

Abstract
The abundance and functional orientation of tumor-infiltrating lymphocytes in breast cancer is associated with distant metastasis-free survival, yet how this association is influenced by tumor phenotypic heterogeneity is poorly understood. Here, a bioinformatics approach defined tumor biological attributes that influence this association, and delineated tumor subtypes that may differ in their ability to sustain durable antitumor immune responses. A large database of breast tumor expression profiles and associated clinical data was compiled, from which the ability of phenotypic markers to significantly influence the prognostic performance of a classification model that incorporates immune cell-specific gene signatures was ascertained. Markers of cell proliferation and intrinsic molecular subtype reproducibly distinguished two breast cancer subtypes that we refer to as immune benefit-enabled (IBE) and immune benefit-disabled (IBD). The IBE tumors, comprised mostly of highly proliferative tumors of the basal-like, HER2-enriched, and luminal B subtypes, could be stratified by the immune classifier into significantly different prognostic groups, while IBD tumors could not, indicating the potential for productive engagement of metastasis-protective immunity in IBE tumors, but not IBD tumors. The prognostic stratification in IBE was independent of conventional variables. Gene network analysis predicted the activation of tumor necrosis factor–α/interferon-γ signaling pathways in IBE tumors and the activation of the transforming growth factor–β pathway in IBD tumors. This supports a model in which breast tumors can be distinguished on the basis of their potential for metastasis-protective immune responsiveness. Whether IBE and IBD represent clinically-relevant contexts for evaluating sensitivity to immunotherapeutic agents warrants further investigation.
Keywords: breast cancer, tumor-infiltrating leukocytes, immune metagenes, prognostic subtypes, distant metastasis-free survival
Introduction
As cancer cells emerge and propagate, they must acquire evasive tactics to escape immune destruction. These tactics must endow malignant cells with the ability to avoid elimination by the immune system early in their formation, establish equilibrium with the immune system as the tumor grows, and eventually escape from immune control enabling tumor progression (1). However, results from both mechanistic and correlative studies indicate that some established tumors, with or without therapeutic intervention, can eventually succumb to immune control by establishment of a helper T cell type 1 (Th1)–type immune response that mediates tumor rejection and gives rise to immunological memory that guards against future metastases (2-4). Consistent with these observations, numerous histological studies have demonstrated that the abundance and functional orientation of tumor-infiltrating effector cells within primary tumors can significantly predict recurrence-free survival of cancer patients (5). This is particularly true of cancers of the breast, colon, ovary, lung, liver, head/neck, skin and brain, whereby abundant tumor-infiltrating lymphocytes (TIL), namely cytotoxic T cells (CTLs), memory T cells, natural killer (NK) and NKT cells, have been associated with the suppression of metastatic recurrence (5-7). Together, these findings suggest that: 1) abundant tumor infiltration by effector cells is indicative of an antitumor immune response, and 2) the induction of tumor-reactive immunity capable of guarding against metastatic progression is a fairly common event in multiple cancer types.
Microarray tumor profiling studies are now providing compelling evidence that the relative abundance of tumor-infiltrating immune cells can be quantified by the surrogate measure of intratumoral transcript levels of immune cell-specific genes that are coordinately-expressed (8-13). Through hierarchical clustering analysis, we and others have demonstrated that these genes self-organize into multiple gene clusters that embody signaling networks fundamental to distinct and identifiable immune cell subpopulations (12-18). These immune gene signatures or “metagenes”, consistent with the effector cell types they reflect, have been shown by multiple groups to be associated with patient outcomes such as disease-free and overall survival (2, 8, 12, 13, 16, 19-21), response to neoadjuvant chemotherapy (22, 23) and adjuvant molecular therapy (anti-Her2/neu) (24) paralleling historical immunohistochemical observations.
We characterized the underlying biology and prognostic ramifications of several distinct immune metagenes in breast cancer (12). The term “metagene” is defined here as a cluster of coordinately-expressed genes that together comprise a single cognate expression vector. By averaging the expression levels of these genes, a composite expression score can be computed for each tumor (25). We showed that the immune metagene scores quantify the relative abundance of distinct effector cell populations, namely cytotoxic T and/or NK cells (the T/NK metagene), antibody-producing plasma B cells (the B/P metagene), and antigen-presenting myeloid/dendritic cells (the M/D metagene). The expression scores of all three metagenes were significantly associated with prolonged distant metastasis-free survival (DMFS) of breast cancer patients and displayed additive prognostic information when considered in multivariate Cox regression models. We have also observed that these same prognostic immune metagenes are predictive of taxane and anthracycline efficacy in the neoadjuvant setting (22).
Although the density of immune effector cell infiltrates represents a significant prognostic marker, it does not comprehensively characterize the immune system's functional orientation towards cancer. For even in the presence of abundant TIL, some tumors still escape immune destruction and progress to metastatic disease. Why effector immune infiltrates equate with protective antitumor immunity in some cancers but not others could depend on one or more immunomodulatory mechanisms acting alone or in concert. A better understanding of which tumors will and will not give rise to productive immunity could serve as a valuable predictor of immunotherapy responsiveness as well as create opportunities to discover and model causal mechanisms of pro- and antitumor immunity in specific cancer types.
In breast cancer, we previously observed that the immune metagenes exhibited clear prognostic power in some tumors, but not others. We found that a gene signature highly correlated with tumor cell mitotic index and Ki67 staining (termed the “proliferation”, or “P” metagene) enabled the stratification of breast tumors into subgroups that either permitted or prohibited prognostication by the different immune metagenes (12). Specifically, in tumors with a high proliferative capacity (defined by the upper tertile of the P metagene scores, denoted as PH), high immune metagene scores equated with significantly prolonged DMFS compared to the poor DMFS associated with low immune metagene scores. By contrast, tumors with intermediate and low proliferative capacities (denoted as the PI and PL tertiles, respectively) could not be stratified by the immune metagenes into significantly different survival groups. Intriguingly, these observations suggest that the successful recognition and elimination of breast cancer by the immune system may be governed, in part, by certain definable tumor characteristics associated with intrinsic tumor immunogenicity.
