

Abstract
The nontemplated addition of single or multiple nucleotides to RNA transcripts is an efficient means to control RNA stability and processing. Cytoplasmic RNA adenylation and the less well-known uridylation are post-transcriptional mechanisms regulating RNA maturation, activity, and degradation. Gld2 is a member of the noncanonical poly(A) polymerases, which include enzymes with varying nucleotide specificity, ranging from strictly ATP to ambiguous to exclusive UTP adding enzymes. Human Gld2 has been associated with transcript stabilizing miRNA monoadenylation and cytoplasmic mRNA polyadenylation. Most recent data have uncovered an unexpected miRNA uridylation activity, which promotes miRNA maturation. These conflicting data raise the question of Gld2 nucleotide specificity. Here, we biochemically characterized human Gld2 and demonstrated that it is a bona fide adenylyltransferase with only weak activity toward other nucleotides. Despite its sequence similarity with uridylyltransferases (TUT4, TUT7), Gld2 displays an 83-fold preference of ATP over UTP. Gld2 is a promiscuous enzyme, with activity toward miRNA, pre-miRNA, and polyadenylated RNA substrates. Apo-Gld2 activity is restricted to adding single nucleotides and processivity likely relies on additional RNA-binding proteins. A phylogeny of the PAP/TUTase superfamily suggests that uridylyltransferases, which are derived from distinct adenylyltransferase ancestors, arose multiple times during evolution via insertion of an active site histidine. A corresponding histidine insertion into the Gld2 active site alters substrate specificity from ATP to UTP.
Keywords: miRNA maturation, miRNA, nucleotidyltransferase, adenylation, uridylation
INTRODUCTION
Since microRNA (miRNA) discovery in the early 1990s (Arasu et al. 1991; Lee et al. 1993), it has become evident that post-transcriptional gene regulation by miRNAs is involved in most biological processes (Grosshans and Slack 2002; Friedman et al. 2009; Ameres and Zamore 2013; Sun and Lai 2013; Takahashi et al. 2014). The latest miRBase release contains 24,521 experimentally validated miRNA genes from 206 species (Kozomara and Griffiths-Jones 2014). Dysfunctional miRNA expression, processing, and degradation have been found in breast cancer (Cammarata et al. 2010), acute myeloid leukemia (Kobayashi et al. 2014), ovarian cancer (Mulrane et al. 2013), and hepatocellular carcinoma (Zhu et al. 2011). Deregulated miRNA processing also contributes to other major diseases such as Hepatitis C (van der Ree et al. 2016) and cardiovascular diseases (Small and Olson 2011). Because miRNAs regulate genes that change cellular fate, miRNAs and proteins involved in miRNA regulation are promising next-generation cancer therapeutic targets and specific components of the RNA processing machinery are current biomarkers for cancer detection (Barh et al. 2010; Di Leva and Croce 2013; Fendler and Jung 2013; Zheng et al. 2013; Moitra 2015).
The generation of miRNAs is a multistage process and translational inhibition by miRNAs is achieved through base-pairing with the 3′UTR of the respective target mRNA, leading to mRNA decay or silencing (Yates et al. 2013). Recent high-throughput sequencing studies revealed the presence of untemplated nucleotide additions to the 3′ termini of nearly 40% of miRNAs (Burroughs et al. 2010; Wyman et al. 2011). Of these, depending on the miRNA species, ∼50% displayed an extra adenine, 25% contained a single additional uridine, and the remaining 25% contained multiple nucleotides appended to the 3′ termini (Wyman et al. 2011). During miRNA maturation and degradation, untemplated uridine (Heo et al. 2009, 2012) and adenine (D'Ambrogio et al. 2012) residues are added to miRNA precursors and mature miRNAs. These untemplated nucleotide additions are an efficient means to control the levels of active miRNAs in the cell. The seemingly innocuous addition of a single nucleotide can initiate miRNA maturation, stabilization, or convert an active miRNA to an inactive form (Thornton et al. 2014). While multiple adenine residues are added to mRNA for stabilization, extending the transcript life span (Norbury 2013), multiple uridine residues mark both miRNA and mRNA for degradation (Mullen and Marzluff 2008; Rissland and Norbury 2009; Lim et al. 2014). The presence of untemplated nucleotides on a variety of miRNAs is now well known; however, the corresponding enzymes have only recently become the focus of biochemical characterization.
Nucleotidyltransferases such as the human terminal uridylyltransferases TUT4 (Zcchc11), TUT7 (Zcchc6) and the minimal homolog Gld2 (TUT2, Papd4) have been shown to play fundamental roles in the regulation and maturation of miRNAs let-7 and mir-122. TUTase homologs are part of the nucleotidyltransferase superfamily of enzymes, and were initially identified as adenylyltransferases associated with miRNA and mRNA adenylation. Gld2 was first described as a cytoplasmic noncanonical poly(A) polymerase involved in Caenorhabditis elegans germline development. C. elegans Gld2 displayed very little activity on its own however, and relies on an additional protein, Gld3, to promote adenylation (Wang et al. 2002; Kwak et al. 2004). In Drosophila, specific depletion of the Gld2 homolog WISPY connected its function with mRNA polyadenylation required for oocyte to egg activation (Cui et al. 2013), as well as long-term memory (Fig. 1A; Kwak et al. 2008). Recent data showed that in addition to mRNA adenylation, WISPY adenylates miRNAs in S2 cells leading to a reduction of miRNA levels (Lee et al. 2014).
FIGURE 1.
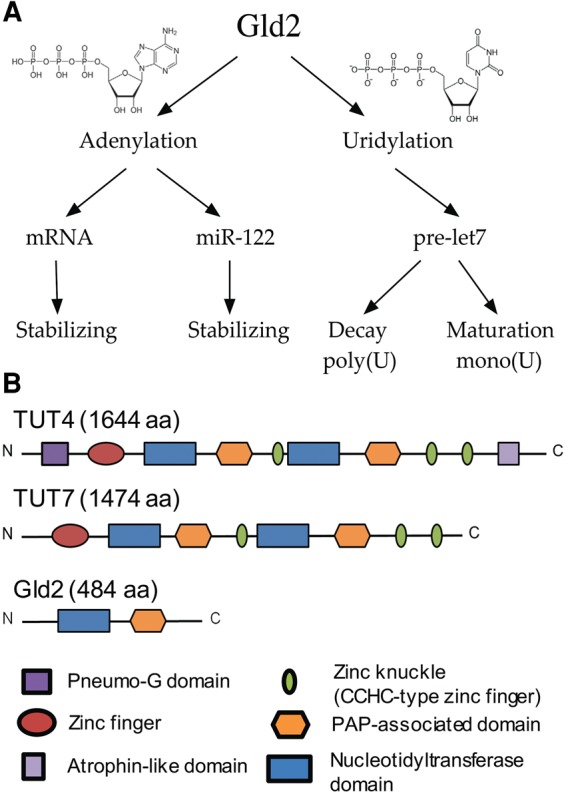
Proposed catalytic activities and domain organization of Gld2. (A) Gld2 has been implicated in nucleotide addition to multiple RNA substrates in different pathways. (B) Domain organization of Gld2 and its homologs TUT4 and TUT7. (Dark purple) Pneumo-G domain; (red) zinc finger domain; (blue) nucleotidyltransferase domain; (orange) poly(A) polymerase associated domain; (green) CCHC type zinc finger domain; (light purple) atrophin-like domain.
