

Abstract
Pro-inflammatory cytokines contribute to pancreatic beta cell apoptosis in type 1 diabetes at least in part by inducing endoplasmic reticulum (ER) stress and the consequent unfolded protein response (UPR). It remains to be determined what causes the transition from “physiological” to “apoptotic” UPR, but accumulating evidence indicates that signaling by the ER transmembrane protein IRE1α is critical for this transition. IRE1α activation is regulated by both intra-ER and cytosolic cues. We evaluated the role for the presently discovered cytokine-induced and IRE1α-interacting protein ubiquitin D (UBD) on the regulation of IRE1α and its downstream targets. UBD was identified by use of a MAPPIT (mammalian protein-protein interaction trap)-based IRE1α interactome screen followed by comparison against functional genomic analysis of human and rodent beta cells exposed to pro-inflammatory cytokines. Knockdown of UBD in human and rodent beta cells and detailed signal transduction studies indicated that UBD modulates cytokine-induced UPR/IRE1α activation and apoptosis. UBD expression is induced by the pro-inflammatory cytokines interleukin (IL)-1β and interferon (IFN)-γ in rat and human pancreatic beta cells, and it is also up-regulated in beta cells of inflamed islets from non-obese diabetic mice. UBD interacts with IRE1α in human and rodent beta cells, modulating IRE1α-dependent activation of JNK and cytokine-induced apoptosis. Our data suggest that UBD provides a negative feedback on cytokine-induced activation of the IRE1α/JNK pro-apoptotic pathway in cytokine-exposed beta cells.
Keywords: apoptosis, c-Jun N-terminal kinase (JNK), cytokinesis, endoplasmic reticulum stress (ER stress), type 1 diabetes, IRE1α, ubiquitin D
Introduction
Endoplasmic reticulum (ER)8 stress and the consequent triggering of the unfolded protein response (UPR) are induced in human pancreatic beta cells by the pro-inflammatory cytokines interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), in combination with interferon-γ (IFN-γ), and probably contribute to beta cell apoptosis in type 1 diabetes (T1D) (1–3). Markers of the UPR are expressed in inflamed islets of both non-obese diabetic (NOD) mice (4, 5) and patients affected by T1D (6).
The UPR is mediated through activation of three ER transmembrane proteins as follows: inositol-requiring protein 1α (IRE1α), protein kinase RNA-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6). These proteins sense the accumulation of unfolded proteins in the ER lumen and activate mechanisms to restore its homeostasis (2, 7). Fine-tuning of the activity of these transmembrane proteins is provided by signals from the cytosol (8). In the case of unresolved ER stress, the persistent stimulation of the UPR triggers apoptosis via activation of C/EBP homologous protein (CHOP), c-Jun N-terminal kinase (JNK), death protein 5 (DP5), and other pro-apoptotic signals (9, 10). What determines the transition from “physiological” to “apoptotic” UPR remains to be clarified (8), but accumulating evidence indicates that the changing nature of IRE1α signaling is critical for this transition (11). In particular, IRE1α-induced JNK activation is crucial for cytokine-induced apoptosis in human pancreatic beta cells (12).
Against this background, we used a high-throughput mammalian two-hybrid technology, MAPPIT (mammalian protein-protein interaction trap) coupled to functional genomic analysis of human and rodent beta cells exposed to IL-1β + IFN-γ to identify novel cytokine-induced IRE1α-interacting proteins that modulate UPR activation in pancreatic beta cells (13). Based on this analysis, we selected two candidates for detailed functional studies: NMI (13) and ubiquitin D (UBD; this study). UBD (also know as FAT10) is expressed in the immune system and in some cancer cells (14–17), and its expression is up-regulated by IFN-γ and TNF-α (18, 19). UBD interacts non-covalently or covalently with different proteins, respectively, modifying their activity (20, 21) or triggering their degradation in the proteasome (22, 23). Of particular interest in the context of T1D, the Ubd gene maps to the telomeric region of the human major histocompatibility complex (MHC), the most important susceptibility locus for T1D (24, 25). Polymorphisms in the region of the Ubd gene have been associated with autoimmune diabetes in rat and human (26–29), but this remains to be confirmed.
We presently show that UBD expression is induced by pro-inflammatory cytokines in rat and human pancreatic beta cells, and it is also present in beta cells of inflamed islets from NOD mice. Of particular importance, we show that UBD interacts with IRE1α in cytokine-treated human and rodent beta cells, providing a negative feedback for IRE1α-induced activation of JNK and consequent apoptosis.
Materials and Methods
Culture of Human Islet Cells, FACS-purified Rat Beta Cells, INS-1E Cells, the Human Beta Cell Line EndoC-βH1, and HEK293T Cells
Human islets from 13 non-diabetic donors were isolated in Pisa using collagenase digestion and density gradient purification (30). The donors (seven women and six men) were 67.1 ± 4.7 years old and had a body mass index of 25.0 ± 1.0 (kg/m2) (Table 1). Beta cell purity, as evaluated by immunofluorescence for insulin, using a specific anti-insulin antibody (Table 2), was 52 ± 5.4%. The islets were cultured as described previously (25, 31).
TABLE 1.
Characteristics of the human islet donors
| Subject | Age | Gender | BMI | Beta cell purity |
|---|---|---|---|---|
| years | kg/m2 | |||
| C1 | 84 | F | 26 | 73% |
| C2 | 59 | M | 25 | 70% |
| C3 | 72 | F | 24 | 62% |
| C4 | 51 | M | NA | 37% |
| C5 | 76 | M | 33 | 68% |
| C6 | 49 | F | 25 | 72% |
| C7 | 66 | F | 20 | 36% |
| C8 | 81 | M | 24 | 29% |
| C9 | 82 | F | 20 | 34% |
| C10 | 75 | F | 29 | 24% |
| C11 | 69 | M | 25 | 85% |
| C12 | 85 | M | 26 | 39% |
| C13 | 23 | F | 23 | 46% |
| X ± S.E. | 67.1 ± 4.7 | 25.0 ± 1.0 | 52 ± 5.4% |
TABLE 2.
Antibodies used in the study
IHC is immunohistochemistry and WB is Western blotting.
| Antibody | Company | Reference | Dilution |
|---|---|---|---|
| Insulin | Sigma, Bornem, Belgium | I2018 | IHC, 1:1000 |
| Donkey anti-mouse IgG rhodamine | Lucron Bioproducts, De Pinte, Belgium | 715-026-156 | IHC, 1:200 |
| IRE1α | Santa Cruz Biotechnology, Santa Cruz, CA | Sc-20790 | IP, 1 mg/ml |
| IRE1α | Cell Signaling, Danvers, MA | 3294 | WB, 1:1000 |
| UBD | Proteintech, Chicago | 13003-2-AP | WB, 1/500 IHC, 1:100 |
| p-SAPK/JNK (T183/Y185) | Cell Signaling | 9251S | WB, 1:1000 |
| SAPK/JNK (56G8) | Cell Signaling | 9258 | WB, 1:1000 |
| JNK1 (2C6) | Cell Signaling | 708S | WB, 1:1000 |
| HRP-conjugated anti-rabbit IgG | Lucron Bioproducts | 711-036-152 | WB, 1:5000 |
| HRP-conjugated anti-mouse IgG | Lucron Bioproducts | 715-036-150 | WB, 1:5000 |
| Alexa Fluor 488 goat anti-mouse | Molecular Probes Life Technologies-Invitrogen | A-11008 | IHC,1/500 |
| Alexa Fluor 546 goat anti-rabbit | Molecular Probes Life Technologies-Invitrogen | A-11030 | IHC, 1/500 |
| Cleaved caspase-3 (D175) (rabbit) | Cell Signaling | 9661S | WB, 1:1000 |
| Cleaved caspase-9 (D353) (rabbit) | Cell Signaling | 9507S | WB, 1:1000 |
| β-Actin | Cell Signaling | 4967 | WB, 1:5000 |
| α-Tubulin | Sigma | T9026 | WB,1 :5000 |
Isolated pancreatic islets of male Wistar rats (Charles River Laboratories, Brussels, Belgium) were dispersed, and beta cells were purified by autofluorescence-activated cell sorting (FACSAria, BD Bioscience, San Jose, CA) (32), with purity >90%. The rat insulin-producing INS-1E cell line (kindly provided by Dr. C. Wollheim, University of Geneva, Switzerland) was cultured in RPMI 1640 GlutaMAX-I medium (Invitrogen, Paisley, UK) (33).