In this study, we address the question of how to optimally stratify breast tumors into subsets that differ in their potential for protective immune responsiveness. Although a number of immunohistochemical and microarray-based studies have analyzed the protective effects of relative levels of immune infiltration in breast cancer, none have addressed the question of how to distinguish tumors where elevated immune involvement does or does not confer protective benefit. We hypothesized that the statistical association between the immune metagenes and DMFS of patients could be used as a means to extrapolate the underlying breast tumor phenotypes that differ in their ability to potentiate long-term, immune-mediated tumor rejection. Using an immune metagene model that combines the prognostic attributes of the B/P, T/NK and M/D metagenes, we define and validate proliferative and intrinsic subtype attributes of breast cancer that indicate the existence of immune benefit-enabled (IBE) and immune benefit-disabled (IBD) tumor subtypes, and shed light on the underlying molecular pathways that may contribute to their differing immunogenic dispositions.
Materials and Methods
Microarray Datasets and Data Processing
All microarray data were obtained from MIAME-compliant (26) data repositories and were associated with institutional review board (IRB)-approved specimen collection protocols compliant with REMARK criteria (27). The meta-cohort #1 (MC1) dataset comprised of 1,954 tumor expression profiles of primary invasive breast cancer generated on the Affymetrix U133 series GeneChip microarrays. Queried transcripts included the 22,268 probe sets common to the U133A, U133A2 and U133 PLUS 2.0 array platforms. Assembly of the dataset is detailed in Nagalla et. al. (12). Briefly, the data represent diverse patient populations spanning 16 medical centers in the U.S., Europe and Asia. Raw data (CEL files) were extracted from the NCBI's Gene Expression Omnibus (GEO) (28) (accessions: GSE1456, GSE2034, GSE5327, GSE12093, GSE7390, GSE6532, GSE9195, GSE2603, GSE7378, GSE8193, GSE4922, GSE11121, and GSE45255), the NCI's caArray database (accession: mille-00271), and the EMBL-EBI's ArrayExpress database (accession: E-TABM-158). The meta-cohort #2 (MC2) dataset consisted of 616 breast tumor expression profiles derived from patients in the U.S., Asia, Europe and New Zealand. Samples were analyzed on the same Affymetrix platforms as MC1, and CEL files were accessed via GEO accessions: GSE19615, GSE20685, GSE31519 and GSE36771. For MC1 and MC2, all CEL files were pre-processed and normalized using the R software package (29) and library files provided by the Bioconductor project. Array data were MAS5.0 normalized using the justMAS function in the simpleaffy library from Bioconductor (30) using a trimmed mean target intensity of 600 without background correction. Non-biological batch effects were corrected using the COMBAT empirical Bayes method (31) with estrogen receptor status used as a categorical covariate. The Cancer Genome Atlas (TCGA) meta-cohort originally included 532 breast tumors derived from 13 TCGA Tissue Source Sites and profiled on the Agilent 244K G4502A-07 custom microarray platform. TCGA Level-3 lowess-normalized data (17,813 genes) were downloaded from the TCGA Data Portal (https://tcga-data.nci.nih.gov/tcga/). Three metastatic tumors (non-primary) and 5 male breast tumors were omitted for a total of 524 cases/profiles. Intrinsic molecular subtype calls for the TCGA data were utilized as previously generated (32). For MC1 and MC2 meta-cohorts, molecular subtype calls were assigned according to (12).
Patient and Tumor Clinical Annotations
De-identified clinical data corresponding to tumor expression profiles and including pathology, demographic, and clinical follow-up data, were compiled from several sources including published supplemental tables, microarray data repositories and through direct inquiry with the original authors. Patient-tumor clinical characteristics are presented in Supplementary Table S1. See Supplementary Methods for further description.
Metagenes, Tertiles, and the Immune Metagene Model
The proliferation and immune metagenes were constructed as described in (12), based on the probe set composition as listed in Additional File 6 of that publication. The immune metagenes, consistent with their transcriptional origins in tumor-infiltrating leukocytes: 1) significantly correlate with histologic TIL abundance in primary breast tumors (12), 2) display immune cell-specific expression patterns in FACS-sorted leukocyte populations, and 3) exhibit negligible and uncorrelated expression in cultured breast cancer cell lines that lack an immune component as illustrated in Supplementary Fig. S1. Tumors were grouped into metagene tertiles by ranking tumors by metagene scores (gene averages) and identifying the 33rd and 66th percentile thresholds. The rationale behind the selection of the tertile combinations used to define the favorable immunogenic disposition (FID), weak immunogenic disposition (WID), and poor immunogenic disposition (PID) subclasses of the Immune Metagene Model is described in Nagalla, et. al. (12). In the MC2 and TCGA cohorts, metagene scores were computed as described for MC1. See Supplementary Methods and Supplementary Fig. S2 for analysis of comparisons of metagene tertile thresholds across the three tumor cohorts.
Survival Analyses
Statistical associations between gene expression levels (metagene scores, probe set signal intensities) and the distant metastasis-free survival (or overall survival) of patients were evaluated by Cox proportional hazards regression or the Kaplan-Meier method using SigmaPlot 11.0 software or R software (survival package). Clinical endpoints and censoring criteria related to distant metastasis-free survival were applied as described in (12). In the multivariable models, covariates were entered as semi-continuous variables. A stepwise covariate selection procedure was used with entry significance level 0.05. Likelihood ratio test P-values were reported. For analysis of cell type-specific marker genes in IBE and IBD populations, Affymetrix probe sets were processed as described in Supplementary Methods.
Differential gene expression analysis
Microarray probe sets (n=22,268) were analyzed for differential gene expression between the IBE-FID (n=179) and IBD-FID (n=135) tumor subtypes. The two-tailed Student's t-test (unequal variance) with false discovery correction (Benjamini-Hochberg) was used to assess the significance of gene differential expression. Probe sets having false discovery rates <1% (q<0.01) and absolute value of log2 fold-change >0.50 were selected for downstream analyses. Pathway activation scores were generated as described previously (33) with further details provided in Supplementary Methods.
