

Abstract
1,N6-Ethenodeoxyadenosine (1,N6-ϵdA) is the major etheno lesion formed in the reaction of DNA with epoxides substituted with good leaving groups (e.g. vinyl chloride epoxide). This lesion is also formed endogenously in DNA from lipid oxidation. Recombinant human DNA polymerase η (hpol η) can replicate oligonucleotide templates containing 1,N6-ϵdA. In steady-state kinetic analysis, hpol η preferred to incorporate dATP and dGTP, compared with dTTP. Mass spectral analysis of incorporation products also showed preferred purine (A, G) incorporation and extensive −1 frameshifts, suggesting pairing of the inserted purine and slippage before further replication. Five x-ray crystal structures of hpol η ternary complexes were determined, three at the insertion and two at the extension stage. Two insertion complexes revealed incoming non-hydrolyzable dATP or dGTP analogs not pairing with but instead in a staggered configuration relative to 1,N6-ϵdA in the anti conformation, thus opposite the 5′-T in the template, explaining the proclivity for frameshift misincorporation. In another insertion complex, dTTP was positioned opposite 1,N6-ϵdA, and the adduct base was in the syn conformation, with formation of two hydrogen bonds. At the extension stage, with either an incorporated dA or dT opposite 1,N6-ϵdA and 2′-deoxythymidine-5′-[(α,β)-imido]triphosphate opposite the 5′-A, the 3′-terminal nucleoside of the primer was disordered, consistent with the tendency not to incorporate dTTP opposite 1,N6-ϵdA. Collectively, the results show a preference for purine pairing opposite 1,N6-ϵdA and for −1 frameshifts.
Keywords: DNA damage, DNA enzyme, DNA polymerase, enzyme kinetics, X-ray crystallography
Introduction
Chemical or physical damage to DNA can lead to replication blockage and miscoding, which in turn can cause death or mutation of cells (1). Such changes can be associated with aging (2), cancer, and other diseases (3–6). Chemical damage to DNA can be the result of either exogenous (e.g. vinyl chloride (7)) or endogenous origin (e.g. oxygen radicals (8)). Some specific types of DNA damage can result from either exogenous or endogenous sources, as exemplified by the etheno (ϵ) adducts (9).
The etheno adducts are so named because they have an extra two carbons attached to DNA bases in an exocyclic arrangement (10). The history goes back to the chemistry of some unusual natural nucleosides found in tRNA, e.g. wybutosine and wyosine (11, 12), and also synthetic work by Leonard and co-workers, who utilized fluorescent etheno analogs to monitor biochemical phenomena (12–15). Barbin et al. (16) and Laib et al. (17) provided evidence that etheno adducts might be involved in the carcinogenicity of vinyl chloride, a known liver carcinogen in humans. Subsequent work showed the presence of etheno DNA adducts in rats that had never been exposed to vinyl chloride (or related vinyl monomers) (18) and that the adducts are the result of lipid peroxidation (19, 20). The reaction of the primary vinyl chloride oxidation product 2-chloroxirane with DNA yields 1,N6-ethenodeoxyadenosine (1,N6-ϵdA)5 as the major etheno adduct (21), although the repair of this adduct is faster than that of some others, such that the N2,3-ethenodeoxyguanosine levels become higher (22).
1,N6-ϵdA has been incorporated into extrachromosomal vectors (23) and studied in cells. The adduct was reported not to be very mutagenic in Escherichia coli (23, 24), but this result may be due to extensive repair by the dioxygenase AlkB, which was later discovered to convert this lesion to deoxyadenosine (25). 1,N6-ϵdA has been reported to be mutagenic in simian COS7 (24) and human HeLa, HCT116, and HEK293 cells (26, 27) and miscoding in human HeLa and XPV cell extracts (28). The bases A, C, and G have all been reported to be misinserted in these systems (24, 26–28) as well as T (i.e. no miscoding). Structural studies (NMR and x-ray) have been reported on oligonucleotides (in the absence of polymerases), with non-planar pairing between 1,N6-ϵdA and dT and anti configurations of both bases (29). 1,N6-ϵdA:G pairing involved a syn configuration of 1,N6-ϵdA and anti configuration of G (30). An x-ray crystallography study of 1,N6-ϵdA:G pairing was interpreted to involve three H-bonds, with distortion of the oligonucleotide backbone to accommodate the pair (31).
Studies of misincorporation at 1,N6-ϵdA have yielded varying results with individual DNA polymerases (pols). Singer et al. (32) reported that E. coli pol I and avian myelovirus reverse transcriptase inserted G, to some extent, but that dTTP incorporation was not blocked. When 1,N6-ϵA was incorporated into an oligoribonucleotide, it was a complete block to avian myelovirus and Moloney murine leukemia virus reverse transcriptases (33). Human pol η (hpol η) was reported to bypass this lesion (34).
Levine et al. (34) reported that hpol η was 100-fold more active than hpol κ in replication past 1,N6-ϵdA. We analyzed incorporation events using steady-state kinetics and LC-MS analysis of primers extended opposite 1,N6-ϵdA by hpol η, in different sequence contexts. The results show hpol η bypass past 1,N6-ϵdA in a highly error-prone manner, with a proclivity for incorporation of purines and generation of frameshifts. We also describe five x-ray crystal structures of hpol η ternary complexes, three containing 1,N6-ϵdA paired with dTTP, dAMPNPP, or dGMPNPP (non-hydrolyzable dATP/dGTP analogs) at the insertion stage and two further structures of complexes at the extension stage, with either dA or dT opposite 1,N6-ϵdA followed by a nascent pair between template A and an incoming dTMPNPP.
Results
Steady-state Kinetics of dNTP Incorporation Opposite A and 1,N6-ϵdA
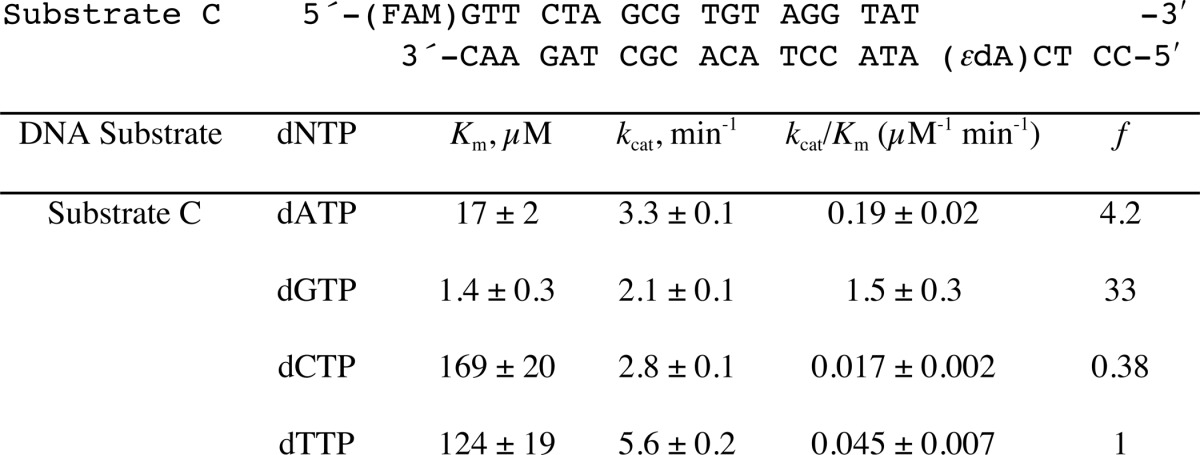
To determine the efficiency and fidelity of translesion synthesis 1,N6-ϵdA adducts by hpol η, steady-state kinetic analysis was performed. With an unmodified template (substrate B, Table 1), hpol η incorporated a single dNTP opposite A in the order of preference T ≫ A > G > C (Table 2), as expected. However, incorporation of dTTP opposite 1,N6-ϵdA was very unfavorable, with a drastically decreased catalytic efficiency (1000-fold) compared with that of dTTP insertion opposite A (Table 2). In this study, dATP and dGTP were inserted more efficiently (3.7- and 2.5-fold, respectively, Table 2) than dTTP opposite 1,N6-ϵdA. These results indicated that hpol η catalyzes translesion synthesis opposite 1,N6-ϵdA in a very error-prone manner. Another sequence context containing 1,N6-ϵdA (substrate C, Table 1), which had been used in a previous study by Levine et al. (34), was also included in our steady-state kinetic studies. Consistently, a high error rate for incorporation opposite 1,N6-ϵdA by hpol η was observed, with preference for dGTP and dATP incorporation compared with dTTP (33- and 4.2-fold higher, respectively) (Table 3 and supplemental Fig. S1).
TABLE 1.
Oligonucleotides used in this study
ϵdA is 1,N6-ϵdA.

TABLE 2.