In humans, Gld2 has been equally associated with miRNA and mRNA adenylation as well as miRNA uridylation. The first reports associated human Gld2 with mRNA polyadenylation (Kwak and Wickens 2007) by monitoring translation of a reporter mRNA tethered to human Gld2 and injected into in Xenopus oocytes. In vivo, RNA polyadenylation requires either artificial RNA tethering (Kwak and Wickens 2007) or accessory RNA-binding proteins such as the cytoplasmic polyadenylation element binding protein (CPEB) in Xenopus (Barnard et al. 2004; Kim and Richter 2006) and Gld3 in C. elegans (Wang et al. 2002). Further studies showed that Gld2-mediated monoadenylation stabilizes miR-122 transcripts in human fibroblasts (D'Ambrogio et al. 2012) and plays a role in translational regulation of p53 (Burns et al. 2011; Glahder and Norrild 2011). Monoadenylation is, in contrast to polyadenylation not entirely dependent on RNA-binding proteins, as purified Gld2 from human cells displayed catalytic activity in vitro. With the discovery of the poly(U) polymerase activity of enzymes previously thought to be poly(A) polymerases, specifically of the human Gld2 homologs TUT4 and TUT7 (Rissland et al. 2007), most recent research has uncovered a previously unknown Gld2-mediated uridylation activity. D'Ambrogio et al. (2012) demonstrated for the first time that human Gld2 is the enzyme responsible for monoadenylation and subsequent stabilization of miRNA-122, but they also reported a weaker uridylation activity. Gld2 has further been shown to catalyze the monouridylation of pre-microRNA let-7a, which is crucial for its maturation (Heo et al. 2012). Flag-tagged human Gld2 purified from HEK293T cells adds a single uridine to pre-let-7a but also displayed catalytic activity adding GTP and ATP, but not CTP in vitro (Heo et al. 2012). Interestingly, Gld2-mediated polyuridylation has been observed on pre-let-7a overhang variants (Kim et al. 2015) in the absence of accessory proteins. Further evidence linking Gld2 to pre-microRNA uridylation stems from knockdown assays, showing that TUT4, TUT7, and Gld2 redundantly control pre-let-7 maturation and are required for let-7 biogenesis (Heo et al. 2012). Gld2 can thus function as either a poly(A) polymerase (PAP) or a TUT in vitro.
Gld2 is composed of two major domains, a PAP associated domain and a nucleotidyltransferase (NT) domain (Fig. 1B). Its closest human homologs, TUT4 and TUT7 are comprised of the same domains but feature additional RNA-binding motifs, such as zinc-finger domains. TUT4 and TUT7 have been characterized in vivo and in vitro as true uridylyltransferases and are involved in multiple processes including miRNA and mRNA uridylation. For example, uridylation of the let-7a precursor by TUT4 can drive processing by Dicer or mark the precursor miRNA for degradation, thus directly controlling let-7a levels in the cell (Heo et al. 2009; Thornton et al. 2012, 2014; Lim et al. 2014). Gld2 has been proposed to carry out a similar function during miRNA maturation (Heo et al. 2012; Kim et al. 2015). While the role of TUT4 and TUT7 in these processes is becoming increasingly clear, the catalytic activity and biological role of the minimal nucleotidyltransferase Gld2 is uncertain. Evidence for both uridylation and adenylation activity of the human enzyme has been shown in in vivo and in vitro experiments, but a conclusive investigation of Gld2 nucleotide preference is lacking.
We here present a biochemical characterization of Gld2, identifying it as a bona fide adenylyltransferase with only weak activity toward UTP and GTP. Conversely, Gld2 displays a wide target RNA substrate range, adenylating multiple RNAs in vitro. The data suggest that Gld2 RNA substrate selectivity may require association with other protein factors in the cell. A detailed phylogenetic analysis shows that uridyl- and adenylyltransferases are closely related, and that uridylyltransferase activity arose independently multiple times during evolution.
RESULTS
Gld2 displays RNA substrate promiscuity
Gld2 has been implicated in multiple pathways of mRNA and miRNA regulation. To test whether Gld2 displays enzymatic activity in vitro, we recombinantly expressed full length human Gld2 in Escherichia coli and purified the enzyme to homogeneity. Gld2 eluted from a gel filtration column as a monomer. To evaluate Gld2 activity, we first assessed its substrate range. Previously, suggested roles for Gld2 activity are in miRNA regulation, including miR-122 adenylation (D'Ambrogio et al. 2012) and the uridylation of pre-let-7a and let-7a (Kim and Richter 2006; Heo et al. 2012). Another study linked Gld2 to mRNA adenylation (Sartain et al. 2011). We therefore included miR-122, pre-let-7a, let-7a, total human mRNA and a poly(A) tail mimic comprised of 15 adenines (15A) in our initial assays and total E. coli tRNA as a control.
Surprisingly, Gld2 displayed adenylation activity on all substrates in vitro (Fig. 2). For miRNAs and the 15A RNA a single band was observed, corresponding to monoadenylation of the substrate RNAs. For the premature miRNA substrate, pre-let-7a, a band around 70 bases was observed, indicating single-nucleotide addition. While the in vitro transcribed pre-let-7a was purified as a single transcript (Supplemental Fig. 1), additional bands of lower molecular weight were observed. These bands are likely due to the adenylation of partially degraded substrate RNA or degradation during the enzymatic reaction. For both total tRNA and total mRNA multiple bands were observed, but due to heterogeneity of the substrates we are unable to confirm that these are single nucleotide additions. Nevertheless, no clusters of ladder-like nucleotide additions were observed in these cases, indicating that the heterogenic substrates were likely monoadenylated.
FIGURE 2.
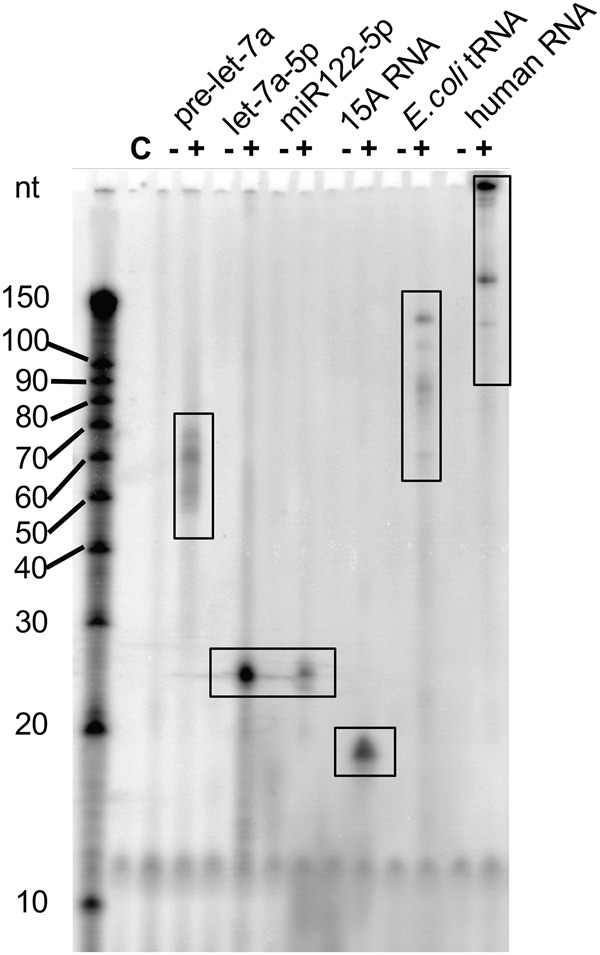
RNA substrate of Gld2. Gld2 was incubated with different RNA substrates and [α-P32]-ATP as indicated. Formation of α-32P labeled RNA products was monitored by electrophoretic separation and subsequent phosphorimaging. Gld2 catalyzed [α-32P]-ATP addition to RNA substrates pre-let-7a (72 nt), mature human miRNAs let-7a-5p (22 nt), miR122 (22 nt), an oligo(A) tail mimic 15A (15 nt), and total E. coli tRNA, and total human mRNA. C is no enzyme control.
Nucleotide preference of recombinant Gld2
We determined the specificity of Gld2 for all of the four nucleotides ATP, CTP, GTP, and UTP. In enzyme assays containing a single-nucleotide species, pre-let-7a and Gld2, the enzyme was active with all NTPs (Fig. 3A). Gld2 can accommodate each NTP in the active site and catalyze their 3′ addition to pre-let-7a. In a competition assay, which included all four nucleotides in equimolar concentrations with only one [α-32P]-labeled nucleotide, only ATP was added to pre-let-7a (Fig. 3B). Thus, while Gld2 displays relaxed specificity toward RNA substrates in vitro, it shows a clear preference for ATP in the presence of all four NTPs. We further investigated whether the 5′ end of the RNA substrates influenced substrate recognition. No difference in enzyme activity was seen when differentially 5′ phosphorylated RNA substrates were assayed with [α-32P]-ATP (Fig. 3C).
FIGURE 3.