Results
Defining phenotypic subclasses of immune responsiveness
We performed a pooled analysis of 1,954 breast tumor expression profiles and corresponding recurrence data (termed Meta-Cohort #1 (MC1); Supplementary Table S1), to derive an optimal classification strategy for distinguishing tumors that are and are not amenable to prognostic stratification by an integrated immune metagene model (IMM). The IMM combines the prognostic attributes of the three immune metagenes (B/P, T/NK, and M/D) into a simplified classification system using unsupervised tertile-based metrics. Specifically, the IMM classifies tumors into one of three outcome-related subclasses according to different combinations of the immune metagene expression-level tertiles (i.e., low, intermediate, and high gene expression; Fig. 1A and B). Based on metastatic recurrence rates in highly proliferative tumors (12), we refer to these subclasses as: favorable immunogenic disposition (FID), weak immunogenic disposition (WID), and poor immunogenic disposition (PID). The FID subclass is associated with excellent long-term DMFS in highly proliferative tumors and comprises tumors ranked into the upper tertiles of all three immune metagenes, reflective of a histopathologic prominent immune infiltrate (Supplementary Fig. S3A and B). In contrast, the PID subclass comprises tumors ranked into the lower tertile of any one or more of the immune metagenes (consistent with an absent or reduced immune infiltrate; Supplementary Fig. S3C and D) and is significantly associated with short interval to distant metastasis. The WID subclass captures tumors of the intermediate/high immune metagene tertiles and corresponds to patient DMFS rates intermediate to that of the FID and PID subclasses.
Figure 1. The immune metagene model has prognostic value in IBE but not IBD breast cancer.
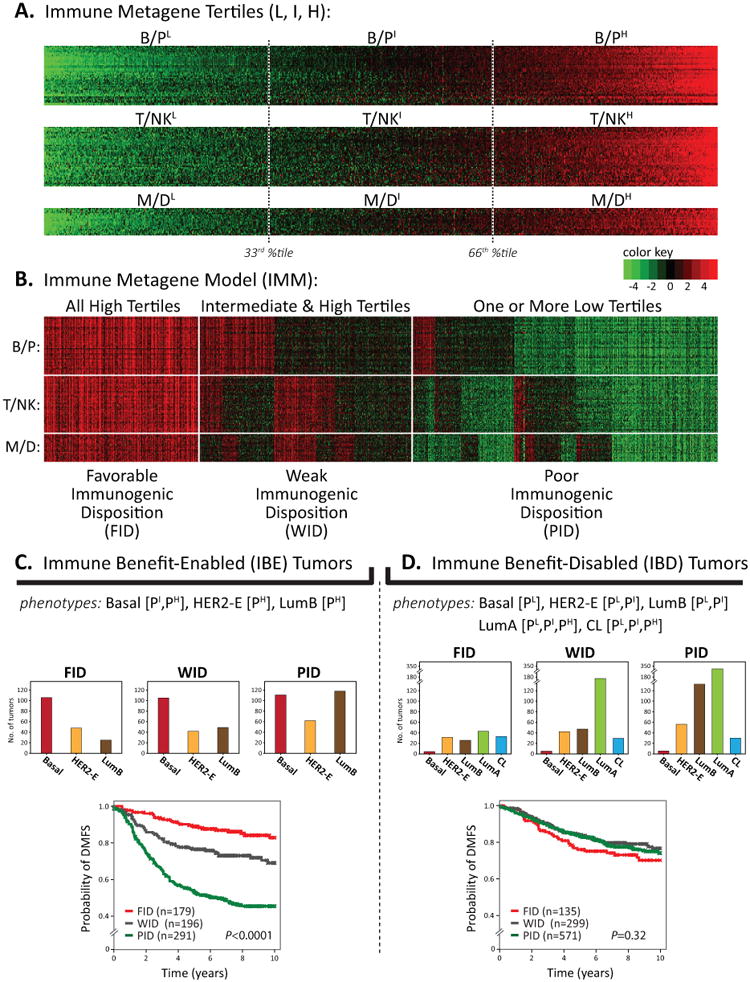
(A) Heatmap of metagene gene expression levels (rows) across 1,954 tumors (columns) as described in (12). Key shows color scale of mean centered, log2-transformed gene signal intensities. For each metagene, tumors are sorted left to right by ascending metagene scores; tertile thresholds are shown (33rd & 66th percentiles) for defining low (L), intermediate (I), and high (H) tertiles. (B) IMM prognostic risk groups are shown. (C, D) IBE and IBD type tumors are shown stratified by IMM sub-classes (FID, WID, PID) in Kaplan-Meier plots of DMFS. The number of tumors (n) in each subclass is shown; the log-rank P-value is reported.
In addition to tumor proliferative capacity, we previously found that the intrinsic molecular subtypes of breast cancer defined by the PAM50 classifier (34) further influenced, to variable degrees, the prognostic potential of the individual immune metagenes (12). Therefore, we employed Cox proportional hazards regression to determine the prognostic relevance of the IMM within each molecular subtype stratified according to proliferative capacity via proliferation metagene tertiles (Supplementary Table S2). The IMM achieved significant associations (P < 0.05, FDR < 10%) with DMFS in four subgroups: intermediate and high proliferating basal-like tumors (designated as Basal [PI, PH]), highly proliferative HER2-enriched tumors (HER2-E [PH]) and highly proliferative luminal B tumors (LumB [PH]). As illustrated in Fig. 1C, we refer to this group as immune benefit-enabled (IBE) in reference to the potential of the FID subclass to exhibit a significantly better DMFS rate as compared to the WID and PID subclasses. In contrast, the IMM was not significantly associated with DMFS in luminal A (LumA) and claudin-low (CL) subtypes, regardless of proliferation tertile, nor was an association observed in low and intermediate proliferating HER2-E and LumB tumors, or low proliferative basal tumors. We refer to these tumors as immune benefit-disabled (IBD) in reference to the inability of the FID subclass to exhibit a significantly better DMFS rate than the WID and PID subclasses (Fig. 1D). As 56% of IBD tumors belong to the LumA subtype, we considered the possibility that LumA tumors may mask an otherwise significant association of the IMM with DMFS in non-LumA IBD tumors. Thus, we analyzed the FID, WID, and PID survival curves in the non-LumA and LumA tumors separately. By Kaplan-Meier analysis, neither the non-LumA IBD (comprised of basal-like, CL, HER2-E, and LumB subtypes) nor the LumA IBD tumors showed a significant stratification of the IMM (Supplementary Fig. S4A and B). This indicates that the LumA subtype is not driving the negligible immune-prognostic characteristic of IBD. Together, these observations suggest that prominent immune infiltrate associated with FID tumors can guard against metastatic progression in some tumor subtypes (IBE), but not others (IBD).
To discern the prognostic value of the IMM in the presence of conventional variables, we fit a stepwise Cox proportional hazards model within IBE and IBD to elucidate independent associations with DMFS (Table 1). In IBE tumors, the IMM and lymph node status were each significant univariably, with only the IMM and tumor size remaining significant after adjusting for other variables. In contrast, in IBD tumors, histologic grade, tumor size, lymph node status, age, and ER status were each individually significant, with histologic grade, tumor size, lymph node status and adjuvant treatment remaining significant after adjustment. These findings suggest that in IBE tumors the IMM adds substantial prognostic information to conventional prognostic markers, and that the significant prognostic stratification conferred by the IMM in IBE tumors is not a spurious event driven by other known associations.