Steady-state kinetics of incorporation of individual dNTPs opposite 1,N6-ϵdA and A (Substrates A and B)
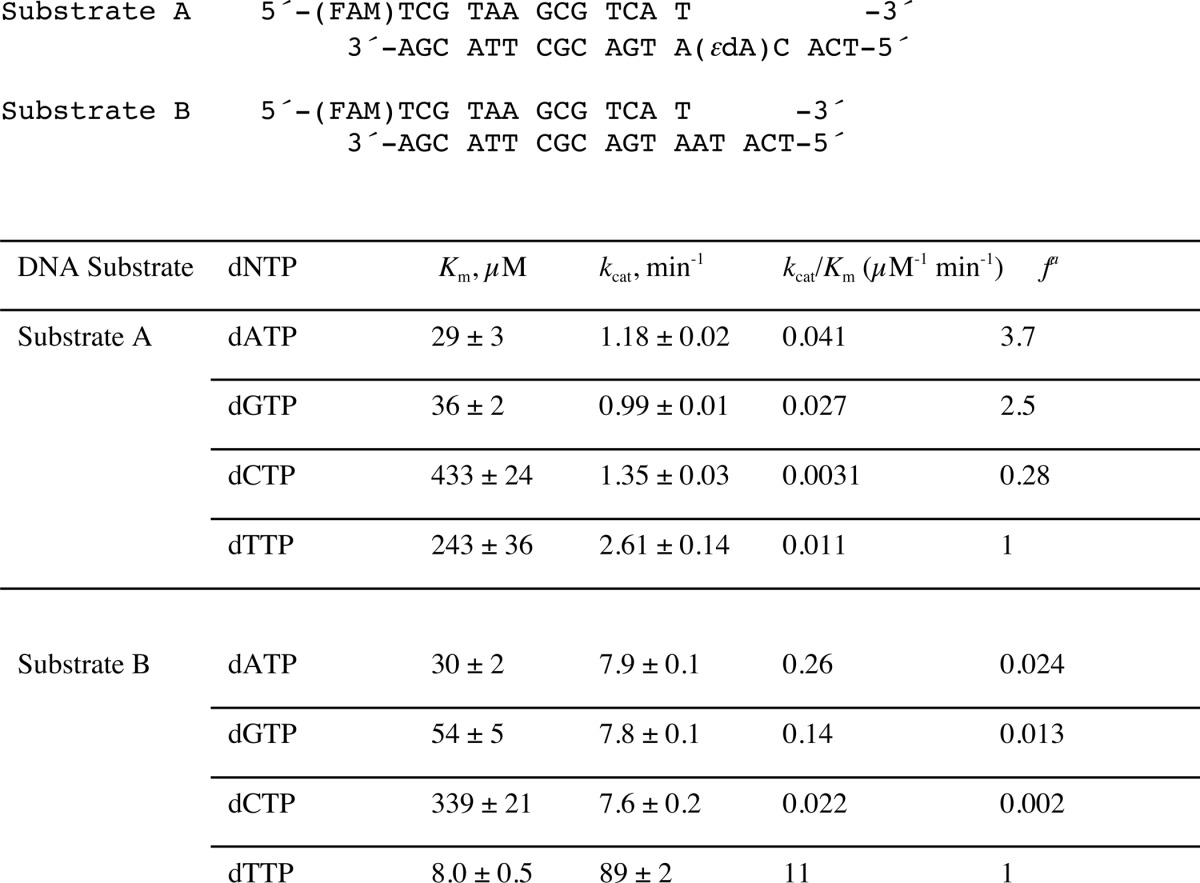
a Misinsertion frequency: f=(kcat/Km)dNTP/(kcat/Km)dTTP. The S.E. (±) is from the fit in Prism software.
TABLE 3.
Steady-state kinetics of incorporation of individual dNTPs opposite 1,N6-ϵdA within an alternate sequence context (Substrate C) (34)
The S.E. (±) is from the fit in Prism software. See Supplemental Data Fig. S1 for plots.
Pre-steady-state Kinetics
dNTP incorporation opposite 1,N6-ϵdA was also examined in the sequence context of substrate A using a rapid quench method. However, no burst phase was observed with any of the four dNTPs, indicating that a step preceding nucleotidyl transfer is rate-limiting in all four cases (supplemental Fig. S2).
LC-MS/MS Analysis of Primer Extension Products
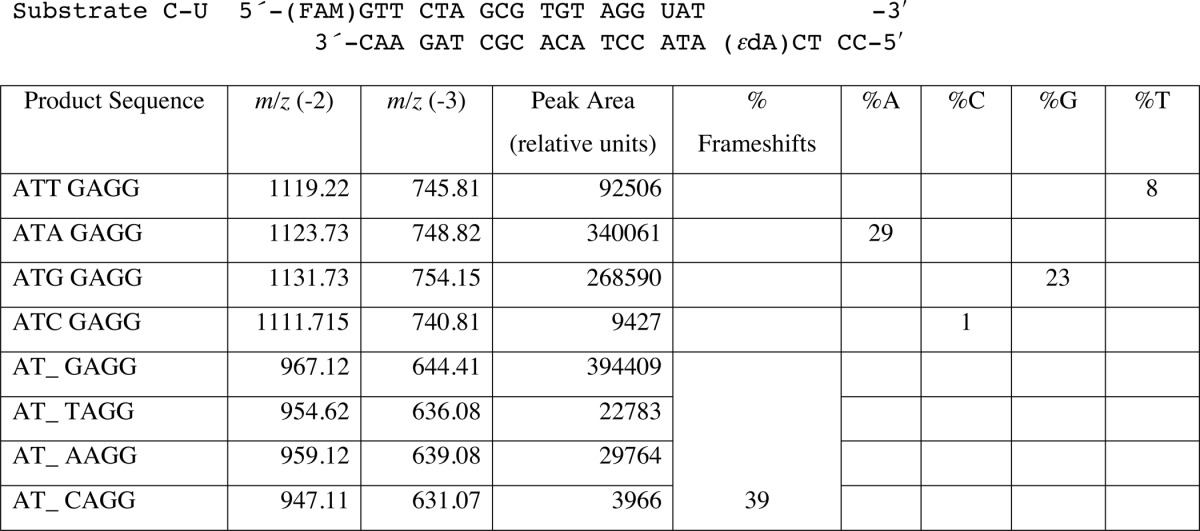
In the steady-state kinetic analysis, hpol η showed a high misinsertion frequency opposite 1,N6-ϵdA during single nucleotide extension (Tables 2 and 3). To gain insight into the ability of hpol η to extend beyond the lesion, an LC-MS/MS method previously developed in this laboratory (34–36) and applied extensively (37–43) was used for sequence analysis of the extension products. dU-containing primers were extended by hpol η in the presence of all four dNTPs, followed by treatment with UDG and piperidine to cleave the fully extended products into shorter fragments for LC-MS/MS analysis (35, 36). Sequences of full-length extension products and relative yields were determined by LC-MS/MS analysis (Fig. 1 and Tables 4–6). The −1 frameshift products accounted for 37% of total products in a sequence with a T positioned 5′ of the 1,N6-ϵdA (substrate D-U, Tables 1 and 4). The fraction of extension products with a G incorporated opposite the lesion was 27% and with an A incorporated opposite the lesion was 25%. Small amounts of products with T (11%) and C (1%) incorporated were also observed in the mass spectra. The formation of the −1 frameshift products may be the result of the incoming dATP skipping the lesion and pairing with the neighboring base T (Table 4).
FIGURE 1.
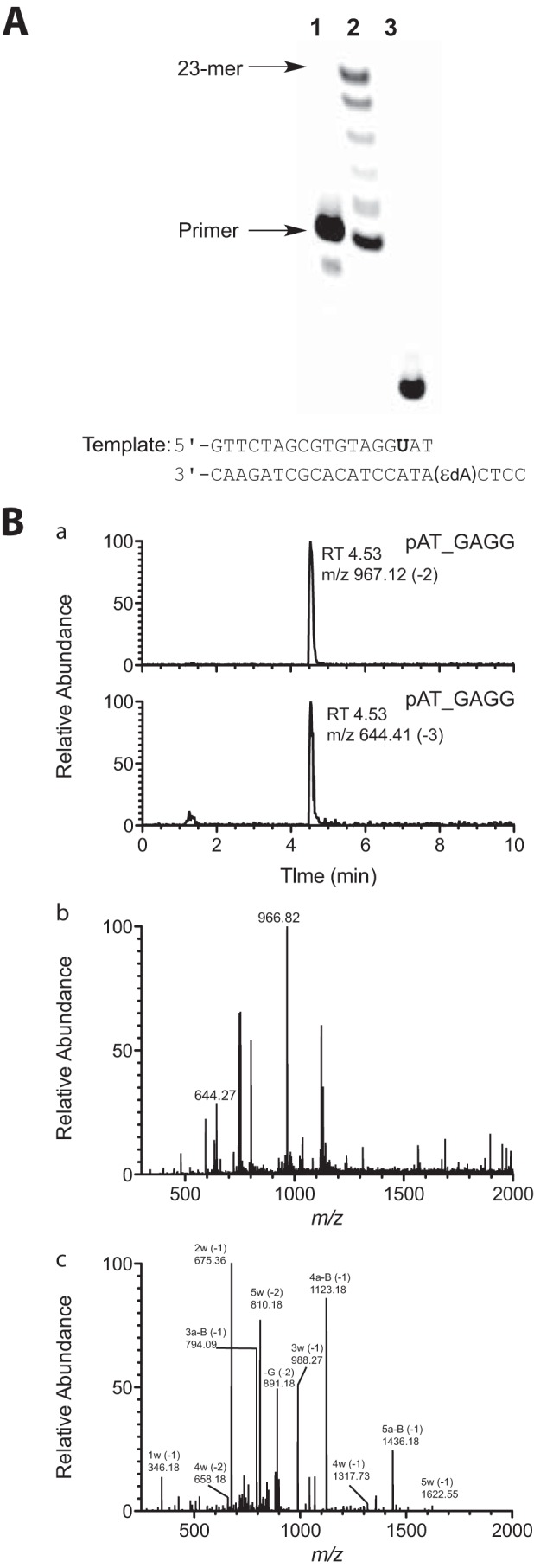
LC-MS analysis of products of extension of primer (opposite template 1,N6-ϵdA, substrate C-U, Table 1) by hpol η in the presence of all four dNTPs. A, denaturing PAGE image showing hpol η extension across the 1,N6-ϵdA adduct: lane 1, 18-mer oligonucleotide primer containing a 5′-FAM label (from substrate C-U, Table 1); lane 2, TLS polymerase extension reaction with hpol η and 18-mer primer/23-mer template duplex (substrate C-U, Table 1); lane 3, cleavage of hpol η extension reaction products with UDG and piperidine. B, mass spectra of frameshift product pAT_GAGG (relative abundance of total ion current measured) (underscore denotes frameshift): panel a, LC chromatograms of product ions m/z 967 (−2 charge) and m/z 644 (−3 charge); panel b, mass spectrum of peak eluted at tR 4.53 min in panel b (product mixture); panel c, CID spectrum of m/z 967. See supplemental Table S1 for assignments.