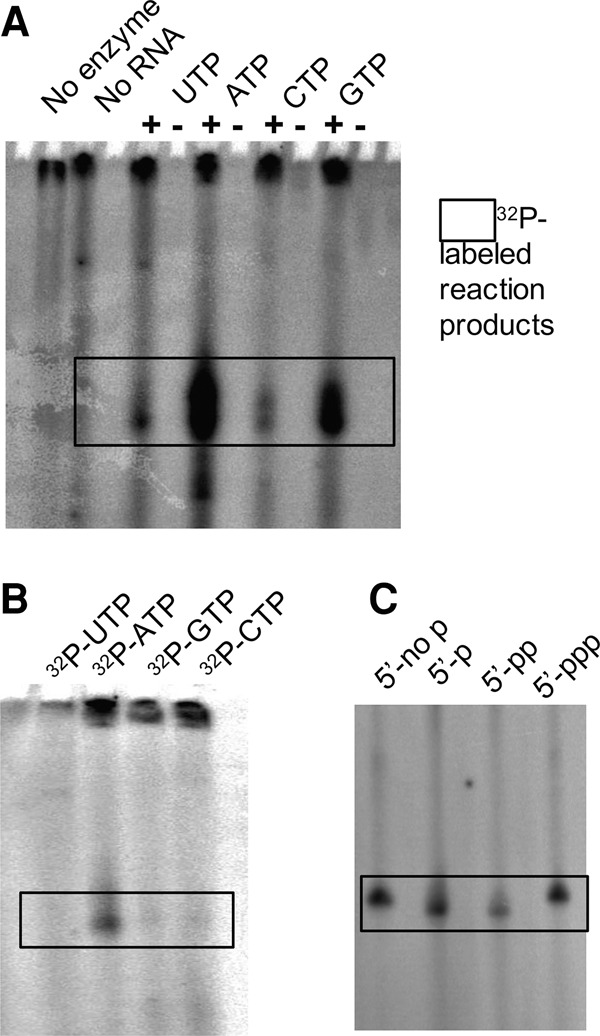
Nucleotide substrates of Gld2. Gld2 was incubated with varying nucleotides and the precursor microRNA let-7a or mature microRNA let-7a-5p. (A) Addition of single [α-32P]-labeled nucleotides to pre-let-7a and Gld2 as indicated. (B) Competitive nucleotide addition: In a competition assay, Gld2 was incubated with pre-let-7a in the presence of all four unlabeled nucleotides in equimolar amounts with a portion of the indicated nucleotide in an [α-32P]-labeled form. (C) 5′ end phosphorylation: Gld2 activity on let-7a-5p substrates with differentially phosphorylated 5′ ends were assayed with [α-32P]-ATP. (5′-no p) unphosphorylated, (5′-p) monophosphate, (5′-pp) diphosphate, (5′-ppp) triphosphate.
Gld2 is an adenylyltransferase
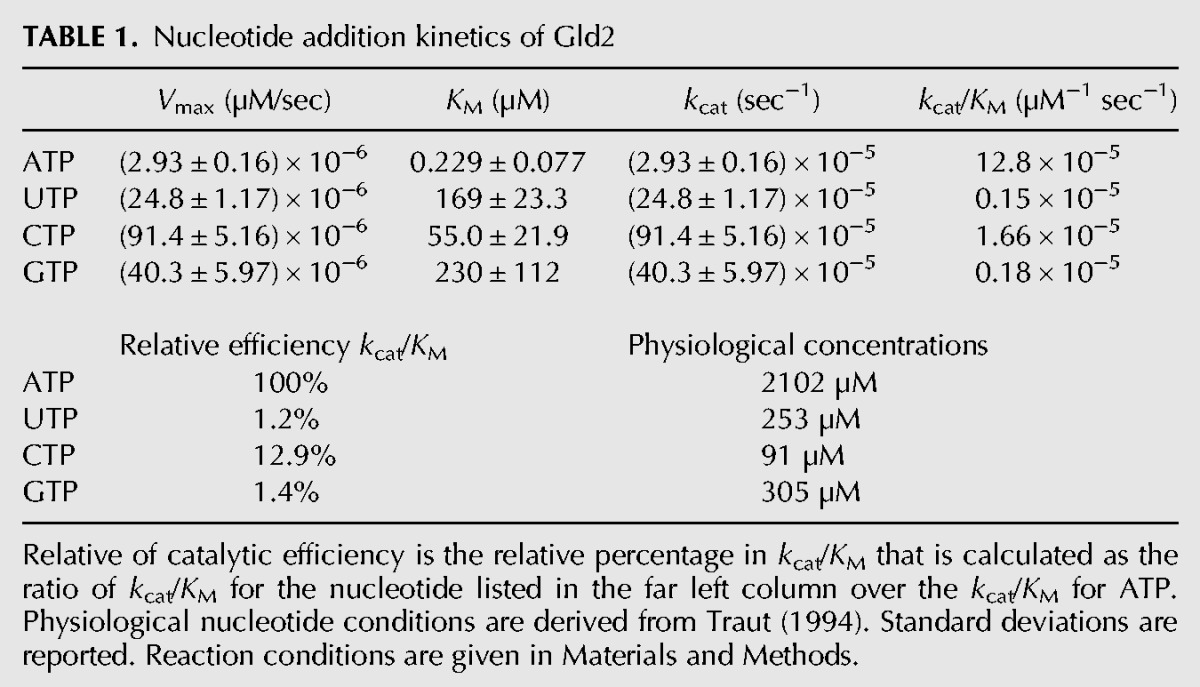
Our initial experiments showed that Gld2 catalyzes the addition of all four nucleotides to pre-let-7a (Fig. 3A). In the subsequent competition assay, a clear preference for ATP was observed (Fig. 3B). This led us to further investigate the nucleotide specificity of Gld2, and we performed a detailed kinetic characterization of Gld2 with all four nucleotides with let-7a (Fig. 4; Table 1). Our kinetic analysis confirmed that Gld2 displays the most affinity for ATP (KM ∼ 0.2 µM) and is most efficient with ATP. Significantly increased KM values for the other nucleotides, suggests far weaker binding affinity. Gld2 showed increases in KM of >700-fold for UTP, 240-fold for CTP, and >1000-fold for GTP. The relative catalytic efficiency for the reactions indicates an 83-fold preference of ATP over UTP and 71% over GTP. Overall catalytic efficiency is greatest for ATP (kcat/KM = 12.8 × 10−5 µM−1 sec−1) with 12.9% relative efficiency for CTP (kcat/KM = 1.66 × 10−5 µM−1 sec−1), and 1.2% for UTP (kcat/KM = 0.15 × 10−5 µM−1 sec−1), and 1.4% for GTP (kcat/KM = 0.18 × 10−5 µM−1 sec−1). Taken together, these data indicate that Gld2 NTP specificity is determined by productive binding of Gld2 to the respective nucleotide. While ATP is preferred and outcompetes all other NTPs, no nucleotide is specifically excluded from the active site.
FIGURE 4.
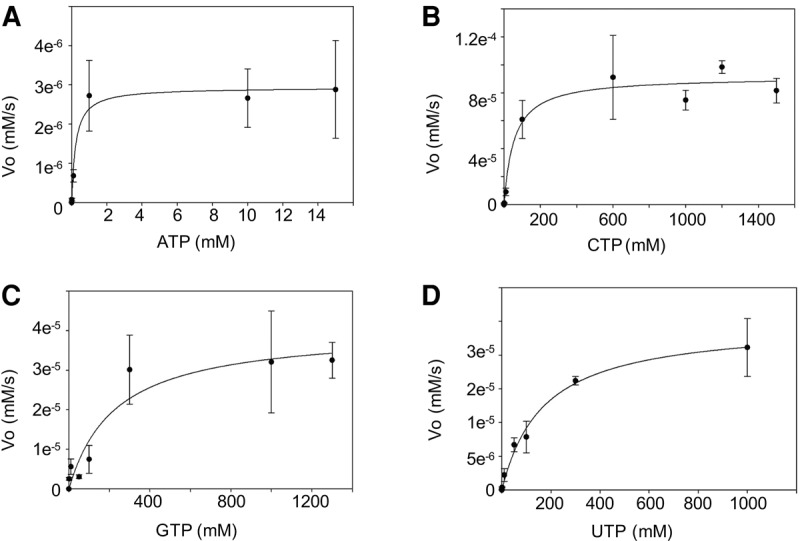
Dependence of the reaction rate on nucleotide concentration. The plot shows the initial velocity of the enzyme reaction plotted against concentration of ATP (A), CTP (B), GTP (C), and UTP (D). Errors show 1 SD.
TABLE 1.
