

Abstract
Herpes simplex virus (HSV) 1 stimulates type I IFN expression through the cGAS–STING–TBK1 signaling axis. Macrophages have recently been proposed to be an essential source of IFN during viral infection. However, it is not known how HSV‐1 inhibits IFN expression in this cell type. Here, we show that HSV‐1 inhibits type I IFN induction through the cGAS–STING–TBK1 pathway in human macrophages, in a manner dependent on the conserved herpesvirus protein ICP27. This viral protein was expressed de novo in macrophages with early nuclear localization followed by later translocation to the cytoplasm where ICP27 prevented activation of IRF3. ICP27 interacted with TBK1 and STING in a manner that was dependent on TBK1 activity and the RGG motif in ICP27. Thus, HSV‐1 inhibits expression of type I IFN in human macrophages through ICP27‐dependent targeting of the TBK1‐activated STING signalsome.
Keywords: herpes simplex virus, immune evasion, innate immunity, type I IFN
Subject Categories: Immunology
Introduction
Herpes simplex virus 1 (HSV‐1) is a human pathogenic herpesvirus belonging to the subfamily of Alphaherpesvirinae. HSV‐1 is widely distributed in human populations and capable of triggering a broad range of pathologies, ranging from the benign cold sores to fatal encephalitis. Primary HSV‐1 infections are initiated by lytic viral replication in mucosal epithelial cells, and from this site, the virus enters into sensory neurons, where a lifelong latent–recurrent infection is established (Roizman et al, 2013). It is well established that the immune status of the host is a central determinant of the clinical outcome of HSV‐1 infections (Paludan et al, 2011).
The innate immune system is the first line of defense against pathogens, including HSV‐1 (Paludan et al, 2011). This arm of the immune system exerts antiviral activity primarily through type I interferons (IFN)s and natural killer cells. In the case of HSV‐1 infections, recent work has established that impaired ability to stimulate type I IFN expression strongly enhances susceptibility to herpes simplex encephalitis (Zhang et al, 2007; Perez de Diego et al, 2010; Sancho‐Shimizu et al, 2011; Herman et al, 2012; Andersen et al, 2015). Crucial for the activation of the antiviral program is the recognition of pathogen‐associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs) (Akira et al, 2006; Paludan & Bowie, 2013). Both RNA and DNA have been reported to stimulate IFN production during HSV‐1 infections. The dsRNA‐sensing TLR3 pathway is central for mounting full IFN responses in different cell types in the CNS, and genetic defects in this pathway lead to susceptibility to herpes simplex encephalitis (Zhang et al, 2007; Perez de Diego et al, 2010; Sancho‐Shimizu et al, 2011; Herman et al, 2012; Andersen et al, 2015). Herpesviral DNA and mitochondrial DNA released in response to infection‐induced stress are detected by the nucleotidyltransferase cGAS in a sequence‐independent fashion (Sun et al, 2013; Wu et al, 2013; Rongvaux et al, 2014; West et al, 2015). DNA sensing stimulates signaling dependent on the adaptor protein stimulator of interferon genes (STING) (Ishikawa et al, 2009). Experiments in mice have shown that the lack of cGAS or STING renders mice highly susceptible to HSV‐1 infections (Ishikawa et al, 2009; Li et al, 2013b). At the mechanistic level, DNA sensing by cGAS leads to production of cyclic GMP‐AMP (cGAMP), which docks on STING to induce a conformational change allowing recruitment of the kinase TANK‐binding kinase 1 (TBK1). This in turn allows recruitment of the transcription factor IFN regulatory factor 3 (IRF3) to the phosphorylated surface of STING, where IRF3 is phosphorylated and activated in a TBK1‐dependent manner, to stimulate IFN‐β gene transcription (Tanaka & Chen, 2012). Interestingly, the phosphorylation of the adaptor protein creates a positively charged surface to which IRF3 can bind. This is a property conserved among all of the three main IFN‐stimulating adaptors in innate immunity, namely STING, TRIF, and MAVS (Liu et al, 2015).
Because viruses have co‐evolved with their hosts, they have gained mechanisms to evade and suppress host immune responses. Different proteins of HSV‐1 are described to target immunological pathways, including the MHC pathway by the infected cell protein (ICP) 47 (York et al, 1994; Oldham et al, 2016). With respect to viral evasion of the type I IFN pathway, most work has concentrated on the viral E3 ubiquitin ligase‐infected cell protein (ICP)0. HSV‐1 deficient for ICP0 induces enhanced type I IFN levels in in vitro cell systems, compared to wild‐type (WT) virus. At the mechanistic level, ICP0 has been reported to target the DNA sensor IFI16 for degradation (Orzalli et al, 2012), but also to employ other mechanisms (Cuchet‐Lourenco et al, 2013). Other HSV‐1 proteins reported to interfere with induction of type I IFN expression include VP16, Us11, ICP34.5, and ICP27 (Melchjorsen et al, 2006; Verpooten et al, 2009; Xing et al, 2012, 2013). Moreover, Vhs, ICP0, and ICP27 have also been reported to inhibit type I IFN‐stimulated signaling through the Jak‐STAT pathway (Mossman et al, 2000; Suzutani et al, 2000; Johnson et al, 2008). ICP27 is a multifunctional protein that is essential for HSV‐1 replication (Rice & Knipe, 1990). The essential function of the protein is supported by the fact that ICP27 is conserved among all herpesviruses (Sandri‐Goldin, 2011). The described functions of ICP27 in the viral replication cycle include transport of intronless HSV‐1 mRNA transcripts from the nucleus to the cytosol in a manner where ICP27 favors the transport of viral transcripts over host mRNAs (Koffa et al, 2001; Chen et al, 2005; Tian et al, 2013). However, the mechanistic basis for inhibition of innate immune responses by ICP27 remains poorly defined.
In recent years, it has emerged that macrophages constitute a central source of type I IFN during infections with viruses, including HSV‐1, and that IFN from this source contributes significantly to antiviral immunity (Eloranta & Alm, 1999; Kumagai et al, 2007; Goritzka et al, 2015). Despite this, there is no mechanistic information on how HSV‐1 evades the type 1 IFN response in human macrophages. In this work, we report that although only very limited productive replication occurs in HSV‐1‐infected human macrophages, ICP27 is expressed and the protein is essential for counteracting the production of type I IFNs. ICP27, which is highly conserved within the Simplexvirus genera of the Herpesvirinae, translocates from the nucleus to the cytoplasm during infection, where it interacts with the activated TBK1–STING signalsome, thus inhibiting IRF3 activation through the cGAS–STING pathway. Our work reveals a novel mechanism of viral inhibition of IFN expression. Moreover, the work demonstrates that viruses must have a broad repertoire of operative immune evasion strategies to counteract host responses even in cell types not supporting productive replication.
Results
HSV‐1 harbors a mechanism to inhibit type I IFN expression, which is dependent on viral gene expression but independent of ICP0
To evaluate the abilities of different HSV‐1 strains to stimulate type I IFN expression, we infected PMA‐differentiated macrophage‐like THP1 cells with four different strains of HSV‐1. Accumulation of type I IFN bioactivity was measured in supernatants harvested at different time points. As shown in Fig 1A, all strains tested induced a modest but significant IFN response at 12 h post‐infection. In supernatants from cells infected with the KOS (or KOS1.1), 17+, and McKrae strains, the IFN bioactivity peaked at the 12 h time point. However, infection with the F strain led to a pronounced further increase in stimulation of IFN bioactivity between 12 and 18 h of infection. The viruses did not affect viability of the cells within the time frame of the experiment (data not shown). These data led us to hypothesize that HSV‐1 harbors a mechanism to inhibit type I IFN expression, which is dependent on viral gene expression, and not activated by the F strain. It should be noted that alternative interpretations of these data cannot formally be excluded, including differential accumulation of non‐productive viral DNA replication intermediates by the HSV‐1 strains being the cause of differential IFN responses. To start testing the hypothesis of viral evasion of IFN expression, we first evaluated whether inhibition of viral replication could augment induction of IFN production by the KOS strain. Interestingly, the blockage of viral replication by UV treatment of virus or acyclovir enabled the KOS strain to induce significantly more IFN than the replication competent virus, whereas the replication capacity of the F strain did not impact on the IFN‐inducing potential (Fig 1B and C, and Appendix Fig S1A and data not shown).
Figure 1. HSV‐1 harbors a mechanism to inhibit type I IFN expression, which is dependent on viral gene expression but independent of ICP0.