TABLE 4.
LC-MS analysis of products of hpol η replication past 1,N6-ϵdA (T 5′-adjacent to adduct, Substrate D-U)
Products were cut at the U and begin at the CAT. An underscore represents a frameshift (deletion).
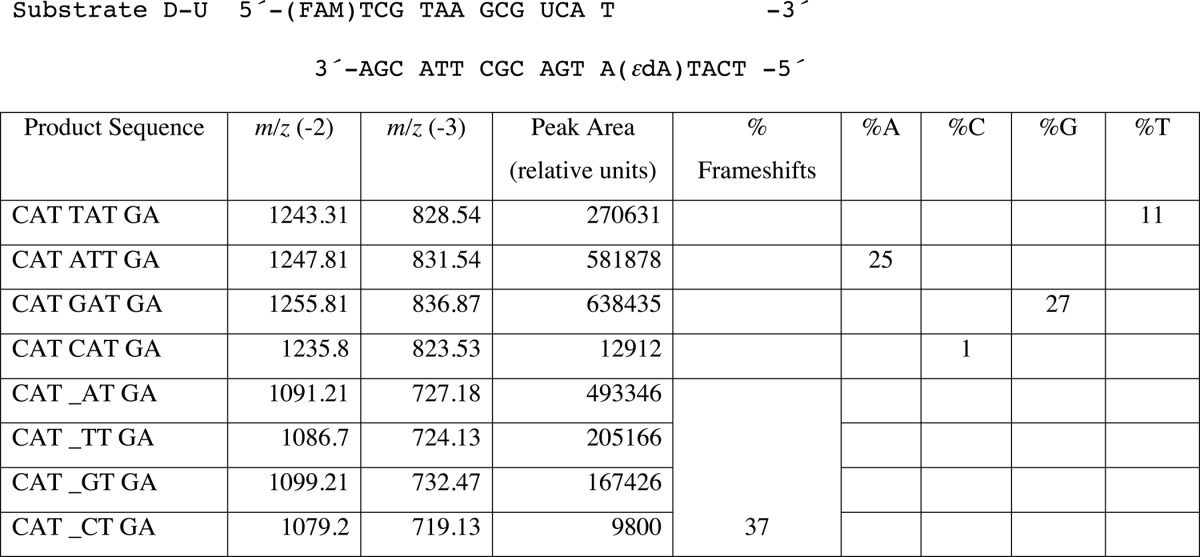
TABLE 5.
LC-MS analysis of products of hpol η replication past template 1,N6-ϵdA (C 5′-adjacent to adduct, Substrate A-U)
Products were cut at the U and begin at the 3' T. An underscore represents a frameshift (deletion). In this case the oligonucleotide pair included an upstream UT mispair. See Supplemental Data Fig. S3 for LC-MS data. The relative units differ from Tables 4 and 6 due to instrumental settings.

TABLE 6.
LC-MS analysis of products of hpol η replication past template 1,N6-ϵdA (T 5′-adjacent to adduct, Substrate C-U) (34)
Products were cut at the U and begin at the 3′ AT. An underscore represents a frameshift (deletion). See Fig. 1 and Supplemental Data Table S1 for LC-MS data.
The T on the 5′ side of 1,N6-ϵdA in the template was replaced with a C, to evaluate the effect of substitution of the (5′) pyrimidine (substrate A-U, Table 1). LC-MS/MS results revealed 14 sequences of full-length products corresponding to nine peaks of M − 2H ions (Table 5). With this template, 70% of the products were due to −1 frameshifts, followed by 16% A and 14% T incorporation opposite the lesion. Extension products containing a G opposite 1,N6-ϵdA were not observed in this setting, presumably because the dGTP skipped 1,N6-ϵdA and paired with the C positioned 5′ to the lesion to yield a frameshift. Human pol η, like many other DNA polymerases (35), can also catalyze blunt-end addition of dATP or dGTP. Some extension oligonucleotides were added to the second nucleotide following the blunt end in this experiment (Table 5).
We also included a template containing 1,N6-ϵdA in another sequence context with a C in the 5′ position (substrate C-U, Table 1), one that had been used in a previous study of 1,N6-ϵdA by Levine et al. (34) (Table 6, Fig. 1B, supplemental Table S1). Again we found −1 frameshifts (39%, dominated by an A in the next position) and incorporation of A (29%) and G (23%) opposite 1,N6-ϵdA, instead of T (8%) and C (<1%), consistent with our results from the steady-state study with this sequence (Table 3 and supplemental Fig. S1).
Crystal Structures of Insertion-stage Ternary Human pol η Complexes
We determined five crystal structures of ternary hpol η-DNA·dNTP complexes with template strands containing 1,N6-ϵdA, three of which were trapped at the insertion stage. The resolution of these three structures varied between 2.12 and 2.26 Å (Table 7), and representative images of the quality of the final electron density are provided in Fig. 2, A–C. The first two complexes are of incoming dAMPNPP and dGMPNPP opposite 1,N6-ϵdA, the nucleoside triphosphates preferentially incorporated according to the steady-state kinetic data (ϵdA:dAMPNPP and ϵdA:dGMPNPP, see Tables 1–3 and 7). The structures revealed that the purine bases of incoming nucleotides adopt a staggered orientation relative to 1,N6-ϵdA in the anti conformation. This arrangement leads to extensive cross-strand stacking between the adducted base and A or G (Fig. 3). Stacking interactions between 1,N6-ϵdA and adenines from the incoming nucleotide and 3′-adjacent template residue appear to be slightly more favorable (Fig. 3B) compared with the structure with incoming G (Fig. 3D). In the latter, the adduct base appears to have shifted slightly into the major groove, whereas the relative positions of the base portions of incoming nucleotides relative to the template A located 3′ to the adduct are very similar in the two structures. In both, Gln-38 forms an H-bond to the sugar O4′ of 1,N6-ϵdA (Fig. 3). In the structure with dGMPNPP, guanine N2 and Gln-38 are also engaged in an H-bond (Fig. 3B), although this interaction is absent in the insertion-stage complex with incoming dAMPNPP (Fig. 3D). Shared properties of the two structures are the conformation of the template nucleotide situated 5′ to 1,N6-ϵdA, in that this T is directed away from the active site and is not engaged in a stacking interaction with the adduct (Fig. 3, A and C). Furthermore, the A:T base pair at the −1 position displays strong buckling in both complexes, with the thymine plane tilted relative to 1,N6-ϵdA. However, this orientation of the 3′-terminal primer nucleotide still leaves its O3′ at an optimal position to carry out a nucleophilic attack at the α-phosphate of the incoming nucleotide (AMPNPP or GMPNPP). The α- and β-phosphate groups of the latter are engaged in an electrostatically favorable interaction with Arg-61 from the hpol η finger domain.
TABLE 7.