Nucleotide addition kinetics of Gld2
Nucleotidyltransferase specificity arose multiple times during evolution
Gld2 is a member of the noncanonical poly(A) polymerases, a diverse group of enzymes with varying RNA and nucleotide preferences. It includes enzymes with nucleotide specificity ranging from strictly ATP to ambiguous ATP or UTP, to exclusive UTP adding enzymes. While some enzymes have a distinct substrate preference, such as the U6 snRNA uridylating enzyme TUT6 (Trippe et al. 2006; Mullen and Marzluff 2008), which is both UTP and U6 snRNA specific, other enzymes are more promiscuous in their substrate specificity. TUT4 and TUT7, for example, have been shown to uridylate miRNAs (Kwak and Wickens 2007; Heo et al. 2012), as well as histone mRNA (Schmidt et al. 2011) and cytoplasmic mRNA (Lim et al. 2014). The Schizosaccharomyces pombe homolog Cid1 protein was initially thought to act as an mRNA adenylating enzyme, but was later characterized as a uridylyltransferase with 1% residual adenylation activity (Rissland and Norbury 2008).
Gld2 was initially annotated as an adenylyltransferase, but recent in vivo (Heo et al. 2012) and in vitro (Kim and Richter 2006; D'Ambrogio et al. 2012; Heo et al. 2012) evidence and its sequence similarity to confirmed TUTs (30% and 32% amino acid sequence identity with TUT4 and TUT7, respectively) suggests a possible uridylyltransferase activity. The similar domain structure of uridyl and adenylyltransferases, as well as a high sequence similarity, suggests that these enzymes evolved from a common ancestor. To elucidate the phylogenetic background evolution of these enzymes, we performed a detailed phylogenetic analysis of the enzyme superfamily to trace the evolutionary origins of NTP specificity. The phylogeny of the nucleotidyltransferase family includes over 400 sequences (Fig. 5; Supplemental data file).
FIGURE 5.

Phylogeny of the TUTase/PAP superfamily. Gld2 is most closely related to the genuine uridylyltransferases TUT4/TUT7. Known enzymatic activities are color-coded. (Purple) UTP preference; (green) ATP preference; (black) unknown. Bootstrap values over 90% are denoted with a star. Sequence data were downloaded from the Integrated Microbial Genomes database. The tree was calculated with PHYML using a BioNJ starting tree and SPR tree search followed by NNI branch swapping to optimize the tree. Bootstrap values were computed according to the Shimodaira–Hasegawa reestimation of log-likelihood test implemented in PHYML.
Our analysis shows that nucleotidyltransferases display a classical star phylogeny, with several distinct subgroups. Interestingly, uridyl and adenylyltransferases do not form two separate clades, but rather TUTs emerge from distinct groups dominated by PAPs. Uridylyltransferases are derived from distinct subfamilies of adenylyltransferases, and TUTase activity, thus, evolved multiple times independently. One group shows the U6 snRNA uridylating enzyme TUT6 (Trippe et al. 2006) evolved from a parent clade composed of known and putative adenylyltransferases. The nucleotide specificity of TUT6 is, however, not restricted to snRNA uridylation, as it was additionally found to adenylate selected mRNAs (Mellman et al. 2008; Li et al. 2012). TUT6 is most closely related to the noncanonical mitochondrial poly(A) polymerase Papd1, which mediates RNA decay by polyadenylation (Bai et al. 2011). A second distinct subgroup includes the uridylyltransferase Cid1, which initiates RNA decay by uridylation (Rissland and Norbury 2009), and Cid14, a poly(A) polymerase that acts on rRNAs (Win et al. 2006) and telomeres (Bah et al. 2012).
Insertion of a histidine residue confers UTP specificity
Gld2 displays clear nucleotide specificity for ATP, as demonstrated by kinetic analysis of Gld2 activity on all four nucleotides and in competition experiments. Our phylogenetic analysis of the nucleotidyltransferase family suggests that uridylyltransferase activity evolved from adenylyltransferases multiple times during evolution (Fig. 5). Previous reports showed that the mutation of the S. pombe Cid1 uridylyltransferase active site histidine (H336, Fig. 6) to asparagine broadens its substrate specificity to include ATP (Lunde et al. 2012). In Gld2, the corresponding amino acid is lacking (Fig. 6A). Consequently, we tested whether an insertion of a histidine residue at the position homologous to Cid1 position H336 (between Gld2 amino acids T439 and N440 [Fig. 6B]) confers UTP specificity over ATP. Purified recombinant Gld2 and Gld2-His (Fig. 7A) were tested with RNA substrates let-7a (Fig. 7B) and 15A (Fig. 7C) and radiolabeled ATP or UTP. As expected, Gld2 displayed activity with ATP and UTP for both substrates in a noncompetitive assay (Fig. 7B panel 1, and 7C panel 1, Fig. 3A). The Gld2-His insertion variant, however, displayed significantly decreased activity with ATP. For let-7a, no activity was observed with ATP, while a band is visible when incubated with UTP. Similarly, we observed no band for the poly(A) tail mimic 15A when incubated with ATP, while incubation with UTP led to a ladder-like addition of nucleotides. In all cases, unlike UTP, ATP was excluded as a substrate. Thus, the insertion of a histidine residue homologous to Cid1 H336 conferred UTP selectivity in Gld2.
FIGURE 6.
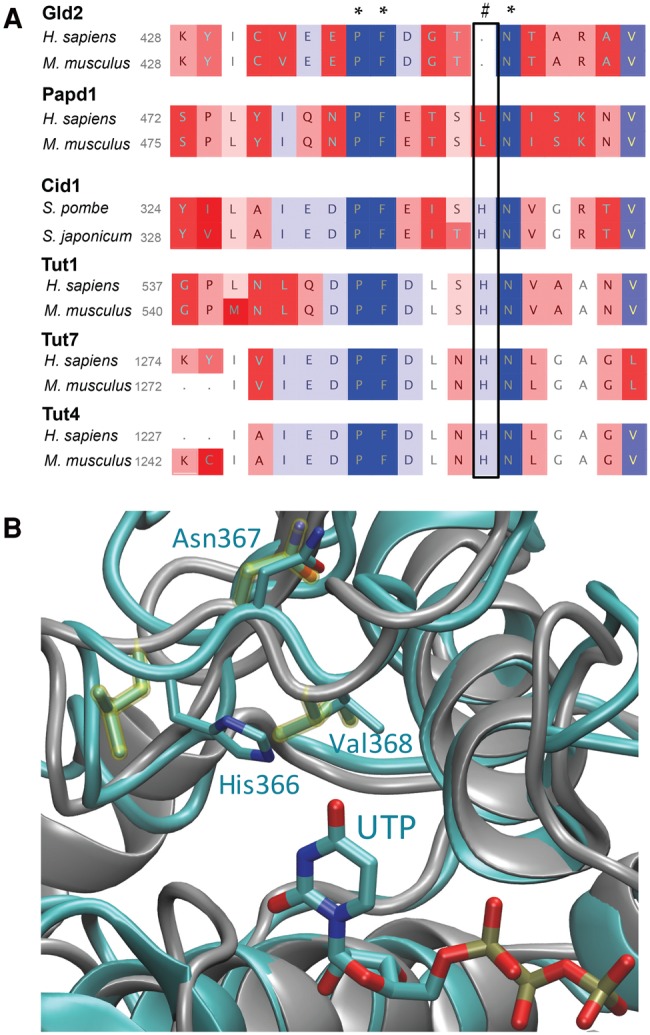
Nucleotide preference is defined by a histidine residue. (A) Excerpt from a multiple sequence alignment of 440 nucleotidyltransferase sequences. After the alignment sequences were grouped into phylogenetic subgroups and two representative sequences are depicted. Sequence similarities within each group were color-coded. Confirmed nucleotidyltransferases Cid1, TUT4, TUT6, and TUT7 and their homologs contain a histidine (#), which confers uridyl selectivity. Adenylyltransferases either substitute histidine with a smaller amino acid (leucine in Papd1) or entirely lack this residue (Gld2). (B) Excerpt of a structural alignment between Cid1 (pdb 4FHP) and Papd1 (pdb 3PQ1). The depicted ribbon diagram shows a structural superposition of human mitochondrial Papd1 (gray) and Cid1 (cyan). Asn367 of Cid1 aligns with the homologous Asn273 of Papd1. Cid1 His336 points into the active site and makes contact with UTP.
FIGURE 7.