Table 1. Cox proportional hazards regression analysis for associations with distant metastasis-free survival in IBE and IBD tumors.
| Univariable | Multivariable | |||
|---|---|---|---|---|
| Variables1 | Hazard Ratio (95% CI)2 | P-value | Hazard Ratio (95% CI) | P-value |
| IBE | ||||
| IMM (PID, WID, FID) | 0.46 (0.38-0.55) | <0.0001 | 0.49 (0.39-0.61) | <0.0001 |
| Histologic Grade (I, II, III) | 1.00 (0.77-1.30) | 0.99 | - | - |
| Tumor Size (T1, T2, T3) | 1.20 (0.93-1.54) | 0.16 | 1.37 (1.02-1.84) | 0.04 |
| LN Status (-, +) | 1.41 (1.05-1.88) | 0.03 | - | - |
| Age (≤40 yrs, >40 yrs) | 0.95 (0.64-1.43) | 0.82 | - | - |
| ER Status (+, -) | 1.15 (0.86-1.53) | 0.34 | - | - |
| Adjuvant Treatment (no, yes) | 0.88 (0.67-1.13) | 0.31 | - | - |
| IBD | ||||
| IMM (PID, WID, FID) | 1.07 (0.89-1.28) | 0.47 | - | - |
| Histologic Grade (I, II, III) | 2.23 (1.76-2.84) | <0.0001 | 2.14 (1.65-2.76) | <0.0001 |
| Tumor Size (T1, T2, T3) | 1.57 (1.24-1.99) | 0.0003 | 1.61 (1.18-2.20) | 0.003 |
| LN Status (-, +) | 1.62 (1.21-2.16) | 0.002 | 2.50 (1.44-4.31) | 0.001 |
| Age (≤40 yrs, >40 yrs) | 0.49 (0.33-0.74) | 0.002 | - | - |
| ER Status (+, -) | 0.49 (0.35-0.68) | <0.0001 | - | - |
| Adjuvant Treatment (no, yes) | 0.89 (0.68-1.16) | 0.39 | 0.56 (0.33-0.96) | 0.03 |
entered as semi-continuous variables: IMM (PID, WID, FID) as 1, 2 or 3; histologic grade (I, II, III) as 1, 2 or 3; tumor size (T1, T2, T3) as 1, 2 or 3; lymph node (LN) status (-, +) as 0 or 1; age (≤40 yrs, >40 yrs) as 0 or 1; ER status (+, -) as 0 or 1; adjuvant treatment (no, yes) as 0 or 1;
95% confidence interval;
likelihood ratio test p-value
IBE and IBD are reproducible disease states
To validate our parameters for defining IBE and IBD, we sought to confirm their existence using microarray datasets from two independent patient populations: MC2 and TCGA. MC2 (Meta-Cohort #2) comprises 616 breast tumors profiled on the Affymetrix U133 series GeneChips, whereas the TCGA cohort comprises 524 breast tumors profiled on an Agilent custom microarray as part of the Cancer Genome Atlas project (32). Using the same procedures employed in MC1, we assigned tumors of MC2 and TCGA to metagene tertiles, IMM subclasses and PAM50 subtypes. We then categorized tumors as IBE or IBD according to MC1-defined thresholds and examined the prognostic performance of the IMM subclasses by Kaplan-Meier analysis. In the MC2 cohort, the prognostic performance of the IMM was highly significant in the IBE-designated tumors, stratifying patients into markedly different DMFS risk groups (Fig. 2A). By contrast, the IMM failed to risk stratify patients in the IBD-designated tumors (Fig. 2B). In the TCGA cohort, the IMM again exhibited statistical significance in the tumors classified as IBE (Fig. 2C), but not IBD (Fig. 2D) despite potential limitations including patient overall survival (OS) as the primary clinical endpoint (rather than DMFS), >75% of patients lost to follow-up at 5 years, and the different microarray technology platform used in the TCGA study (Agilent versus Affymetrix). Together, these findings confirm the reproducibility of the parameters used to distinguish the IBE and IBD subtypes and demonstrate the robust association of the IMM with both distant recurrence and overall survival in IBE tumors, but not IBD tumors.
Figure 2. Validation of IBE and IBD subtypes in independent cohorts.

Kaplan-Meier plots of patients stratified by the IMM subclasses (FID, WID, PID) are shown for (A) IBE tumors (MC2), (B) IBD tumors (MC2), (C) IBE tumors (TCGA), and (D) IBD tumors (TCGA).
The IMM classifies tumors according to a combination of immune-derived gene expression patterns. To better understand how common immunological mechanisms relate to the IBE and IBD designations, we investigated how individual genes with more clearly defined roles in antitumor immunity associate with clinical outcome in IBE and IBD. CTLs and NK cells are the primary effectors of cell-mediated tumor rejection. Expression of the CD8 transmembrane receptor contributes to synaptic stability during tumor-responsive antigenic activation and is a defining feature of CTLs. Expression of the CD16 (FCGR3A/B) transmembrane receptor is a defining feature of NK cells (though it may also be expressed by neutrophils and macrophages) and contributes to antibody-dependent cell mediated cytotoxicity (ADCC) through binding of the Fc portion of IgG type antibodies bound to tumor-associated antigens expressed on cancer cells. Ativated CTLs and NK cells alike secrete cytolytic proteins that induce apoptosis in their cancer cell targets. Central proteins of this class include GZMB, GNLY, and PRF1. Finally, professional antigen-presenting cells—including dendritic cells, macrophages, and B cells—present tumor antigens to T cell effectors (e.g., helper T cells) through MHC (HLA) class II molecules (e.g., HLA-DR) and provide costimulatory signals necessary for T-cell activation through expression of B7-1/B7-2 type I membrane proteins (e.g., CD86). DMFS survival curves based on expression tertiles of the genes coding for CD8, CD16, GZMB, HLA-DR, and CD86 are shown in Fig. 3. In all instances, significant positive associations were observed in IBE tumors, but not in corresponding IBD tumors. In contrast, some genes such as GZMB, CD16, and CD86, which showed positive associations with DMFS in IBE tumors consistent with antitumorigenic functions, displayed small but statistically significant negative associations with DMFS in IBD tumors – consistent with possible pro-tumorigenic tendencies in IBD.
