

Abstract
Improvements in screening and preventive measures have led to an increased detection of early stage colorectal cancers (CRC) where patients undergo treatment with a curative intent. Despite these efforts, a high proportion of patients are diagnosed with advanced stage disease that is associated with poor outcomes, as CRC remains one of the leading causes of cancer-related deaths in the world. The development of next generation sequencing and collaborative multi-institutional efforts to characterize the cancer genome has afforded us with a comprehensive assessment of the genomic makeup present in CRC. This knowledge has translated into understanding the prognostic role of various tumor somatic variants in this disease. Additionally, the awareness of the genomic alterations present in CRC has resulted in an improvement in patient outcomes, largely due to better selection of personalized therapies based on an individual’s tumor genomic makeup. The benefit of various treatments is often limited, where recent studies assessing the genomic diversity in CRC have identified the development of secondary tumor somatic variants that likely contribute to acquired treatment resistance. These studies have begun to alter the landscape of treatment for CRC that include investigating novel targeted therapies, assessing the role of immunotherapy and prospective, dynamic assessment of changes in tumor genomic alterations that occur during the treatment of CRC.
Keywords: Colorectal cancer, KRAS mutation, BRAF mutation, Genomic diversity, Tumor DNA
Core tip: Tumor somatic variants have a prognostic role, in addition to treatment selection in patients with solid tumor malignancies, including colorectal cancer (CRC). The application of this knowledge in the development of novel, targeted therapies has resulted in improved patient outcomes in this disease. Our objective is to provide an overview of the genomic alterations present in CRC and its role in treatment implications, in addition to providing an overview of ongoing and future clinical trials.
INTRODUCTION
Colorectal cancer (CRC) is the fourth most common cancer in the world, leading to more than 500000 deaths annually[1,2]. An increased awareness of the genomic makeup of CRC has allowed us to understand the prognostic role of certain tumor genomic alterations. This knowledge and its incorporation into the treatment of metastatic CRC has translated to significant improvements in patient outcomes, where patients’ median overall survival has approached 3 years[3-5]. The incorporation of our knowledge about the genomic landscape of CRC into treatment decisions with selected targeted agents has led to an improvement in patient outcomes. With this increased understanding, clinical trials are now being designed to assign treatment selection with novel therapies based upon identified specific tumor somatic variants in each individual. Herein we review the genomic landscape of CRC, its current role in treatment selection, and its integration in ongoing and future studies.
GENOMIC ALTERATIONS IN DOWNSTREAM SIGNALING PATHWAYS IN CRC
RAS Mutations in CRC
The Kirsten Ras (KRAS) oncogene encodes for a guanosine triphosphate (GTP)/guanosine diphosphate binding protein downstream of the extracellular epidermal growth factor receptor in the RAS/RAF/MAPK signaling pathway. Activating KRAS exon 2 mutations occur in up to 45% of all CRC, and are involved in initiation, proliferation and progression of CRC[6-10]. While initial studies suggested a possible clinical benefit from anti-epidermal growth factor receptor (EGFR) therapy for patients whose tumors express KRAS codon 13 (G13D) mutations, a meta-analysis comprised of several large phase III trials failed to demonstrate benefit from panitumumab, an anti-EGFR monoclonal antibody, in CRC patients whose tumor harbored a KRAS codon 13 mutation[11]. In addition to KRAS exon 2, an approximate additional 10% of patients with other RAS mutations have been identified in CRC, including NRAS or non-exon 2 KRAS mutations[6]. In patients who exhibited activating non-exon 2 KRAS and NRAS mutations, an absence of clinical benefit, and perhaps a negative effect, was seen from the addition of anti-EGFR therapy in combination with several chemotherapy regimens in various treatment settings[6,12,13]. On this basis, anti-EGFR therapy should not be given to any patient with CRC exhibiting a RAS mutation.
While mutations in RAS as a predictive biomarker to anti-EGFR therapy has been recognized, its relevance as a therapeutic target is unknown. Given the high incidence of RAS in CRC and its importance as an oncogene, targeting RAS represents an ideal and promising strategy. Developing strategies to directly block oncogenic RAS activity has remained a challenge due to several factors, including the high binding affinity of the oncoprotein to the GTP-bound “on” state, as well as the lack of accessibility to active sites within KRAS to bind[14]. One alternative approach includes targeting pathways and its effectors downstream of RAS. The clinical benefit from targeting single pathways is often limited due to mechanisms of resistance including communication between signaling pathways and its resulting downstream effector activation and inhibition through a feedback loop mechanism[15,16]. Alternatively, secondary treatment resistance to anti-EGFR treatment may result from the development of RAS mutations during a course of anti-EGFR therapy. Several studies have demonstrated up to 96% of patients who initially had RAS and BRAF wild-type CRC were later identified to have acquired activating RAS (KRAS or NRAS) or BRAF mutations on repeat tumor genomic assessment at the time of disease progression on anti-EGFR therapy[17-19].