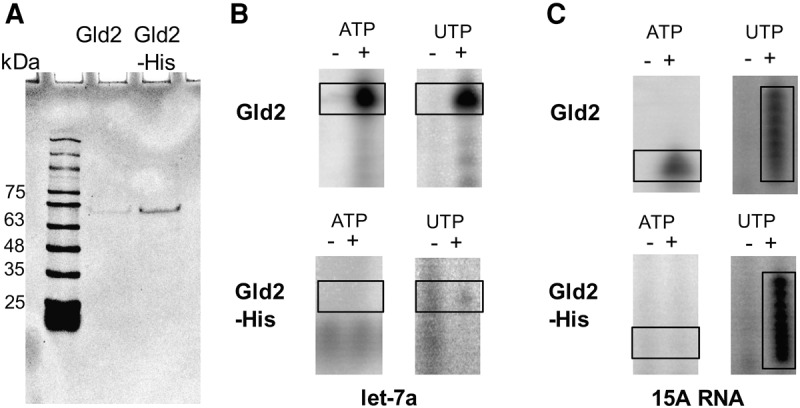
Insertion of an active site histidine into Gld2 alters nucleotide specificity. (A) Gld2 and Gld2-His purification. Recombinant His-tagged Gld2 and the Gld2-His mutant were purified via Ni-NTA chromatography to apparent homogeneity as judged by SDS-PAGE. (B,C) Nucleotide preference of Gld2 and Gld2-His. Recombinant enzymes were incubated with and without RNA substrates and radiolabeled ATP or UTP. Formation of α-32P labeled RNA products was monitored by electrophoretic separation and subsequent phosphorimaging. RNA substrates were let-7a (B) and 15A (C).
DISCUSSION
Gld2 is an adenylyltransferase
Previous studies presented evidence associating Gld2 with monoadenylation (D'Ambrogio et al. 2012) or monouridylation (Kim and Richter 2006; D'Ambrogio et al. 2012; Heo et al. 2012) of miRNAs in humans. While monoadenylation confers increased miRNA stability, monouridylation is a required step in biogenesis of group II miRNAs. During maturation, group II miRNAs are processed into pre-miRNAs with a single nucleotide 3′ overhang. TUT4, TUT7, and potentially Gld2 add an essential uridyl residue to 1-nt 3′ pre-miRNA to yield a 2-nt overhang, which is a prerequisite for processing by Dicer (Heo et al. 2012; Kim et al. 2015). In Xenopus laevis and Caenorhabditis elegans, Gld2 is required for polyadenylation of specific mRNAs, aided by RNA-binding proteins such as the CPEB and Gld3, respectively (Wang et al. 2002; Kim and Richter 2006).
To elucidate the nucleotide preference of human Gld2, we performed a detailed enzyme kinetic analysis. Using homogenous purified enzyme, we demonstrated that Gld2 is an adenylyltransferase preferentially adding single nucleotides to small RNAs. The catalytic efficiency of Gld2 is reduced 83-fold for UTP in comparison to ATP. Conversely, the uridylyltransferase Cid1 displays a 100-fold higher specificity for UTP over ATP (Lunde et al. 2012; Yates et al. 2012). Our data show a clear preference of ATP over all other nucleotides. The rate-limiting step is most likely the NTP-binding event. The KM for ATP is 0.23 µM, which is about 10,000-fold lower than the cellular ATP concentration of 2.1 mM (Traut 1994). Thus, Gld2 encounters a vast excess of ATP in the cell and will attain maximal substrate turnover. Cellular UTP, GTP, and CTP concentrations are overall lower than ATP concentrations (Traut 1994). In addition, we measured KM values in the 102 μM range for the other NTPs. Physiological concentrations for UTP and GTP are between 250 and 300 μM (Traut 1994), which is between one- and sixfold higher than the respective KM. The overall higher KM of Gld2 for UTP, CTP, and GTP combined with a much higher cellular ATP concentration further shifts enzymatic activity toward adenylation. Interestingly, at a CTP concentration 1000-fold in excess of the physiological concentration, the catalytic turnover for CTP is 30-fold higher than for ATP. Once NTP binding occurs, the other NTPs are ligated to the substrate RNA more rapidly than ATP (Table 1). Nevertheless, the low affinities of Gld2 for nucleotides other than ATP and the fact that the other NTPs fail to outcompete ATP (Fig. 3B) indicate that Gld2 activity is shifted to adenylation under physiological conditions.
Our data indicate that Gld2 may not have evolved to function exclusively with ATP, as we could show that Gld2 is active with UTP as well as GTP and CTP, albeit with low efficiency. While we cannot exclude that post-translational modifications or interactions with other proteins may influence nucleotide specificity, our data suggest that the observed NTP specificity is only in part determined by binding constants. Gld2 nucleotide specificity alone provides an 83-fold preference for ATP over UTP, which is combined with a cellular environment that has 10-fold excess of ATP compared to the other NTPs. Gld2 substrate preference and the cellular nucleotide concentrations together increase Gld2 selectivity toward ATP to >800-fold, enhancing the enzyme's specificity without evolving a precisely selective adenylyltransferase.
Gld2 monoadenylates small RNA substrates
In in vitro activity assays, Gld2 monoadenylates a variety of small RNAs, and does not specifically discriminate between different substrate RNAs. Our data show that Gld2 displays activity on tRNAs, miRNAs, pre-miRNAs, and mRNA alike, with a slight preference for miRNAs (Fig. 3). This agrees with earlier studies of X. laevis and C. elegans Gld2 homologs, which showed a role for Gld2 in miRNA metabolism (Kwak et al. 2004). The RNA substrate promiscuity that we observed with human Gld2 may be related to the fact that this minimal adenylyltransferase lacks RNA-binding domains, which are thought to confer substrate specificity in Gld2's closest human homologs, TUT4 and TUT7 (Fig. 1). Interestingly, we found no evidence of polyadenylation activity on any of the employed substrates. For processive polyadenylation, human Gld2 most likely requires the assistance of RNA binding proteins in vivo, which may confer specificity or activate elongation. Intriguingly, an extended incubation of Gld2 or Gld2-His with 15A RNA and UTP lead to a ladder-like addition of nucleotides (Fig. 7C). While UTP is not the natural Gld2 substrate, it is possible that polyuridylation, in contrast to polyadenylation, does not require accessory proteins.
Several studies from nonhuman Gld2 homologs show that in the presence of RNA-binding proteins such as CPEB (Kim and Richter 2006) and Gld3 (Wang et al. 2002) in X. laevis and C. elegans, respectively, Gld2 can processively add multiple adenine residues. To date, no such interaction has been shown for Gld2, but the fact that Gld2 alone hardly discriminates between several RNA substrates suggests the regulation of substrate specificity by additional RNA-binding proteins. While humans lack a Gld3 homolog, several CPEB homologs are encoded in the genome. In X. laevis, an additional poly(A) polymerase Gld4 polyadenylates p53 mRNA in a CPEB-dependent manner. In this case, Gld2 is not associated with CPEB directly, but regulates its expression via miR-122 adenylation (Burns et al. 2011). In C. elegans the RNA-binding protein Gld3 stimulates Gld2 catalyzed polyadenylation by increasing its affinity to the substrate RNA (Kwak et al. 2004). Similarly, TUT7 can be triggered to polyuridylate RNA substrates in association with the RNA-binding protein Lin28A (Hagan et al. 2009; Heo et al. 2009). Potential Gld2 associated proteins, however, remain to be identified.
Convergent evolution of TUTase activity by histidine insertion in the PAP active site
Recent biochemical and structural data on fission yeast Cid1 (Lunde et al. 2012; Yates et al. 2012) and X. laevis TUT7 (Lapointe and Wickens 2013) suggested that the nucleotide preference of nucleotidyltransferases is determined by a single histidine near the active site (Fig. 6A,B). In Cid1, histidine 336, which is located on a flexible loop near the catalytic site, sterically excludes ATP from the active site. Prior experiments have shown that mutation of this histidine to a smaller amino acid broadens nucleotide specificity in uridylyltransferases to include ATP, concluding that an asparagine to histidine mutation confers ATP specificity (Lunde et al. 2012; Yates et al. 2012; Lapointe and Wickens 2013). Our multiple sequence alignment (Fig. 6B; Supplemental File 1) and structural superposition of Cid1 and human mitochondrial Papd1 (Fig. 6B) show that the Asn337 in Cid1 is homologous to Asn273 residue of Papd1. This Asn is strictly conserved in all members of the nucleotidyltransferase superfamily. The structural superposition clearly shows that Cid1 His336 is an inserted residue relative to the Papd1 homolog. His336 points directly into the active site, making contact with the UTP substrate, while the Asn337/Asn273 residue is oriented away from the active site (Fig. 6B). The Asn conformation is structurally conserved between TUTases and PAPs. Consequently, UTP selectivity of Gld2, which is phylogenetically derived from Tut4/7 (Fig. 5), is conferred by a histidine insertion, rather than an Asn to His mutation as previously suggested (Lunde et al. 2012; Yates et al. 2012; Lapointe and Wickens 2013).