The human beta cell line EndoC-βH1 (kindly provided by Dr. R. Scharfmann, University of Paris, France) (34) was cultured as described previously (12). The human embryonic kidney cells HEK293T were cultured in DMEM containing 25 mm glucose, 5% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 100× sodium pyruvate (Invitrogen).
Cell Treatment and NO Measurement
Cells were exposed to the following cytokine concentrations, based on previous dose-response experiments performed by our group (31, 35–37): recombinant human IL-1β (R&D Systems, Abingdon, UK) 10 units/ml for INS-1E cells or 50 units/ml for human islet cells, primary rat beta cells, and the EndoC-βH1 cells; recombinant rat IFN-γ (R&D Systems) 100 units/ml for INS-1E cells or 500 units/ml for primary rat beta cells; and human IFN-γ (PeproTech, London, UK) 1000 units/ml for human islet cells or the EndoC-βH1 cells. Lower cytokine concentration and shorter time points were used in the rodent experiments because rat beta cells are more sensitive to cytokine-induced damage than human islets (38, 39). Culture medium was collected for nitrite determination (nitrite is a stable product of NO oxidation) by the Griess method.
ArrayMAPPIT and Binary MAPPIT
To identify IRE1α-interacting proteins, an ArrayMAPPIT screen was performed as described (40), using as MAPPIT bait the cytoplasmic portion of IRE1α (amino acids 571–977) (13). The same bait was also used in the binary MAPPIT analyses, and it was generated based on the original pSEL MAPPIT bait construct (41). MAPPIT preys were cloned in the previously described pMG1 vector (42). Briefly, HEK293T cells were transfected with bait, prey, and STAT3 reporter plasmids (Table 3), and luciferase activity was measured 48 h after transfection using the luciferase assay system kit (Promega, Leiden, The Netherlands) on a TopCount illuminometer. Cells were stimulated with erythropoietin (5 ng/ml) 24 h after transfection. To provide further support for the specificity of the IRE1α/UBD interaction, we performed additional MAPPIT two-hybrid analyses, using irrelevant protein baits in addition to the empty bait. From the Cytokine Receptor Laboratory bait collection, we randomly selected a number of baits that were cloned in the same vector backbone (pSEL + 2L) as the IRE1α bait. In addition, because PERK, like IRE1α, is an ER stress sensor protein with a very similar topology to that of IRE1α, we generated a MAPPIT bait consisting of the PERK cytosolic portion, similar to the IRE1α bait that was used to show the UBD interaction.
TABLE 3.
Plasmids used in the study
| Plasmids | Encoding protein |
|---|---|
| pSEL-IRE1α bait | Cytosolic domain of IRE1α |
| pSEL-IRE1K599A bait | Cytosolic domain of IRE1α with K599A mutation |
| pSEL-IRE1Kin bait | Kinase domain of IRE1α |
| pSEL-IRE1Ken bait | Endonuclease domain of IRE1α |
| pSEL-empty bait | Empty |
| pMG1-UBD prey | UBD |
| pMG1-empty prey | Empty |
| pXP2d2-rPAP1 | Luciferase STAT3 reporter plasmid |
| pMG1-REM2 | RAS (RAD and GEM)-like GTP-binding 2 protein (REM2) |
RNA Interference
The siRNAs (30 nm) used in this study are described in Table 4. They were transfected into beta cells using previously defined optimal conditions (31, 43). After 16 h of transfection, cells were cultured for a 24- or 48-h recovery period before exposure to cytokines.
TABLE 4.
siRNAs used in the study
| Name | Type | Distributors | Sequence |
|---|---|---|---|
| siCTRL | Allstar negative control siRNA | Qiagen, Venlo, The Netherlands | Sequence not provided |
| Rat siUBD UbdRSS353947 (siU1) | BLOCK-iT StealthTM select siRNA | Life Technologies-Invitrogen | 1, 5′-CAAUGGCCAUUAAUGACCUUUGACA-3′ |
| 2, 5′-UGUCAAAGGUCAUUAAUGGCCAUUG-3′ | |||
| Rat siUBD UbdRSS353946 (siU2) | BLOCK-iT StealthTM select siRNA | Life Technologies-Invitrogen | 1, 5′-CCUAAAGGUGGUGAAGCCCAGUGAU-3′ |
| 2, 5′-AUCACUGGGCUUCACCACCUUUAGG-3′ | |||
| Rat siUBD UbdRSS353945 (siU3) | BLOCK-iT StealthTM select siRNA | Life Technologies-Invitrogen | 1, 5′-GUUCCCAAACCAAGGUCUCUGUGCA-3′ |
| 2, 5′-UGCACAGAGACCUUGGUUUGGGACC-3′ | |||
| Human si-hU1 | BLOCK-iT StealthTM select siRNA | Life Technologies-Invitrogen | Sequence not provided |
| Human si-hU2 | BLOCK-iT StealthTM select siRNA | Life Technologies-Invitrogen | Sequence not provided |
| Rat siIRE1α | ON-TARGETplus | Thermo Scientific, Chicago, IL | 1, CUGAUGACUUCGUGCGCUA |
| 2, GGCCAGCAGAUUAGCGAAU | |||
| 3, CAGACAAGCACGAGGACGU | |||
| 4, AAGAUGGACUGGCGGGAGA | |||
| Rat siJNK1 | BLOCK-iT StealthTM select siRNA | Life Technologies-Invitrogen | Sequence not provided |
Assessment of Cell Viability
The percentage of viable, apoptotic, and necrotic cells was determined after staining the cells with the DNA-binding dyes propidium iodide (5 μg/ml; Sigma, Bornem, Belgium) and Hoechst dye 33342 (5 μg/ml; Sigma). A minimum of 600 cells was counted for each experimental condition by two independent observers, with one of them unaware of sample identity. The agreement between findings obtained by the two observers was always >90%. This fluorescence assay for single cells is quantitative and has been validated by systematic comparisons against electron microscopy observations, ladder formation, and caspase 3/9 activation (31, 44–46).
Western Blot, mRNA Extraction, and Real Time PCR
For Western blot, cells were washed with cold PBS and lysed using Laemmli Sample Buffer. Total protein was extracted and resolved by 8–10% SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted with the specific antibodies for the protein of interest (Table 2) as described (31). The densitometric values were corrected by the housekeeping proteins α-tubulin or β-actin.
Poly(A)+ mRNA was isolated from cultured cells using the Dynabeads mRNA DIRECTTM kit (Invitrogen) and reverse-transcribed as described previously (47, 48). The real time PCR amplification reactions were done using iQ SYBR Green Supermix on a LightCycler instrument (Roche Diagnostics, Vilvoorde, Belgium) and on a Rotor-Gene Q (Qiagen, Venlo, Netherlands), and the concentration of the gene of interest was calculated as copies per μl using the standard curve method (47, 49). Gene expression values in human and rat cells were corrected by the housekeeping genes β-actin and Gapdh, respectively. Expression of these genes was not affected by cytokine treatment (31, 50). The primers used in this study are provided in the Table 5.
TABLE 5.