Crystal data, data collection parameters, and structure refinement statistics
| Complex | ϵdA·dTTP | ϵdA·dAMPNPP | ϵdA·dGMPNPP | ϵdA·dT extension | ϵdA·dA extension |
|---|---|---|---|---|---|
| Data collection | |||||
| Wavelength (Å) | 0.97856 | 0.97872 | 0.97872 | 0.97856 | 0.97856 |
| Space group | P61 | P61 | P61 | P61 | P61 |
| Resolution (Å) | 50–2.26 (2.30–2.26)a | 50–2.12 (2.16–2.12) | 50–2.15 (2.19–2.15) | 50–2.30 (2.34–2.30) | 50–1.79 (1.82–1.79) |
| Unit cell a = b, c (Å) | 99.04, 81.83 | 99.13, 81.40 | 98.93, 81.71 | 98.42, 81.97 | 98.66, 82.00 |
| Unique reflections | 21,572 (1,058) | 25,919 (1,305) | 24,805 (1,213) | 20,156 (1,004) | 42,790 (2,112) |
| Completeness (%) | 100 (99.8) | 100 (100) | 100 (100) | 99.9 (99.9) | 99.7 (99.8) |
| I/σ(I) | 12.57 (1.70) | 15.90 (1.81) | 15.00 (1.70) | 11.80 (1.96) | 19.52 (1.83) |
| Wilson B-factor (Å2) | 18.5 | 26.7 | 25.7 | 15.1 | 17.6 |
| R-merge | 0.165 (0.893) | 0.135 (0.921) | 0.136 (0.996) | 0.169 (0.879) | 0.102 (0.902) |
| Redundancy | 7.3 (5.1) | 7.6 (7.2) | 7.6 (7.4) | 5.6 (5.1) | 7.5 (7.3) |
| Refinement | |||||
| R-work | 0.156 (0.196) | 0.175 (0.200) | 0.170 (0.221) | 0.156 (0.197) | 0.164 (0.204) |
| R-free | 0.212 (0.285) | 0.239 (0.271) | 0.223 (0.296) | 0.233 (0.295) | 0.212 (0.260) |
| No. of atoms protein/DNA | 3,387/390 | 3,395/390 | 3,397/390 | 3,367/374 | 3,429/374 |
| dNMPNPP/Mg2+ | 29/2 (Ca2+) | 30/2 | 31/2 | 29/2 | 29/2 |
| Water/solute | 351/1 | 308/3 | 343/1 | 332/0 | 532/2 |
| Protein residues | 434 | 436 | 436 | 431 | 447 |
| B-factor (Å2) | |||||
| Average | 29.7 | 35.1 | 33.9 | 27.4 | 23.7 |
| Protein/DNA | 28.9/33.4 | 34.3/37.7 | 32.9/38.5 | 26.3/33.9 | 21.7/28.5 |
| dNMPNPP/Mg2+ | 20.6/20.1 | 29.8/28.6 | 38.3/36.3 | 16.6/14.5 | 12.4/10.1 |
| Water/glycerol | 34.1/25.0 | 39.7/36.9 | 38.1/22.0 | 31.4/ - | 32.5/21.5 |
| Root mean square deviations | |||||
| Bonds (Å) | 0.010 | 0.009 | 0.009 | 0.008 | 0.011 |
| Angles (degree) | 1.2 | 1.1 | 1.2 | 1.1 | 1.1 |
| Ramachandran | |||||
| Favored (%) | 97 | 97 | 97 | 97 | 98 |
| Allowed (%) | 3 | 2.5 | 2.8 | 2.5 | 1.5 |
| Outliers (%) | 0 | 0.5 | 0.2 | 0.5 | 0.5 |
| Protein Data Band code | 5DG7 | 5DG8 | 5DG9 | 5DGA | 5DGB |
a Statistics for the highest resolution shell are shown in parentheses.
FIGURE 2.

Quality of the final models of ternary hpol η complexes with 1,N6-ϵdA-adducted DNA template strands. Fourier (2Fo − Fc) sum electron density drawn at the 1σ threshold (green meshwork) around the active site region. A, staggered arrangement of dAMPNPP and 1,N6-ϵdA; insertion stage (ϵdA:dAMPNPP, Tables 1 and 7). B, staggered arrangement of dGMPNPP and 1,N6-ϵdA; insertion stage (ϵdA:dGMPNPP, Tables 1 and 7). C, dTTP opposite syn 1,N6-ϵdA; insertion stage (ϵdA:dTTP, Tables 1 and 7). D, anti 1,N6-ϵdA opposite conformationally disordered primer dA, followed by dTMPNPP opposite template dA; extension stage (ϵdA:dA extension, Tables 1 and 7). E, anti 1,N6-ϵdA opposite conformationally disordered primer dT, followed by dTMPNPP opposite template dA; extension stage (ϵdA:dT extension, Tables 1 and 7). Selected active site residues are colored by atom with carbon atoms shown in maroon (1,N6-ϵdA), orange (incoming nucleotide), light blue (template nucleotide 5′-adjacent to 1,N6-ϵdA; D and E), purple (Arg-61 and Gln-38 from the finger domain), or magenta (Asp/Glu coordinating to Mg2+ or Ca2+). Base pairs at the −1 (A–C) or −2 (D and E) positions are shown in yellow, primer dA and dT in idealized stacked positions (that would result in a clash with anti 1,N6-ϵdA) in D and E, respectively, are shown in dark gray, and Mg2+ and Ca2+ ions are cyan and pink spheres, respectively.
FIGURE 3.

Staggered arrangements of incoming purine nucleoside triphosphates and 1,N6-ϵdA in two hpol η insertion stage complexes. A, active site conformation in the complex with dAMPNPP opposite 1,N6-ϵdA, viewed into the DNA major groove; B, rotated by 90° and viewed perpendicular to the adenine plane (ϵdA:dAMPNPP, Tables 1 and 7). C, active site conformation in the complex with dGMPNPP opposite 1,N6-ϵdA, viewed into the DNA major groove; and D, rotated by 90° and viewed perpendicular to the guanine plane (ϵdA:dGMPNPP, Tables 1 and 7). Selected active site residues are colored by atom with carbon atoms shown in maroon (1,N6-ϵdA), orange (incoming nucleotide), purple (Arg-61 and Gln-38 from the finger domain), or magenta (Asp/Glu coordinating to Mg2+; cyan spheres).
An additional structure of an insertion stage hpol η complex was determined for incoming dTTP opposite 1,N6-ϵdA (ϵdA:dTTP, Tables 1 and 7). In this complex, thymine and the adduct base are coplanar, with the former in the standard anti configuration and the adduct flipped into the syn conformation (Fig. 4). Unlike the structures with incoming purine nucleoside triphosphates, the −1 base pair is devoid of buckling and the T 5′-adjacent to the adduct is now rotated into the active site and stacked on 1,N6-ϵdA. However, the syn orientation of the latter results in diminished overlap with the 3′-adjacent template A compared with the above complexes with incoming AMPNPP or GMPNPP (Figs. 3 and 4). The syn-1,N6-ϵdA:anti-dT base pair features virtually equidistant spacings between the (ϵdA)N7···(H)N3(T) and (ϵdA)N6(H)···O4(T) atom pairs. In the first contact, N7 of 1,N6-ϵdA is an acceptor, and N3 of T is a donor. Conversely, N6 of 1,N6-ϵdA and O4 of T are both acceptors. Rather than a clash between the two latter functionalities, the 2.8-Å separation might be indicative of protonation at N6 of 1,N6-ϵdA. The pKa of N6 in 1,N6-ϵdA is 4.1 (44, 45), and under the conditions used for crystallization (pH 5.5), the N6 nitrogen of the adduct may be at least partially protonated, consistent with the geometry of the nascent base pair at the active site.
FIGURE 4.

Active site conformation in the ternary hpol η insertion step complex with dTTP opposite 1,N6-ϵdA. A, view into the DNA major groove; B, rotated by 90° and viewed perpendicular to the thymine and adduct base planes (ϵdA:dTTP, Tables 1 and 7). Selected active site residues are colored by atom with carbon atoms shown in maroon (1,N6-ϵdA), orange (incoming dTTP), purple (Arg-61 and Gln-38 from the finger domain), or magenta (Asp/Glu coordinating to Ca2+; pink spheres).
Crystal Structures of Extension-stage Ternary hpol η Complexes
Structures of ternary complexes with 1,N6-ϵdA opposite either primer dA or dT were determined at resolutions of 1.79 and 2.30 Å, respectively (ϵdA:dA extension and ϵdA:dT extension, Tables 1 and 7; Fig. 2, D and E). Unlike the structure of the insertion complex with dTTP opposite the adduct, the latter adopts the energetically more favorable anti conformation in both extension complexes (Fig. 5). This orientation results in extensive stacking within the template strand between 1,N6-ϵdA and the base portions of 5′- and 3′-adjacent A and T nucleotides. In this fashion, the adduct is bracketed by Watson-Crick A:T(MPNPP) pairs at the 0 and −2 positions. As in the insertion complexes, Gln-38 establishes an H-bond to the sugar O4′ of the adducted nucleotide, but the Arg-61 side chain has swung away from the phosphates of the incoming nucleotide, and its guanidino moiety is directed into the major groove (Fig. 5). Similar to the dTTP insertion complex, the stacked template nucleotides include the T 5′-adjacent to Ap(1,N6-ϵdA) that is next in line to be replicated. The anti conformation of 1,N6-ϵdA leaves insufficient room for either dA or dT from the extended primer to be accommodated in the standard anti conformation (modeled nucleotides colored in gray in Fig. 5). Indeed, the electron density maps show good density for the phosphate group of the 3′-terminal primer nucleotide in the structures of the two extension-stage complexes but only partial density for its sugar moiety and no density even at the 0.7 σ level for the nucleobase portion (Fig. 2, D and E). The disordered state of the 3′-terminal T is consistent with the relatively low proclivity by hpol η to insert T (or A when the 5′-adjacent template nucleotide is not T) opposite the 1,N6-ϵdA adduct and its preference for purines (Tables 2–6) with concomitant −1 frameshifts (Tables 4–6).