The phylogenetic analysis presented here shows that the presence/absence of the TUT-specific histidine is consistent within all nucleotidyltransferase groups and coincides with the divergence of uridylyltransferases from adenylyltransferases. While previous studies alluded to a point mutation from histidine (denoted with [#] in Fig. 6A) to asparagine (Lunde et al. 2012), conferring ATP over UTP selectivity, our sequence alignment clearly shows that the respective asparagine is part of a highly conserved motif (denoted with a [*] in Fig. 6A) found in all nucleotidyltransferases. Thus a histidine insertion rather than a mutation confers uridine specificity. In agreement with our biochemical data, the respective residue is absent from the Gld2 amino acid sequence, which vacates the active site for the larger ATP. Similarly, Cid14 is also lacking the respective histidine residue, conferring a preference of ATP over UTP. Consequently, a histidine insertion into Gld2 switched the nucleotide preference from ATP to UTP, excluding the larger ATP from the active site. Gld2-His shows no activity toward ATP, but is active on multiple substrates with UTP (Fig. 7B,C). These data clearly show that a single amino acid insertion can change nucleotide specificity in nucleotidyltransferases.
The human uridylyltransferases TUT4 and TUT7 are closely grouped within the phylogeny and are likely the result of recent gene duplication in the chordata linage. Consistent with their amino acid sequence similarity, Gld2 is most closely related to the TUT4/7 phylogenetic group, although Gld2 has the conserved TUTase histidine deleted. In adenylyltransferases, such as human Papd1 and its homologs, leucine replaces histidine. How a leucine insertion impacts enzyme activity and substrate specificity remains to be elucidated. Considering that uridylyltransferase activity has evolved multiple times, the flexible loop which harbors histidine 336 in Cid1 can be denoted as a preferred spot for mutations and insertions, allowing for facile alterations in substrate specificity.
The phylogeny, supported by our mutational analysis of Gld2, shows that uridylyltransferase activity diverged from adenylyltransferase activity multiple times during evolution and prior to the split of slime molds and bilateria. Interestingly, noncanonical nucleotidyltransferases are very prevalent in Fungi, which include several nucleotidyltransferase clades (e.g., Cid16, Cid11/13) not found in other organisms. These homologs appear to result from initial gene duplication, giving rise to Cid16 and Cid 11/13 groups, with a more recent duplication leading to the divergence of Cid11 and Cid13. The nuclear poly(A) polymerase Cid11 (Saitoh et al. 2002) and the Cid13 homolog encode an Arg, while Cid16 homologs a Lys in position His336 of Cid1. The biological function of these enzymes is not entirely clear, but the small positively charged side chain suggests a role in RNA adenylation, as ATP could still be spatially accommodated in the active site. One nucleotidyltransferase clade of entirely unknown function contains Papd1-like proteins with homologs in Drosophilidae and plants. No data are available on nucleotide specificity or biological function of these proteins. This group is diverse in its active site constituents. Interestingly, members of the Drosophilidae encode two Papd1-like homologs. One homolog encodes an arginine, and one a histidine, suggesting a recent gene duplication to allow for both uridylyltransferase and adenylyltransferase activity. These homologs could potentially share a specific RNA substrate range or interacting protein partners, while differing in nucleotide preference, thus fulfilling distinct biological functions.
MATERIALS AND METHODS
Gld2 cloning and site-directed mutagenesis
Total RNA was extracted from HeLa cells using the GeneJET RNA purification Kit (Thermo Scientific) and reverse transcription was performed with SuperScript II Reverse Transcriptase (Invitrogen) using an oligo(dT)16 primer. PCR was performed on the cDNA with gene specific primers (Gld2EcoR1for 5′-GAATTCGATGTTCCCAAACTCAATTTTGGG-3′ and Gld2Xho1rev 5′-CTCGAGTCTTTTCAGGACAGCAGCTC-3′) and the cDNA ligated into pET20b with EcoRI/XhoI. Quickchange Site-directed mutagenesis (Agilent) was used according to manufacturer's instructions to generate a Gld2-His insertion variant, using primers Tut2Hisfor: 5′-GAACCTTTTGATGGAACACATAATACAGCCAGAGCAGTGC; and Tut2Hisrev: 5′-GCACTGCTCTGGCTGTATTATGTGTTCCATCAAAAGGTTC. The construct and mutation were verified by DNA sequencing (Genewiz).
Gld2 expression and purification
Escherichia coli BL21 (DE3) RIL cells (Agilent) were transformed with pET20b-Gld2 and grown in LB medium containing ampicillin (100 µg/mL) and chloramphenicol (34 µg/mL) at 37°C until OD600 = 0.6. The temperature was lowered to 20°C and isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 250 µM to induce protein expression. Cells were harvested after 19 h by centrifugation and suspended in Buffer A (10 mM Tris–HCl, pH 8.0, 200 mM KCl, and 5 mM MgCl2). Cells were broken by the addition of lysozyme followed by sonication on ice. The cell lysate was centrifuged for 45 min at 15,000g and 4°C. Cell-free extract was loaded onto HisPur Ni-NTA resin (Thermo Scientific) equilibrated with Buffer A. The resin was washed with Buffer B (10 mM Tris–HCl, pH 8.0, 200 mM KCl, 5 mM MgCl2, and 10 mM imidazole) and proteins were eluted with Buffer C (10 mM Tris–HCl [pH 8.0], 200 mM KCl, 5 mM MgCl2, and 250 mM imidazole). The elution fractions containing Gld2 were dialyzed against Buffer D (10 mM Tris–HCl, pH 8.0, 200 mM KCl, 5 mM MgCl2, and 10% glycerol) overnight at 4°C with gentle mixing and stored at −80°C. Gld2 was purified to apparent homogeneity and the concentration determined by a Bradford test.
Size exclusion chromatography
A 200 µL sample containing Gld2 in Buffer A was passed through an ENrich SEC 650 high-resolution size exclusion column (Bio-Rad) equilibrated with Buffer A. The flow rate was 0.75 mL/min and 1 mL fractions were collected upon injection of the sample. The fractions that corresponded to the peaks on the absorbance graph were subjected to ammonium sulfate precipitation and analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on a 15% polyacrylamide gel. For the purposes of calibration, vitamin B12 (Mr ≈ 1.3 kDa), bovine insulin (Mr ≈ 6 kDa), cytochrome c (Mr ≈ 13.6 kDa) bovine carbonic anhydrase (Mr ≈ 30 kDa), ovalbumin (Mr ≈ 43 kDa), BSA (Mr ≈ 67 kDa), ferritin (Mr ≈ 440 kDa), and thyroglobulin (Mr ≈ 669 kDa) were used as marker components and chromatographed under identical conditions.
RNA substrate preparation
The following RNA substrates were purchased from Sigma-Aldrich: monophosphorylated human let-7a-5p [5′(p)-UGAGGUAGUAGGUUGUAUAGUU-3′], unphosphorylated human let-7a-0P (5′-UGAGGUAGUAGGUUGUAUAGUU-3′), diphosphorylated human let-7a-5p-2p [5′(pp)-UGAGGUAGUAGGUUGUAUAGUU-3′], triphosphorylated human let-7a-5p-3p [5′(ppp)-UGAGGUAGUAGGUUGUAUAGUU-3′], the poly(A) tail mimic 15ARNA [5′(p)-AAAAAAAAAAAAAAA-3′], human microRNA miR-122-5p [5′(p)-UGGAGUGUGACAAUGGUGUUUG-3′], and total E. coli tRNA. The coding sequence for a ribozyme-pre-let-7a with a T7 Promoter sequence was cloned into pUC19 for in vitro transcription with T7 RNA polymerase using the following primers (pre-let7 5′-ctagaTAATACGACTCACTATAGGGAGACTACTACCTCACTGATGAGTCCGTGAGGACGAAACGGTACCCGGTATAGGTTGTATAGTTTTAGGGTCACACCCACCACTGGGAGATAACTATACAATCTACTGTCTTTCGAA-3′ and pre-let-7rev 5′-ATCCTTCGAAAGACAGTAGATTGTATAGTTATCTCCCAGTGGTGGGTGTGACCCTAAAACTATACAACCTACTACGGGTACCGTTTCGTCCTCACGGACTCATCAGTGAGGTAGTAGTCTCCCTATAGTGAGTCGTATTAT-3′). Primers were phosphorylated, annealed, and cloned into pUC19 using XbaI/BamHI. For in vitro transcription, the DNA template was amplified using primers; the forward (5′-GTTGGGAAGGGCGATCGGTG-3′) and let-7PCRrev (5′-GAAAGACAGTAGATTGTATAG-3′). The PCR product was purified by phenol chloroform extraction, and RNA was transcribed with T7 RNA polymerase as described previously (Milligan et al. 1987). Upon transcription, the ribozyme constructs auto-cleaves into ribozyme and pre-let-7a. The transcripts were separated by electrophoresis on a 12% denaturing polyacrylamide gel, the band corresponding to self-cleaved pre-let-7a excised from the gel and eluted as described previously (Köhrer and Rajbhandary 2008).