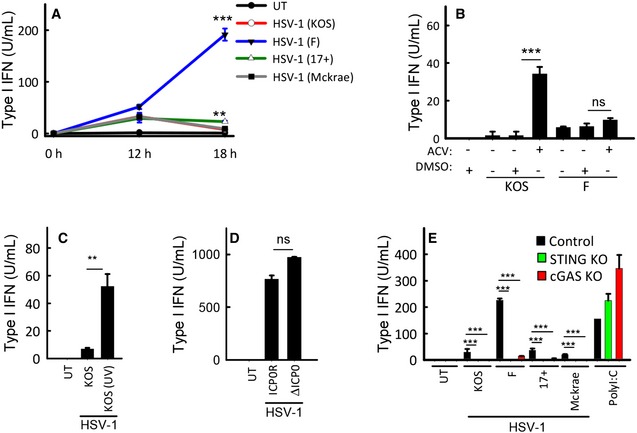
- THP1 cells were infected with the shown strains of HSV‐1 (MOI 3). Supernatants were harvested from untreated cultures or cells infected for 12 or 18 h for measurement of type I IFN bioactivity.
- THP1 cells were treated with 0.1 μg/ml of acyclovir (ACV) and infected with the KOS and F HSV‐1 strains (MOI 3). Supernatants were harvested 18 hpi for measurement of type I IFN bioactivity.
- THP1 cells were treated with infectious or UV‐inactivated HSV‐1 (strain KOS). Supernatants were harvested 18 hpi for measurement of type I IFN bioactivity.
- MDMs were infected with ICP0‐deficient or revertant HSV‐1 (strain KOS, MOI 3). Supernatants were harvested 18 hpi for measurement of type I IFN bioactivity.
- THP1‐derived cells deficient for cGAS or STING were infected with the shown strains of HSV‐1 (MOI 3) or stimulated with poly(I:C) (2 μg/ml). Supernatants were harvested 18 hpi for measurement of type I IFN bioactivity.
HSV‐1 ICP0 is an ubiquitin E3 ligase, which has been reported to be central for viral evasion of type I IFN responses in human fibroblasts (Mossman et al, 2001; Eidson et al, 2002; Orzalli et al, 2012, 2013). We confirmed the previously published data by the finding that supernatants from human foreskin fibroblasts (HFF)s infected with HSV‐1 7134 ICP0‐deficient KOS‐derived HSV‐1 did indeed lead to strongly elevated IFN induction (Appendix Fig S1B). However, there is no information on the role of ICP0 in the inhibition of innate responses in human macrophages. Therefore, we infected primary human monocyte‐derived macrophages (MDM)s with the HSV‐1 7134 ICP0‐deficient mutant virus as well as the 7134R rescued virus. In contrast to what was seen in HFFs, lack of ICP0 did not affect the ability of the virus to induce type I IFN production (Fig 1D). Likewise, ICP0 deficiency did not affect HSV‐1‐induced expression of IFN‐α or IFN‐β mRNA in peripheral blood mononuclear cells (Appendix Fig S1C and D). Finally, we were interested in evaluating whether the elevated IFN induction by the HSV‐1F strain proceeded through the same innate immune pathways as for the other strains tested. Therefore, CRISPR/Cas9‐generated THP1‐derived cells lacking cGAS or STING were infected with different virus strains and IFN bioactivity in the supernatants was measured. For all virus strains, the stimulated IFN bioactivity was abrogated in cells lacking cGAS or STING (Fig 1E). Collectively, these data demonstrate that HSV‐1 harbors a mechanism to inhibit type I IFN expression, which is dependent on viral gene expression, but independent of ICP0. This viral immune evasion strategy targets component(s) in the cGAS–STING pathway.
Expression of HSV‐1 ICP27 in macrophages correlates inversely with induction of type I IFN
Given the dependence on viral replication products for inhibition of type I IFN production, we used quantitative MS‐based proteomics to map the HSV‐1 proteome in the cytoplasm of infected MDMs. Altogether, we identified 17 of the 77 annotated HSV‐1 proteins. Among the viral proteins identified, 10 were virion proteins and 2 were involved in viral assembly and egress. In addition, we identified three proteins with functions in viral genome replication, and finally, the 2 proteins ICP4 and ICP27 involved in early viral gene expression (Fig 2A and Table S1). While ICP4 is mainly described to regulate viral transcription, ICP27 is a multifunctional protein with many identified protein interaction partners (Sandri‐Goldin, 2011). The expression of ICP27 in HSV‐1‐infected macrophages was confirmed by Western blotting on lysates from MDMs, and interestingly, we observed lower expression of ICP27 in cells infected with the F strain as compared to the KOS strain at 12 hpi (Fig 2B). Despite the detectable expression of viral proteins in MDMs, the virus was not able to produce progeny virus in this cell type (Fig 2C), potentially suggesting alternative functions for HSV‐1 proteins in MDMs. We therefore compared expression of ICP27 in THP1 cells after infection with the four HSV‐1 strains evaluated for IFN stimulation in Fig 1A. Interestingly, the KOS and McKrae strains, which induced less type I IFN after 18 h of infection, stimulated strong ICP27 expression, whereas infection with the 17+ strain led to a modest but clear expression of ICP27 (Fig 2D). In contrast to this, the F strain did not express ICP27 in the THP1 cells. Thus, ICP27 is part of a small subset of non‐structural HSV‐1 proteins expressed during infection in macrophages, and its expression correlated inversely with expression of type I IFNs.
Figure 2. HSV‐1 does not replicate productively in macrophages, but ICP27 expression correlates inversely with type I IFN production.
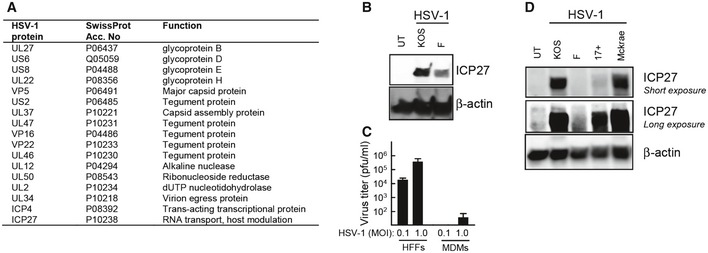
- MDMs were infected for 18 h with HSV‐1 (KOS, MOI 1). Cytosolic viral proteins were detected by mass spectrometry using iTRAQ labeling. Identified viral proteins are shown.
- MDMs were infected with the KOS or F strains of HSV‐1 (MOI 3). Cell lysates were harvested 12 hpi, and levels of ICP27 and β‐actin were determined by Western blotting.
- HFFs and MDMs were infected with HSV‐1 (KOS, MOI 0.1, and 1.0). Culture medium was replaced 3 hpi and isolated 24 hpi for viral plaque assay. Data are presented as means of triplicates ± SD.
- THP1 cells were infected with the indicated strains of HSV‐1 (MOI 3) for 12 h. Cell lysates were isolated, and levels of ICP27 and β‐actin were determined by Western blotting.
Source data are available online for this figure.
HSV‐1 ICP27 is a negative regulator of IFN induction through the cGAS–STING pathway in macrophages
It has been reported that the strain F has 961‐bp changes relative to the strain 17+, resulting in 310 amino acid differences between strains F and 17+, affecting 63 out of 77 viral proteins (Szpara et al, 2010). Thus, given the difference between HSV‐1 strains, we further wanted to test ICP27 as a candidate immune evasion protein. First, to evaluate the role of ICP27 in modulation of the type I IFN response in human macrophages, we measured type I IFN bioactivity in supernatants from THP1 cells infected with either WT or ICP27‐deficient KOS. At 12 h post‐infection, both WT and ICP27‐deficient virus induced modest but significant IFN production, with the ICP27‐deficient strain inducing slightly more type I IFN than the WT strain (Fig 3A). However, while the IFN levels in supernatants from cells infected with the WT strain decreased after the 12‐h time point, the IFN levels in supernatant of cells infected with ICP27‐deficient virus increased very strongly in this time interval (Fig 3A). This was also seen when measuring levels of the IFN‐stimulated gene CXCL10 (Appendix Fig S2A). Similar to the observation in THP1 cells, ICP27‐deficient HSV‐1 also stimulated type I IFN bioactivity to much higher levels than WT virus in MDMs (Fig 3B). The WT and ICP27‐deficient viruses did not affect cell viability within the time frame of the experiments (data not shown). The potent induction of type I IFNs by the ICP27‐deficient mutant was not limited to human macrophages, because it was also seen in HFFs and murine macrophages (Appendix Fig S2B and C). The observed elevated IFN induction by ICP27‐deficient HSV‐1 was unlikely to be explained merely by the impairment of the replication cycle, since both ICP0‐deficient and ICP27‐deficient viruses failed to induce expression of the major capsid protein VP5 (Appendix Fig S2D), but only the latter virus induced elevated levels of IFN. To examine whether the elevated IFN response induced by the ICP27‐deficient virus proceeded through the cGAS–STING pathway, THP1‐derived KO cell lines were infected with HSV‐1 KOS or ΔICP27 virus. Measurement of IFN bioactivity in the supernatants revealed that the elevated IFN production in cells infected with the ΔICP27 virus was dependent on cGAS and STING (Fig 3C), thus suggesting that ICP27 is required for targeting of the cGAS–STING pathway in the context of HSV‐1 infection. To investigate whether ICP27 is sufficient to inhibit the cGAS–STING pathway, HEK293T cells were transfected with STING, ICP27, and an ISRE‐driven luciferase promoter. STING transfection alone stimulated luciferase expression, but ICP27 strongly inhibited this response (Fig 3D).