Figure 3. Prognostic associations of cell type-specific markers in IBE and IBD breast cancers.

Gene expression tertiles for cell type-specific gene markers were used to stratify patients into survival groups (low, mid, and high tertiles) for Kaplan-Meier analysis. (A-J) Kaplan-Meier plots are shown for each gene marker in IBE and IBD tumor subtypes within cohorts MC1 and MC2.
Immunomodulatory pathways distinguish IBE and IBD
The FID subclass is defined as the subset of tumors that sort into the upper tertiles of all three immune metagenes, simultaneously. Yet, IBE-FID and IBD-FID tumors reflect different DMFS associations relative to the WID and PID subclasses. This observation is consistent with the biological hypothesis of a differential metastasis-protective potential between IBE-FID and IBD-FID, with IBE-FID tumors potentiating a metastasis-protective immune phenotype whereas IBD-FID tumors do not. Moreover, patients with IBD-FID tumors exhibit a worse prognosis than those with IBE-FID (Supplementary Fig. S4C and D), despite the lower proliferative capacity of IBD tumors in general. Thus, we investigated the transcriptional differences that distinguish IBE-FID from IBD-FID. First, we considered the expression of a pre-defined panel of genes with known roles in tumor immunology (Table 2). Central to this panel were the genes comprising the immunologic constant of rejection (ICR) (2), a group of genes repeatedly observed to participate in different forms of immune-mediated tissue destruction (2, 35-37). Consistent with the prognostic observations, a number of these genes were found to be statistically significantly overexpressed in IBE-FID tumors, including genes involved in Th1 polarization (IFNG, IRF1, STAT1), cell-mediated cytotoxicity (GZMB, GNLY, PRF1, GZMH), antitumor chemoattraction (CXCL9, CXCL10, CX3CL1, CCL5) and leukocyte adhesion (ICAM1). Positive regulators of CTL activation—including CTLA4, CD80, and CD86—were also overexpressed in IBE-FID. Conversely, we observed that TGFB1, encoding the immunosuppressive cytokine TGF-beta, was overexpressed in IBD-FID tumors. Using an unsupervised approach, we applied Gene Ontology (GO) enrichment analysis to the genes significantly differentially expressed between IBE-FID and IBD-FID using the DAVID Bioinformatics Resource (38, 39). Controlling for false discoveries, we identified 460 and 569 probe sets significantly overexpressed in IBE-FID and IBD-FID tumors, respectively. As anticipated, we found that many genes overexpressed in IBE-FID tumors were significantly associated with the GO terms cell cycle transit, cell division and proliferation, consistent with the IBE subtype comprising mostly of highly proliferative tumors. Specifically, we found that 38% of the IBE-FID–overexpressed probe sets were correlated with the proliferation metagene at R>0.5. After adjusting for these cell cycle-associated genes and analyzing for GO enrichment, one annotation cluster of significantly enriched terms was related to immunological processes, namely chemokine activity (P = 7.3 × 10-7) and defense response (P = 2.9 × 10-5). In addition to the previously-described ICR genes, these terms comprised a number of additional chemokine ligands (CCL7, CCL8, CCL13, CCL18, CXCL11, CXCL13, XCL1) as well as genes involved in acute phase responses (ORM1, DEFB1, LBP). In contrast, genes overexpressed in the IBD-FID tumors were associated with highly significant enrichment of GO terms such as secreted (P < 0.0001), extracellular matrix (P < 0.0001), skeletal system development (P < 0.0001), and cell adhesion (P < 0.0001). Within these categories were genes representing more distinct biological processes such as angiogenesis (BMP4, CTGF, SLIT2, SLIT3, LEPR, EMCN, FGF18, PLXDC1, EGFL7, CXCL12), proteolysis (ADAM12, ADAM23, MMP28, PLAT, CPE, CPZ, C4A, C7, CFD, HTRA1, AEBP1, PCSK5, TPSAB1), TGF-beta signaling (TGFBR3, BMP4, BMPR1B, THBS4, DCN, CHRD, COMP) and response to wounding (C7, C4A, CFD, CTGF, EPHA3, IGF1, IGF2, IGFB4, LAMB2, LYVE1, MMRN1, THBD, TIMP3, VWF).
Table 2. Differential expression of markers of immune functional orientation in IBE-FID and IBD-FID tumors.
| Marker Name/Functional Type | IBE-FID vs. IBD-FID | |
|---|---|---|
| mean log difference1 | P-value2 | |
| ICR: Th1 Polarization | ||
| IFNG | 210354_at | 0.52 | 7.6E-07 |
| IRF1 | 202531_at | 0.24 | 7.6E-05 |
| STAT1 | 209969_s_at | 0.56 | 4.3E-07 |
| IL12B | 207901_at | 0.03 | 8.5E-01 |
| TBX21 | 220684_at | 0.13 | 9.6E-02 |
| IL12A | 207160_at | 0.09 | 5.2E-01 |
| ICR: Cytotoxic Mechanisms | ||
| GZMB | 210164_at | 0.96 | 1.3E-12 |
| GNLY | 205495_s_at | 0.65 | 3.4E-06 |
| PRF1 | 214617_at | 0.36 | 4.8E-04 |
| GZMH | 210321_at | 0.52 | 4.2E-03 |
| GZMA | 205488_at | 0.22 | 8.9E-02 |
| ICR: Tissue Rejection Chemokines | ||
| CXCL10 | 204533_at | 0.90 | 7.6E-11 |
| CXCL9 | 203915_at | 0.83 | 2.7E-08 |
| CX3CL1 | 203687_at | 0.46 | 3.5E-03 |
| CCL5 | 1405_i_at | 0.43 | 6.9E-04 |
| CCL2 | 216598_s_at | 0.20 | 9.5E-02 |
| ICR: Adhesion Molecules | ||
| ICAM1 | 202637_s_at, 215485_s_at | 0.18 | 1.2E-02 |
| VCAM1 | 203868_s_at | 0.14 | 1.4E-01 |
| MADCAM1 | 208037_s_at | -0.12 | 2.4E-01 |
| CTL Activation and Regulatory Markers | ||
| CD8A | 205758_at | 0.15 | 2.8E-01 |
| CD8B | 207979_s_at, 215332_s_at | 0.11 | 4.1E-01 |
| CD28 | 206545_at | 0.02 | 9.0E-01 |
| CD86 | 205686_s_at | 0.25 | 8.3E-04 |
| CD80 | 207176_s_at | 0.15 | 2.1E-02 |
| CTLA4 | 221331_x_at | 0.29 | 1.7E-02 |
| PDCD1 | 207634_at | 0.03 | 8.1E-01 |
| FOXP3 | 221333_at | -0.15 | 2.5E-01 |
| NK Cell Activation and Regulatory Markers | ||
| KLRK1 (NKG2D) | 205821_at | 0.02 | 9.0E-01 |
| MICA | 205904_at | -0.08 | 5.0E-01 |
| MICB | 206247_at | -0.08 | 5.2E-01 |
| ULBP1 | 221323_at | 0.05 | 7.9E-01 |
| ULBP2 | 221291_at | -0.02 | 9.0E-01 |
| Pro-tumorigenic Cytokines | ||
| TGFB1 |203085_s_at | -0.25 | 1.4E-03 |
| IL8 | 202859_x_at | 0.36 | 1.0E-01 |
| IL10 | 207433_at | -0.13 | 4.2E-01 |
| TGFB2 | 220407_s_at | 0.12 | 4.1E-01 |
positive values reflect log fold increase in IBE-FID relative to IBD-FID;
adjusted for false discovery