Determination of enzymatic activity and substrate range
Ten microliter reactions were performed containing 100 nM Gld2 in Buffer D and 1 µM of the respective RNA substrates. Dithiothreitol (DTT) and MgCl2 were added for a final concentration of 1 mM and 3.2 mM, respectively. [α-32P]-UTP or [α-32P]-ATP (PerkinElmer) was used as indicated at a final concentration of 0.33 µM. All reactions were incubated at 37°C for 20 min and stopped by the addition of 2× RNA loading dye (95% v/v formamide, 0.1% w/v xylene xyanol, 0.1% w/w bromophenol blue, and 10 mM EDTA). Reactions were analyzed via gel electrophoresis on a 12% denaturing polyacrylamide gel and visualized with a phosphorimager (Storm 860 Molecular Imager). The radiolabeled RNA Decade marker (Ambion) was used as reference.
Determination of enzyme kinetics
Twenty microliter reactions were performed containing 100 nM Gld2 in Buffer D and 2 µM of let-7a. DTT and MgCl2 were added to a final concentration of 1 mM and 3.2 mM, respectively. Separate reactions contained one of the four NTPs at various concentrations. Three technical replicates were performed for each nucleotide concentration. Higher nucleotide concentrations were achieved by using a mixture of [α-32P]-labeled and unlabeled nucleotides. Nucleotide concentrations were adjusted so that in an 8-min time course the reaction progressed linearly and <10% of the total substrate RNA was converted to product. The following nucleotide concentrations were used: ATP 0–15 µM (0, 0.01, 0.1, 1, 10, and 15 µM); UTP 0–1000 µM (0, 0.01, 0.1, 1, 10, 50, 100, 300, and 1000 µM); GTP 0–1300 µM (0, 1, 10, 50, 100, 300, 1000, and 1300 µM); CTP 0–1500 µM (0, 1, 10, 100, 600, 1000, 1200, and 1500 µM). All reactions were incubated at 37°C for 30 sec before Gld2 was added (at t = 0) and 5 µL samples were then taken out at 2, 4, 6, and 8 min. The reactions were stopped with the addition of 2× RNA loading dye. Reactions were analyzed via gel electrophoresis on a 12% denaturing polyacrylamide gel and visualized by phosphorimaging overnight. To quantify product formation, a strip of Whatman filter paper was spotted with different known concentrations for each [α-32P]-labeled nucleotide and imaged on the same phosphorimaging screen. Spot intensity was quantified using ImageJ. Kinetic constants were derived from plotting initial velocity (vo) against nucleotide concentration. Kinetics were fitted to the standard Michaelis–Menten curve using Kaleida Graph 3.1 (Synergy Software) and SigmaPlot (Systat Software). Error bars represent 1 SD from three replicates.
Phylogenetic analysis
Sequences were downloaded from the National Center for Biotechnology Information (NCBI). Sequence alignment and alignment editing were performed using Muscle (Edgar 2004) and the Multiseq alignment editor from VMD 1.8.7 (Roberts et al. 2006). A maximum likelihood phylogeny for Thg1 sequences was determined using PhyML (Guindon and Gascuel 2003). The starting tree was generated with BioNJ, and the tree space was searched with the SPR followed by the NNI algorithm to find the best tree. The JTT+Γ model with four rate categories was applied. Likelihood parameters were initially estimated from the alignment, Shimodaira–Hasegawa bootstrap values were computed as implemented in PhyML.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada to I.U.H. (RGPIN 04776-2014) and a Seed Grant for CIHR success from The University of Western Ontario. We thank Julia Jaramillo, Mathura Vithyananthan, Riddhi Desai, and Kunmo Kim for their support, Susanna George, Nileeka Balasuria, and Yumin Bi for their advice, and Patrick O'Donoghue and Murray Junop for critical discussions.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.056077.116.
REFERENCES
- Ameres SL, Zamore PD. 2013. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol 14: 475–488. [DOI] [PubMed] [Google Scholar]
- Arasu P, Wightman B, Ruvkun G. 1991. Temporal regulation of lin-14 by the antagonistic action of two other heterochronic genes, lin-4 and lin-28. Genes Dev 5: 1825–1833. [DOI] [PubMed] [Google Scholar]
- Bah A, Wischnewski H, Shchepachev V, Azzalin CM. 2012. The telomeric transcriptome of Schizosaccharomyces pombe. Nucleic Acids Res 40: 2995–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Srivastava SK, Chang JH, Manley JL, Tong L. 2011. Structural basis for dimerization and activity of human PAPD1, a noncanonical poly(A) polymerase. Mol Cell 41: 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barh D, Malhotra R, Ravi B, Sindhurani P. 2010. MicroRNA let-7: an emerging next-generation cancer therapeutic. Curr Oncol 17: 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard DC, Ryan K, Manley JL, Richter JD. 2004. Symplekin and xGLD-2 are required for CPEB-mediated cytoplasmic polyadenylation. Cell 119: 641–651. [DOI] [PubMed] [Google Scholar]
- Burns DM, D'Ambrogio A, Nottrott S, Richter JD. 2011. CPEB and two poly(A) polymerases control miR-122 stability and p53 mRNA translation. Nature 473: 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AM, Ando Y, de Hoon MJ, Tomaru Y, Nishibu T, Ukekawa R, Funakoshi T, Kurokawa T, Suzuki H, Hayashizaki Y, et al. 2010. A comprehensive survey of 3′ animal miRNA modification events and a possible role for 3′ adenylation in modulating miRNA targeting effectiveness. Genome Res 20: 1398–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarata G, Augugliaro L, Salemi D, Agueli C, La Rosa M, Dagnino L, Civiletto G, Messana F, Marfia A, Bica MG, et al. 2010. Differential expression of specific microRNA and their targets in acute myeloid leukemia. Am J Hematol 85: 331–339. [DOI] [PubMed] [Google Scholar]
- Cui J, Sartain CV, Pleiss JA, Wolfner MF. 2013. Cytoplasmic polyadenylation is a major mRNA regulator during oogenesis and egg activation in Drosophila. Dev Biol 383: 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrogio A, Gu W, Udagawa T, Mello CC, Richter JD. 2012. Specific miRNA stabilization by Gld2-catalyzed monoadenylation. Cell Rep 2: 1537–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Leva G, Croce CM. 2013. miRNA profiling of cancer. Curr Opin Genet Dev 23: 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendler A, Jung K. 2013. MicroRNAs as new diagnostic and prognostic biomarkers in urological tumors. Crit Rev Oncog 18: 289–302. [DOI] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP. 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahder JA, Norrild B. 2011. Involvement of hGLD-2 in cytoplasmic polyadenylation of human p53 mRNA. APMIS 119: 769–775. [DOI] [PubMed] [Google Scholar]
- Grosshans H, Slack FJ. 2002. Micro-RNAs: small is plentiful. J Cell Biol 156: 17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52: 696–704. [DOI] [PubMed] [Google Scholar]
- Hagan JP, Piskounova E, Gregory RI. 2009. Lin28 recruits the TUTase Zcchc11 to inhibit let-7 maturation in mouse embryonic stem cells. Nat Struct Mol Biol 16: 1021–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo I, Joo C, Kim YK, Ha M, Yoon MJ, Cho J, Yeom KH, Han J, Kim VN. 2009. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell 138: 696–708. [DOI] [PubMed] [Google Scholar]
- Heo I, Ha M, Lim J, Yoon MJ, Park JE, Kwon SC, Chang H, Kim VN. 2012. Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell 151: 521–532. [DOI] [PubMed] [Google Scholar]
- Kim JH, Richter JD. 2006. Opposing polymerase-deadenylase activities regulate cytoplasmic polyadenylation. Mol Cell 24: 173–183. [DOI] [PubMed] [Google Scholar]