Alternative treatment strategies in RAS mutant CRC include the combination of various therapeutic agents targeting several genes involved in the MAPK pathway that would cause sufficient suppression of activated RAS activity. Combining small molecule inhibitors of MEK to anti-EGFR therapies have demonstrated the reversal of acquired anti-EGFR therapy resistance, prompting ongoing clinical trials investigating the clinical utility of the combination of multiple signaling pathway inhibitors in the first-line setting, as well as in salvage therapy for refractory disease (Table 1). Targeting multiple signaling pathways may also be effective in overcoming resistance from secondary activation of parallel signaling pathways[20]. Alternatively, administering anti-EGFR therapies in a pulsatile manner instead of to the point of clinical progression may prolong anti-EGFR therapy efficacy. A recent study that performed dynamic monitoring of tumor somatic variants through circulating tumor DNA demonstrated a decrease in drug resistant KRAS clones upon withdraw of cetuximab, allowing for the reemergence of drug sensitivity to anti-EGFR therapy[21]. This suggests a rationale for intermittent EGFR blockade, and explains for the seldom efficacy seen with re-challenging anti-EGFR therapies. Lastly, inhibitors of the MAPK and PI3K/Akt/mTOR pathway are considered to cause G1 cell cycle arrest through the suppression of D-type cyclins and the upregulation of cell cycle inhibitors[22,23]. Pre-clinical studies have demonstrated potent inhibition of G1/S transition and phosphorylation of retinoblastoma (Rb) protein with the inhibition of the MAPK and PI3K/Akt pathway[24]. The combination of MEK and CDK inhibitors may be considered a potential strategy in treating RAS activated CRC or in tumors harboring mutations in CDK.
Table 1.
Ongoing combination targeted therapy trials for colorectal cancer
| Agent(s) | Class of agent | Phase | Trial number1 | Misc |
| MEK162 + Panitumumab | MEK tyrosine kinase inhibitor, anti-EGFR mAb | Ib/II | NCT01927341 | mCRC with mutant or wild-type RAS tumors |
| Dabrafenib + Trametinib + Panitumumab + 5-Fluorouracil | BRAF tyrosine kinase inhibitor, MEK tyrosine kinase inhibitor, anti-EGFR mAb | I/II | NCT01750918 | BRAF-V600E mutant + and in pts with secondary resistance to anti-EGFR mAb |
| LGX818 + Cetuximab ± BYL719 | BRAF tyrosine kinase inhibitor, anti-EGFR mAb, PI3K tyrosine kinase inhibitor | I/II | NCT01719380 | BRAF mutant mCRC |
| Irinotecan + Cetuximab ± Vemurafenib | anti-EGFR mAb, BRAF tyrosine kinase inhibitor | II | NCT02164916 | BRAF mutant mCRC |
| Neratinib + Cetuximab | HER-2 tyrosine kinase inhibitor, anti-EGFR mAb | I/II | NCT01960023 | KRAS, NRAS, BRAF, PIK3CA wild type |
Utilizing the NCT number, clinical trial information can be obtained at https://clinicaltrials.gov. mAb: Monoclonal antibody; mCRC: Metastatic colorectal cancer; EGFR: Epidermal growth factor receptor; PIK3CA: Phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha; BRAF: v-Raf murine sarcoma viral oncogene homolog B.
BRAF mutations in CRC
BRAF V600E mutations occur in up to 5% of metastatic CRC[25,26] and are often associated with a more aggressive phenotype (with the exception in microsatellite instability high CRC, where the effect is attenuated). Patients with BRAF V600E mutations tend to respond poorly to conventional chemotherapy and have worse outcomes[6,27-31]. The constitutive activation of BRAF evading any upstream inhibition of EGFR may explain the limited clinical benefit seen with anti-EGFR therapies in BRAF mutant metastatic CRC[6,32,33].
In metastatic melanoma, inhibition of BRAF with small molecule inhibitors has led to improvement in clinical outcomes in patients whose tumors exhibit mutations in BRAF[34-37]. However, the clinical efficacy from single agent BRAF inhibition has not translated to patients with BRAF mutant metastatic CRC[36,38]. The lack of anti-tumor activity may be a result of insufficient inhibition of the MAPK pathway as a result of a feedback loop mechanism, resulting in the persistent activation of the MAPK signaling pathway[16,39] (Figure 1). To overcome this compensatory mechanism, the combination of tyrosine kinase inhibitors aimed at inhibiting the MAPK pathway has resulted in an improvement in treatment efficacy in comparison to single-agent BRAF small molecule inhibitor in metastatic melanoma[40]. Based on these results, a phase II study investigating the clinical activity from the combination of BRAF and MEK inhibition with dabrafenib and trametinib was conducted in patients with BRAF mutant metastatic CRC[41]. While modest clinical activity was observed with the combination, with 12% of patients experiencing either a partial or complete response, correlative laboratory studies suggest an inhibition in MAPK signaling in patients receiving the combination[37]. The absence of significant anti-tumor activity may be attributed to insufficient suppression of MAPK signaling, which may be due to upstream activation of EGFR, leading to reactivation of MAPK and other integral signaling pathways[15]. Based on this rationale, ongoing studies are investigating the combination of anti-EGFR therapies with BRAF tyrosine kinase inhibitors in patients with BRAF mutant mCRC (Table 1)[42-45].
Figure 1.
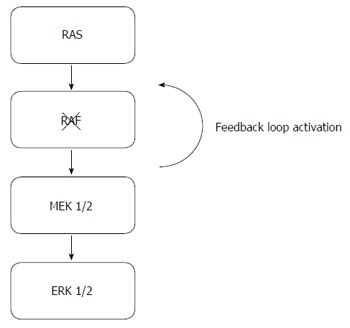
RAS/RAF/MEK/ERK pathway and mechanisms of resistance to BRAF inhibition. The figure above shows the MAPK pathway and the compensatory feedback loop activation (arrow) despite BRAF inhibition, resulting in the upstream reactivation of the MAPK pathway.
