3 equiv). The mixture was stirred at room temperature for 10 min. The reaction flask was then sealed with a septum. The atmosphere above the solution was removed with gentle vacuum, and SO2F2 gas (sulfuryl fluoride, Vikane) from a balloon was introduced through a three-way valve joint. For large scale reactions, depletion of the sulfuryl fluoride from the balloon was easily observed, and more SO2F2 gas was introduced with a fresh balloon when required. The reaction mixture was vigorously stirred at room temperature for 2–24 hours, monitoring by TLC. After completion, the solvent was removed by rotary evaporation. The residue was dissolved in EtOAc, washed with saturated sodium bicarbonate and brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel to give heteroaryl fluorosulfates 1.
General procedures for Pd-catalyzed Suzuki reactions
Method A. General Suzuki procedure associate with (aqueous) alcohol solution
Het-OSO2F (1.0 mmol, 1.0 equiv), (hetero)arylboronic acid (1.5 mmol, 1.5 equiv), Pd-catalyst (1–10 mol%), base (3.0 mmol, 3.0 equiv) were successively added to a 50 mL flask equipped with a stirring bar and a thermometer. The air in the flask was replaced by nitrogen gas by vacuum, and then (aqueous) alcohol solution (with different ratios) (1–20mL) was injected into the flask through a rubber plug. The reaction mixture was placed into an oil bath, heated to the corresponding temperature and stirred for between 1–24 hours as determined by TLC or HPLC analysis. After completion, most of organic solvent was removed by rotary evaporation. The residue was dissolved in EtOAc, washed with saturated brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography over silica gel to afford the coupling product.
Method B. General Suzuki procedure associate with aqueous NaHCO3 solution in 1,4-dioxane at room temperature
Het-OSO2F (1.0 mmol, 1.0 equiv), (hetero)arylboronic acid (1.5 mmol, 1.5 equiv), Pd(PPh3)4 (10 mol%) were successively added to a 50 mL flask equipped with a stirring bar and a thermometer. The air in the flask was replaced by nitrogen gas by vacuum, and then 1,4-dioxane (10 mL) was injected into the flask through a rubber plug. The reaction mixture was stirred for 10 min and then NaHCO3 (3.0 mmol, 3 equiv) in 10 mL water was injected into the flask. The reaction mixture was stirred at room temperature for 3–24 hours as monitored by HPLC. After completion, the mixture was diluted with EtOAc (20mL), washed with saturated brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel to afford the coupling product.
Method C. General Suzuki procedure associate with KF in toluene or 1,4-dioxane at refluxing temperature
Het-OSO2F (1.0 mmol, 1.0 equiv), arylboronic acid (1.5 mmol, 1.5 equiv), Pd(PPh3)4 (10 mol%), KF (3.0 mmol, 3.0 equiv) were successively added to a 50 mL flask equipped with a stirring bar and a thermometer. The air in the flask was replaced by nitrogen gas by vacuum, and then toluene or 1,4-dioxane (10 mL) was injected into the flask through a rubber plug. The reaction mixture was placed into an oil bath, heated to reflux for between 3–10 hours as determined by HPLC analysis. After completion, the mixture was diluted with EtOAc (20 mL), washed with saturated brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography over silica gel to afford the coupled product.
Supplementary Material
Acknowledgments
We thank Asymchem Laboratories Inc. and the NIH (P50 GM103368, R01 GM117145 to K.B.S.) for financial support for this project. S. Li was partially supported by the Skaggs Institute of Chemical Biology. We thank the AC department at Asymchem Laboratories Inc. for access to instrumentation, Dr. Hao Hong and James R. Gage at Asymchem Laboratories Inc. for helpful disucssions and support. We thank Prof. Jin-Quan Yu at The Scripps Research Institute for helpful discussions.
Footnotes
Supporting Information: Full experimental details and characterization data for all new compounds, and copies of 1H and 13C NMR spectra.
Contributor Information
Enxuan Zhang, Email: zhangenxuan@asymchem.com.cn.
K. Barry Sharpless, Email: sharples@scripps.edu.
References
- 1.Fang AG, Mello JV, Finney NS. Tetrahedron. 2004;60:11075–11087. [Google Scholar]
- 2.Petty MC, Bryce MR, Bloor D. In: Introduction to Molecular Electronics. Siebbeles LDA, Grozema FC, editors. Oxford University Press; New York: 1995. [Google Scholar]
- 3.Doebelin C, Wagner P, Bertin I, Simonin F, Schmitt M, Bihel F, Bourguignon J-J. RSC Adv. 2013;3:10296–10300. [Google Scholar]
- 4.Clarke R, Derry S, Moore RA. Cochrane database Syst Rev. 2014;5:CD004309. doi: 10.1002/14651858.CD004309.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dube D, Fortin R, Friesen R, Gauthier JY, Wang Z. 9803484A1. WO. 1998
- 6.Henke BR, Drewry DH, Jones SA, Stewart EL, Weaver SL, Wiethe RW. Bioorg Med Chem Lett. 2001;11:1939–1942. doi: 10.1016/s0960-894x(01)00321-3. [DOI] [PubMed] [Google Scholar]
- 7.Eastwood P, Gonzalez J, Paredes S, Nueda A, Domenech T, Alberti J, Vidal B. Bioorg Med Chem Lett. 2010;20:1697–1700. doi: 10.1016/j.bmcl.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 8.Kingsberg E, editor. Chemistry of Heterocyclic Compounds: Pyridine and its derivatives. John Wiley & Sons, Inc; Hoboken, NJ, USA: 1960. [Google Scholar]
- 9.Abramovitch RA, editor. Pyridine and its derivatives. John Wiley & Sons, Inc; Hoboken, NJ, USA: 1972. [Google Scholar]
- 10.Newkome GR, editor. Pyridine and its derivatives. John Wiley & Sons, Inc; Hoboken, NJ, USA: 1984. [Google Scholar]
- 11.For reviews and leading references, see: Miyaura N, Yamada K, Suzuki A. Tetrahedron Lett. 1979;20:3437–3440.Suzuki A. Angew Chem. 2011;123:6854–6869.Angew Chem Int Ed. 2011;50:6722–6737. doi: 10.1002/anie.201101379.