Figure 3. ICP27 inhibits the cGAS–STING pathway in macrophages.
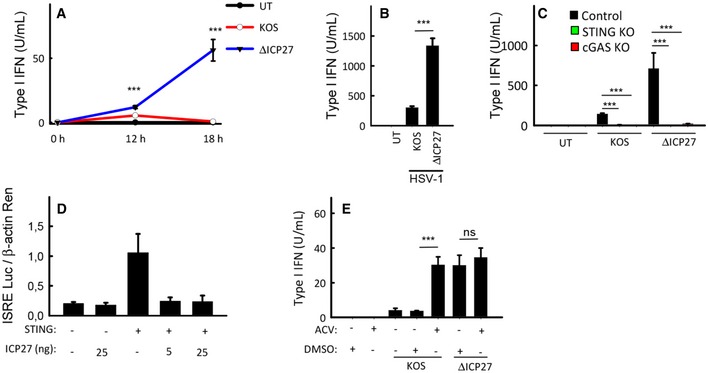
- THP1 cells were infected with HSV‐1 KOS or an ICP27‐deficient mutant (ΔICP27) on KOS genetic background (MOI 3). Supernatants were harvested from untreated cultures or cells infected for 12 or 18 h for measurement of type I IFN bioactivity.
- MDMs were infected with HSV‐1 KOS or ΔICP27 (MOI 3). Supernatants were harvested 18 hpi for measurement of type I IFN bioactivity.
- THP1‐derived cells deficient for cGAS or STING were infected with HSV‐1 KOS or ΔICP27 (MOI 3). Supernatants were harvested 18 hpi for measurement of type I IFN bioactivity.
- HEK293T cells were transfected with 12 ng STING plasmid DNA, 5 or 25 ng ICP27 plasmid DNA, and reporter gene constructs as indicated (2 × 104 cells per well). Reporter gene activity was measured in lysates isolated 24 h post‐transfection.
- THP1 cells were infected with HSV‐1 KOS or ΔICP27 (MOI 3) in the presence or absence of acyclovir (ACV). Supernatants were isolated 18 hpi for measurement of type I IFN bioactivity.
ICP27 is an immediate‐early protein, but is also produced at late time points during infection, and reported to be packed into viral particles as a tegument protein (Maringer & Elliott, 2010). Western blotting of viral stocks confirmed the presence of ICP27 in virions (Appendix Fig S2E). Therefore, to examine whether ICP27 was responsible for the replication‐dependent nature of the evasion of IFN responses, we treated cells with acyclovir prior to infection with WT or ΔICP27 HSV‐1. Acyclovir treatment inhibited late but not early ICP27 expression in THP1 cells (Appendix Fig S2F), thus suggesting early ICP27 expression in macrophages to occur as an immediate‐early or gene, and the late ICP27 expression to exhibit features of a late gene. As also observed in Fig 1B, inhibition of viral replication enabled WT HSV‐1 to induce high levels of IFN (Fig 3E). By contrast, for the ΔICP27 virus, which induced much more IFN than the WT virus, IFN levels were not affected by acyclovir treatment. Collectively, these data suggest that de novo produced ICP27 targets the STING pathway in immortalized and primary cells to inhibit production of type I IFN.
ICP27 inhibits the cGAS–STING pathway downstream of TBK1 phosphorylation but upstream of IRF3 phosphorylation
The ICP27 protein is reported to have specific functions in both nuclear and cytosolic compartments, enabled through a shuttling mechanism, which is independent of other HSV‐I proteins (Mears & Rice, 1998). To start characterization of the mechanism through which ICP27 inhibits the cGAS–STING pathway, we first determined the subcellular distribution of ICP27 in macrophages and its development over time. Cytosolic and nuclear fractions of KOS‐stimulated THP1 cells were analyzed by Western blot. ICP27 was found to be expressed in the nucleus within the first hours of infection and started also to accumulate in the cytosol after ~8 h (Fig 4A). A similar pattern was observed by confocal microscopy (Fig 4B). Next, we investigated how the cellular localization of ICP27 affects the inhibition of IFN expression. For this purpose, we used mutated HSV‐1 virus strains, containing deletions of either the nuclear export signal (ΔNES) or the major nuclear localization signal (ΔNLS) of ICP27 (Fig 4C). In THP1 cells infected with the ICP27 ΔNES virus mutant, very low levels of ICP27 were expressed at the late time points where we observed increased IFN induction by the ΔICP27 virus (data not shown), thus preventing us from using this virus in our studies. By contrast, in cells infected with the ICP27 ΔNLS, ICP27 accumulated to higher levels in the cytoplasm at early time points, and to lower levels in the nucleus (Appendix Fig S3A), consistent with the first description of this virus mutant (Mears et al, 1995). Interestingly, the ICP27 ΔNLS mutant virus retained the ability to suppress induction of type I IFNs (Fig 4D). This indicates that full inhibition of the IFN response by HSV‐1 is dependent on cytosolic location of ICP27.
Figure 4. ICP27 inhibits the cGAS–STING pathway downstream of TBK1 phosphorylation but upstream of IRF3 phosphorylation.
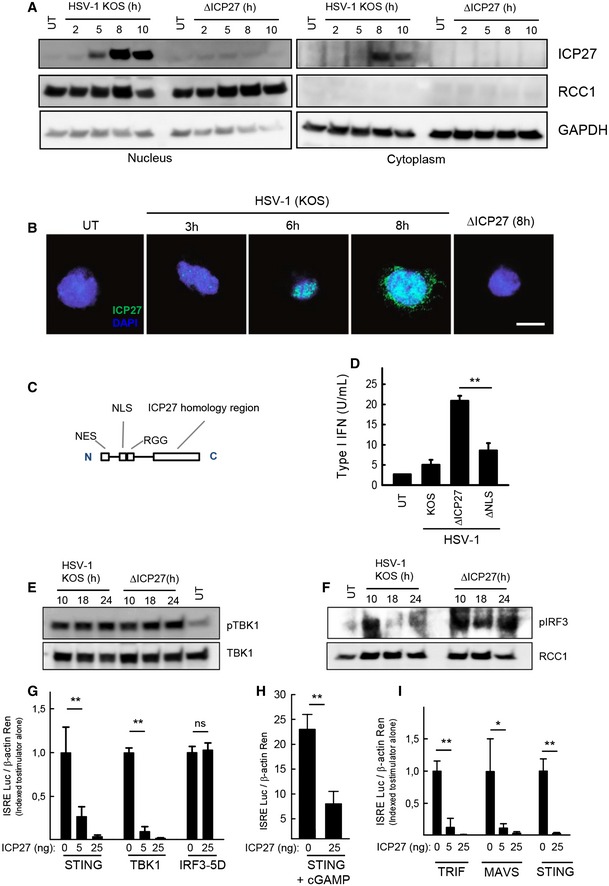
-
ATHP1 cells were infected with HSV‐1 KOS or ΔICP27 (MOI 3). Cytoplasmic and nuclear extracts were isolated at the indicated time points post‐infection, and levels of ICP27, RCC1, and GAPDH were determined by Western blotting.
-
BTHP1 cells were infected with HSV‐1 KOS or ΔICP27 (MOI 10). The cells were fixed at the indicated time points post‐infection, stained with DAPI and an antibody against ICP27, and visualized by confocal microscopy. Scale bar, 10 μm.
-
C, DTHP1 cells were infected with HSV‐1 KOS, ΔICP27, or HSV‐1 ΔNLS. The localization of the major NLS is illustrated in (C). Supernatants were harvested 18 hpi for measurement of type I IFN bioactivity.
-
E, FTHP1 cells were infected with HSV‐1 KOS or ΔICP27 (MOI 3). Cytoplasmic (E) and nuclear extracts (F) were isolated at the indicated time points post‐infection. Western blotting was used to determine levels of phospho‐ and total TBK1 were determined in the cytoplasmic fractions and levels of phospho‐IRF3 and RCC1 in the nuclear fractions.
-
G–IHEK293T cells were (G) transfected with STING, TBK1, IRF3‐5D, and ICP27, (H) transfected with STING and stimulated for 16 h with cGAMP (4 μg/ml) or (I) transfected with TRIF, MAVS, STING, and ICP27, together with the reporter gene constructs indicated. Reporter gene activity was measured 24 h post‐transfection.