GENOMIC DIVERSITY AND ITS ROLE IN DEVELOPING PERSONALIZED THERAPIES WITH TARGETED AGENTS IN CRC
While RAS mutations are the most common genomic alteration in CRC, recent efforts have allowed us to understand the genomic diversity and identify potential therapeutic targets of interest in this disease[46,47]. Through the efforts of The Cancer Genome Atlas (TCGA), 224 CRC cases underwent extensive molecular characterization to describe the genomic landscape present in CRC[46]. 24 genes were significantly mutated, where most were an actionable mutation, including ERBB2 (HER-2/neu) mutations, a therapeutic target in HER-2 positive gastric and breast cancer, were identified in 19% of tumors[46]. This comprehensive assessment allowed us to have a better understanding of the genomic landscape, in addition to identifying several potential targetable genes of interest that are essential for tumor growth and carcinogenesis that we will discuss in further detail below (Table 2).
Table 2.
Tumor genomic variants and potential targeted therapies of interest
| Gene | Agents of Interest | Mechanism of action | Phase | Trial number1 | Comment |
| FGFR (FGFR1, FGFR2, FGFR3, FGFR4) | Ponatinib | Multi-kinase small molecule inhibitor | II | NCT02272998 | |
| BGJ398 | Pan FGFR small molecule inhibitor | I | NCT01928459 | ||
| RET | Cabozantinib | Multi-kinase small molecule inhibitor | I | NCT02008383 | Cabozantinib + panitumumab |
| Vandetanib | Multi-kinase small molecule inhibitor | I | NCT01582191 | ||
| Apatinib | Multi-kinase small molecule inhibitor | ||||
| Ponatinib | Multi-kinase small molecule inhibitor | II | NCT02272998 | ||
| RXDX-105 | RET and BRAF small molecule inhibtor | I | NCT01877811 | ||
| Sunitinib | Multi-kinase small molecule inhibitor | ||||
| Sorafenib | Multi-kinase small molecule inhibitor | I | NCT01531361 | ||
| HER-2 | AZD8931 | Small molecule inhibitor of EGFR, HER-2, HER-3 | I/II | NCT01862003 | AZD8931 + FOLFIRI |
| Neratinib | Small molecule inhibitor of EGFR, HER-2, HER-3 | II | NCT01953926 | ||
| HER-2 vaccine | B cell peptide vaccine | I | NCT01376505 | ||
| T-DM1 | Antibody-drug conjugate of traztuzumab and DM1 | II | HERACLES-RESCUE | At the time of traztumab failure | |
| Pertuzumab | Anti HER-2 monoclonal antibody | II | HERACLES | Pertuzumab + trastuzumab | |
| Lapatinib | Anti HER-2 small molecule inhibitor | II | HERACLES | Lapatinib + trastuzumab | |
| c-MET (MET, HGFR) | Crizotinib | Multi-kinase small molecule inhibitor | NCT02510001 | Crizotinib + PD-0325901 | |
| Tivantinib | c-MET inhibitor | NCT01892527 | Tivantinib + cetuximab | ||
| Cabozantinib | c-MET and VEGFR2 inhibitor | ||||
| INC280 | Small molecule inhibitor of c-MET | II | NCT2205398 | INC280 + cetuximab | |
| AMG102 | HGF inhibitor | ||||
| AV299 | HGF inhibitor |
Utilizing the NCT number, clinical trial information can be obtained at https://clinicaltrials.gov. FGFR: Fibroblast growth factor receptor; EGFR: Epidermal growth factor receptor; BRAF: v-Raf murine sarcoma viral oncogene homolog B; HER-2: Human growth factor receptor 2; HGFR: Hepatocyte growth factor receptor; TDM-1: Ado-trastuzumab emtansine; VEGFR: Vascular endothelial growth factor receptor.
Fibroblast growth factor receptor in CRC
The fibroblast growth factor receptors (FGFR) comprise a group of highly conserved tyrosine kinase receptors consisting of four members (FGFR1, 2, 3 and 4). These receptors bind to one of 18 secreted glycoprotein ligands, or fibroblast growth factors (FGFs), to their extracellular domain[48]. Binding of the appropriate ligand results in FGFR dimerization, autophosphorylation and activation of downstream signaling pathways that include the MAPK, PI3K/Akt and signaling transducer and activator of transcription or STAT pathway, inducing cell differentiation, growth and survival[49]. FGFR overexpression has been identified in CRC samples, where the presence of FGFR signaling has been identified as playing an important role in the tumor microenvironment, with a correlation in FGFR overexpression with tumor invasion, advanced stage disease and chemotherapy resistance[50-53]. Pre-clinical studies have demonstrated the reversal of chemotherapy resistance by combining small molecule FGFR inhibitors with chemotherapy in CRC cell lines, confirming the importance of targeting this receptor and representing a potential strategy in overcoming treatment resistance in CRC[54].
C-MET (MET)
Hepatocyte growth factor receptor, also known as c-MET, is a proto-oncogene that encodes the tyrosine kinase receptor for hepatocyte growth factor (HGF). Abnormal expression of c-MET through somatic mutations or overexpression has been identified in up to 66.7% of CRC samples and its microenvironment and is a negative prognostic marker related to tumor oncogenesis, invasiveness, local recurrence, and chemotherapy resistance[55-58]. MET activation confers acquired anti-EGFR therapy resistance, through re-activation of anti-apoptotic signaling pathways, including the PI3K and MAPK pathways[59-62]. Thus, inhibiting c-MET represents an emerging target of interest in the development of novel agents in CRC. Pre-clinical studies have demonstrated that the blockade of c-MET inhibits tumor growth in CRC cell lines[63,64], thus agents targeting MET, including small molecule multi-kinase inhibitors (e.g., crizotinib, tivantinib), HGF inhibitors (e.g., AMG102, AV299) and immunotherapeutic agents are of interest and under investigation in the treatment of CRC.