FIGURE 5.

Active site conformations in two ternary hpol η extension step complexes with primer dA or dT opposite 1,N6-ϵdA, followed by dTMPNPP across template dA. A, ternary extension stage complex with dA opposite 1,N6-ϵdA, viewed into the major groove, and B, rotated by 90° and viewed perpendicular to the best plane through the nascent base pair (ϵdA:dA extension, Tables 1 and 7). C, ternary extension stage complex with dT opposite 1,N6-ϵdA viewed into the major groove; D, rotated by 90° and viewed perpendicular to the best plane through the nascent base pair (ϵdA:dT extension, Tables 1 and 7). The 3′-terminal primer nucleoside (dA/dT) is disordered in both complexes. Residues shown with dark gray bonds represent idealized stacked positions of primer dA (A and B) and primer dT (C and D). The conformational disorder of these residues in the two complex structures might arise from a clash between dA or dT in the modeled orientation with anti 1,N6-ϵdA. Selected active site residues are colored by atom with carbon atoms shown in maroon (1,N6-ϵdA), orange (incoming nucleotide), light blue (template nucleotide 5′-adjacent to 1,N6-ϵdA), purple (Arg-61 and Gln-38 from the finger domain), or magenta (Asp/Glu coordinating to Mg2+; cyan spheres).
Discussion
The kinetic results presented here for insertion by hpol η at the 1,N6-ϵdA adduct demonstrate that this lesion is significantly miscoding (Tables 2 and 3; supplemental Fig. S1). Under steady-state conditions, incorporation of T opposite 1,N6-ϵdA is strongly attenuated relative to insertion of T opposite A, and both purines are preferred over T (Tables 2 and 3). However, all four possible insertions opposite the adduct lack a burst phase as evidenced by our pre-steady-state analysis (supplemental Fig. S2). Therefore, it is unnecessary to invoke impediments during the extension step as the main reason for the low efficiency of bypass past the 1,N6-ϵdA adduct by hpol η, and the LC-MS/MS results (Tables 4–6) are consonant with the kinetic insertion results (Tables 2 and 3).
Our misincorporation results may be compared with previous literature. Incorporation of A, C, and G opposite 1,N6-ϵdA has been reported in different mammalian cell lines and extracts, in varying ratios (24, 26–28). T incorporation is reflected in a lack of mutation, but the mutagenic frequency is a function of the DNA repair status of each cell line. Pandya and co-workers (24, 27) reported a 70% mutation frequency for 1,N6-ϵdA in (monkey) COS7 cells, utilizing a single-stranded vector. Of these mutations, 63% corresponded to C incorporation (i.e. A → G transition), 6% to A incorporation, and 1% to G incorporation. No frameshift analysis was reported. In 2000, the same laboratory (26) utilized a double-stranded vector with HeLa, HCT116, and (HEK) 293 cells, derived from human cervical cancer, colon cancer, and embryonic kidney epithelium, respectively. High percentages of large deletions were reported in the 1,N6-ϵdA-plasmid cells (16–89%), with deletion of several hundred to 2000 bases. The number of targeted single mutations was low in the HTC116 cells and higher in the HeLa cells (HEK293 not reported). Of the 25 HeLa cell mutants analyzed with 1,N6-ϵdA, 36% corresponded to G insertion, 16% correspond to C insertion, and 48% corresponded to A insertion. One- and two-base frameshifts were not reported. In 2008 Tolentino et al. (28) used a double-stranded vector containing 1,N6-ϵdA with extracts of HeLa cells and XPV cells. The misincorporation frequency was very low, and with the HeLa cells only 2–3 mutants were seen for A, G, and C insertion, making any conclusions about preferences untenable.
At the time that some of the previous cellular mutation results were reported (24, 26, 27), the repair of 1,N6-ϵdA by glycosylases (46, 47) was known but repair by AlkB (25) and its mammalian orthologs was not recognized yet. The compositions of the DNA polymerase pools in these cell lines are largely uncharacterized, and the assignment of actions of the individual polymerases is not possible based on that information. As Levine et al. (26) state in comparing their results with cells of human origin with (monkey) COS7 cells (24): “…there are several differences in the design of these two studies, including single-stranded versus double-stranded vector, sequence context, location of the DNA adduct relative to replication origin, and host cells. One or several of these factors could have contributed to the differences observed.” Ultimately, the contribution of hpol η to mutations should be evaluated in cells equivalent in all respects except hpol η (and the characterization of other DNA polymerases and repair enzymes established).
Our results with several sequences, including that used by Levine et al. (34), clearly show that hpol η bypasses 1,N6-ϵdA in a highly error-prone manner, dominated by purine incorporations or generation of −1 frameshifts instead of insertion of T. This conclusion is based on both the kinetic analyses (Tables 2 and 3; supplemental Fig. S1) and LC-MS/MS analysis of the extended products (Tables 4–6), including sequences used previously by others (substrate C, Table 1) (34). These misincorporation patterns are also consistent with the results obtained in mammalian cells (26–28), although there is considerable variation among cell lines (26). Although A, C, and G insertions have all been reported opposite 1,N6-ϵdA in many systems, the presence of frameshifts in cells replicating past 1,N6-ϵdA has not been addressed. Levine et al. (26) listed a −1 deletion probe to be used for detecting frameshifts in cells but did not report any analyses for such events. In that study, large deletions (several hundred to 2000 bp) were observed in 16–89% of the mutants, depending on the cell line (26).
We found that the higher rate of incorporation of purine nucleoside triphosphates is in large part dependent on the availability of the matching template nucleotide 5′-adjacent to 1,N6-ϵdA (i.e. T and C for incoming dATP and dGTP, respectively). This purine incorporation is related in part to −1 frameshifts (Tables 4–6). The structures of insertion-stage complexes with incoming non-hydrolyzable dNTP analogs (dAMPNPP and dGMPNPP) in the presence of Mg2+ reveal that the 1,N6-ϵdA and adenine (guanine) bases are offset at the active site and, instead of direct pairing, engaged in stacking interactions across the template and primer strands (Fig. 3). Thus, unlike the structures of isolated oligonucleotide duplexes (30, 31), where the adduct switches into a syn orientation to accommodate pairing with a purine, 1,N6-ϵdA remains in the favored anti orientation and the incoming purines adopt the observed staggered orientation.
From the states seen in the two insertion-stage complex structures, the replication process can proceed in two distinct ways. (i) The incoming purine is accommodated opposite the adduct (Tables 4 and 6). (ii) Alternatively, the template T(C) 5′-adjacent to the adduct, seen outside the active site in the structures (Fig. 3), rotates inward and above the adduct and then pairs with dAMPNPP (dGMPNPP), thus resulting in the −1 frameshift products that constituted the majority of the extended oligonucleotides according to the LC-MS/MS analysis. The actual configurations by which A or G are accommodated opposite 1,N6-ϵdA in the template·primer duplex remain unclear from the two structures at the insertion stage, although it is possible that the adduct and purine nucleotides are stacked in a cross-strand fashion.
The structure of the extension complex with dA opposite 1,N6-ϵdA is useful in addressing this question. Rather than in the staggered configuration, the 1,N6-ϵdA:A pair is wedged between A:T(TP) pairs at the −2 and 0 positions that are separated by ∼7 Å in an axial direction (Fig. 5, A and C). The staggered orientation following insertion can thus be discarded, but the nucleobase portion of primer dA is completely disordered, suggesting a position of the adduct partner either inside the major or the minor groove.
Even the smaller thymine moiety of dT is disordered in the second extension-stage complex (Fig. 5, C and D). However, the anti orientation of 1,N6-ϵdA is common to both complexes and is likely preferred because it allows for more optimal stacking interactions with flanking bases, even if it results in a suboptimal arrangement of the pairing partner and loss of H-bonds. This conclusion is supported by the structural and kinetic data for T incorporation opposite 1,N6-ϵdA. The structure of the corresponding insertion complex shows the adduct in the syn conformation and therefore has diminished stacking with the 3′-adjacent template base (A; Fig. 5). The incoming T and 1,N6-ϵdA are in the same plane, and it is possible, based on the relative orientation and taking into account pKa data for the adduct and the acidic pH of the crystallization solution, that two H-bonds are established between the pairing partners. However, the 1000-fold lower efficiency of T incorporation opposite 1,N6-ϵdA relative to T opposite A (steady-state results, Table 2) renders it unnecessary to envision an optimal structural context for the 1,N6-ϵdA:T pair at the insertion step. Moreover, the incoming C can form a bifurcated H-bond with its N4 amino group to the N6 and N7 acceptors of the adduct or, alternatively (envisioning a protonated state of C (pKa 4.6) under pH 5.5 crystallization conditions), two H-bonds. However, these assumptions are mute as the efficiency of C incorporation is further reduced compared with T (4-fold) and considerably below the levels of efficiency for A or G incorporation and frameshifting.