To examine at which level ICP27 targets the STING pathway, the phosphorylation status of STING, TBK1, and IRF3 in KOS‐ and ΔICP27‐infected THP1 cells was evaluated. Phosphorylation of STING has been demonstrated to occur in response to DNA and to generate platforms for interaction with TBK1 and IRF3 (Zhong et al, 2008; Liu et al, 2015). Consistent with this, DNA stimulation induced a slower migrating STING band on the immunoblot and also a stronger signal when probing with the anti‐pS366 STING antibody (Appendix Fig S3B and C). For the lysates from the cells infected with virus, we were not able to reproducibly detect the slow‐migrating band of phospho‐STING, which emerged after stimulation with synthetic DNA (Appendix Fig S3B). Moreover, we did not detect an increase of signal with the anti‐pS366 STING antibody when analyzing lysates from HSV‐1‐infected macrophages (Appendix Fig S3B and C). Together, this suggests that phosphorylation of STING occurs at only low level during HSV‐1 infection and that the assays used were not sensitive enough to conclusively establish how HSV‐1 infection impacts on STING phosphorylation. Despite this, TBK1 was activated during HSV‐1 infection, and this was independent of ICP27 expression (Fig 4E). By contrast, while phosphorylation of IRF3 was suppressed in HSV‐infected cells at late time points after infection, this signal was retained in cells infected with ΔICP27 HSV‐1 (Fig 4F). In support of ICP27 acting at a step between TBK1 and IRF3 in the STING pathway, we found that ICP27 was able to inhibit an ISRE‐driven luciferase promoter stimulated by STING or TBK1 but not constitutively active IRF3 (Fig 4G). The ability of ICP27 to inhibit STING‐driven signaling was also observed when cells were stimulated with cGAMP, the natural ligand for STING (Fig 4H). Interestingly, the ability of ICP27 to inhibit the ISRE reporter gene was also observed when the transcription was stimulated by TRIF and MAVS, the adaptor proteins for TLR3 and RIG‐I‐like receptors, respectively (Fig 4I). By contrast, ICP27 did not affect NF‐κB‐driven transcription stimulated by IKKβ, nor the modest activation of this reporter gene by TBK1 (Appendix Fig S3D). Thus, ICP27 inhibits HSV‐1‐induced IFN expression by targeting a step between activation of TBK1 and IRF3 in the cGAS–STING pathway.
ICP27 interacts with the active STING–TBK1 signalsome
It is well established that DNA stimulation leads to recruitment of TBK1 to STING (Saitoh et al, 2009; Tanaka & Chen, 2012). We confirmed the inducible nature of the interaction between endogenous TBK1 and STING in lysates from cells infected with the HSV‐1 KOS strain (Fig 5A). Importantly, ICP27 was co‐immunoprecipitated with the STING–TBK1 complex (Fig 5A), and ICP27 exhibited co‐localization with both pTBK1 and STING by confocal microscopy (Fig 5B and C). ICP27 was also co‐immunoprecipitated with antibodies against the TBK1‐related kinase IKKε (Appendix Fig S4A), demonstrating that the viral protein interacts with both of the IKK‐related kinases. By contrast, ICP27 did not co‐immunoprecipitate with AKT, and another kinase involved in the cellular response to HSV infections (Appendix Fig S4B) (Cheshenko et al, 2014; Seo et al, 2015). Immunoprecipitation of TBK1 in lysates from HEK293T cells transfected with ICP27 and STING revealed that ICP27 was able to engage in the STING–TBK1 complex in the absence of other viral proteins (Fig 5D).
Figure 5. ICP27 interacts with the active STING–TBK1 signalsome.
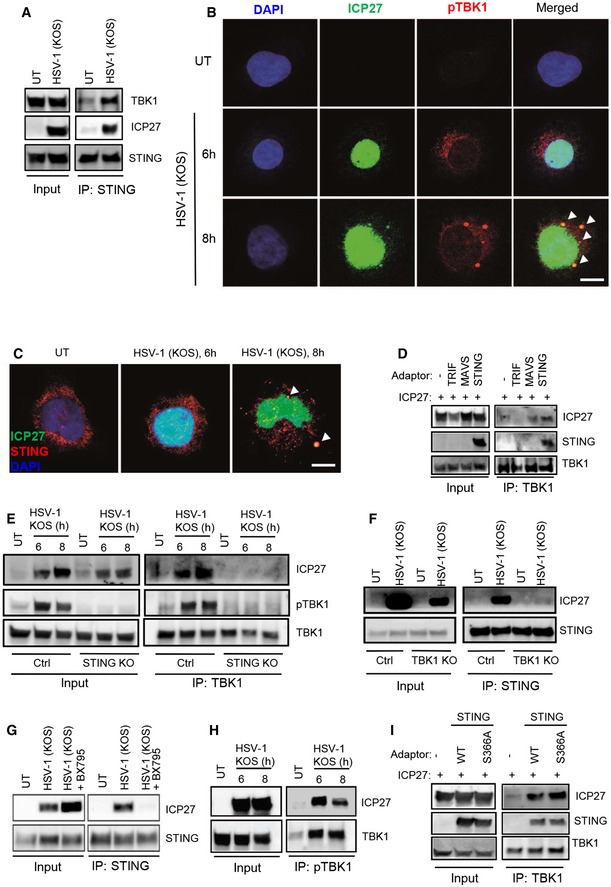
-
ALysates from THP1 cells infected with HSV‐1 KOS (MOI 10) were subjected to immunoprecipitation using an anti‐STING antibody, and the presence of TBK1, STING, and ICP27 in the precipitates was detected by Western blotting.
-
B, CTHP1 cells were infected with HSV‐1 KOS (MOI 10) for 6 and 8 h. The cells were fixed and stained with DAPI and anti‐ICP27 together with anti‐pTBK1 (B), or anti‐STING (C). The stainings were visualized by confocal microscopy. Arrowheads, co‐localizations between ICP27 and pTBK1/STING. Scale bar, 10 μm.
-
DTBK1 was immunoprecipitated from whole‐cell lysates from HEK293T cells transfected with ICP27 and each of the adaptor proteins TRIF, MAVS, and STING. The precipitates were immunoblotted for TBK1, ICP27, and STING.
-
ETHP1 control and STING KO cells were infected with HSV‐1 KOS (MOI 10) for 6 and 8 h, and total lysates were generated. TBK1 was immunoprecipitated and subjected to Western blotting using antibodies against ICP27, phospho‐TBK1 (pTBK1), and TBK1.
-
FTHP1 control and TBK1 KO cells were infected with HSV‐1 KOS (MOI 10) for 8 h, and total lysates were generated. STING was immunoprecipitated and subjected to Western blotting using antibodies against ICP27 and STING.
-
GTHP1 cells were infected with HSV‐1 KOS (MOI 10) for 8 h in the presence or absence of the TBK1 inhibitor BX795 (BX, 200 nM). Total lysates were generated and STING was immunoprecipitated and subjected to Western blotting using antibodies against ICP27 and STING.
-
HTHP1 cells were infected with HSV‐1 KOS (MOI 10) for 6 and 8 h, and total lysates were generated. Phospho‐TBK1 was immunoprecipitated and subjected to Western blotting using antibodies against ICP27 and total TBK1.
-
ITBK1 was immunoprecipitated from whole‐cell lysates from HEK293T cells transfected with ICP27 and STING (either WT or S366A). The precipitates were immunoblotted for TBK1, ICP27, and STING.
Source data are available online for this figure.
To decipher the requirements for the interaction between ICP27 and the STING–TBK1 complex, we immunoprecipitated TBK1 from HSV‐1‐infected control and STING KO THP1 cells. Interestingly, ICP27 was co‐immunoprecipitated with TBK1 in lysates from control cells but not from STING‐deficient cells (Fig 5E). Likewise, we did not co‐precipitate ICP27 with STING in TBK1‐deficient cells (Fig 5F). This demonstrates a dependency for both STING and TBK1 to enable ICP27 to engage in the interaction. Assembly of the STING signalsome upon recognition of cytosolic DNA is critically dependent on the kinase activity of TBK1, which undergoes autophosphorylation (Larabi et al, 2013; Tu et al, 2013), and also phosphorylates both STING and IRF3 (Liu et al, 2015). We next examined whether the kinase activity of TBK1 was required for the interaction of ICP27 with the STING–TBK1 complex. As expected, the TBK1 inhibitor did block induction of type I IFN production by HSV‐1 and the ΔICP27 mutant (Appendix Fig S4C). Strikingly, the readily detectable interaction between ICP27 and STING in cells infected with HSV‐1 KOS was totally abrogated if TBK1 activation was blocked (Fig 5G). Although the interaction between ICP27 and the TBK1–STING complex was dependent on the kinase activity of TBK1, the protein–protein interactions seemed not to involve the phosphorylated serine 172 on TBK1, because the epitope recognized by the anti‐pTBK1 antibody was able to co‐immunoprecipitate ICP27 together with phosphorylated TBK1 (Fig 5H). Moreover, the co‐immunoprecipitation of STING, TBK1, and ICP27 was independent of the STING serine 366, which is essential for recruitment of IRF3 and transcriptional activation (Fig 5I and Appendix Fig S4D) (Liu et al, 2015). This suggests that ICP27 is recruited to the TBK1‐activated STING signalsome, to inhibit TBK1‐mediated phosphorylation of IRF3 and downstream expression of type I IFN genes.