RET
RET is located on chromosome 10q11.2 and encodes a transmembrane receptor tyrosine kinase that has three unique isoforms[65]. Four ligands can bind and activate RET, leading to the aberrant activity of several signaling pathways including PI3K/Akt and MAPK pathway[66]. While the aberrant expression of RET may function as an oncogene in certain solid tumor malignancies including papillary and medullary thyroid cancers[67], in colorectal cancer RET has been identified as a tumor suppressor and as an oncogene. Studies have shown that the hypermethylation and mutational inactivation of RET, as well as RET fusions, promote colorectal cancer formation[68-70]. In several studies, RET mutations were identified in up to 7% of mCRC samples[46,71,72]. Regorafenib, an approved multi-target small molecule inhibitor in metastatic CRC[73], has demonstrated tumor growth inhibition in RET mutant cancer cells lines[70,74], and may explain part of its efficacy in this disease. Further studies investigating its activity in RET mutant CRC is warranted as it may provide further benefit in this specific cohort of CRC patients.
HER-2/Neu
Human growth factor receptor 2, also known as HER-2 or HER-2/neu, is part of the human epidermal growth factor receptor family and has been identified as an oncogene in several solid tumor malignancies, including CRC. Its overexpression has been a poor prognostic biomarker in breast cancer and is an effective therapeutic target in breast and gastric cancer[75-77]. Studies evaluating HER-2 overexpression in CRC have identified HER-2 somatic mutations and amplification in 7% of patients with CRC[46], where pre-clinical studies have demonstrated its role in inducing resistance to anti-EGFR therapy, and its inhibition showing durable tumor regression with anti-HER-2 therapy[78]. Based on these findings, the HERACLES trial, a phase II study investigated dual anti-HER-2 therapy blockade in patients with HER-2 amplified (immunohistochemistry staining 3+ or 2+ with FISH positive (HER2:CEP17 ratio > 2) in > 50% of tumor cells) mCRC with previous anti-EGFR therapy. Patients received the combination of trastuzumab, an anti HER-2 monoclonal antibody with either lapatinib, a small molecule inhibitor of HER-2 or pertuzumab, an anti HER-2 monoclonal antibody. Early results from the lapatinib and trastuzumab arm demonstrated a 35% response rate and median progression free survival of 5.5 mo, despite being heavily pre-treated after failing multiple lines of therapy[79]. Based on these findings, anti HER-2 therapy may be an effective treatment option in a pre-selected patient population with mCRC.
DNA mismatch repair genes and their therapeutic relevance in advanced CRC
Mismatch repair (MMR) genes function to remove erroneous DNA nucleotides during mitosis. With deficient MMR activity, altered DNA nucleotides are incorporated into cells that increase their risk in forming into a neoplastic, hypermutated makeup[80], resulting in microsatellite instability. Deficient MMR activity is found in approximately 15% of CRC, in which 3% is attributed to Lynch Syndrome through germline mutations in MLH1, MSH2, MSH6, PMS2 or EPCAM3 genes[81,82]. The remaining 12% is due to sporadic inactivation of MLH1. While microsatellite instability (MSI) is a driver for tumor formation and proliferation in CRC, recent studies have demonstrated its relevance for potential novel therapeutic agents in this cohort of CRC. Tumors expressing MSI have been characterized by an intense immune infiltration, likely related to a high density of mutations, creating numerous neo-antigens and targets for immune therapies. Based on this rationale, a single-arm phase II study was conducted to assess the clinical efficacy of pembrolizumab, a programmed cell death protein (PD)-1 inhibitor, in patients with treatment resistant, metastatic cancer that did or did not express mismatch-repair deficiency[83]. Individuals with deficient MMR colorectal cancer experienced a response rate of 40% and immune-related PFS of 78% at 20 wk. Interestingly, a lack of activity was associated with mismatch repair-proficient CRC patients, confirming that immunotherapeutic agents may be beneficial in only certain cohorts of CRC, unless alternative approaches can be developed to transform tumors into an immune-responsive phenotype[83]. Based on these findings, several ongoing clinical trials are investigating various novel, immunotherapeutic agents in the treatment of CRC that include studies in patients with MSI high tumors and those with positive PDL-1 expression (Table 3). While current trials will substantiate the role of immune therapy in CRC, a better understanding of mechanisms of acquired resistance, optimal duration of necessary treatment and predictive biomarkers associated with treatment efficacy are paramount.
Table 3.
Current ongoing immune therapy trials for colorectal cancer
| Agent | Class of agent | Phase | Trial number1 | Comment |
| MK-3475 | Anti-PD-1 mAb | II | NCT01876511 | MSI-high tumors |
| MEDI4736 | Anti-PD-L1 mAb | I/II | NCT01693562 | |
| Nivolumab ± ipiliumumab | Anti-PD-1 mAb/anti-CTLA-4 mAb | I/II | NCT02060188 | Recurrent and metastatic CRC |
| MK-3475 + mFOLFOX6 | Anti-PD-1 mAb | II | NCT02375672 | |
| Tremelimumab + MEDI4736 | Anti-CTLA-4 mAb + anti-PD-L1 mAb | I | NCT01975831 |
Utilizing the NCT number, clinical trial information can be obtained at https://clinicaltrials.gov. mAb: Monoclonal antibody; CRC: Colorectal cancer; MSI: Microsatellite instability; CTLA: Cytotoxic T associated lymphocyte protein; PD-1: Program cell death protein 1; PD-L1: Programmed cell death protein ligand 1; FOLFOX: Combination of 5-Flurouracil, folinic acid and oxaliplatin chemotherapy regimen.