The structures of incoming dAMPNPP and dGMPNPP in a staggered orientation relative to 1,N6-ϵdA and the kinetic data manifesting preferred incorporation of purines opposite the adduct, accompanied with frameshifts offer some parallels to our observations of hpol η-catalyzed bypass reactions opposite an abasic site. Specifically, we invoke a “purine rule,” meaning that dATP and dGTP are preferred over dCTP and dTTP by this polymerase opposite an abasic lesion (48). Structures of the preferred dNTPs opposite the abasic site in ternary hpol η complexes offered insight into this preference, in that the longer purine bases could be linked to the phosphate group of the abasic residue via water bridges. By comparison, the smaller pyrimidines were at a disadvantage, both because of the reduced level of stacking interactions and the inability to tether them to the template strand via solvent molecules in the active site. Similarly, purines opposite the 1,N6-ϵdA adduct at the hpol η active site exploit cross-strand stacking interactions that result in facilitated error-prone bypass. In addition, at least in the case of incoming dGMPNPP, the side chain of Gln-38 mediates an interaction between guanine (N2) and the backbone of the template strand (sugar ring of the adduct, Fig. 3, C and D). Finally, the structure of the third insertion-stage complex with dTTP opposite 1,N6-ϵdA and with the adduct adopting a syn orientation (Fig. 5) is reminiscent of the pairing between this lesion and incoming dTTP observed in the crystal structure of a ternary hpol ι complex (49). However, this Y-family pol is more efficient and less error prone than hpol η in regard to bypass of 1,N6-ϵdA and incorporates a T opposite the lesion with only about 10-fold reduced efficiency relative to T opposite A, consistent with a more narrow active site that forces base pairs into the normally less favorable Hoogsteen configuration.
In summary, bypass of the 1,N6-ϵdA lesion by hpol η proceeds with relatively low efficiency and in an error-prone fashion by misinsertion of A and G or generation of −1 frameshifts. Thus, the outcomes of hpol η-catalyzed bypass of the bulky 1,N6-ϵdA lesion and an abasic site (48) are surprisingly similar.
Experimental Procedures
Materials
The catalytic core (amino acids 1–432) of hpol η was expressed and purified as described previously (50). Unlabeled dNTPs, T4 polynucleotide kinase, and UDG were purchased from New England Biolabs (Ipswich, MA). A mixture of four dNTPs was purchased from Invitrogen. All non-hydrolyzable dNMPNPPs were obtained from Jena Bioscience (Jena, Germany). [γ-32P]ATP (specific activity 3000 Ci/mmol) was purchased from PerkinElmer Life Sciences. Biospin columns were purchased from Bio-Rad. All oligonucleotides (purified by HPLC by the manufacturers) were obtained from Midland Certified Reagent Co. (Midland, TX), Integrated DNA Technologies (Coralville, IA), or TriLink Biotechnologies (San Diego). The oligonucleotides containing 1,N6-ϵdA were from TriLink.
Steady-state Kinetics
A 13-mer primer was 5′-5/6-carboxyfluorescein (FAM)-labeled and annealed to an 18-mer template at a 1:1.2 molar ratio (Table 1). The same procedure was used for the 18/23-mer pair, annealing the primer and template in a 1:1 molar ratio. Enzyme concentrations and reaction times were optimized using time course assays to ensure ≤20% conversion of substrate to product. A typical assay solution included 4–55 nm hpol η, 5 μm primer·template duplex, 40 mm Tris-HCl buffer (pH 7.5), 5 mm MgCl2, 10 mm DTT, 100 mm KCl, 5% glycerol (v/v), 100 μg/ml bovine serum albumin, and varying concentrations of a single dNTP (0–2 mm). Reactions were incubated at 37 °C for 5–20 min and were terminated with 9 volumes of a quench solution (20 mm EDTA, 95% formamide (v/v), bromphenol blue, and xylene cyanol). Products were resolved by electrophoresis on 18% (w/v) polyacrylamide gels containing 7.5 m urea. The gels were scanned by a Typhoon Scanner (GE Healthcare) and quantified by fluorescence intensity using ImageJ software (National Institutes of Health). Data were fit to the Michaelis-Menten equation using Prism software (GraphPad, La Jolla, CA) to estimate kcat and Km values (non-linear regression).
Pre-steady-state Kinetics
Rapid quench experiments were performed using a model RQF-3 KinTek quench flow apparatus (KinTek, Austin, TX). The 13-mer primer was 32P-labeled at the 5′ end by T4 polynucleotide kinase/[γ-32P]ATP and annealed to the 18-mer templates. Reactions were initiated by rapid mixing of 32P-primer·template/polymerase mixtures with dNTP and Mg2+ at 37 °C. The final concentrations of the reactants were 50 nm hpol η, 500 nm 32P-labeled primer·template complex, and 0.5 mm dNTP. Other reaction conditions are the same as described for steady-state kinetics. Reactions were quenched with 0.5 m EDTA at times varying from 5 ms to 5 s. Products were separated on 18% (w/v) polyacrylamide gels and scanned and quantitated using a PhosphorImaging system (Bio-Rad, Molecular Imager FX) and Quantity One software as described previously (35).
LC-MS/MS Analysis of Full-length Extended Products
A 5′-FAM-labeled primer containing an appropriately positioned deoxyuridine (dU) (opposite A in some but not all cases) (Table 1) was annealed to the 18- or 23-mer templates at a 1:1 molar ratio. Reaction conditions were similar to those used in steady-state kinetics assays, except that the final concentrations were 3 μm hpol η and 25 μm primer·template duplex, in a total volume of 80 μl. Reactions were initiated by addition of a mixture of 1 mm each of dNTPs (A, G, C, and T) and terminated by spin-column separation to remove dNTP and Mg2+ after 1 h of incubation (37 °C). The resulting products were treated with 50 units of UDG and 0.25 m hot piperidine, following a previous protocol (35, 36). The extent of the reaction was monitored by denaturing PAGE (Fig. 1A) prior to LC-MS analysis. The cleavage solution was lyophilized and reconstituted in 60 μl of H2O.
LC-MS/MS analysis was performed on an Acquity ultraperformance liquid chromatography (UPLC) system (Waters Associates) coupled to a Thermo Finnigan LTQ mass spectrometer (Thermo Scientific, San Jose, CA) with an electrospray ionization source. Samples were separated on an Acquity UPLC BEH octadecylsilane (C18) column (1.7 μm, 2.1 × 100 mm) at a flow rate of 0.3 ml/min. The column temperature was maintained at 50 °C. Eluent A contained 10 mm NH4CH3CO2 in 98% H2O, 2% CH3CN (v/v), and eluent B consisted of 10 mm NH4CH3CO2 in 90% CH3CN, 10% H2O (v/v). A gradient program was run as follows: 0–3% B over 3 min, 3–20% B over 2 min, 20–100% B over 1 min, held at 100% B for 2 min, 100–0% B over 2 min, and held at 0% B for 3 min (all v/v). MS data were acquired in the negative mode and controlled by Xcalibur 2.1 software (Thermo). Electrospray ionization settings were as follows: source voltage 4 kV, source current 100 A, capillary voltage −49 V, capillary temperature 350, tube lens voltage −90 V. The most abundant species (−2 or −3 charged) were fragmented in the ion trap by collision-induced dissociation (CID) with a normalized collision energy of 35%. An activation Q of 0.25 and activation time of 30 ms were used. Oligonucleotide sequences can be identified by comparing the observed CID spectra and theoretical spectra of candidate oligonucleotide sequences calculated using Mongo Oligo Calculator 2.0 software (University of Utah, Salt Lake City). The relative yields of various DNA extension products were based on their respective peak areas in the extracted ion chromatograms.
Crystallization
Crystals were obtained by the hanging drop vapor diffusion technique at 18 °C. 1,N6-ϵdA-modified DNA templates and primer sequences used in the crystallization experiments are listed in Table 1. DNA solutions were prepared by mixing template and primer strands in a 1:1 molar ratio and annealing the mixture in the presence of 10 mm sodium HEPES buffer (pH 8.0), 0.1 mm EDTA, and 50 mm NaCl at 85 °C for 10 min, followed by slow cooling to room temperature. hpol η protein was mixed with the DNA duplex in a 1:1.2 molar ratio in the presence of 50 mm Tris-HCl (pH 7.5) containing 450 mm KCl and 3 mm DTT, followed by addition of either 5 μl of 100 mm MgCl2 or 5 μl of 100 mm CaCl2. Using a spin concentrator with an Amicon cutoff filter (Millipore, Billerica, MA), the complex was concentrated to a final concentration of ∼2 mg of protein/ml. Either dTTP or one of the non-hydrolyzable nucleoside triphosphate analogs was added to the concentrated mixtures containing Ca2+ or Mg2+. The ternary complex solution was mixed with an equal volume of reservoir solution containing 0.10 m sodium MES (pH 5.5), 5 mm MgCl2, and 16–21% (w/v) PEG 2000 monomethyl ether and equilibrated against 500-μl reservoir solutions. Crystals typically appeared after overnight incubation and were allowed to grow for 1 week. They were transferred to cryoprotectant solution containing reservoir solution along with 25% glycerol (v/v) and then frozen in liquid nitrogen for data collection.