ICP27 from the Simplexvirus genera of Alphaherpesvirinae inhibits IFN production in a manner dependent on the RGG box
ICP27 is conserved among herpesviruses. We performed a sequence alignment of human herpesvirus ICP27 homologs to more precisely determine the degree of homology and to identify the regions in the proteins exhibiting most homology. As seen in Fig 6A, although clearly homologous, the herpesvirus ICP27 homologs differ substantially, with the core ICP27 homology box being the most conserved between alpha‐, beta‐, and gamma‐herpesviruses. Within the family of alpha‐herpesviruses, HSV‐2 ICP27 is closely related to HSV‐1 ICP27 with all known domains and motifs being conserved, while only the ICP27 homology domain is conserved in varicella zoster virus (VZV) ICP27 (ORF4). We therefore decided to make a more detailed comparison of ICP27 from different alpha‐herpesviruses. This family can be subdivided into the Ilto, mardi, varicello, and simplex genera, and we found that although the ICP27 homology domain was conserved, significant differences were observed between the genera (Fig 6B). In particular, we observed that lack of the amino‐terminal part of ICP27, which was conserved in the simplex genera of alpha‐herpesviruses, was a general phenomenon for alpha‐herpesviruses of the varicello genera. To examine whether ICP27 of other alpha‐herpesvirus also contributed to viral inhibition of type I IFN expression, we focused on ICP27 of HSV‐2 and the UL54 of varicello alpha‐herpesvirus pseudorabies virus (PRV). First, we examined an HSV‐1 ΔICP27 mutant reconstituted with HSV‐2 ICP27. Similar to HSV‐1, HSV‐2 was a weak inducer of IFN production in macrophages, and introduction of HSV‐2 ICP27 into the HSV‐1 ΔICP27 mutant enabled the virus to strongly suppress IFN production (Fig 6C). In contrast to this, the ability of PRV to induce IFN production was not affected by the lack of UL54 (Fig 6D).
Figure 6. ICP27 from the simplex virus genera of alpha‐herpesviruses inhibits IFN production in a manner dependent on the RGG box.
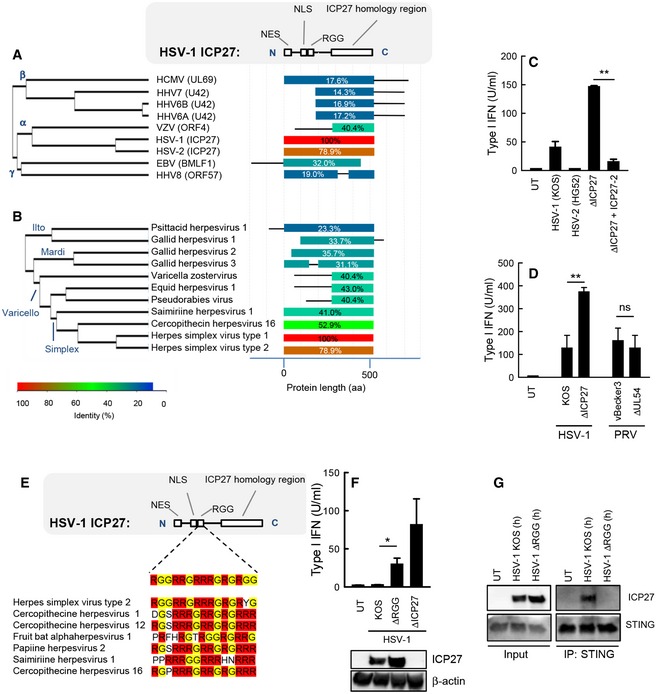
-
A, BAlignment of the ICP27 homologs of the human herpesviruses (A) and a series of alpha‐herpesviruses (B). The diagram to the right illustrates the degree of similarity with HSV‐1 ICP27 and the distribution within the molecules.
-
CTHP1 cells were infected with HSV‐1 (KOS), HSV‐2 (HG52), HSV‐1 ΔICP27, or HSV‐1 ΔICP27 rescued with HSV‐2 ICP27 (MOI 3). Supernatants were harvested 18 hpi for measurement of type I IFN bioactivity.
-
DRAW264.7 cells were infected with HSV‐1 (KOS), PRV (vBecker3) and the corresponding mutants lacking ICP27 (UL54 in the case of PRV). Supernatants were harvested 18 hpi for measurement of type I IFN bioactivity.
-
EAlignment of the RGG box of in ICP27 proteins of the simplex virus genera of alpha‐herpesviruses.
-
FTHP1 cells were infected with HSV‐1 (KOS), ΔRGG, or ΔICP27 (MOI 3). Supernatants were harvested 18 hpi for measurement of type I IFN bioactivity. ICP27 expression in the infected cells was determined by Western blotting.
-
GTHP1 cells were infected with HSV‐1 KOS or HSV‐1 ΔRGG (MOI 10) for 8 h, and total lysates were generated. STING was immunoprecipitated and subjected to Western blotting using antibodies against ICP27 and STING.
Within the amino‐terminal domain of ICP27 from the Simplexvirus genera is found an RGG box, which is involved in both RNA binding and protein–protein interactions (Mears & Rice, 1996; Corbin‐Lickfett et al, 2010; Sandri‐Goldin, 2011). The RGG box is well conserved among ICP27 proteins of the simplex genera (Fig 6E). We therefore wanted to examine whether this motif contributed to the ability of ICP27 to inhibit HSV‐1‐induced expression of type I IFNs. Cells were infected with a virus strain carrying an ICP27 ΔRGG mutant, and IFN production was compared to what was induced by HSV‐1 KOS or the ΔICP27 mutant. Interestingly, the supernatants from cells infected with the ICP27 ΔRGG mutant contained significantly more type I IFN than cultures from cells infected with HSV‐1 KOS (Fig 6F), and co‐immunoprecipitation revealed that ICP27 ΔRGG failed to interact with the STING signalsome (Fig 6G). Collectively, these data demonstrate that the ability of ICP27 to inhibit type I IFN production is conserved among viruses of the simplex genera of alpha‐herpesviruses and is mediated by targeting of the TBK‐1 activated STING signalsome to prevent activation of the central IFN‐inducing transcription factor IRF3.
Discussion
The ability of viruses to evade and modulate the host immune response is of central importance for successful establishment and maintenance of infection. The innate immune system constitutes the first line of defense against infection, and particularly the type I IFN system is important for early control of viruses (McNab et al, 2015). Therefore, viruses have evolved sophisticated strategies to inhibit and evade the IFN system, and knowledge of these strategies will greatly advance our understanding of the pathogenesis of specific viral diseases (Bowie & Unterholzner, 2008). To identify HSV‐1 IFN evasion proteins, we used a system where the IFN induction potential of different HSV‐1 strains was compared and combined with mass spectrometric analysis for viral gene expression in cells infected with a low‐inducing strain. Identified candidates were subsequently tested further. The key criteria for the validations were as follows: (i) the viral candidate protein should be expressed in lower levels in human primary macrophages infected with the F strain as compared to the KOS strain, (ii) a viral mutant on KOS background lacking the candidate protein should induce significantly more protein than the WT virus, (iii) expression of the viral candidate protein alone should reduce induction of IFN‐β expression. We report that HSV‐1 inhibits the STING–TBK1–IRF3 pathway by directing ICP27 to the activated STING–TBK1 signalsome, thus preventing phosphorylation and activation of the transcription factor IRF3 (Appendix Fig S5).
Macrophages are often resident at or recruited to sites of viral infection, but are rarely the major source of viral production. Despite this, data suggest that the IFN derived from this cell lineage is very important for the ability of an organism to control viral infections (Eloranta & Alm, 1999; Kumagai et al, 2007; Goritzka et al, 2015). However, there are limited data as to how viruses seek to evade IFN expression in macrophages and in general in cell types where many viruses replicate inefficiently. We found that HSV‐1 infection in human macrophages led to detectable expression of only a subset of viral genes and no or only very limited productive replication. Despite this, the virus had the capacity to both stimulate and inhibit the STING–TBK1 pathway. Entry of HSV involves a spectrum of broadly expressed receptors, and consequently, this class of viruses can enter into many cell types (Heldwein & Krummenacher, 2008), whereas highly efficient replication mainly occurs in epithelial cells, fibroblasts, and neurons (Roizman et al, 2013). The present data suggest that viral gene expression and protein production in cells not productively infected are not merely dead‐end activities by the virus, but may serve the purpose of modulating host responses, including IFN production. In cell types where the virus replicates productively, such as HFFs, ICP27‐deficient HSV‐1 also stimulated more IFN production than WT virus, demonstrating that the mechanism described in this work is also operative in permissive cell types. However, consistent with the literature on ICP0 as a central immune evasion protein in fibroblasts, viruses lacking ICP0 were even more potent inducers of IFN in HFFs (Mossman et al, 2001; Eidson et al, 2002; Orzalli et al, 2012, 2013). These data suggest that HSV uses cell type‐specific mechanisms to counteract the production of type I IFN.