Tumor genomic assessment at the time of acquired therapy resistance in mCRC
While anti-tumor activity from targeted therapies can lead to dramatic responses, the clinical benefit is often limited due to inherent and acquired resistance through the acquisition of new tumor genomic alternations, including the oncogenic activation of MET or acquiring RAS mutations, as described above. While tumor genomic alterations are usually assessed through tumor samples, obtaining tissue can be challenging due to insufficient material from tumors only accessible through fine-needle aspirates, as well as the invasive nature of such procedures.
Tumor circulating free DNA can be non-invasively assessed in peripheral blood, a “liquid biopsy,” through the assessment of circulating tumor DNA (ctDNA) and tumor cells (CTCs) and are present in advanced malignancies[84,85]. CtDNA and CTCs can provide dynamic assessments of tumor specific mutations that may arise during the course of therapy. Although previous methodologies demonstrate low rates of sensitivity and concordance, the incorporation of new technology, including real time digital PCR has increased sensitivity (87.2%) and specificity (99.2%) in identifying tumor specific mutations responsible for treatment resistance in patients who initially responded to targeted therapies[18]. While there are limitations, including false negative results as well as the inability to identify genomic alterations from central nervous system (CNS) lesions, this non-invasive tool can monitor patients for resistance-conferring mutations as well as assessing all tumors concurrently, as heterogeneity can exist between different foci of disease.
CONCLUSION
Advancements in genomic sequencing have resulted in our increased understanding of the genomic landscape of CRC, allowing us to develop and tailor personalized therapies for patients. Despite these improvements, future studies are needed to characterize and understand the functionality of the various different mutations of each gene, including mutational assessment at the time of treatment failure. This will allow us to improve therapies for patients with CRC by assessing the prognostic and potential therapeutic implication of genes of interest, and to identify predictive biomarkers of response and resistance.
Footnotes
Conflict-of-interest statement: None of the authors have any potential conflicts of interest, and no financial support was used for the work of this paper.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Manuscript source: Invited manuscript
Peer-review started: March 22, 2016
First decision: May 12, 2016
Article in press: June 13, 2016
P- Reviewer: Cecchin E, Li YY, Meshikhes AW S- Editor: Gong ZM L- Editor: A E- Editor: Wang CH
References
- 1.Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014;64:104–117. doi: 10.3322/caac.21220. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 3.Tol J, Nagtegaal ID, Punt CJ. BRAF mutation in metastatic colorectal cancer. N Engl J Med. 2009;361:98–99. doi: 10.1056/NEJMc0904160. [DOI] [PubMed] [Google Scholar]
- 4.Loupakis F, Cremolini C, Masi G, Lonardi S, Zagonel V, Salvatore L, Cortesi E, Tomasello G, Ronzoni M, Spadi R, et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N Engl J Med. 2014;371:1609–1618. doi: 10.1056/NEJMoa1403108. [DOI] [PubMed] [Google Scholar]
- 5.Loupakis F, Cremolini C, Lonardi S, Tomasello G, Ronzoni M, Zaniboni A, Tonini G, Valsuani C, Chiara S, Boni C, et al. Subgroup analyses in RAS mutant, BRAF mutant and all-wt mCRC pts treated with FOLFOXIRI plus bevacizumab (bev) or FOLFIRI plus bev in the TRIBE study. J Clin Oncol. 2014;32:3519. [Google Scholar]
- 6.Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–1034. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 7.Van Cutsem E, Köhne CH, Láng I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29:2011–2019. doi: 10.1200/JCO.2010.33.5091. [DOI] [PubMed] [Google Scholar]
- 8.Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, André T, Chan E, Lordick F, Punt CJ, et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol. 2010;28:4706–4713. doi: 10.1200/JCO.2009.27.6055. [DOI] [PubMed] [Google Scholar]
- 9.Maughan TS, Adams RA, Smith CG, Meade AM, Seymour MT, Wilson RH, Idziaszczyk S, Harris R, Fisher D, Kenny SL, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet. 2011;377:2103–2114. doi: 10.1016/S0140-6736(11)60613-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tveit KM, Guren T, Glimelius B, Pfeiffer P, Sorbye H, Pyrhonen S, Sigurdsson F, Kure E, Ikdahl T, Skovlund E, et al. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: the NORDIC-VII study. J Clin Oncol. 2012;30:1755–1762. doi: 10.1200/JCO.2011.38.0915. [DOI] [PubMed] [Google Scholar]
- 11.Peeters M, Douillard JY, Van Cutsem E, Siena S, Zhang K, Williams R, Wiezorek J. Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: assessment as prognostic and predictive biomarkers of response to panitumumab. J Clin Oncol. 2013;31:759–765. doi: 10.1200/JCO.2012.45.1492. [DOI] [PubMed] [Google Scholar]
- 12.Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, Heintges T, Lerchenmüller C, Kahl C, Seipelt G, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1065–1075. doi: 10.1016/S1470-2045(14)70330-4. [DOI] [PubMed] [Google Scholar]