Sequence of the primers used in the study
| Primer | Forward sequence | Reverse sequence |
|---|---|---|
| Ubd (rat) | 5′-GGACCAGATCCTTCTGCTAGA-3′ | 5′-ACCTTTAGGGTGAGGTGGATAG-3′ |
| UBD (human) | 5′-CCATCCACCTTACCCTGAAA-3′ | 5′-TTTCACTTGTGCCACTGAGC-3′ |
| β-Actin (human) | 5′-CTGTACGCCAACACAGTGCT-3′ | 5′-GCTCAGGAGGAGCAATGATC-3′ |
| Ins-2 (rat) | 5′-TGTGGTTCTCACTTGGTGGA-3′ | 5′-CTCCAGTTGTGCCACTTGTG-3′ |
| Gapdh (rat) | 5′-AGTTCAACGGCACAGTCAAG-3′ | 5′-TACTCAGCACCAGCATCACC-3′ |
| Xbp1s (rat) | 5′-GAGTCCGCAGCAGGTG-3′ | 5′-GTGTCAGAGTCCATGGGA-3′ |
| Ccl-2 (rat) | 5′-CTTGTGGGCCTGTTGTTCA-3′ | 5′-CCAGCCACTCATTGGGATCA-3′ |
Immunoprecipitation
Proteins from the EndoC-βH1 cells were collected with cold lysis buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 1% Triton X-100, 2 mm MgCl2, 25 mm NaF, 2 mm Na3VO4, 2 mm Na4P2O7, 1 mm sodium Gly-phosphate, and Complete Protease inhibitor mixture, Roche Diagnostics) and then pre-cleared for 1 h at 4 °C with Dynabeads Protein G (Novex by Life Technologies, Inc., Oslo, Norway). The same amounts of protein were then incubated overnight at 4 °C, either with the anti-IRE1α antibody (Table 2) or normal rabbit IgG (Santa Cruz Biotechnology, sc-2027) as negative controls. After incubation for 1 h with Dynabeads Protein G, the complexes were washed five times with cold lysis buffer and then resuspended in 5× Laemmli Sample Buffer. Immunoprecipitates and total proteins were run in 8–12% SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted with specific antibodies for the protein of interest (Table 2).
Histology
Pancreas from NOD-SCID (control) mice and diabetes-prone NOD mice were collected at 8, 9, and 13 weeks of age for NOD-SCID and 3, 8, and 12 weeks for NOD mice and processed and stained as described previously (13). After overnight incubation at 4 °C with the primary antibody against UBD, slides were double-stained for insulin. Alexa Fluor fluorescent secondary antibodies (Molecular Probes-Invitrogen) were applied for 1 h at room temperature (Table 2). After nuclear staining with Hoechst, pancreatic sections were mounted with fluorescence mounting medium (DAKO, Glostrup, Denmark). Immunofluorescence was visualized on a Zeiss microscope (Axio ImagerA1, Zeiss-Vision, Munich, Germany) equipped with a camera (AxioCAM Zeiss) (6).
Ethic Statements
Human islet collection and handling were approved by the local Ethical Committee in Pisa, Italy. Male Wistar rats were housed and used according to the guidelines of the Belgian Regulations for Animal Care. All experiments were approved by the local Ethical Committee. NOD mice have been housed at the KULeuven animal facility since 1989. NOD-SCID mice were also from a stock colony at the KULeuven. All experiments in mice were approved and performed in accordance with the Ethics Committee of the KULeuven, Leuven, Belgium.
Statistical Analysis
Data are expressed as means ± S.E. A significant difference between experimental conditions was assessed by one-way ANOVA followed by a paired Student's t test with Bonferroni correction. p values < 0.05 were considered statistically significant. The figures are shown as a box plot indicating lower quartile, median, and higher quartile, with whiskers representing the range of the remaining data points, when the number of experiments is ≥4 for each conditions. Alternatively, data are represented as points indicating individual experiments plus the average and the S.E. or bar graph with indicated S.E., when the number of experiments is <4.
Results
UBD Interacts with IRE1α
Candidate proteins that interact with IRE1α and are modified by pro-inflammatory cytokine treatment in pancreatic beta cells were identified using ArrayMAPPIT (13). UBD was chosen for detailed signal transduction studies following additional selection based on the review of the literature.
A binary MAPPIT analysis confirmed the interaction between UBD and IRE1α (Fig. 1A). HEK293T cells were co-transfected with an erythropoietin (EPO) receptor-based MAPPIT bait protein containing the cytoplasmic portion of IRE1α (pSEL-IRE1α), a FLAG-tagged UBD prey (pMG1-UBD), and a STAT3-responsive luciferase reporter (pXP2d2-rPAP1). After stimulation with EPO, the UBD prey induced a 10-fold increase in luciferase signal as compared with cells co-transfected with the empty prey vector. The binary MAPPIT analysis was also used to assess the ability of UBD to bind to the kinase-defective K599A IRE1α mutant (51) and to the different domains of IRE1α (Fig. 1A, kinase domain (Kin) and endoribonuclease domain (Ken)). After stimulation with EPO, the luciferase signals observed in both kinase-defective K599A IRE1α mutant and IRE1α Kin were comparable with the signal observed with the wild type IRE1α when combined with the UBD prey (Fig. 1A). However, the signal obtained with the IRE1α Ken was significantly reduced compared with the IRE1α wild type, suggesting that the endoribonuclease domain of IRE1α is less important for the interaction between IRE1α and UBD. These results indicate that the UBD-IRE1α interaction is independent of the phosphorylated state of IRE1α.
FIGURE 1.

IRE1α interacts with UBD. The IRE1α/UBD interaction was confirmed by binary MAPPIT (A) and endogenous co-immunoprecipitation in human EndoC-βH1 cells (B and C). For Binary MAPPIT HEK293T cells were transiently transfected with plasmids encoding IRE1α (IRE1), kinase-defective K599A IRE1α mutant (K599), the kinase domain of IRE1α (Kin), the endoribonuclease domain of IRE1α bait (Ken), or empty bait (Emp), together with an empty vector (gray bars) or pMG1-UBD prey construct (white bars), combined with the pXP2d2-rPAP1-luciferase reporter. After 24 h, cells were left untreated or stimulated with erythropoietin (EPO) for 24 h. Luciferase counts of triplicate measurements are expressed as fold induction versus non-stimulated. Results are represented as a box plot indicating lower quartile, median, and higher quartile, with whiskers representing the range of the remaining data points, n = 4 (A). INS-1 E cells (B) and EndoC-βH1 cells (C and D) were lysed and proteins collected for immunoprecipitation (IP) with anti-IRE1α antibodies. Nonspecific rabbit IgG was used as a negative control. IPs and total proteins were analyzed by Western blot (WB) using anti-IRE1α, anti-UBD, and anti-α-tubulin antibodies, as indicated. The figures shown are representative of three independent experiments (B and C). The means ± S.E. of the optical density analysis of 2–3 independent experiments of UBD-specific binding to the IRE1α IP compared with IgG is shown in D. Ab LC, antibody light chain. @, p < 0.05 IgG versus IRE1α; ***, p < 0.001 IRE1α-UBD versus IRE1α-empty; ###, p < 0.001 K599-UBD versus K599-empty; †††, p < 0.001 Kin-UBD versus Kin-empty; $$, p < 0.01 as indicated by bar; one-way ANOVA (A) and unpaired Student's t test (D).
The interaction between UBD and IRE1α was further validated by endogenous co-immunoprecipitation experiments in INS-1E cells (Fig. 1B) and in human EndoC-βH1 cells (Fig. 1, C and D). Using an anti-IRE1α antibody, we detected UBD in the immunoprecipitate (IP), as shown by Western blot analysis (Fig. 1, B and C). In the EndoC-βH1 lysate, UBD bound weakly also to the rabbit IgG (Fig. 1C), but the amount of UBD binding to IRE1α was 2-fold higher compared with that bound by the negative control rabbit IgG (Fig. 1D). To confirm the specificity of the IRE1α/UBD interaction, we have performed additional MAPPIT two-hybrid analyses using irrelevant protein baits in addition to the empty bait (Fig. 2). Using the 10-fold cutoff value that we routinely apply for selecting specific MAPPIT interactions, only PPP3CA would be considered as a specific UBD interactor in addition to IRE1α. To assess whether the strong signal obtained with IRE1α does not result from better bait expression, we measured the interaction of the different baits with UBD in parallel to their interaction with the REM2 protein. The REM2 interaction signal is a measure of the expression level of each individual bait, as it interacts with the cytokine backbone of the two-hybrid bait. The REM2 signal was not stronger for IRE1α as compared with the other baits, indicating that IRE1α more potently interacts with UBD than any of the other baits tested.