- 12.Littke AF, Dai C, Fu GC. J Am Chem Soc. 2000;122:4020–4028. [Google Scholar]
- 13.Doebelin C, Wagner P, Bihel F, Humbert N, Kenfack CA, Mely Y, Bourguignon JJ, Schmitt M. J Org Chem. 2014;79:908–918. doi: 10.1021/jo402200q. [DOI] [PubMed] [Google Scholar]
- 14.The use of fluorosulfates have also been described in Negishi and Stille cross-couplings and palladium-catalyzed alkoxy carbonylation reactions was originally investigated by the process group at Bristol-Myers Squibb: Roth GP, Fuller CE. J Org Chem. 1991;56:3493–3496.McGuire MA, Sorenson E, Owings FW, Resnick TM, Fox M, Baine NH. J Org Chem. 1994;59:6683–6686.Roth GP, Thomas JA. Tetrahedron Lett. 1992;33:1959–1962.Roth GP, Sapino C. Tetrahedron Lett. 1991;32:4073–4076.Pridgen LN, Huang GK. Tetrahedron Lett. 1998;39:8421–8424.
- 15.For early published syntheses of aryl fluorosulfates, see: Lange W, Müller E. Ber Dtsch Chem Ges B. 1930;63:2653–2657.Cramer R, Coffman DD. J Org Chem. 1961;26:4164–4165.Firth WCJ. J Polym Sci B. 1972;10:637–641.Stang PJ, Hanack M, Subramanian L. Synthesis. 1982:85–126.Boudakian MM, Hyde GA, Kongpricha S. J Org Chem. 1971;36:940–942.
- 16.Dong J, Krasnova L, Finn MG, Sharpless KB. Angew Chem Int Ed. 2014;53:9430–9448. doi: 10.1002/anie.201309399. [DOI] [PubMed] [Google Scholar]
- 17.Liang Q, Xing P, Huang Z, Dong J, Sharpless KB, Li X, Jiang B. Org Lett. 2015;17:1942–1945. doi: 10.1021/acs.orglett.5b00654. [DOI] [PubMed] [Google Scholar]
- 18.SO2F2 (ca. $1/Kg) is significantly less expensive than triflic anhydride; the atom economy for triflate formation is also very poor compared to the fluorosulfate, with loss of the expensive F3CSO2O− compared to a single F−.
- 19.Hanley PS, Ober MS, Krasovskiy AL, Whiteker GT, Kruper WJ. ACS Catal. 2015;5:5041–5046. [Google Scholar]
- 20.As a fumigant, sulfuryl fluoride (SO2F2) has been produced annually at more than 3 million kilograms per year since 2000, with a price as low as $1/kg. see Andersen MPS, Blake DR, Rowland FS, Hurley MD, Wallington TJ. Environ Sci Technol. 2009;43:1067–1070. doi: 10.1021/es802439f.
- 21.Hopkins GC, Jonak JP, Minnemeyer HJ, Tieckelmann H. J Org Chem. 1967;32:4040–4044. [Google Scholar]
- 22.For other synthetic routes to Etoricoxib, see: Friesen RW, Brideau C, Chan CC, Charleson S, Deschênes D, Dubé D, Ethier D, Fortin R, Gauthier JY, Girard Y, Gordon R, Greig GM, Riendeau D, Savoie C, Wang Z, Wong E, Visco D, Xu LJ, Young RN. Bioorg Med Chem Lett. 1998;8:2777–2782. doi: 10.1016/s0960-894x(98)00499-5.Marcoux JF, Corley EG, Rossen K, Pye P, Wu J, Robbins MA, Davies IW, Larsen RD, Reider PJ. Org Lett. 2000;2:2339–2341. doi: 10.1021/ol006097b.Davies IW, Marcoux JF, Corley EG, Journet M, Cai DW, Palucki M, Wu J, Larsen RD, Rossen K, Pye PJ, DiMichele L, Dormer P, Reider PJ. J Org Chem. 2000;65:8415–8420. doi: 10.1021/jo000870z.Sorbera LA, Castañer RM, Silvestre J, Castañer J. Drugs Future. 2001;26:346–353.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.