At the mechanistic level, ICP27 interferes with events in the STING signalsome occurring between TBK1 activation and phosphorylation of IRF3. This part of the STING signaling pathway has recently been elucidated in some details (Tanaka & Chen, 2012; Liu et al, 2015). Notably, active TBK1 phosphorylates STING at least at two residues, namely S358 and S366, which serve as docking sites for TBK1 and IRF3, respectively (Zhong et al, 2008; Liu et al, 2015). Key findings of the present study are as follows: (i) ICP27 did not impact on phosphorylation of TBK1, but led to reduced phosphorylation and activation of IRF3, (ii) ICP27 interacted with TBK1 and STING in a manner dependent on all factors being expressed, (iii) the recruitment of ICP27 to the STING signalsome was dependent on TBK1 activity, (iv) the recruitment of ICP27 to the STING signalsome was dependent on the RGG motif in ICP27, but independent of S366 on STING, (v) ICP27 inhibited ISRE transcription by all three major IFN‐stimulating innate adaptor proteins. Based on these findings, our data are consistent with two models, which are not mutually exclusive. In the first model, ICP27 interacts directly with active TBK1. If this is the case, the interaction most likely does not involve the phosphorylation site on TBK1, since we were able to co‐immunoprecipitate ICP27 with an anti‐pTBK1 antibody, but must occur through a different surface. In this respect, it is interesting that TBK1 undergoes a conformational change upon activation and exposes surfaces which are hidden in the inactive kinase (Ma et al, 2012). We did observe co‐immunoprecipitation of TBK1 and ICP27 in HEK293T cells not expressing STING, and this was amplified by co‐expression of STING. In the second model, ICP27 interacts with activated STING in the STING–TBK1 complex. Following phosphorylation on S366, STING harbors a negatively charged surface, which is conserved on TRIF and MAVS, and interacts with IRF3 to position it for TBK1‐dependent phosphorylation (Liu et al, 2015). ICP27 could interact with this surface through the positively charged RGG box to prevent recruitment and activation of IRF3. However, we did observe recruitment of ICP27 to the STING–TBK1 complex in cells expressing the S366A mutant, thus arguing against S366 on STING to be the binding site for ICP27 on STING. Therefore, based on the data presented, we favor a model that combines the two models described above: ICP27 has low basal affinity for non‐activated TBK1. Upon DNA stimulation, ICP27 binds strongly to the TBK1–STING complex. This strong interaction is either due to increased affinity of ICP27 for activated TBK1, or ICP27 interacting with both TBK1 and STING in the signalsome. Future work should further characterize the interaction between ICP27 and TBK1/STING and evaluate whether this can be mechanistically separated from the role of ICP27 in viral replication. Such knowledge would pave the way for generation of viruses that are selectively defective in ICP27‐mediated targeting of TBK1–STING, and thus allow evaluation of the impact of this mechanism on the pathophysiology of HSV‐1 in vivo.
There is now an emerging literature on inhibition of DNA‐stimulated IFN responses by herpesviruses. In particular, KSHV has been demonstrated to target these responses with several proteins. The KSHV tegument protein ORF52, which is conserved among gammaherpesviruses, has also been reported to antagonize the cGAS–STING pathway by preventing cGAS‐mediated cGAMP production (Wu et al, 2015). This involves a dual mechanism with ORF52 binding both cGAS and DNA. Moreover, a cytoplasmic isoform of the latency‐associated nuclear antigen interacts with cGAS and prevents phosphorylation of STING (Zhang et al, 2016). Finally, the KSHV protein vIRF1 inhibits the cGAS–STING pathway by preventing recruitment of TBK1 to STING (Ma et al, 2015). For HCMV, the pUL83 protein targets IFI16 (Li et al, 2013a), which is a DNA‐binding protein that is essential for DNA‐stimulated cGAS‐ and STING‐dependent IFN production (Jakobsen et al, 2013; Hansen et al, 2014). Finally, we recently reported that the deubiquitinase activity of the conserved large herpesvirus tegument protein contributes to control of type I IFN production in macrophages, through a mechanism involving prevention of viral DNA release into the cytoplasm (Sun et al, 2015). Thus, herpesviruses have evolved conserved as well as virus‐specific mechanisms to prevent innate DNA sensing and signaling.
In conclusion, we have identified a mechanism through which HSV‐1 inhibits IFN production in macrophages through ICP27‐mediated interaction with the active STING signalsome. ICP27 prevented IRF‐driven signaling downstream of not only STING but also MAVS and TRIF. Therefore, ICP27 could potentially inhibit IFN signaling from all known HSV‐sensing PRRs, including TLR3, which is essential for control of HSV‐1 replication in the CNS in humans (Zhang et al, 2007). Finally, since we found that ICP27 did not inhibit TBK1 phosphorylation, we cannot exclude that the STING–TBK1 complex remains active, and is redirected toward other substrates, which would favor viral infection. The concept of viral exploitation of existing cellular signaling platforms has been suggested (Chaurushiya et al, 2012) and deserves more attention in the context of immune evasion.
Materials and Methods
Cells
PBMCs were isolated from healthy blood donors (Finnish Red Cross Blood Transfusion Service) and differentiated into MDMs in the presence macrophage‐SFM medium (Life Technologies) supplemented with GM‐CSF. PBMCs were isolated from heparin‐stabilized blood by a Ficoll density gradient and cultured in RPMI 1640 media (BioWhittaker; Lonza/Biowest) containing 10% heat‐inactivated FBS (Life Technologies) and 1% l‐glutamine (Life Technologies; Medium+). THP1 cells and CRISPR‐generated knock cell lines were maintained in RPMI (Lonza), supplemented with 10% FCS (Sigma), 600 μg/ml l‐glutamine (Sigma), 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco). For experiments, the THP1 cells were differentiated with 150 nM phorbol 12‐myristate 13‐acetate (PMA) (Sigma‐Aldrich). Further details can be found in the Appendix Supplementary Experimental Procedures.
Viruses
The virus strains used were HSV‐1 KOS, HSV‐1 McKrae, HSV‐1F, HSV‐2 HG52, HSV‐1 K2F1 (a HSV‐1 mutant that encodes HSV‐2 ICP27 in place of HSV‐1 ICP27), HSV‐1 d27‐1, HSV‐1 dLeu, HSV‐1 d3‐4, HSV‐1 d4‐5, HSV‐1 ICP0‐7134, ICP0‐7134R PRV‐vBecker3, and PRV vJSΔ54N. The viruses were propagated in parental Vero or U2OS cells or derived cell lines engineered to express ICP27. Further details can be found in the Appendix Supplementary Experimental Procedures.
Type I IFN bioassay
Bioactive type I IFN was measured on cell supernatants by use of HEK‐Blue™ IFN‐α/β cells as reporter cells according to the manufacturer instructions (Invivogen). For details, see Appendix Supplementary Experimental Procedures.
ELISA
CXCL10 protein levels were measured by ELISA using the hCXCL10/IP‐10 DuoSet (R&D Systems). Further details can be found in the Appendix Supplementary Experimental Procedures.
Western blotting
Whole‐cell extracts, cytoplasmic fractions, nuclear fractions, stocks of viral particles, and Co‐IP samples were analyzed by Western blot. Samples were diluted in XT sample buffer and XT reducing agent and ran on a SDS–PAGE (Criterion™ TGX™). Trans‐Blot Turbo™ Transfer System® was used for the transfer of proteins to PVDF membranes (all reagents Bio‐Rad). The membrane was blocked in 5% Difco™ skim milk (BD) or 5% bovine serum albumin (BSA) (Sigma). For further information on sample preparation and antibodies, see Appendix Supplementary Experimental Procedures.
Immunoprecipitation
Cleared cell lysates were incubated with antibodies against pTBK1 (1:50), TBK1 (1:50), IKKε (1:50), STING (1:50), or pan‐AKT (1:50) antibody (all Cell Signaling Technology) overnight at 4°C. Protein G Dynabeads® (Novex by Life Technologies) were added for the last 2 h (0.9 mg per IP reaction in 240 μl). Immunoprecipitated complexes were eluted from the beads with a glycine buffer (200 mM glycine, pH 2,5), and protein expression was evaluated by Western blotting.