FIGURE 2.
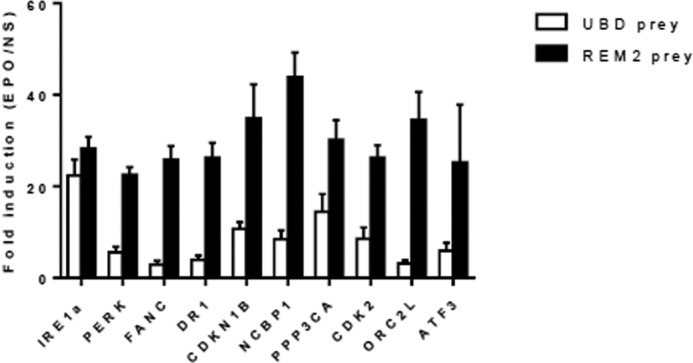
UBD specifically interacts with IRE1α in MAPPIT mammalian two-hybrid analysis. HEK293T cells seeded in 96-well plates were co-transfected with different bait proteins (gene symbols indicated on the x axis, all cloned in the pSEL(+2L) expression vector) along with the UBD or REM2 prey proteins and a STAT3 luciferase-based reporter gene. Twenty four hours after transfection, cells were stimulated with vehicle (non-stimulated) or EPO (20 ng/ml) to activate the two-hybrid system. After 18 h cells were lysed, and luciferase activity was measured. Data are presented as fold induction (EPO-stimulated over vehicle-stimulated luciferase values). Average and standard deviation of triplicate measurements are shown.
Inflammatory Signals Increase UBD Expression in Pancreatic Islet Cells
We confirmed by real time PCR (RT-PCR) our previous microarray findings (52, 53) indicating that pro-inflammatory cytokines induce UBD mRNA expression in rat insulin-producing cells. There was a peak of UBD expression after 16 or 24 h of IL-1β + IFN-γ exposure in INS-1E cells (Fig. 3A) and FACS-purified rat beta cells (Fig. 3C), respectively, with subsequent decrease. This induction was mostly dependent on IFN-γ, with only a minor contribution by IL-1β (Fig. 3B). These findings were reproduced in both human islets and in the human beta cell line EndoC-βH1 exposed for 24–48 h to IL-1β + IFN-γ (Fig. 3, D and E), confirming our previous RNA sequencing data (25). The observed cytokine-induced increase in UBD mRNA expression was confirmed at the protein level in EndoC-βH1 cells (Fig. 3, F and G).
FIGURE 3.

IL-1β + IFN-γ induce UBD expression in rodent and human beta cells. The expression of UBD was assessed by RT-PCR (A–E) and by Western blot (F and G) in INS-1E cells (A and B), FACS-purified rat beta cells (C), human islet cells (D), and the human EndoC-βH1 cells (E–G), and normalized by the housekeeping gene Gapdh (A–C) or β-actin (D–G). Cells were left untreated or treated with IL-1β + IFN-γ (IL+IFN) at different times, as indicated (A and C–G) or left untreated or treated with IL-1β (white bar), IFN-γ (gray bar), and IL-1β + IFN-γ (IL+IFN; black bar) for 24 h (B). Results are represented as a box plot indicating lower quartile, median, and higher quartile, with whiskers representing the range of the remaining data points (A and C–E). One representative Western blot for UBD (F) and optical density analysis of 6–7 independent experiments are shown (G). Data were normalized against the highest value (considered as 1) in each independent experiment (G). *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus 0 h or control (ctrl); paired Student's t test. Data shown are mean ± S.E. of 3–7 independent experiments.
To test whether increased UBD expression occurs during beta cell inflammation in vivo, we evaluated UBD expression in islets from diabetes-prone NOD mice. Immunofluorescence staining of pancreatic sections from NOD mice (Fig. 4A) showed UBD expression in insulin-positive cells of immune-infiltrated islets at 8 and 12 weeks (Fig. 4A, panels h–l), although UBD was not detected in islets from age-matched, genetically similar but not diabetes-prone NOD-SCID mice (Fig. 4B, panels b, c, e, f, h, and i). There was also no detectable UBD expression in islets from BALB/c mice examined at 4, 9 and 13 weeks of age (data not shown). The observed UBD expression in NOD mouse islets seems a direct consequence of islet inflammation (insulitis), because it was not detected in NOD mouse islets at 3 weeks of age (Fig. 4A, panels b and c), an age when islets are not yet invaded by immune cells, and in insulitis-free islets of NOD mice at 8 weeks of age (Fig. 4A, panels e and f).
FIGURE 4.

Increased expression of UBD in inflamed islets from diabetes-prone NOD mice. Pancreatic sections from 3-, 8-, and-12 week-old NOD mice were stained for insulin (panels a, d, g, and j, green), UBD (panels b, e, h, and k, red), and Hoechst for nuclear staining (panels c, f, i, and l, blue). Infiltrated lymphocytes, a sign of insulitis, are indicated by white arrowheads (panels i and l). Insulitis is present at 8 and 12 weeks of age but not at 3 weeks. The immunofluorescence analysis shows UBD expression in insulin-positive cells in NOD mice at 8 and 12 weeks (panels i and l, yellow) but not at 3 weeks (panels b and c) or at 8 weeks when insulitis is not present (panels e and f). The images shown are representative of 16 ± 3 islet sections from 3 to 4 different mice per age (A). Pancreatic sections from 8-, 9-, and 13-week-old NOD-SCID mice were stained with antibodies specific for insulin (panels a, d, and g, green), UBD (panels b, e, and h, red), and with Hoechst for nuclear staining (panels c, f, and i, blue). Immunofluorescence analysis indicates absence of UBD expression in insulin-positive cells in NOD-SCID mice (panels c, f, and i, merge). The images shown are representative of 12 ± 3 islet sections from 1 to 3 mice per age (B). Bars, 50 μm.
UBD Inhibition Does Not Affect IRE1α Endonuclease Activity in Rat and Human Pancreatic Beta Cells
To understand the function of cytokine-induced UBD expression on IRE1α activity, we knocked down (KD) UBD with three different specific siRNAs. The effects of two of them, siU2 and siU3, are shown in Fig. 5A; both siRNAs induced a >70% inhibition of UBD expression following cytokine exposure. UBD KD in INS-1E cells (Fig. 5, A–C) did not modify cytokine-induced IRE1α endonuclease activity as evaluated by Xbp1 splicing (Xbp1s) (Fig. 5B) and Ins-2 mRNA degradation (Fig. 5C). These data were confirmed in human beta cells, where UBD KD (60–80% inhibition; Fig. 5D) did not change cytokine-induced Xbp1 splicing (Fig. 5E).
FIGURE 5.