- 13.Schwartzberg LS, Rivera F, Karthaus M, Fasola G, Canon JL, Hecht JR, Yu H, Oliner KS, Go WY. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J Clin Oncol. 2014;32:2240–2247. doi: 10.1200/JCO.2013.53.2473. [DOI] [PubMed] [Google Scholar]
- 14.Gysin S, Salt M, Young A, McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer. 2011;2:359–372. doi: 10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 16.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diaz LA, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schellens J, Van Geel R, Bendell J, Spreafico A, Schuler M, Yoshino T, Delord J, Yamada Y, Lolkema MP, Faris JE, et al. Final biomarker analysis of the pahse I study of the selective BRAF V600 inhibitor encorafenib (LGX818) combined with cetuximab with or without the α-specific PI3K inhibitor alpelisib (BYL719) in patients with advanced BRAF-mutant colorectal cancer. Presented at the 106th Annual Meeting of the American Association for Cancer Research. Philadelphia, PA, April 18-22; 2015. [Google Scholar]
- 21.Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, Ponzetti A, Cremolini C, Amatu A, Lauricella C, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:827. doi: 10.1038/nm0715-827b. [DOI] [PubMed] [Google Scholar]
- 22.Leontieva OV, Demidenko ZN, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Differ. 2013;20:1241–1249. doi: 10.1038/cdd.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu B, Fang M, Lu Y, Mendelsohn J, Fan Z. Fibroblast growth factor and insulin-like growth factor differentially modulate the apoptosis and G1 arrest induced by anti-epidermal growth factor receptor monoclonal antibody. Oncogene. 2001;20:1913–1922. doi: 10.1038/sj.onc.1204277. [DOI] [PubMed] [Google Scholar]
- 24.Paternot S, Roger PP. Combined inhibition of MEK and mammalian target of rapamycin abolishes phosphorylation of cyclin-dependent kinase 4 in glioblastoma cell lines and prevents their proliferation. Cancer Res. 2009;69:4577–4581. doi: 10.1158/0008-5472.CAN-08-3260. [DOI] [PubMed] [Google Scholar]
- 25.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 26.Tie J, Gibbs P, Lipton L, Christie M, Jorissen RN, Burgess AW, Croxford M, Jones I, Langland R, Kosmider S, et al. Optimizing targeted therapeutic development: analysis of a colorectal cancer patient population with the BRAF(V600E) mutation. Int J Cancer. 2011;128:2075–2084. doi: 10.1002/ijc.25555. [DOI] [PubMed] [Google Scholar]
- 27.Samowitz WS, Sweeney C, Herrick J, Albertsen H, Levin TR, Murtaugh MA, Wolff RK, Slattery ML. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res. 2005;65:6063–6069. doi: 10.1158/0008-5472.CAN-05-0404. [DOI] [PubMed] [Google Scholar]
- 28.Yokota T, Ura T, Shibata N, Takahari D, Shitara K, Nomura M, Kondo C, Mizota A, Utsunomiya S, Muro K, et al. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br J Cancer. 2011;104:856–862. doi: 10.1038/bjc.2011.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Price TJ, Hardingham JE, Lee CK, Weickhardt A, Townsend AR, Wrin JW, Chua A, Shivasami A, Cummins MM, Murone C, et al. Impact of KRAS and BRAF Gene Mutation Status on Outcomes From the Phase III AGITG MAX Trial of Capecitabine Alone or in Combination With Bevacizumab and Mitomycin in Advanced Colorectal Cancer. J Clin Oncol. 2011;29:2675–2682. doi: 10.1200/JCO.2010.34.5520. [DOI] [PubMed] [Google Scholar]
- 30.Morris V, Overman MJ, Jiang ZQ, Garrett C, Agarwal S, Eng C, Kee B, Fogelman D, Dasari A, Wolff R, et al. Progression-free survival remains poor over sequential lines of systemic therapy in patients with BRAF-mutated colorectal cancer. Clin Colorectal Cancer. 2014;13:164–171. doi: 10.1016/j.clcc.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tran B, Kopetz S, Tie J, Gibbs P, Jiang ZQ, Lieu CH, Agarwal A, Maru DM, Sieber O, Desai J. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117:4623–4632. doi: 10.1002/cncr.26086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28:4697–4705. doi: 10.1200/JCO.2009.27.4860. [DOI] [PubMed] [Google Scholar]
- 33.Seymour MT, Brown SR, Middleton G, Maughan T, Richman S, Gwyther S, Lowe C, Seligmann JF, Wadsley J, Maisey N, et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncol. 2013;14:749–759. doi: 10.1016/S1470-2045(13)70163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, Infante JR, Millward M, Pavlick AC, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379:1893–1901. doi: 10.1016/S0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH, Kaempgen E, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 38.Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Maru D, Morris V, Janku F, Dasari A, Chung W, et al. Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF-Mutated Colorectal Cancer. J Clin Oncol. 2015;33:4032–4038. doi: 10.1200/JCO.2015.63.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate AJ, Settleman J, Engelman JA. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal. 2010;3:ra84. doi: 10.1126/scisignal.2001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, Hamid O, Messersmith WA, Daud A, Kurzrock R, et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J Clin Oncol. 2015;33:4023–4031. doi: 10.1200/JCO.2015.63.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yaeger R, Cercek A, O’Reilly EM, Reidy DL, Kemeny N, Wolinsky T, Capanu M, Gollub MJ, Rosen N, Berger MF, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res. 2015;21:1313–1320. doi: 10.1158/1078-0432.CCR-14-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hong DS, Morris VK, Fu S, Overman MJ, Piha-Paul SA, Kee BK, Zinner R, Fogelman DR, Mistry R, Shureiqi I, et al. Phase 1B study of vemurafenib in combination with irinotecan and cetuximab in patients with BRAF-mutated advanced cancers and metastatic colorectal cancer. J Clin Oncol. 2014;32:3516. [Google Scholar]