X-ray Diffraction Data Collection, Structure Determination, and Refinement
X-ray diffraction data were collected at 100 K either on the 21-ID-F or the 21-ID-G beamline of the Life Sciences Collaborative Access Team at the Advanced Photon Source, Argonne National Laboratory (Argonne, IL). All data were processed with the program HKL2000 (51). (Data collection statistics are summarized in Table 7.) All structures were determined by molecular replacement in MOLREP (52, 53), using the coordinates of the complex between hpol η and native DNA (Protein Data Bank code 4O3N) as the search model. Structures were refined using PHENIX (54), and model building was carried out in COOT (55). Model statistics and geometric parameters are summarized in Table 7. Illustrations were generated with the program UCSF Chimera (56).
Author Contributions
A. P. crystallized the protein complexes and solved the structures; Q. Z. was responsible for the pre-steady-state and part of the steady-state kinetic analysis and part of the LC-MS measurements; Y. S. purified the enzyme and carried out part of the steady-state kinetic analysis and preparation of extended primers for some of the extended primers for LC-MS analysis; K. M. J. did part of the LC-MS measurements, and F. P. G. and M. E. conceived the studies and wrote the paper, along with Y. S.
Supplementary Material
Acknowledgments
We thank L. Lei for help with protein expression and K. Trisler for assistance in the preparation of the manuscript. Vanderbilt University is a member institution of the Life Sciences Collaborative Access Team at sector 21 of the Advanced Photon Source, Argonne, IL. Use of the Advanced Photon Source at Argonne National Laboratory was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 ES010375 (to F. P. G. and M. E.), P01 CA160032 (to M. E.), R01 ES010546 (to F. P. G.), T32 CA009582 (to K. M. J.), and P30 ES000267 (to F. P. G. and M. E.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Table S1 and Figs. S1–S3.
- 1,N6-ϵdA
- 1,N6-ethenodeoxyadenosine
- CID
- collision-induced dissociation
- dNMPNPP
- 2′-deoxynucleoside-5′-[(α,β)-imido]triphosphate
- dAMPNPP
- 2′-deoxyadenosine-5′-[(α,β)-imido]triphosphate
- dGMPNPP
- 2′-deoxyguanosine-5′-[(α,β)-imido]triphosphate
- dTMPNPP
- 2′-deoxythymidine-5′-[(α,β)-imido]triphosphate
- FAM
- 6-carboxyfluorescein
- h
- human
- pol
- DNA polymerase
- UDG
- uracil DNA glycosylase
- UPLC
- ultraperformance liquid chromatography.
References
- 1. Friedberg E. C., Walker G. C., Siede W., Wood R. D., Schultz R. A., and Ellenberger T. (2006) DNA Repair and Mutagenesis. 2nd Ed., American Society for Microbiology, Washington, D. C. [Google Scholar]
- 2. Shigenaga M. K., Hagen T. M., and Ames B. N. (1994) Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. U.S.A. 91, 10771–10778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McCann J., Choi E., Yamasaki E., and Ames B. N. (1975) Detection of carcinogens as mutagens in the Salmonella/microsome test: assay of 300 chemicals. Proc. Natl. Acad. Sci. U.S.A. 72, 5135–5139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Erickson R. P. (2003) Somatic gene mutation and human disease other than cancer. Mutat. Res. 543, 125–136 [DOI] [PubMed] [Google Scholar]
- 5. Weismann C. G., and Gelb B. D. (2007) The genetics of congenital heart disease: a review of recent developments. Curr. Opin. Cardiol. 22, 200–206 [DOI] [PubMed] [Google Scholar]
- 6. Wessels M. W., and Willems P. J. (2010) Genetic factors in non-syndromic congenital heart malformations. Clin. Genet. 78, 103–123 [DOI] [PubMed] [Google Scholar]
- 7. Malaveille C., Bartsch H., Barbin A., Camus A. M., Montesano R., Croisy A., and Jacquignon P. (1975) Mutagenicity of vinyl chloride, chloroethylene oxide, chloroacetaldehyde and chloroethanol. Biochem. Biophys. Res. Commun. 63, 363–370 [DOI] [PubMed] [Google Scholar]
- 8. DeMott M. S., and Dedon P. C. (2010) in The Chemical Biology of DNA Damage (Geacintov N. E., and Broyde S., eds) pp. 21–51, Wiley-VCH Verlag GmbH, KGaA Weinheim, Germany [Google Scholar]
- 9. Swenberg J. A., Lu K., Moeller B. C., Gao L., Upton P. B., Nakamura J., and Starr T. B. (2011) Endogenous versus exogenous DNA adducts: their role in carcinogenesis, epidemiology, and risk assessment. Toxicol. Sci. 120, S130–S145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leonard N. J. (1992) Etheno-bridged nucleotides in structural diagnosis and carcinogenesis. Chemtracts-Biochem. Mol. Biol. 3, 273–297 [Google Scholar]
- 11. Dudock B. S., Katz G., Taylor E. K., and Holley R. W. (1969) Primary structure of wheat germ phenylalanine transfer RNA. Proc. Natl. Acad. Sci. U.S.A. 62, 941–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sattsangi P. D., Leonard N. J., and Frihart C. R. (1977) 1,N2-Ethenoguanine and N2,3-ethenoguanine. Synthesis and comparison of the electronic spectral properties of these linear and angular triheterocycles related to the Y bases. J. Org. Chem. 42, 3292–3296 [DOI] [PubMed] [Google Scholar]
- 13. Secrist J. A. 3rd., Barrio J. R., Leonard N. J., and Weber G. (1972) Fluorescent modification of adenosine-containing coenzymes. Biological activities and spectroscopic properties. Biochemistry 11, 3499–3506 [DOI] [PubMed] [Google Scholar]
- 14. Leonard N. J. (1984) Etheno-substituted nucleotides and coenzymes: fluorescence and biological activity. CRC Crit. Rev. Biochem. 15, 125–199 [DOI] [PubMed] [Google Scholar]
- 15. Leonard N. J. (1993) Etheno-bridged nucleotides in enzyme reactions and protein binding. Chemtracts-Biochem. Mol. Biol. 4, 251–284 [Google Scholar]
- 16. Barbin A., Brésil H., Croisy A., Jacquignon P., Malaveille C., Montesano R., and Bartsch H. (1975) Liver-microsome-mediated formation of alkylating agents from vinyl bromide and vinyl chloride. Biochem. Biophys. Res. Commun. 67, 596–603 [DOI] [PubMed] [Google Scholar]
- 17. Laib R. J., Gwinner L. M., and Bolt H. M. (1981) DNA alkylation by vinyl chloride metabolites: etheno derivatives or 7-alkylation of guanine? Chem. Biol. Interact. 37, 219–231 [DOI] [PubMed] [Google Scholar]
- 18. Fedtke N., Boucheron J. A., Walker V. E., and Swenberg J. A. (1990) Vinyl chloride-induced DNA adducts. II. Formation and persistence of 7-(2′-oxoethyl)guanine and N2,3-ethenoguanine in rat tissue DNA. Carcinogenesis 11, 1287–1292 [DOI] [PubMed] [Google Scholar]
- 19. Rindgen D., Nakajima M., Wehrli S., Xu K., and Blair I. A. (1999) Covalent modifications to 2′-deoxyguanosine by 4-oxo-2-nonenal, a novel product of lipid peroxidation. Chem. Res. Toxicol. 12, 1195–1204 [DOI] [PubMed] [Google Scholar]
- 20. Carvalho V. M., Asahara F., Mascio P. D., de Arruda Campos I. P., Cadet J., and Medeiros M. H. (2000) Novel 1,N6-etheno-2′-deoxyadenosine adducts from lipid peroxidation products. Chem. Res. Toxicol. 13, 397–405 [DOI] [PubMed] [Google Scholar]
- 21. Guengerich F. P., and Raney V. M. (1992) Formation of etheno adducts of adenosine and cytidine from 1-halooxiranes. Evidence for a mechanism involving initial reaction with the endocyclic nitrogens. J. Am. Chem. Soc. 114, 1074–1080 [Google Scholar]
- 22. Swenberg J. A., Fedtke N., Ciroussel F., Barbin A., and Bartsch H. (1992) Etheno adducts formed in DNA of vinyl chloride-exposed rats are highly persistent in liver. Carcinogenesis 13, 727–729 [DOI] [PubMed] [Google Scholar]
- 23. Basu A. K., Niedernhofer L. J., and Essigmann J. M. (1987) Deoxyhexanucleotide containing a vinyl chloride induced DNA lesion, 1,N6-ethenoadenine: synthesis, physical characterization, and incorporation into a duplex bacteriophage M13 genome as part of an amber codon. Biochemistry 26, 5626–5635 [DOI] [PubMed] [Google Scholar]