UBD KD does not affect IRE1 endonuclease activity in IL-1β + IFN-γ-treated rodent and human beta cells. INS-1E cells (A–C) and EndoC-βH1 cells (D and E) were transfected with siControl (siC) or two different siRNAs targeting rat UBD (siU2 and siU3, A–C) or human UBD (si-hU1 and si-hU2, D and E). 48 h later, cells were left untreated or treated with IL-1β + IFN-γ (IL+IFN) for 24 h (A–E). The expression levels of Ubd (A and D), Xbp1s (B and E), and Ins-2 (C) were assessed by RT-PCR and normalized by the housekeeping gene Gapdh (A–C) and β-actin (D and E). The results are represented as points that indicate single experiments plus the average and the S.E. (A–C) or as a box plot indicating lower quartile, median, and higher quartile, with whiskers representing the range of the remaining data points (D and E). *, p < 0.05; **, p < 0.01; and ***, p < 0.001 IL + IFN versus siC; #, p < 0.05, and ##, p < 0.01 as indicated by bars; one-way ANOVA followed by Student's paired t test with Bonferroni correction. Data shown are mean ± S.E. of 3–5 independent experiments.
UBD Modulates IRE1α-dependent JNK Activation
We next evaluated whether UBD KD interferes with IRE1α-dependent JNK phosphorylation in beta cells. A decrease of more than 70% in UBD protein expression was obtained with three different siRNAs (Fig. 6, A and B). The KD of UBD significantly increased cytokine-induced JNK phosphorylation in INS-1E cells after 2 h (Fig. 6, C and D) and 8 h of exposure (Fig. 6, E and F). Double KD of UBD and IRE1α reversed the stimulatory effect of UBD silencing on JNK phosphorylation (Fig. 7, A–D), confirming that the UBD effects on JNK are IRE1α-dependent. This was confirmed by quantification of the area under the curve for phospho-JNK expression (Fig. 7D) and was reproduced using a second siRNA targeting UBD (data not shown).
FIGURE 6.

UBD KD increases cytokine-dependent JNK phosphorylation. INS-1E cells were transfected with siControl (siC) or three different siRNAs targeting UBD (siU1, siU2, and siU3, A–F). 48 h later, cells were left untreated or treated with IL-1β + IFN-γ (IL+IFN) for 16 h (A and B). The phosphorylation of JNK was measured after 2 h (C and D) and 8 h (E and F) of IL-1β + IFN-γ (IL+IFN) treatment. Representative Western blots for UBD (A) and phospho-JNK (C and D) and the optical density analysis of 3–7 independent experiments (B, D, and F) are shown. α-Tubulin expression was used as loading control. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus siC untreated; #, p < 0.05; ##, p < 0.01 as indicated by bars. Paired Student's t test. Data shown are mean ± S.E. of 3–7 independent experiments.
FIGURE 7.

Effect of UBD on JNK phosphorylation is IRE1α-dependent. INS-1E cells were transfected with siControl (siC), siIRE1α (siIRE1), siUBD (siU2) or co-transfected with siIRE1 + siU2. 48 h later, cells were incubated with IL-1β + IFN-γ (IL+IFN) and analyzed by Western blot with phospho-JNK (phoJNK), total JNK, and IRE1α antibodies at the indicated time points. One representative Western blot of five independent experiments (A) and the means ± S.E. of the optical density measurements of the Western blots (B) are shown. α-Tubulin expression was used as loading control. The means ± S.E. of JNK expression in cytokine-treated cells are shown in C, and the quantitative analysis of the area under the curves (A.U.C.) is shown in D. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 versus siC untreated; @, p < 0.05 versus siC IL + IFN; #, p < 0.05, and ##, p < 0.01 as indicated by bars; one-way ANOVA followed by Student's paired t test with Bonferroni correction.
UBD-dependent JNK Modulation Regulates Cytokine-induced Apoptosis in Pancreatic Beta Cells
JNK has an important role in cytokine-induced beta cell apoptosis (12, 54–58). In line with this, the KD of UBD with two different siRNAs (Fig. 8, A, C, and E) significantly increased apoptosis induced by cytokines in INS-1E cells (Fig. 8B) and rat beta cells (Fig. 8, D and F), as evaluated by nuclear dyes. KD of UBD increased basal apoptosis in INS-1E cells (Fig. 8B), but this was not confirmed in primary rat beta cells (Fig. 8, D and F) or human beta cells (see below). UBD KD increased expression of cleaved caspases 3 and 9, suggesting activation of the intrinsic pathway of apoptosis (Fig. 8, G–I). Importantly, KD of UBD by two independent siRNAs (Fig. 9, A and C) also augmented cytokine-induced apoptosis in human islet cells (Fig. 9B) and in the EndoC-βH1 cells (Fig. 9D).
FIGURE 8.

UBD KD increases cytokine-induced apoptosis and caspase cleavage in INS-1E and FACS-purified rat beta cells. INS-1E cells (A, B, and G–I) and FACS-purified rat beta cells (C–F), were transfected with siControl (siC) or three different siUBDs (siU1, siU2, and siU3) as indicated. 48 h later, cells were left untreated or treated with IL-1β + IFN-γ (IL+IFN) as indicated. UBD KD was assessed by RT-PCR (A, C, and E) and normalized by the housekeeping gene Gapdh. Apoptosis was evaluated after 16 h of treatment in INS-1E (B) and after 24 h (D) or 48 h (F) in FACS-purified rat beta cells. Cleaved caspases (Casp) 3 and 9 were measured by Western blotting after 16 h of cytokine treatment in INS-1E cells (G–I). One representative Western blot of five independent experiments (G) and the densitometry results for cleaved caspase 3 (H) and cleaved caspase 9 (I) are shown. α-Tubulin expression was used as a loading control. Results are represented as a box plot indicating lower quartile, median, and higher quartile, with whiskers representing the range of the remaining data points in A, B, F, H, and I and as points indicating single experiments, plus the average and the S.E. in C–E. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 versus siC untreated; #, p < 0.05; ##, p < 0.01; and ###, p < 0.001; @@, p < 0.01 as indicated by bars; one-way ANOVA followed by Student's paired t test with Bonferroni correction.
FIGURE 9.

UBD KD increases cytokine-induced apoptosis in human islets and human EndoC-βH1 cells. Human islet cells (A and B) and the human beta cell line EndoC-βH1 (C and D) were transfected with siC and two different siUBDs (si-hU1 and si-hU2). 48 h later, cells were left untreated or treated with IL-1β + IFN-γ (IL+IFN) for 24 h. UBD KD was evaluated by RT-PCR and normalized by the housekeeping gene β-actin (A and C), and apoptosis was analyzed by propidium iodide/Hoechst staining (B and D). Results are represented as a box plot indicating lower quartile, median, and higher quartile, with whiskers representing the range of the remaining data points in B–D and as box plot indicating the single experiments, the average, and the S.E. in A. *, p < 0.05, and ***, p < 0.001 versus siC untreated; #, p < 0.05; ##, p < 0.01; and ###, p < 0.001 as indicated by bars; one-way ANOVA followed by Student's paired t test with Bonferroni correction. n = 3–7 independent experiments.
In additional experiments we observed that KD of IRE1α (Figs. 10, A and B, and 11A) or JNK1 (Figs. 10, C and D, and 11B) reversed the pro-apoptotic effect of UBD KD in both control and cytokine-treated INS-1E cells (Fig. 10, E and F) and FACS-purified rat beta cells (Fig. 11, E and F), confirming that cell death induced by UBD silencing in the context of cytokine exposure is mediated by up-regulation of the IRE1α/JNK1 pathway.
FIGURE 10.

KD of IRE1α or JNK1 protects INS-1E cells from cytokine-induced apoptosis in the context of UBD deficiency. INS-1E cells were transfected with siC, siIRE1, siU2, or co-transfected with siIRE1 + siU2 (A, B, and E). 48 h later, cells were left untreated or incubated with IL-1β + IFN-γ (IL+IFN) for 16 h. The KD of IRE1α and UBD was evaluated by Western blot (A) and apoptosis by propidium iodide/Hoechst staining (E). One representative Western blot of 4–7 independent experiments (A) and the densitometry results for IRE1α expression (B) are shown. INS-1E cells were transfected with siC, siJNK1, siU2, or co-transfected with siJNK1 + siU2 (C, D, and F). 48 h later, cells were left untreated or incubated with IL + IFN for 16 h. The KD of JNK1 was evaluated by Western blot (C and D) and apoptosis by propidium iodide/Hoechst staining (F). One representative Western blot of five independent experiments (C) and the densitometry results for JNK1 expression (D) are shown. α-Tubulin expression was used as loading control (A–D). Results are represented as a box plot indicating lower quartile, median, and higher quartile, with whiskers representing the range of the remaining data points in B, D, E, and F. *, p < 0.05, and ***, p < 0.001 versus siC untreated; #, p < 0.05; ##, p < 0.01, and ##, p < 0.001 as indicated by bars. One-way ANOVA followed by Student's paired t test with Bonferroni correction.