- Kim B, Ha M, Loeff L, Chang H, Simanshu DK, Li S, Fareh M, Patel DJ, Joo C, Kim VN. 2015. TUT7 controls the fate of precursor microRNAs by using three different uridylation mechanisms. EMBO J 34: 1801–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Salomon C, Tapia J, Illanes SE, Mitchell MD, Rice GE. 2014. Ovarian cancer cell invasiveness is associated with discordant exosomal sequestration of Let-7 miRNA and miR-200. J Transl Med 12: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhrer C, Rajbhandary UL. 2008. The many applications of acid urea polyacrylamide gel electrophoresis to studies of tRNAs and aminoacyl-tRNA synthetases. Methods 44: 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. 2014. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42: D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak JE, Wickens M. 2007. A family of poly(U) polymerases. RNA 13: 860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak JE, Wang L, Ballantyne S, Kimble J, Wickens M. 2004. Mammalian GLD-2 homologs are poly(A) polymerases. Proc Natl Acad Sci 101: 4407–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak JE, Drier E, Barbee SA, Ramaswami M, Yin JC, Wickens M. 2008. GLD2 poly(A) polymerase is required for long-term memory. Proc Natl Acad Sci 105: 14644–14649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe CP, Wickens M. 2013. The nucleic acid-binding domain and translational repression activity of a Xenopus terminal uridylyl transferase. J Biol Chem 288: 20723–20733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. 1993. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854. [DOI] [PubMed] [Google Scholar]
- Lee M, Choi Y, Kim K, Jin H, Lim J, Nguyen TA, Yang J, Jeong M, Giraldez AJ, Yang H, et al. 2014. Adenylation of maternally inherited microRNAs by Wispy. Mol Cell 56: 696–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Laishram RS, Ji Z, Barlow CA, Tian B, Anderson RA. 2012. Star-PAP control of BIK expression and apoptosis is regulated by nuclear PIPKIα and PKCδ signaling. Mol Cell 45: 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Ha M, Chang H, Kwon SC, Simanshu DK, Patel DJ, Kim VN. 2014. Uridylation by TUT4 and TUT7 marks mRNA for degradation. Cell 159: 1365–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde BM, Magler I, Meinhart A. 2012. Crystal structures of the Cid1 poly (U) polymerase reveal the mechanism for UTP selectivity. Nucleic Acids Res 40: 9815–9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman DL, Gonzales ML, Song C, Barlow CA, Wang P, Kendziorski C, Anderson RA. 2008. A PtdIns4,5P2-regulated nuclear poly(A) polymerase controls expression of select mRNAs. Nature 451: 1013–1017. [DOI] [PubMed] [Google Scholar]
- Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. 1987. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res 15: 8783–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moitra K. 2015. Overcoming multidrug resistance in cancer stem cells. Biomed Res Int 2015: 635745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen TE, Marzluff WF. 2008. Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5′ to 3′ and 3′ to 5′. Genes Dev 22: 50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulrane L, McGee SF, Gallagher WM, O'Connor DP. 2013. miRNA dysregulation in breast cancer. Cancer Res 73: 6554–6562. [DOI] [PubMed] [Google Scholar]
- Norbury CJ. 2013. Cytoplasmic RNA: a case of the tail wagging the dog. Nat Rev Mol Cell Biol 14: 643–653. [DOI] [PubMed] [Google Scholar]
- Rissland OS, Norbury CJ. 2008. The Cid1 poly(U) polymerase. Biochim Biophys Acta 1779: 286–294. [DOI] [PubMed] [Google Scholar]
- Rissland OS, Norbury CJ. 2009. Decapping is preceded by 3′ uridylation in a novel pathway of bulk mRNA turnover. Nat Struct Mol Biol 16: 616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissland OS, Mikulasova A, Norbury CJ. 2007. Efficient RNA polyuridylation by noncanonical poly(A) polymerases. Mol Cell Biol 27: 3612–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts E, Eargle J, Wright D, Luthey-Schulten Z. 2006. MultiSeq: unifying sequence and structure data for evolutionary analysis. BMC Bioinformatics 7: 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh S, Chabes A, McDonald WH, Thelander L, Yates JR, Russell P. 2002. Cid13 is a cytoplasmic poly(A) polymerase that regulates ribonucleotide reductase mRNA. Cell 109: 563–573. [DOI] [PubMed] [Google Scholar]
- Sartain CV, Cui J, Meisel RP, Wolfner MF. 2011. The poly(A) polymerase GLD2 is required for spermatogenesis in Drosophila melanogaster. Development 138: 1619–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MJ, West S, Norbury CJ. 2011. The human cytoplasmic RNA terminal U-transferase ZCCHC11 targets histone mRNAs for degradation. RNA 17: 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small EM, Olson EN. 2011. Pervasive roles of microRNAs in cardiovascular biology. Nature 469: 336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Lai EC. 2013. Adult-specific functions of animal microRNAs. Nat Rev Genet 14: 535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RU, Miyazaki H, Ochiya T. 2014. The role of microRNAs in the regulation of cancer stem cells. Front Genet 4: 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton JE, Chang HM, Piskounova E, Gregory RI. 2012. Lin28-mediated control of let-7 microRNA expression by alternative TUTases Zcchc11 (TUT4) and Zcchc6 (TUT7). RNA 18: 1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton JE, Du P, Jing L, Sjekloca L, Lin S, Grossi E, Sliz P, Zon LI, Gregory RI. 2014. Selective microRNA uridylation by Zcchc6 (TUT7) and Zcchc11 (TUT4). Nucleic Acids Res 42: 11777–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traut TW. 1994. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem 140: 1–22. [DOI] [PubMed] [Google Scholar]
- Trippe R, Guschina E, Hossbach M, Urlaub H, Lührmann R, Benecke BJ. 2006. Identification, cloning, and functional analysis of the human U6 snRNA-specific terminal uridylyl transferase. RNA 12: 1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ree MH, van der Meer AJ, van Nuenen AC, de Bruijne J, Ottosen S, Janssen HL, Kootstra NA, Reesink HW. 2016. Miravirsen dosing in chronic hepatitis C patients results in decreased microRNA-122 levels without affecting other microRNAs in plasma. Aliment Pharmacol Ther 43: 102–113. [DOI] [PubMed] [Google Scholar]
- Wang L, Eckmann CR, Kadyk LC, Wickens M, Kimble J. 2002. A regulatory cytoplasmic poly(A) polymerase in Caenorhabditis elegans. Nature 419: 312–316. [DOI] [PubMed] [Google Scholar]
- Win TZ, Draper S, Read RL, Pearce J, Norbury CJ, Wang SW. 2006. Requirement of fission yeast Cid14 in polyadenylation of rRNAs. Mol Cell Biol 26: 1710–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman SK, Knouf EC, Parkin RK, Fritz BR, Lin DW, Dennis LM, Krouse MA, Webster PJ, Tewari M. 2011. Post-transcriptional generation of miRNA variants by multiple nucleotidyl transferases contributes to miRNA transcriptome complexity. Genome Res 21: 1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates LA, Fleurdépine S, Rissland OS, De Colibus L, Harlos K, Norbury CJ, Gilbert RJ. 2012. Structural basis for the activity of a cytoplasmic RNA terminal uridylyl transferase. Nat Struct Mol Biol 19: 782–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates LA, Norbury CJ, Gilbert RJ. 2013. The long and short of microRNA. Cell 153: 516–519. [DOI] [PubMed] [Google Scholar]
- Zheng H, Liu JY, Song FJ, Chen KX. 2013. Advances in circulating microRNAs as diagnostic and prognostic markers for ovarian cancer. Cancer Biol Med 10: 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XM, Wu LJ, Xu J, Yang R, Wu FS. 2011. Let-7c microRNA expression and clinical significance in hepatocellular carcinoma. J Int Med Res 39: 2323–2329. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.