Next, we utilized the Ingenuity Pathway Analysis (IPA) Upstream Regulator Analysis tool to identify potential transcriptional regulators that may explain the differential expression of genes observed between IBE-FID and IBD-FID. This tool leverages the Ingenuity Knowledge Base™ of regulatory relationships among genes, proteins, microRNAs, protein complexes and chemicals to statistically rank potential transcriptional regulators causally involved in activation and inhibition of differentially expressed genes (40). Table 3 shows the top two most significant protein regulator candidates identified as activated in IBE-FID or IBD-FID. In IBE-FID tumors, tumor necrosis factor–α (TNF; TNFα) and interferon-γ (IFNG; IFNγ) were identified as the top two most significant gene regulators with largest activation scores. Biologically, TNFα and IFNγ are pro-inflammatory cytokines known to play central roles in the activation of Th1-type tissue rejection and antitumor immune responses. The many differentially expressed genes that support the activation z-scores of TNFα and IFNγ are shown in the network schematic of Supplementary Fig. S5A. By contrast, among the top most significant regulator to emerge in IBD-FID with greatest activation z-score was transforming growth factor β1. TGF-beta is a potent immunosuppressive cytokine that antagonizes Th1-type immune responses. Shown in Supplementary Fig. S5B are the genes contributing to the significant TGF β activation z-score. The identification of estrogen receptor α -1 (ESR1) as a top regulator may be explained by the significant over-representation of ER-positive (luminal) tumors comprising the IBD subtype. To further corroborate the IPA findings of TNFα/IFNγ and TGFβ activity in IBE-FID and IBD-FID, respectively, we investigated the corresponding pathway activation signatures previously defined by Gatza, et. al. (33). Briefly, these signatures were empirically deduced from expression profiles of cell culture experiments and utilize a binary regression algorithm that measures the probability of pathway activation for each sample in a microarray dataset. Consistent with the IPA Upstream Regulator analysis results, we found that the IBE-FID tumors were characterized by significantly higher activation scores for TNFα (P = 9.2 × 10-05) and IFNγ (3.0 × 10-07), whereas IBD-FID tumors displayed significantly higher scores for TGFβ pathway activation (P = 3.5 × 10-06). Together, these findings suggest that major regulators of pro- and anti-Th1 immune responses distinguish the IBE and IBD breast tumor subtypes, which could explain their differential metastasis-protective associations.
Table 3. Ingenuity Pathway Analysis: Top predicted upstream regulators in IBE-FID and IBD-FID.
| Upstream Regulator | Molecule Type | Activation z-score | P-value |
|---|---|---|---|
| Activated in IBE-FID | |||
| TNF | cytokine | 4.68 | 7. 4E-16 |
| IFNG | cytokine | 4.15 | 3.1E-11 |
| Activated in IBD-FID | |||
| TGFB1 | growth factor | 3.40 | 1.6E-14 |
| ESR1 | ligand-dependent nuclear receptor | 2.62 | 1.5E-15 |
Discussion
The contemporary view of breast cancer as poorly immunogenic is predicated on the absence of an observed increase in breast cancer incidence in immunocompromised patients and lack of responsiveness to otherwise clinically effective immunotherapy (41). Nonetheless, mounting evidence from studies of breast cancer-specific immune responses suggest variable immunogenic activity (42, 43). Recently, the immune contexture of breast cancer has gained broad acceptance as an important clinical correlate in both patient prognosis and therapy prediction. An understanding of how the prognostic value of immune infiltrates varies across the spectrum of breast tumor subtypes is relevant to disease management and can provide much needed biological insight into the underlying variation in tumor immunogenicity. To this point, we hypothesized that breast tumor immunogenicity, like other biological properties of cancer, is subject to phenotypic heterogeneity, varying from tumor to tumor according to molecular cues intrinsic to cancer cells or to other components of the tumor microenvironment. Our findings suggest that the immunogenic heterogeneity of human breast cancer may be resolved, to a substantial degree, by the molecular classification of breast tumors into two discernible subtypes that vary on both clinical and biological scales. We show that these subtypes, termed immune benefit-enabled (IBE) and immune benefit-disabled (IBD), can be distinguished by phenotype and differ widely with respect to the ability of an immune-dependent model to stratify patients into prognostic subclasses with significantly different DMFS. Our data suggest the IMM subclasses (FID, WID and PID) reflect a differential ability of the immune system to mount durable antitumor immune responses protective of distant recurrence in IBE tumors, but not IBD tumors. Furthermore, we show that the distinguishing biological features of IBE and IBD are differential measures of proliferative capacity and composition of intrinsic molecular subtype, and as such provide a reproducible classification system for delineating IBE and IBD (Fig. 2).
Our data indicate that IBD are least likely to be controlled by immune infiltrates perhaps because their functional orientation is less clearly polarized towards a Th1/effector phenotype as supported by evidence in Tables 2 and 3. Expression of CD8 did not significantly differ between IBE and IBD (Table 2), suggesting that the functional molecular orientation is the key determinant of these opposite phenotypes. The results of a gene network analysis suggest that some of the phenotypic characteristics may be related to the production by IBD tumors of potent immune suppressant factors, such as TGFβ, that may alter the functional status of the immune response in the tumor microenvironment, though this hypothesis remains to be confirmed. Recent new evidence suggests that in some breast tumors, TGFβ signaling (in association with Smad3) contributes to the maintenance of an anti-proliferative effect manifesting as a reduced tumor proliferative capacity (44). Thus, the potential duality of TGFβ function, as an intrinsic tumor suppressor and potent immunosuppressive cytokine could explain, in part, the low-proliferative phenotype of IBD tumors in general, and an impaired immune response associated with IBD-FID, specifically.