FIGURE 11.

KD of IRE1α or JNK1 protects rat beta cells from cytokine-induced apoptosis in the context of UBD-deficiency. FACS-purified rat beta cells were transfected with siControl (siC), siIRE1, siU2, or co-transfected with siIRE1 + siU2 (A, C, and E). 48 h later, cells were left untreated or incubated with IL-1β + IFN-γ (IL+IFN) for 48 h. The KD of IRE1α (A) and UBD (C) was assessed by RT-PCR and normalized by the housekeeping gene Gapdh. Apoptosis was evaluated by propidium iodide/Hoechst staining (E). FACS-purified rat beta cells were transfected with siC, siJNK1, siU2, or co-transfected with siJNK1 + siU2 (B, D, and F). 48 h after the transfection, the cells were left untreated or incubated with IL + IFN for 48 h. The KD of JNK1 (B) and UBD (D) was assessed by RT-PCR and normalized by the housekeeping gene Gapdh. Apoptosis was evaluated by propidium iodide/Hoechst staining (F). Results were normalized against the highest value in each independent experiment, considered as 1 (A–D). *, p < 0.05; **, p < 0.01, and ***, p < 0.001 versus siC untreated; #, p < 0.05; ##, p < 0.01, and ###, p < 0.001 versus siC treated; @, p < 0.05; @@, p < 0.01, and @@@, p < 0.001 as indicated by bars. One-way ANOVA followed by Student's paired t test with Bonferroni correction. Data shown are mean ± S.E. of six independent experiments.
UBD Inhibition Does Not Affect NO Production or Expression of the Chemokine Ccl-2
To investigate whether UBD deficiency inhibits cytokine-induced NF-κB activation and reduces the induction of NF-κB-regulated genes as has been described in other cell types (61), we measured two NF-κB-dependent phenomena, namely induction of NO formation (measured as nitrite accumulation in the medium) and expression of the chemokine Ccl-2 (3). Neither nitrite accumulation nor Ccl-2 expression was modulated by UBD KD in cytokine-treated INS-1E cells (Fig. 12), arguing against a role for NF-κB inhibition in the above described effects of UBD inhibition.
FIGURE 12.
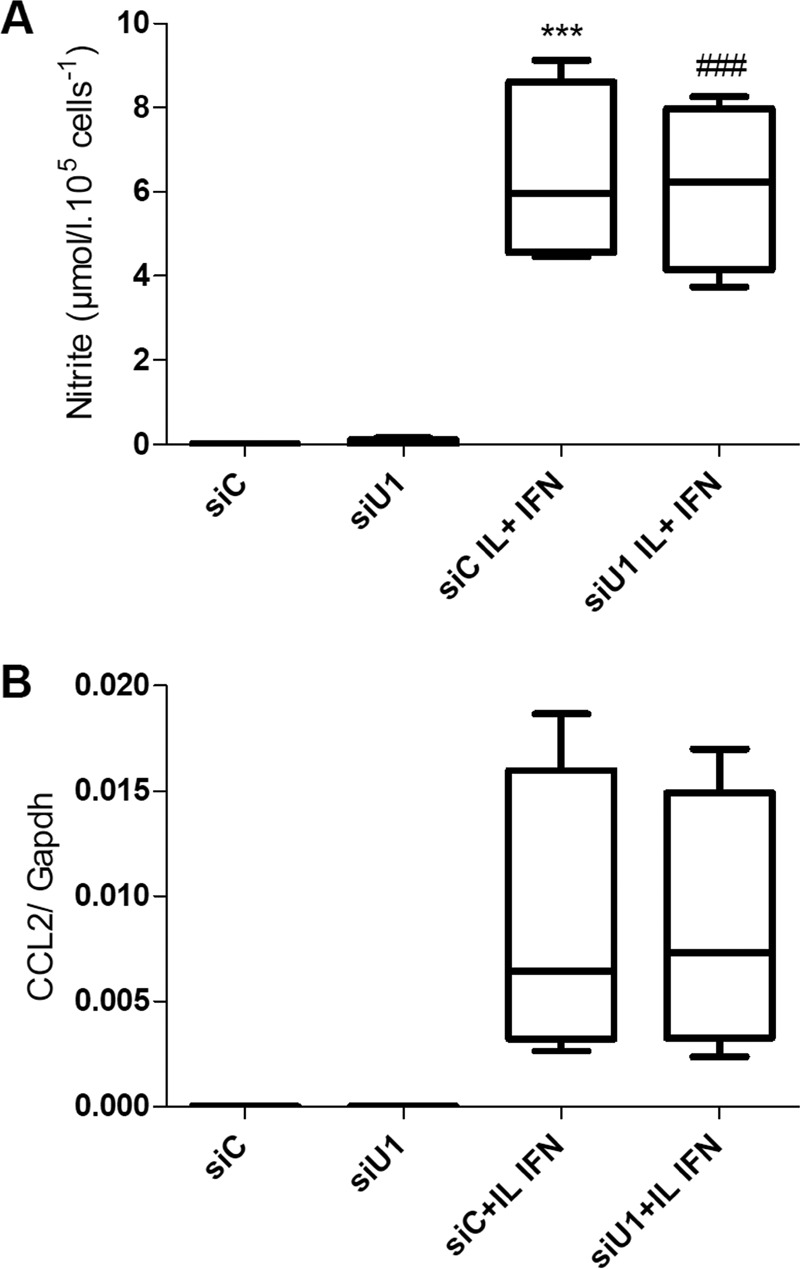
UBD KD does not affect nitrite accumulation in the medium or Ccl2 expression. INS-1E cells were transfected with siControl (siC) or an siRNA targeting UBD (siU1). 48 h later, cells were left untreated or treated with IL-1β + IFN-γ (IL+IFN). Supernatants from the cells were collected after 24 h of cytokine treatment, and nitrite accumulation in the culture medium was evaluated by the Griess method (A). The expression of CCL2 (B) was assessed by RT-PCR after 8 h of treatment and corrected for the housekeeping gene Gapdh. ***, p < 0.001 versus siC; ###, p < 0.001 versus siU1. ANOVA followed by Student's paired t test with Bonferroni correction. Data shown are mean ± S.E. of four independent experiments.
Discussion
IRE1α and its downstream targets XBP1s and JNK are potential regulatory molecules in the cross-talk between cytokine-induced ER stress and beta cell pro-inflammatory responses and apoptosis (3, 12, 13, 59). The activity of IRE1α is regulated by both signals generated from inside the ER and the cytosol (8, 11, 60). Against this background, we aim to identify novel cytokine-induced IRE1α-interacting proteins that modulate UPR activation in pancreatic beta cells and thus contribute to inflammation and beta cell death in early T1D. For this purpose, we previously combined a high-throughput mammalian two-hybrid technology, MAPPIT with functional genomic analysis of human and rodent beta cells exposed to pro-inflammatory cytokines (13). This allowed the identification of two proteins of particular interest, namely NMI (13) and UBD (this study).