Confocal microscopy
PMA‐differentiated THP1 cells were seeded on coverslips and stimulated as indicated. The cells were fixed with 4% formaldehyde (Sigma), permeabilized with 0.1% Triton X‐100 (Sigma), and blocked with 1% BSA (Sigma). Cells were stained with primary anti‐ICP27 (P1113) (1:200) and anti‐pTBK1 (1:50) or anti‐STING (1:50) for 2 h and with secondary antibody (all 1:500, Alexa Fluor)(Invitrogen) for 1 h. Cell nucleai were stained with DAPI (Invitrogen). Images were acquired on Zeiss LSM 710 confocal microscope using a 63× 1.4 oil‐immersion objective and processed with the Zen2010 (Zeiss) and ImageJ software.
Mutagenesis
The S366A mutation was introduced into STING by site‐directed mutagenesis using the pcDNA3/STING vector as template and forward primer 5′‐GGGCTTTTCCATTCCAGCGATGAGGAGCTCAGGC‐3′ and reverse primer 5′‐GCCTGAGCTCCTCATCGCTGGAATGGAAAAGCCC‐3′. The PCR was made by using Pfu Ultra II buffer and Pfu Ultra II Fusion HS DNA polymerase (Agilent), 100 ng template, and primers at a concentration of 1.25 ng/μl in a total volume of 100 μl.
Reporter gene assay
HEK293T cells were transfected as indicated with polyethylenimine and left for 24 h before analysis of luciferase activity. The firefly and renilla luciferase signals were developed with Dual Glo® luciferase assay (Promega) and read on Luminoskan Ascent (Labsystems). Further details can be found in the Appendix Supplementary Experimental Procedures.
Real‐time qPCR
PBMCs were treated as described for 6 h, and total RNA was isolated with High Pure RNA Isolation Kit (Roche). IFN‐β and IFN‐α2 mRNA expressions were determined by real‐time qPCR, using TaqMan detection systems (Thermo scientific). mRNA expressions were normalized to β‐actin. Primer‐probe kits: IFN‐β, Hs01077958_s1; IFN‐α2, Hs00265051_s1; and β‐actin, Hs99999903_m1 (all Thermo Scientific).
Virus plaque assay
MDMs and HFFs were infected with HSV‐1 KOS at MOI 0.1 and 1.0. Supernatants were harvested 24 h later, and the virus yield was quantified using the plaque titration assay on Vero cells as described previously (Reinert et al, 2012).
Mass spectrometry
The cytosolic proteins were identified and quantified using iTRAQ labeling combined with liquid chromatography–tandem mass spectrometry (LC‐MS/MS) analysis of the resulting peptides. Further details can be found in the Appendix Supplementary Experimental Procedures.
Statistical analysis and reproducibility of results
The data are shown as means of biological replicates ± SD. Statistically significant differences between groups were determined using two‐tailed Student's t‐test when the data exhibited normal distribution and Wilcoxon rank‐sum test when the data set did not pass the normal distribution test. The data shown are from single experiments. All experiments were performed at least 3 times with similar results.
Author contributions
MHC, SBJ, TAN, SM, and SRP designed the study. MHC, SBJ, JJM, SL, TP, and LSR performed the experiments. TM, ZJC, DMK, RMS‐G, LWE, THM, and SAR provided reagents. RH, TAN, SM, and SRP supervised experiments. All authors were involved in the discussion of results. MHC and SRP wrote the manuscript, and all authors commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank Kirsten S Petersen for technical assistance and Prof. Andrew Bowie for generous donation of reagents. This work was funded by The Danish Medical Research Council (grants no: 12‐124330 and 11‐107588), The Novo Nordisk Foundation, The Lundbeck Foundation (grant no R34‐3855), Aarhus University Research Foundation (all to SRP), and by NIH grant AAI106934 to DMK. SL was supported by a scholarship from The Boehringer Ingelheim Fonds.
The EMBO Journal (2016) 35: 1385–1399
References
- Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124: 783–801 [DOI] [PubMed] [Google Scholar]
- Andersen LL, Mork N, Reinert LS, Kofod‐Olsen E, Narita R, Jorgensen SE, Skipper KA, Honing K, Gad HH, Ostergaard L, Orntoft TF, Hornung V, Paludan SR, Mikkelsen JG, Fujita T, Christiansen M, Hartmann R, Mogensen TH (2015) Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J Exp Med 212: 1371–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie AG, Unterholzner L (2008) Viral evasion and subversion of pattern‐recognition receptor signalling. Nat Rev Immunol 8: 911–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaurushiya MS, Lilley CE, Aslanian A, Meisenhelder J, Scott DC, Landry S, Ticau S, Boutell C, Yates JR III, Schulman BA, Hunter T, Weitzman MD (2012) Viral E3 ubiquitin ligase‐mediated degradation of a cellular E3: viral mimicry of a cellular phosphorylation mark targets the RNF8 FHA domain. Mol Cell 46: 79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen IH, Li L, Silva L, Sandri‐Goldin RM (2005) ICP27 recruits Aly/REF but not TAP/NXF1 to herpes simplex virus type 1 transcription sites although TAP/NXF1 is required for ICP27 export. J Virol 79: 3949–3961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheshenko N, Trepanier JB, Gonzalez PA, Eugenin EA, Jacobs WR Jr, Herold BC (2014) Herpes simplex virus type 2 glycoprotein H interacts with integrin alphavbeta3 to facilitate viral entry and calcium signaling in human genital tract epithelial cells. J Virol 88: 10026–10038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin‐Lickfett KA, Souki SK, Cocco MJ, Sandri‐Goldin RM (2010) Three arginine residues within the RGG box are crucial for ICP27 binding to herpes simplex virus 1 GC‐rich sequences and for efficient viral RNA export. J Virol 84: 6367–6376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuchet‐Lourenco D, Anderson G, Sloan E, Orr A, Everett RD (2013) The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J Virol 87: 13422–13432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eidson KM, Hobbs WE, Manning BJ, Carlson P, DeLuca NA (2002) Expression of herpes simplex virus ICP0 inhibits the induction of interferon‐stimulated genes by viral infection. J Virol 76: 2180–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eloranta ML, Alm GV (1999) Splenic marginal metallophilic macrophages and marginal zone macrophages are the major interferon‐alpha/beta producers in mice upon intravenous challenge with herpes simplex virus. Scand J Immunol 49: 391–394 [DOI] [PubMed] [Google Scholar]
- Goritzka M, Makris S, Kausar F, Durant LR, Pereira C, Kumagai Y, Culley FJ, Mack M, Akira S, Johansson C (2015) Alveolar macrophage‐derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J Exp Med 212: 699–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen K, Prabakaran T, Laustsen A, Jorgensen SE, Rahbaek SH, Jensen SB, Nielsen R, Leber JH, Decker T, Horan KA, Jakobsen MR, Paludan SR (2014) Listeria monocytogenes induces IFNbeta expression through an IFI16‐, cGAS‐ and STING‐dependent pathway. EMBO J 33: 1654–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldwein EE, Krummenacher C (2008) Entry of herpesviruses into mammalian cells. Cell Mol Life Sci 65: 1653–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman M, Ciancanelli M, Ou YH, Lorenzo L, Klaudel‐Dreszler M, Pauwels E, Sancho‐Shimizu V, Perez de Diego R, Abhyankar A, Israelsson E, Guo Y, Cardon A, Rozenberg F, Lebon P, Tardieu M, Heropolitanska‐Pliszka E, Chaussabel D, White MA, Abel L, Zhang SY et al (2012) Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med 209: 1567–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN (2009) STING regulates intracellular DNA‐mediated, type I interferon‐dependent innate immunity. Nature 461: 788–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen MR, Bak RO, Andersen A, Berg RK, Jensen SB, Tengchuan J, Laustsen A, Hansen K, Ostergaard L, Fitzgerald KA, Xiao TS, Mikkelsen JG, Mogensen TH, Paludan SR (2013) IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV‐1 replication. Proc Natl Acad Sci USA 110: E4571–E4580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KE, Song B, Knipe DM (2008) Role for herpes simplex virus 1 ICP27 in the inhibition of type I interferon signaling. Virology 374: 487–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffa MD, Clements JB, Izaurralde E, Wadd S, Wilson SA, Mattaj IW, Kuersten S (2001) Herpes simplex virus ICP27 protein provides viral mRNAs with access to the cellular mRNA export pathway. EMBO J 20: 5769–5778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E, Aozasa K, Kawai T, Akira S (2007) Alveolar macrophages are the primary interferon‐alpha producer in pulmonary infection with RNA viruses. Immunity 27: 240–252 [DOI] [PubMed] [Google Scholar]
- Larabi A, Devos JM, Ng SL, Nanao MH, Round A, Maniatis T, Panne D (2013) Crystal structure and mechanism of activation of TANK‐binding kinase 1. Cell Rep 3: 734–746 [DOI] [PubMed] [Google Scholar]
- Li T, Chen J, Cristea IM (2013a) Human cytomegalovirus tegument protein pUL83 inhibits IFI16‐mediated DNA sensing for immune evasion. Cell Host Microbe 14: 591–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ (2013b) Pivotal roles of cGAS‐cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341: 1390–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, Chen ZJ (2015) Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347: aaa2630 [DOI] [PubMed] [Google Scholar]
- Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, Bowman KK, Starovasnik MA, Dueber EC (2012) Molecular basis of Tank‐binding kinase 1 activation by transautophosphorylation. Proc Natl Acad Sci USA 109: 9378–9383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Jacobs SR, West JA, Stopford C, Zhang Z, Davis Z, Barber GN, Glaunsinger BA, Dittmer DP, Damania B (2015) Modulation of the cGAS‐STING DNA sensing pathway by gammaherpesviruses. Proc Natl Acad Sci USA 112: E4306–E4315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maringer K, Elliott G (2010) Recruitment of herpes simplex virus type 1 immediate‐early protein ICP0 to the virus particle. J Virol 84: 4682–4696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNab F, Mayer‐Barber K, Sher A, Wack A, O'Garra A (2015) Type I interferons in infectious disease. Nat Rev Immunol 15: 87–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears WE, Lam V, Rice SA (1995) Identification of nuclear and nucleolar localization signals in the herpes simplex virus regulatory protein ICP27. J Virol 69: 935–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears WE, Rice SA (1996) The RGG box motif of the herpes simplex virus ICP27 protein mediates an RNA‐binding activity and determines in vivo methylation. J Virol 70: 7445–7453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears WE, Rice SA (1998) The herpes simplex virus immediate‐early protein ICP27 shuttles between nucleus and cytoplasm. Virology 242: 128–137 [DOI] [PubMed] [Google Scholar]
- Melchjorsen J, Siren J, Julkunen I, Paludan SR, Matikainen S (2006) Induction of cytokine expression by herpes simplex virus in human monocyte‐derived macrophages and dendritic cells is dependent on virus replication and is counteracted by ICP27 targeting NF‐kappaB and IRF‐3. J Gen Virol 87: 1099–1108 [DOI] [PubMed] [Google Scholar]
- Mossman KL, Saffran HA, Smiley JR (2000) Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J Virol 74: 2052–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossman KL, Macgregor PF, Rozmus JJ, Goryachev AB, Edwards AM, Smiley JR (2001) Herpes simplex virus triggers and then disarms a host antiviral response. J Virol 75: 750–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham ML, Hite RK, Steffen AM, Damko E, Li Z, Walz T, Chen J (2016) A mechanism of viral immune evasion revealed by cryo‐EM analysis of the TAP transporter. Nature 529: 537–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, DeLuca NA, Knipe DM (2012) Nuclear IFI16 induction of IRF‐3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci USA 109: E3008–E3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, Conwell SE, Berrios C, DeCaprio JA, Knipe DM (2013) Nuclear interferon‐inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc Natl Acad Sci USA 110: E4492–E4501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paludan SR, Bowie AG, Horan KA, Fitzgerald KA (2011) Recognition of herpesviruses by the innate immune system. Nat Rev Immunol 11: 143–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paludan SR, Bowie AG (2013) Immune sensing of DNA. Immunity 38: 870–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez de Diego R, Sancho‐Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, Herman M, Cardon A, Durandy A, Bustamante J, Vallabhapurapu S, Bravo J, Warnatz K, Chaix Y, Cascarrigny F, Lebon P, Rozenberg F, Karin M, Tardieu M, Al‐Muhsen S et al (2010) Human TRAF3 adaptor molecule deficiency leads to impaired Toll‐like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity 33: 400–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinert LS, Harder L, Holm CK, Iversen MB, Horan KA, Dagnaes‐Hansen F, Ulhoi BP, Holm TH, Mogensen TH, Owens T, Nyengaard JR, Thomsen AR, Paludan SR (2012) TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of CNS infection in mice. J Clin Invest 122: 1368–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice SA, Knipe DM (1990) Genetic evidence for two distinct transactivation functions of the herpes simplex virus alpha protein ICP27. J Virol 64: 1704–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman B, Knipe DM, Whitley RJ (2013) Herpes simplex viruses In Fields Virology, Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B. (eds), pp 1823 Philadelphia, PA: Lippincott Williams & Wilkins; [Google Scholar]
- Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY, Taniguchi T, Shadel GS, Chen ZJ, Iwasaki A, Flavell RA (2014) Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159: 1563–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, Yamamoto N, Kawai T, Ishii K, Takeuchi O, Yoshimori T, Akira S (2009) Atg9a controls dsDNA‐driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA 106: 20842–20846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho‐Shimizu V, Perez de Diego R, Lorenzo L, Halwani R, Alangari A, Israelsson E, Fabrega S, Cardon A, Maluenda J, Tatematsu M, Mahvelati F, Herman M, Ciancanelli M, Guo Y, AlSum Z, Alkhamis N, Al‐Makadma AS, Ghadiri A, Boucherit S, Plancoulaine S et al (2011) Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Invest 121: 4889–4902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri‐Goldin RM (2011) The many roles of the highly interactive HSV protein ICP27, a key regulator of infection. Future Microbiol 6: 1261–1277 [DOI] [PubMed] [Google Scholar]
- Seo GJ, Yang A, Tan B, Kim S, Liang Q, Choi Y, Yuan W, Feng P, Park HS, Jung JU (2015) Akt kinase‐mediated checkpoint of cGAS DNA sensing pathway. Cell Rep 13: 440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ (2013) Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Schattgen SA, Pisitkun P, Jorgensen JP, Hilterbrand AT, Wang LJ, West JA, Hansen K, Horan KA, Jakobsen MR, O'Hare P, Adler H, Sun R, Ploegh HL, Damania B, Upton JW, Fitzgerald KA, Paludan SR (2015) Evasion of innate cytosolic DNA sensing by a gammaherpesvirus facilitates establishment of latent infection. J Immunol 194: 1819–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzutani T, Nagamine M, Shibaki T, Ogasawara M, Yoshida I, Daikoku T, Nishiyama Y, Azuma M (2000) The role of the UL41 gene of herpes simplex virus type 1 in evasion of non‐specific host defence mechanisms during primary infection. J Gen Virol 81: 1763–1771 [DOI] [PubMed] [Google Scholar]
- Szpara ML, Parsons L, Enquist LW (2010) Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J Virol 84: 5303–5313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Chen ZJ (2012) STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 5: ra20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Devi‐Rao G, Golovanov AP, Sandri‐Goldin RM (2013) The interaction of the cellular export adaptor protein Aly/REF with ICP27 contributes to the efficiency of herpes simplex virus 1 mRNA export. J Virol 87: 7210–7217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu D, Zhu Z, Zhou AY, Yun CH, Lee KE, Toms AV, Li Y, Dunn GP, Chan E, Thai T, Yang S, Ficarro SB, Marto JA, Jeon H, Hahn WC, Barbie DA, Eck MJ (2013) Structure and ubiquitination‐dependent activation of TANK‐binding kinase 1. Cell Rep 3: 747–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verpooten D, Ma Y, Hou S, Yan Z, He B (2009) Control of TANK‐binding kinase 1‐mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J Biol Chem 284: 1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury‐Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520: 553–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ (2013) Cyclic GMP‐AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339: 826–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JJ, Li W, Shao Y, Avey D, Fu B, Gillen J, Hand T, Ma S, Liu X, Miley W, Konrad A, Neipel F, Sturzl M, Whitby D, Li H, Zhu F (2015) Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe 18: 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Wang S, Lin R, Mossman KL, Zheng C (2012) Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG‐I and MDA‐5. J Virol 86: 3528–3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Ni L, Wang S, Wang K, Lin R, Zheng C (2013) Herpes simplex virus 1‐encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF‐kappaB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J Virol 87: 9788–9801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- York IA, Roop C, Andrews DW, Riddell SR, Graham FL, Johnson DC (1994) A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell 77: 525–535 [DOI] [PubMed] [Google Scholar]
- Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho‐Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, von Bernuth H, Ku CL, Casrouge A, Zhang XX, Barreiro L et al (2007) TLR3 deficiency in patients with herpes simplex encephalitis. Science 317: 1522–1527 [DOI] [PubMed] [Google Scholar]
- Zhang G, Chan B, Samarina N, Abere B, Weidner‐Glunde M, Buch A, Pich A, Brinkmann MM, Schulz TF (2016) Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc Natl Acad Sci USA 113: E1034–E1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB (2008) The adaptor protein MITA links virus‐sensing receptors to IRF3 transcription factor activation. Immunity 29: 538–550 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6