- 44.Tabernero J, Chan E, Baselga J, Blay J-Y, Chau I, Hyman D, Raje N, Wolf J, Sirzen F, Veronese L, et al. VE-BASKET, a Simon 2-stage adaptive design, phase II, histology-independent study in nonmelanoma solid tumors harboring BRAF V600 mutations (V600m): Activity of vemurafenib (VEM) with or without cetuximab (CTX) in colorectal cancer (CRC) J Clin Oncol. 2014;32:3518. [Google Scholar]
- 45.Atreya CE, Van Cutsem E, Bendell JC, Schellens JHM, Gordon MS, McRee A, O’Dwyer PJ, Muro K, Tabernero J, Van Geel R, et al. Updated efficacy of the MEK inhibitor trametinib (T), BRAF inhibitor dabrafenib (D), and anti-EGFR antibody panitumumab (P) in patients (pts) with BRAF V600E mutated (BRAFm) metastatic colorectal cancer (mCRC) J Clin Oncol. 2015;33:103. [Google Scholar]
- 46.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.El-Deiry WS, Vijayvergia N, Xiu J, Scicchitano A, Lim B, Yee NS, Harvey HA, Gatalica Z, Reddy S. Molecular profiling of 6,892 colorectal cancer samples suggests different possible treatment options specific to metastatic sites. Cancer Biol Ther. 2015;16:1726–1737. doi: 10.1080/15384047.2015.1113356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Belov AA, Mohammadi M. Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a015958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger J. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell. 1997;89:693–702. doi: 10.1016/s0092-8674(00)80252-4. [DOI] [PubMed] [Google Scholar]
- 50.Stevenson L, Allen WL, Turkington R, Jithesh PV, Proutski I, Stewart G, Lenz HJ, Van Schaeybroeck S, Longley DB, Johnston PG. Identification of galanin and its receptor GalR1 as novel determinants of resistance to chemotherapy and potential biomarkers in colorectal cancer. Clin Cancer Res. 2012;18:5412–5426. doi: 10.1158/1078-0432.CCR-12-1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bange J, Prechtl D, Cheburkin Y, Specht K, Harbeck N, Schmitt M, Knyazeva T, Müller S, Gärtner S, Sures I, et al. Cancer progression and tumor cell motility are associated with the FGFR4 Arg(388) allele. Cancer Res. 2002;62:840–847. [PubMed] [Google Scholar]
- 52.Liu R, Li J, Xie K, Zhang T, Lei Y, Chen Y, Zhang L, Huang K, Wang K, Wu H, et al. FGFR4 promotes stroma-induced epithelial-to-mesenchymal transition in colorectal cancer. Cancer Res. 2013;73:5926–5935. doi: 10.1158/0008-5472.CAN-12-4718. [DOI] [PubMed] [Google Scholar]
- 53.Henriksson ML, Edin S, Dahlin AM, Oldenborg PA, Öberg Å, Van Guelpen B, Rutegård J, Stenling R, Palmqvist R. Colorectal cancer cells activate adjacent fibroblasts resulting in FGF1/FGFR3 signaling and increased invasion. Am J Pathol. 2011;178:1387–1394. doi: 10.1016/j.ajpath.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Turkington RC, Longley DB, Allen WL, Stevenson L, McLaughlin K, Dunne PD, Blayney JK, Salto-Tellez M, Van Schaeybroeck S, Johnston PG. Fibroblast growth factor receptor 4 (FGFR4): a targetable regulator of drug resistance in colorectal cancer. Cell Death Dis. 2014;5:e1046. doi: 10.1038/cddis.2014.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Al-Maghrabi J, Emam E, Gomaa W, Saggaf M, Buhmeida A, Al-Qahtani M, Al-Ahwal M. c-MET immunostaining in colorectal carcinoma is associated with local disease recurrence. BMC Cancer. 2015;15:676. doi: 10.1186/s12885-015-1662-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Di Renzo MF, Olivero M, Giacomini A, Porte H, Chastre E, Mirossay L, Nordlinger B, Bretti S, Bottardi S, Giordano S. Overexpression and amplification of the met/HGF receptor gene during the progression of colorectal cancer. Clin Cancer Res. 1995;1:147–154. [PubMed] [Google Scholar]
- 57.Osada S, Matsui S, Komori S, Yamada J, Sanada Y, Ihawa A, Tanaka Y, Tokuyama Y, Okumura N, Nonaka K, et al. Effect of hepatocyte growth factor on progression of liver metastasis in colorectal cancer. Hepatogastroenterology. 2010;57:76–80. [PubMed] [Google Scholar]
- 58.Gayyed MF, Abd El-Maqsoud NM, El-Hameed El-Heeny AA, Mohammed MF. c-MET expression in colorectal adenomas and primary carcinomas with its corresponding metastases. J Gastrointest Oncol. 2015;6:618–627. doi: 10.3978/j.issn.2078-6891.2015.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A, Zecchin D, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3:658–673. doi: 10.1158/2159-8290.CD-12-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liska D, Chen CT, Bachleitner-Hofmann T, Christensen JG, Weiser MR. HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin Cancer Res. 2011;17:472–482. doi: 10.1158/1078-0432.CCR-10-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Troiani T, Martinelli E, Napolitano S, Vitagliano D, Ciuffreda LP, Costantino S, Morgillo F, Capasso A, Sforza V, Nappi A, et al. Increased TGF-α as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res. 2013;19:6751–6765. doi: 10.1158/1078-0432.CCR-13-0423. [DOI] [PubMed] [Google Scholar]