We presently confirmed by quantitative RT-PCR and Western blot that IL-1β + IFN-γ induce UBD expression in rat INS-1E cells, FACS-purified rat beta cells, human islet cells, and in the human beta cell line EndoC-βH1. Induction of UBD is mostly an effect of IFN-γ, with a minor contribution by IL-1β. Expression of the protein in both clonal rat and human beta cells indicates that cytokine-induced UBD expression takes place at least in part at the beta cell level. Immunofluorescence staining of pancreatic sections from NOD mice showed UBD expression in insulin-positive cells at 8 and 12 weeks, although the protein was not detected in islets from age-matched NOD-SCID mice or pre-insulitis NOD mice, confirming up-regulation of UBD in vivo as a consequence of local inflammation (insulitis).
It has been described in other cell types that UBD deficiency abrogates cytokine-induced NF-κB activation and reduces the induction of NF-κB-regulated genes (61). It is, however, unlikely that these effects explain the present observations, because KD of UBD in beta cells does not affect the expression of downstream targets of NF-κB, such as inducible nitric oxide synthase and the chemokine (C-C motif) ligand 2 (CCL2) CCL2 (Fig. 12). Moreover, down-regulation of NF-κB has a protective effect in cytokine-induced beta cell apoptosis (62), whereas we presently observed that KD of UBD increases apoptosis in beta cells.
UBD effects seem to be independent of the phosphorylated state of IRE1α because UBD interacts with the K599 kinase-defective mutant IRE1α in the binary MAPPIT. UBD KD in INS-1E cells and human EndoC-βH1 cells does not modify cytokine-induced IRE1α endonuclease activity as evaluated by Xbp1 splicing (Xbp1s) and Ins-2 mRNA degradation. This last result is in line with the limited capacity of UBD to interact with the endoribonuclease domain (Ken) of IRE1α in the binary MAPPIT (Fig. 1A). The main effect induced by UBD KD was to increase JNK activity, with consequent augmentation of apoptosis. Thus, UBD KD increased cytokine-induced JNK phosphorylation after 2 and 8 h of exposure. Double KD of UBD and IRE1α reversed the up-regulation of JNK phosphorylation and the increase in apoptosis, confirming the regulatory function of UBD on IRE1α-dependent JNK activation in beta cells. Of interest, our previous findings indicate that another cytosolic IRE1α regulator, namely NMI, also inhibits JNK induction. The discovery that two cytosolic regulators of IRE1α provide specific negative feedback against IRE1α-induced JNK activation in beta cells (present data and see Ref. 13) confirms the key biological role of JNK for ER stress-induced beta cell apoptosis.
Induction of ER stress in human beta cells has basic mechanistic differences as compared with rat beta cells (12). Thus, cytokine-induced ER stress in rat beta cells involves inducible NOS expression, nitric oxide (NO) production, and consequent inhibition of the ER Ca2+-transporting ATPase SERCA2b (1). However, inhibition of NO formation does not prevent cytokine-induced ER stress activation and apoptosis in human islets (12). Moreover, the expression of SERCA-2b is not modified by cytokines in human islets and in the human EndoC-βH1 cells. Cytokines induce a biphasic JNK activation in human EndoC-βH1 cells, with peaks at 0.5 and 8 h and a parallel activation of IRE1α, which is more marked at 8–16 h. KD of IRE1α decreases JNK phosphorylation in EndoC-βH1 cells after both 0.5 and 8 h of IL-1β + IFN-γ treatment (12) and in INS-1E after both 2 and 8 h (this study) indicating that the IRE1α pathway contributes to both the early and late JNK activation in cytokine-exposed beta cells. Suppression of JNK1 expression by siRNAs partially protects both rat INS-1E and primary beta cells (this study) and human EndoC-βH1 cells against cytokine-induced apoptosis, confirming the key role for JNK in cytokine toxicity in human beta cells (12).
The concomitant activation of both JNK and IRE1α after cytokine exposure in human beta cells, the decrease on JNK phosphorylation after IRE1α KD, and the modulation of the IRE1α-induced JNK pathway by two cytokine-induced IRE1α-binding proteins (this study and see Ref. 13), all point to the conclusion that JNK is the major IRE1α-regulated pathway in the cross-talk between ER-stressed/apoptotic beta cells and the immune system.
The Ubd gene lies in the telomeric region of MHC class I, a major susceptibility locus for T1D. Polymorphisms in the Ubd gene have been associated with autoimmune diabetes in rat and human (26–29), but the role of these polymorphisms in the expression of UBD remains to be clarified. This study indicates that UBD provides a negative feedback to a key pro-apoptotic branch of the UPR in the beta cells, namely the activation of JNK. During a protracted autoimmunity assault, this protective feedback will be eventually overrun, but it is conceivable that both UBD (this study) and NMI (13) play an important protective role during putative transient periods of mild inflammation in the islets, preventing excessive beta cell lost.
Author Contributions
F. B., S. G., S. L., F. A. G., J. T., and D. L. E. conceived and designed the experiments. F. B., S. G., S. L., A. B., M. J., C. G., and M. B. performed the experiments. F. B., S. G., F. A. G., M. J., J. T., and D. L. E. analyzed the data. F. B., S. G., S. L., C. G., P. M., J. T., C. M., and D. L. E. contributed reagents/materials/analysis tools. F. B. and D. L. E. wrote the paper. All authors revised the manuscript.
Acknowledgments
We thank A. Musuaya, M. Pangerl, S. Mertens, and R. Lemma for excellent technical support; Dr. Guy Bottu and Dr. Jean-Valery Turatsinze for the bioinformatics support. We are grateful to the Flow Cytometry Facility of the Erasmus Campus of the Universitè Libre de Bruxelles and Christine Dubois for the cell sorting. We are greatly indebted to Drs. Marc Vidal and David Hill, Center for Cancer Systems Biology (CCSB) at Dana-Farber Cancer Institute (Boston), for making the CCSB ORFeome collection 5.1 available.
This work was supported in part by grants from the Juvenile Diabetes Research Foundation International Grant 17-2013-515 and NIDDK, Human Islet Research, National Institutes of Health Network Consortium Grant 1UC4DK104166-01 (to D. L. E.), the Fund for Scientific Research Flanders (FWO Grant 1522313N), European Union (Seventh Framework Projects of the European Union. NAIMIT and BetaBat), Actions de Recherche Concertée de la Communauté Française, and the Fonds National de la Recherche Scientifique, Belgium (to D. L. E.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article.
- ER
- endoplasmic reticulum
- T1D
- type 1 diabetes
- UPR
- unfolded protein response
- UBD
- ubiquitin D
- NOD
- non-obese diabetic
- MAPPIT
- mammalian protein-protein interaction trap
- ANOVA
- analysis of variance
- EPO
- erythropoietin
- IP
- immunoprecipitate
- PERK
- protein kinase RNA-like endoplasmic reticulum kinase.