- 24. Pandya G. A., and Moriya M. (1996) 1,N6-Ethenodoexyadenosine, a DNA adduct highly mutagenic in mammalian cells. Biochemistry 35, 11487–11492 [DOI] [PubMed] [Google Scholar]
- 25. Delaney J. C., Smeester L., Wong C., Frick L. E., Taghizadeh K., Wishnok J. S., Drennan C. L., Samson L. D., and Essigmann J. M. (2005) AlkB reverses etheno DNA lesions caused by lipid oxidation in vitro and in vivo. Nat. Struct. Mol. Biol. 12, 855–860 [DOI] [PubMed] [Google Scholar]
- 26. Levine R. L., Yang I.-Y., Hossain M., Pandya G. A., Grollman A. P., and Moriya M. (2000) Mutagenesis induced by a single 1,N6-ethenodeoxyadenosine adduct in human cells. Cancer Res. 60, 4098–4104 [PubMed] [Google Scholar]
- 27. Moriya M., Pandya G. A., Johnson F., and Grollman A. P. (1999) in Exocyclic DNA Adducts in Mutagenesis and Carcinogenesis (Singer B., and Bartsch H., eds) IARC Sci. Publication No. 150, pp. 263–270, Int. Agency Res. Cancer Scientific Publications, Lyon, France: [PubMed] [Google Scholar]
- 28. Tolentino J. H., Burke T. J., Mukhopadhyay S., McGregor W. G., and Basu A. K. (2008) Inhibition of DNA replication fork progression and mutagenic potential of 1,N6-ethenoadenine and 8-oxoguanine in human cell extracts. Nucleic Acids Res. 36, 1300–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kouchakdjian M., Eisenberg M., Yarema K., Basu A., Essigmann J., and Patel D. J. (1991) NMR studies of the exocyclic 1,N6-ethenodeoxyadenosine adduct (ϵdA) opposite thymidine in a DNA duplex. Nonplanar alignment of ϵdA(anti) and dT(anti) at the lesion site. Biochemistry 30, 1820–1828 [DOI] [PubMed] [Google Scholar]
- 30. de los Santos C., Kouchakdjian M., Yarema K., Basu A., Essigmann J., and Patel D. J. (1991) NMR studies of the exocyclic 1,N6-ethenodeoxyadenosine adduct (ϵdA) opposite deoxyguanosine in a DNA duplex. ϵdA(syn)·dG(anti) pairing at the lesion site. Biochemistry 30, 1828–1835 [DOI] [PubMed] [Google Scholar]
- 31. Leonard G. A., McAuley-Hecht K. E., Gibson N. J., Brown T., Watson W. P., and Hunter W. N. (1994) Guanine-1,N6-ethenoadenine base pairs in the crystal structure of d(CGCGAATT(ϵdA)GCG). Biochemistry 33, 4755–4761 [DOI] [PubMed] [Google Scholar]
- 32. Singer B., Abbott L. G., and Spengler S. J. (1984) Assessment of mutagenic efficiency of two carcinogen-modified nucleosides, 1,N6-ethenodeoxyadenosine and O4-methyldeoxythymidine, using polymerases of varying fidelity. Carcinogenesis 5, 1165–1171 [DOI] [PubMed] [Google Scholar]
- 33. Calabretta A., and Leumann C. J. (2013) Base pairing and miscoding properties of 1,N6-ethenoadenine- and 3,N4-ethenocytosine-containing RNA oligonucleotides. Biochemistry 52, 1990–1997 [DOI] [PubMed] [Google Scholar]
- 34. Levine R. L., Miller H., Grollman A., Ohashi E., Ohmori H., Masutani C., Hanaoka F., and Moriya M. (2001) Translesion DNA synthesis catalyzed by human pol η and pol κ across 1,N6-ethenodeoxyadenosine. J. Biol. Chem. 276, 18717–18721 [DOI] [PubMed] [Google Scholar]
- 35. Zang H., Goodenough A. K., Choi J. Y., Irimia A., Loukachevitch L. V., Kozekov I. D., Angel K. C., Rizzo C. J., Egli M., and Guengerich F. P. (2005) DNA adduct bypass polymerization by Sulfolobus solfataricus DNA polymerase Dpo4: analysis and crystal structures of multiple base pair substitution and frameshift products with the adduct 1,N2-ethenoguanine. J. Biol. Chem. 280, 29750–29764 [DOI] [PubMed] [Google Scholar]
- 36. Christov P. P., Angel K. C., Guengerich F. P., and Rizzo C. J. (2009) Replication past the N5-methyl-formamidopyrimidine lesion of deoxyguanosine by DNA polymerases and an improved procedure for sequence analysis of in vitro bypass products by mass spectrometry. Chem. Res. Toxicol. 22, 1086–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Choi J. Y., Chowdhury G., Zang H., Angel K. C., Vu C. C., Peterson L. A., and Guengerich F. P. (2006) Translesion synthesis across O6-alkylguanine DNA adducts by recombinant human DNA polymerases. J. Biol. Chem. 281, 38244–38256 [DOI] [PubMed] [Google Scholar]
- 38. Eoff R. L., Irimia A., Egli M., and Guengerich F. P. (2007) Sulfolobus solfataricus DNA polymerase Dpo4 is partially inhibited by “wobble” pairing between O6-methylguanine and cytosine, but accurate bypass is preferred. J. Biol. Chem. 282, 1456–1467 [DOI] [PubMed] [Google Scholar]
- 39. Yuan B., and Wang Y. (2008) Mutagenic and cytotoxic properties of 6-thioguanine, S6-methylthioguanine, and guanine-S6-sulfonic acid. J. Biol. Chem. 283, 23665–23670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao L., Christov P. P., Kozekov I. D., Pence M. G., Pallan P. S., Rizzo C. J., Egli M., and Guengerich F. P. (2012) Replication of N2,3-ethenoguanine by DNA polymerases. Angew. Chem. Int. Ed. Engl. 51, 5466–5469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chowdhury G., and Guengerich F. P. (2011) Liquid chromatography-mass spectrometry analysis of DNA polymerase reaction products. Curr. Protoc. Nucleic Acid Chem. Chapter 7, Unit 7.16.1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wickramaratne S., Boldry E. J., Buehler C., Wang Y. C., Distefano M. D., and Tretyakova N. Y. (2015) Error-prone translesion synthesis past DNA-peptide cross-links conjugated to the major groove of DNA via C5 of thymidine. J. Biol. Chem. 290, 775–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chowdhury G., and Guengerich F. P. (2016) Mass Spectrometry of Nucleic Acids, in Encyclopedia of Analytical Chemistry (Meyers R. A., ed) Wiley Interscience, New York, in press [Google Scholar]
- 44. Spencer R. D., Weber G., Tolman G. L., Barrio J. R., and Leonard N. J. (1974) Species responsible for the fluorescence of 1:N6-ethenoadenosine. Eur. J. Biochem. 45, 425–429 [DOI] [PubMed] [Google Scholar]
- 45. Inoue Y., and Kuramochi T. (1979) Protonation and quaternization of 1,N6-ethenoadenosine: what are the species responsible for the fluorescence of 1,N6-ethenoadenosine? Biopolymers 18, 2175–2194 [Google Scholar]
- 46. Singer B., Antoccia A., Basu A. K., Dosanjh M. K., Fraenkel-Conrat H., Gallagher P. E., Kuśmierek J. T., Qiu Z. H., and Rydberg B. (1992) Both purified human 1,N6-ethenoadenine-binding protein and purified human 3-methyladenine-DNA glycosylase act on 1,N6-ethenoadenine and 3-methyladenine. Proc. Natl. Acad. Sci. U.S.A. 89, 9386–9390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Saparbaev M., Kleibl K., and Laval J. (1995) Escherichia coli, Saccharomyces cerevisiae, rat and human 3-methyladenine DNA glycolases repair 1,N6-ethenoadenine when present in DNA. Nucleic Acids Res. 23, 3750–3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Patra A., Zhang Q., Lei L., Su Y., Egli M., and Guengerich F. P. (2015) Structural and kinetic analysis of nucleoside triphosphate incorporation opposite an abasic site by human translesion DNA polymerase η. J. Biol. Chem. 290, 8028–8038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nair D. T., Johnson R. E., Prakash L., Prakash S., and Aggarwal A. K. (2006) Hoogsteen base pair formation promotes synthesis opposite the 1,N6-ethenodeoxyadenosine lesion by human DNA polymerase ι. Nat. Struct. Mol. Biol. 13, 619–625 [DOI] [PubMed] [Google Scholar]
- 50. Biertümpfel C., Zhao Y., Kondo Y., Ramón-Maiques S., Gregory M., Lee J. Y., Masutani C., Lehmann A. R., Hanaoka F., and Yang W. (2010) Structure and mechanism of human DNA polymerase η. Nature 465, 1044–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 52. Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 53. Collaborative Computational Project No. 4 (1994) The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 54. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., et al. (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 56. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.