- 62.Yonesaka K, Satoh T, Ueda S, Yoshida T, Takeda M, Shimizu T, Okamoto I, Nishio K, Tamura T, Nakagawa K. Circulating hepatocyte growth factor is correlated with resistance to cetuximab in metastatic colorectal cancer. Anticancer Res. 2015;35:1683–1689. [PubMed] [Google Scholar]
- 63.Sun Y, Sun L, An Y, Shen X. Cabozantinib, a Novel c-Met Inhibitor, Inhibits Colorectal Cancer Development in a Xenograft Model. Med Sci Monit. 2015;21:2316–2321. doi: 10.12659/MSM.893590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Song EK, Tai WM, Messersmith WA, Bagby S, Purkey A, Quackenbush KS, Pitts TM, Wang G, Blatchford P, Yahn R, et al. Potent antitumor activity of cabozantinib, a c-MET and VEGFR2 inhibitor, in a colorectal cancer patient-derived tumor explant model. Int J Cancer. 2015;136:1967–1975. doi: 10.1002/ijc.29225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Runeberg-Roos P, Saarma M. Neurotrophic factor receptor RET: structure, cell biology, and inherited diseases. Ann Med. 2007;39:572–580. doi: 10.1080/07853890701646256. [DOI] [PubMed] [Google Scholar]
- 66.Arighi E, Borrello MG, Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 2005;16:441–467. doi: 10.1016/j.cytogfr.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 67.Phay JE, Shah MH. Targeting RET receptor tyrosine kinase activation in cancer. Clin Cancer Res. 2010;16:5936–5941. doi: 10.1158/1078-0432.CCR-09-0786. [DOI] [PubMed] [Google Scholar]
- 68.Luo Y, Tsuchiya KD, Il Park D, Fausel R, Kanngurn S, Welcsh P, Dzieciatkowski S, Wang J, Grady WM. RET is a potential tumor suppressor gene in colorectal cancer. Oncogene. 2013;32:2037–2047. doi: 10.1038/onc.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mokarram P, Kumar K, Brim H, Naghibalhossaini F, Saberi-firoozi M, Nouraie M, Green R, Lee E, Smoot DT, Ashktorab H. Distinct high-profile methylated genes in colorectal cancer. PLoS One. 2009;4:e7012. doi: 10.1371/journal.pone.0007012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Le Rolle AF, Klempner SJ, Garrett CR, Seery T, Sanford EM, Balasubramanian S, Ross JS, Stephens PJ, Miller VA, Ali SM, et al. Identification and characterization of RET fusions in advanced colorectal cancer. Oncotarget. 2015;6:28929–28937. doi: 10.18632/oncotarget.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–664. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brannon AR, Vakiani E, Sylvester BE, Scott SN, McDermott G, Shah RH, Kania K, Viale A, Oschwald DM, Vacic V, et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014;15:454. doi: 10.1186/s13059-014-0454-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, Humblet Y, Bouché O, Mineur L, Barone C, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:303–312. doi: 10.1016/S0140-6736(12)61900-X. [DOI] [PubMed] [Google Scholar]
- 74.Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schütz G, Thierauch KH, Zopf D. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129:245–255. doi: 10.1002/ijc.25864. [DOI] [PubMed] [Google Scholar]
- 75.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh DY, Diéras V, Guardino E, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–1791. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 77.Janjigian YY, Werner D, Pauligk C, Steinmetz K, Kelsen DP, Jäger E, Altmannsberger HM, Robinson E, Tafe LJ, Tang LH, et al. Prognosis of metastatic gastric and gastroesophageal junction cancer by HER2 status: a European and USA International collaborative analysis. Ann Oncol. 2012;23:2656–2662. doi: 10.1093/annonc/mds104. [DOI] [PubMed] [Google Scholar]
- 78.Kavuri SM, Jain N, Galimi F, Cottino F, Leto SM, Migliardi G, Searleman AC, Shen W, Monsey J, Trusolino L, et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015;5:832–841. doi: 10.1158/2159-8290.CD-14-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Siena S, Sartore-Bianchi A, Lonardi S, Trusolino L, Martino C, Bencardino K, Leone F, Zagonel V, Valtorta V, Torri V, et al. Trastuzumab and lapatinib in HER2-amplified metastatic colorectal cancer patients (mCRC): The HERACLES trial. J Clin Oncol. 2015;33:3508. [Google Scholar]
- 80.Parsons R, Li GM, Longley MJ, Fang WH, Papadopoulos N, Jen J, de la Chapelle A, Kinzler KW, Vogelstein B, Modrich P. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell. 1993;75:1227–1236. doi: 10.1016/0092-8674(93)90331-j. [DOI] [PubMed] [Google Scholar]
- 81.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352:1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 82.Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, Lee TY, Bodmer D, Hoenselaar E, Hendriks-Cornelissen SJ, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3’ exons of TACSTD1. Nat Genet. 2009;41:112–117. doi: 10.1038/ng.283. [DOI] [PubMed] [Google Scholar]
- 83.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fleischhacker M, Schmidt B. Circulating nucleic acids (CNAs) and cancer--a survey. Biochim Biophys Acta. 2007;1775:181–232. doi: 10.1016/j.bbcan.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 85.Alix-Panabières C, Schwarzenbach H, Pantel K. Circulating tumor cells and circulating tumor DNA. Annu Rev Med. 2012;63:199–215. doi: 10.1146/annurev-med-062310-094219. [DOI] [PubMed] [Google Scholar]