In contrast, our findings suggest that IBE tumors are endowed with the potential to achieve a metastasis-protective immune response (in the FID subclass) that may be facilitated by a proclivity toward Th1 polarization (Table 2) via activation of TNFα and/or IFNγ signaling (Table 3). However, as indicated by the reduced DMFS associated with WID and PID tumors, this immunogenic potential may be negatively regulated by the selection of oncogenic mechanisms that induce immune tolerance or block effector cell trafficking into the tumor mass. Indeed, this hypothesis is consistent with recent reports that breast and other solid tumors evade immune destruction and the control of metastatic spread through expression of secreted factors that fortify the tumor endothelial barrier thereby limiting tumor infiltration by effector cells and impeding immunotherapeutic efficacy in vivo (45, 46).
In reality, the causality for these distinct phenotypes of breast cancer remains elusive as it is for other types of cancer such as colon cancer and melanoma. Although the presence of a Th1 polarization appears to be relevant for improved survival in IBE, it remains unclear why some tumors are endowed with this immune phenotype independent of their ontogeny (47). Genetic determinants of intratumoral immune reactions are only now beginning to be elucidated (48).
Our findings suggest that biologic characteristics of tumor cells, and in particular their proliferative connotation, may be related to the immune phenotype. This may be of particular interest because this observation suggests that intrinsic characteristics of tumor cells may be driving the recruitment, and shaping the function, of leukocyte populations in the tumor microenvironment. This proliferation component may complement the notion of genetic instability of cancer cells as a potential lead to the production of mutated protein products recognized as foreign by the immune system leading, therefore, to higher immunogenicity (49). This hypothesis, on the other hand, would be in contrast with the alternative explanation that the genetic background of the host determines tumor immune responsiveness, which in turn would preferentially explain why such immune phenotypes are seen in tumors of different histology as recently reviewed (5). To date, there remains no valid explanation for this phenomenon, and this study may provide a clue toward dissecting the genetic and epigenetic mechanisms leading to immune responsiveness.
Independent of the causality, however, the current sub-classification strategy may provide the practical benefit of allowing a better prognostic stratification of patients if the association with better prognosis will prove to also reflect a phenotype predictive of responsiveness to immunotherapy, a hypothesis that warrants clinical testing. This would not be completely surprising since the expression of ICR genes that was observed in IBE-FID tumors has been shown to span a continuum of immune surveillance that includes immune responsiveness (prediction of response), prognostic significance (improved survival independent of treatment) and has mechanistic implications (activation of genes during active rejection) (2). This leads to the hypothesis that all such phenomena related to immune surveillance are part of a same process and only quantitative variations determine the ultimate outcome (2).
In conclusion, this work suggests a new immunogenic perspective for breast cancer, in which tumor proliferative capacity and intrinsic subtype are organizing principles that distinguish a tumor's capacity for clinically beneficial immunogenicity. Future studies aimed at optimizing the IBE/IBD tumor classification, as well as the prognostication of patients using more comprehensive measures of immune responsiveness, should increasingly improve our contextual understanding of breast tumor immunogenicity and complement ongoing efforts to standardize histopathological assessment of TIL in breast cancer (50). In addition, such studies should potentiate a better understanding of the factors that regulate antitumor immunity with the potential to spark novel therapeutic strategies that target specific immunogenic subtypes.
Supplementary Material
Acknowledgments
Financial Support: This work was supported in part by the American Cancer Society (RSG-12-198-01-TBG) (to L.D. Miller) and the Mary Kirkpatrick Professorship for Breast Cancer Research (to L.D. Miller). The Cancer Genomics Shared Resource and the Biostatistics and Bioinformatics Shared Resource of the Comprehensive Cancer Center of Wake Forest University (supported by NCI CCSG P30CA012197) provided resources for this study. Breast Cancer Cure NZ provided funds for tissue collection and array analysis of the New Zealand cohort.
Footnotes
Financial Interests: the authors have no conflicts of interest to disclose
References
- 1.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–8. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 2.Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity. 2013;39(1):11–26. doi: 10.1016/j.immuni.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 3.Jessy T. Immunity over inability: The spontaneous regression of cancer. J Nat Sci Biol Med. 2011;2(1):43–9. doi: 10.4103/0976-9668.82318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. 2007;117(5):1137–46. doi: 10.1172/JCI31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2011;12(4):298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 6.Ascierto ML, Idowu MO, Zhao Y, Khalak H, Payne KK, Wang XY, et al. Molecular signatures mostly associated with NK cells are predictive of relapse free survival in breast cancer patients. J Transl Med. 2013;11(1):145. doi: 10.1186/1479-5876-11-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Senovilla L, Vacchelli E, Galon J, Adjemian S, Eggermont A, Fridman WH, et al. Trial watch: Prognostic and predictive value of the immune infiltrate in cancer. Oncoimmunology. 2012;1(8):1323–43. doi: 10.4161/onci.22009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ascierto ML, Kmieciak M, Idowu MO, Manjili R, Zhao Y, Grimes M, et al. A signature of immune function genes associated with recurrence-free survival in breast cancer patients. Breast Cancer Res Treat. 2012;131(3):871–80. doi: 10.1007/s10549-011-1470-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denkert C, Loibl S, Noske A, Roller M, Muller BM, Komor M, et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol. 2010;28(1):105–13. doi: 10.1200/JCO.2009.23.7370. [DOI] [PubMed] [Google Scholar]
- 10.Denkert C, von Minckwitz G, Brase JC, Sinn BV, Gade S, Kronenwett R, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol. 2015;33(9):983–91. doi: 10.1200/JCO.2014.58.1967. [DOI] [PubMed] [Google Scholar]
- 11.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–45. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagalla S, Chou JW, Willingham MC, Ruiz J, Vaughn JP, Dubey P, et al. Interactions between immunity, proliferation and molecular subtype in breast cancer prognosis. Genome Biol. 2013;14(4):R34. doi: 10.1186/gb-2013-14-4-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rody A, Holtrich U, Pusztai L, Liedtke C, Gaetje R, Ruckhaeberle E, et al. T-cell metagene predicts a favorable prognosis in estrogen receptor-negative and HER2-positive breast cancers. Breast Cancer Res. 2009;11(2):R15. doi: 10.1186/bcr2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desmedt C, Haibe-Kains B, Wirapati P, Buyse M, Larsimont D, Bontempi G, et al. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res. 2008;14(16):5158–65. doi: 10.1158/1078-0432.CCR-07-4756. [DOI] [PubMed] [Google Scholar]
- 15.Panelli MC, Wang E, Phan G, Puhlmann M, Miller L, Ohnmacht GA, et al. Gene-expression profiling of the response of peripheral blood mononuclear cells and melanoma metastases to systemic IL-2 administration. Genome Biol. 2002;3(7) doi: 10.1186/gb-2002-3-7-research0035. RESEARCH0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmidt M, Bohm D, von Torne C, Steiner E, Puhl A, Pilch H, et al. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008;68(13):5405–13. doi: 10.1158/0008-5472.CAN-07-5206. [DOI] [PubMed] [Google Scholar]
- 17.Spivey TL, De Giorgi V, Zhao Y, Bedognetti D, Pos Z, Liu Q, et al. The stable traits of melanoma genetics: an alternate approach to target discovery. BMC Genomics. 2012;13:156. doi: 10.1186/1471-2164-13-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang E, Miller LD, Ohnmacht GA, Mocellin S, Perez-Diez A, Petersen D, et al. Prospective molecular profiling of melanoma metastases suggests classifiers of immune responsiveness. Cancer Res. 2002;62(13):3581–6. [PMC free article] [PubMed] [Google Scholar]
- 19.Rody A, Karn T, Liedtke C, Pusztai L, Ruckhaeberle E, Hanker L, et al. A clinically relevant gene signature in triple negative and basal-like breast cancer. Breast Cancer Res. 2011;13(5):R97. doi: 10.1186/bcr3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teschendorff AE, Caldas C. A robust classifier of high predictive value to identify good prognosis patients in ER-negative breast cancer. Breast Cancer Res. 2008;10(4):R73. doi: 10.1186/bcr2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teschendorff AE, Miremadi A, Pinder SE, Ellis IO, Caldas C. An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol. 2007;8(8):R157. doi: 10.1186/gb-2007-8-8-r157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alistar A, Chou JW, Nagalla S, Black MA, D'Agostino R, Jr, Miller LD. Dual roles for immune metagenes in breast cancer prognosis and therapy prediction. Genome Med. 2014;6(10):80. doi: 10.1186/s13073-014-0080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stoll G, Enot D, Mlecnik B, Galon J, Zitvogel L, Kroemer G. Immune-related gene signatures predict the outcome of neoadjuvant chemotherapy. Oncoimmunology. 2014;3(1):e27884. doi: 10.4161/onci.27884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perez EA, Thompson EA, Ballman KV, Anderson SK, Asmann YW, Kalari KR, et al. Genomic analysis reveals that immune function genes are strongly linked to clinical outcome in the north central cancer treatment group n9831 adjuvant trastuzumab trial. J Clin Oncol. 2015;33(7):701–8. doi: 10.1200/JCO.2014.57.6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dave SS, Wright G, Tan B, Rosenwald A, Gascoyne RD, Chan WC, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004;351(21):2159–69. doi: 10.1056/NEJMoa041869. [DOI] [PubMed] [Google Scholar]
- 26.Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, et al. Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nat Genet. 2001;29(4):365–71. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- 27.McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM. REporting recommendations for tumour MARKer prognostic studies (REMARK) Eur J Cancer. 2005;41(12):1690–6. doi: 10.1016/j.ejca.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 28.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.R. Development. Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2010. [Google Scholar]
- 30.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–27. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 32.Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gatza ML, Lucas JE, Barry WT, Kim JW, Wang Q, Crawford MD, et al. A pathway-based classification of human breast cancer. Proc Natl Acad Sci U S A. 2010;107(15):6994–9. doi: 10.1073/pnas.0912708107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–7. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spivey TL, Uccellini L, Ascierto ML, Zoppoli G, De Giorgi V, Delogu LG, et al. Gene expression profiling in acute allograft rejection: challenging the immunologic constant of rejection hypothesis. J Transl Med. 2011;9:174. doi: 10.1186/1479-5876-9-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang E, Bedognetti D, Marincola FM. Prediction of response to anticancer immunotherapy using gene signatures. J Clin Oncol. 2013;31(19):2369–71. doi: 10.1200/JCO.2013.49.2157. [DOI] [PubMed] [Google Scholar]
- 37.Wang E, Worschech A, Marincola FM. The immunologic constant of rejection. Trends Immunol. 2008;29(6):256–62. doi: 10.1016/j.it.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 38.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4(5):3. [PubMed] [Google Scholar]
- 39.Huang da W, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35(Web Server issue):W169–75. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kramer A, Green J, Pollard J, Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30(4):523–30. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Criscitiello C, Curigliano G. Immunotherapeutics for breast cancer. Curr Opin Oncol. 2013;25(6):602–8. doi: 10.1097/CCO.0000000000000020. [DOI] [PubMed] [Google Scholar]
- 42.Criscitiello C. Tumor-associated antigens in breast cancer. Breast Care (Basel) 2012;7(4):262–6. doi: 10.1159/000342164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Piura E, Piura B. Autoantibodies to tumor-associated antigens in breast carcinoma. J Oncol. 2010;2010:264926. doi: 10.1155/2010/264926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sato M, Kadota M, Tang B, Yang HH, Yang YA, Shan M, et al. An integrated genomic approach identifies persistent tumor suppressive effects of transforming growth factor-beta in human breast cancer. Breast Cancer Res. 2014;16(3):R57. doi: 10.1186/bcr3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008;14(1):28–36. doi: 10.1038/nm1699. [DOI] [PubMed] [Google Scholar]
- 46.Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014 doi: 10.1038/nm.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang E, Bedognetti D, Tomei S, Marincola FM. Common pathways to tumor rejection. Ann N Y Acad Sci. 2013;1284:75–9. doi: 10.1111/nyas.12063. [DOI] [PubMed] [Google Scholar]
- 48.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1-2):48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–99. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salgado R, Denkert C, Demaria S, Sirtaine N, Klauschen F, Pruneri G, et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs Working Group 2014. Ann Oncol. 2014 doi: 10.1093/annonc/mdu450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.