References
- 1. Cardozo A. K., Ortis F., Storling J., Feng Y. M., Rasschaert J., Tonnesen M., Van Eylen F., Mandrup-Poulsen T., Herchuelz A., and Eizirik D. L. (2005) Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta cells. Diabetes 54, 452–461 [DOI] [PubMed] [Google Scholar]
- 2. Eizirik D. L., Cardozo A. K., and Cnop M. (2008) The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 29, 42–61 [DOI] [PubMed] [Google Scholar]
- 3. Eizirik D. L., Miani M., and Cardozo A. K. (2013) Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia 56, 234–241 [DOI] [PubMed] [Google Scholar]
- 4. Tersey S. A., Nishiki Y., Templin A. T., Cabrera S. M., Stull N. D., Colvin S. C., Evans-Molina C., Rickus J. L., Maier B., and Mirmira R. G. (2012) Islet beta cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 61, 818–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Engin F., Yermalovich A., Nguyen T., Ngyuen T., Hummasti S., Fu W., Eizirik D. L., Mathis D., and Hotamisligil G. S. (2013) Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Sci. Transl. Med. 5, 211ra156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marhfour I., Lopez X. M., Lefkaditis D., Salmon I., Allagnat F., Richardson S. J., Morgan N. G., and Eizirik D. L. (2012) Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 55, 2417–2420 [DOI] [PubMed] [Google Scholar]
- 7. Walter P., and Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 8. Eizirik D. L., and Cnop M. (2010) ER stress in pancreatic beta cells: the thin red line between adaptation and failure. Sci. Signal. 3, e7. [DOI] [PubMed] [Google Scholar]
- 9. Fonseca S. G., Gromada J., and Urano F. (2011) Endoplasmic reticulum stress and pancreatic beta cell death. Trends Endocrinol. Metab. 22, 266–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gurzov E. N., and Eizirik D. L. (2011) Bcl-2 proteins in diabetes: mitochondrial pathways of beta cell death and dysfunction. Trends Cell Biol. 21, 424–431 [DOI] [PubMed] [Google Scholar]
- 11. Woehlbier U., and Hetz C. (2011) Modulating stress responses by the UPRosome: a matter of life and death. Trends Biochem. Sci. 36, 329–337 [DOI] [PubMed] [Google Scholar]
- 12. Brozzi F., Nardelli T. R., Lopes M., Millard I., Barthson J., Igoillo-Esteve M., Grieco F. A., Villate O., Oliveira J. M., Casimir M., Bugliani M., Engin F., Hotamisligil G. S., Marchetti P., and Eizirik D. L. (2015) Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia 58, 2307–2316 [DOI] [PubMed] [Google Scholar]
- 13. Brozzi F., Gerlo S., Grieco F. A., Nardelli T. R., Lievens S., Gysemans C., Marselli L., Marchetti P., Mathieu C., Tavernier J., and Eizirik D. L. (2014) A combined “Omics” approach identifies N-Myc interactor as a novel cytokine-induced regulator of IRE1α protein and c-Jun N-terminal kinase in pancreatic beta cells. J. Biol. Chem. 289, 20677–20693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee C. G., Ren J., Cheong I. S., Ban K. H., Ooi L. L., Yong Tan S., Kan A., Nuchprayoon I., Jin R., Lee K. H., Choti M., and Lee L. A. (2003) Expression of the FAT10 gene is highly upregulated in hepatocellular carcinoma and other gastrointestinal and gynecological cancers. Oncogene 22, 2592–2603 [DOI] [PubMed] [Google Scholar]
- 15. Lukasiak S., Schiller C., Oehlschlaeger P., Schmidtke G., Krause P., Legler D. F., Autschbach F., Schirmacher P., Breuhahn K., and Groettrup M. (2008) Proinflammatory cytokines cause FAT10 upregulation in cancers of liver and colon. Oncogene 27, 6068–6074 [DOI] [PubMed] [Google Scholar]
- 16. Ebstein F., Lange N., Urban S., Seifert U., Krüger E., and Kloetzel P. M. (2009) Maturation of human dendritic cells is accompanied by functional remodelling of the ubiquitin-proteasome system. Int. J. Biochem. Cell Biol. 41, 1205–1215 [DOI] [PubMed] [Google Scholar]
- 17. Buerger S., Herrmann V. L., Mundt S., Trautwein N., Groettrup M., and Basler M. (2015) The ubiquitin-like modifier FAT10 is selectively expressed in medullary thymic epithelial cells and modifies T cell selection. J. Immunol. 195, 4106–4116 [DOI] [PubMed] [Google Scholar]
- 18. Raasi S., Schmidtke G., de Giuli R., and Groettrup M. (1999) A ubiquitin-like protein which is synergistically inducible by interferon-γ and tumor necrosis factor-α. Eur. J. Immunol. 29, 4030–4036 [DOI] [PubMed] [Google Scholar]
- 19. Lisak R. P., Nedelkoska L., Studzinski D., Bealmear B., Xu W., and Benjamins J. A. (2011) Cytokines regulate neuronal gene expression: differential effects of Th1, Th2 and monocyte/macrophage cytokines. J. Neuroimmunol. 238, 19–33 [DOI] [PubMed] [Google Scholar]
- 20. Liu Y. C., Pan J., Zhang C., Fan W., Collinge M., Bender J. R., and Weissman S. M. (1999) A MHC-encoded ubiquitin-like protein (FAT10) binds noncovalently to the spindle assembly checkpoint protein MAD2. Proc. Natl. Acad. Sci. U.S.A. 96, 4313–4318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu X., Liu X., Liu T., Hong K., Lei J., Yuan R., and Shao J. (2012) Identification of a novel binding protein of FAT10: eukaryotic translation elongation factor 1A1. Dig. Dis. Sci. 57, 2347–2354 [DOI] [PubMed] [Google Scholar]
- 22. Aichem A., Kalveram B., Spinnenhirn V., Kluge K., Catone N., Johansen T., and Groettrup M. (2012) The proteomic analysis of endogenous FAT10 substrates identifies p62/SQSTM1 as a substrate of FAT10ylation. J. Cell Sci. 125, 4576–4585 [DOI] [PubMed] [Google Scholar]
- 23. Schmidtke G., Aichem A., and Groettrup M. (2014) FAT10ylation as a signal for proteasomal degradation. Biochim. Biophys. Acta 1843, 97–102 [DOI] [PubMed] [Google Scholar]
- 24. Pociot F., and McDermott M. F. (2002) Genetics of type 1 diabetes mellitus. Genes Immun. 3, 235–249 [DOI] [PubMed] [Google Scholar]
- 25. Eizirik D. L., Sammeth M., Bouckenooghe T., Bottu G., Sisino G., Igoillo-Esteve M., Ortis F., Santin I., Colli M. L., Barthson J., Bouwens L., Hughes L., Gregory L., Lunter G., Marselli L., et al. (2012) The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 8, e1002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aly T. A., Baschal E. E., Jahromi M. M., Fernando M. S., Babu S. R., Fingerlin T. E., Kretowski A., Erlich H. A., Fain P. R., Rewers M. J., and Eisenbarth G. S. (2008) Analysis of single nucleotide polymorphisms identifies major type 1A diabetes locus telomeric of the major histocompatibility complex. Diabetes 57, 770–776 [DOI] [PubMed] [Google Scholar]
- 27. Blankenhorn E. P., Cort L., Greiner D. L., Guberski D. L., and Mordes J. P. (2009) Virus-induced autoimmune diabetes in the LEW.1WR1 rat requires Iddm14 and a genetic locus proximal to the major histocompatibility complex. Diabetes 58, 2930–2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baschal E. E., Sarkar S. A., Boyle T. A., Siebert J. C., Jasinski J. M., Grabek K. R., Armstrong T. K., Babu S. R., Fain P. R., Steck A. K., Rewers M. J., and Eisenbarth G. S. (2011) Replication and further characterization of a Type 1 diabetes-associated locus at the telomeric end of the major histocompatibility complex. J. Diabetes 3, 238–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cort L., Habib M., Eberwine R. A., Hessner M. J., Mordes J. P., Blankenhorn E. P. (2014) Diubiquitin (Ubd) is a susceptibility gene for virus-triggered autoimmune diabetes in rats. Genes Immun. 15, 168–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marchetti P., Bugliani M., Lupi R., Marselli L., Masini M., Boggi U., Filipponi F., Weir G. C., Eizirik D. L., and Cnop M. (2007) The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 50, 2486–2494 [DOI] [PubMed] [Google Scholar]
- 31. Moore F., Colli M. L., Cnop M., Esteve M. I., Cardozo A. K., Cunha D. A., Bugliani M., Marchetti P., and Eizirik D. L. (2009) PTPN2, a candidate gene for type 1 diabetes, modulates interferon-γ-induced pancreatic beta cell apoptosis. Diabetes 58, 1283–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marroqui L., Masini M., Merino B., Grieco F. A., Millard I., Dubois C., Quesada I., Marchetti P., Cnop M., and Eizirik D. L. (2015) Pancreatic alpha cells are resistant to metabolic stress-induced apoptosis in type 2 diabetes. EBioMedicine 2, 378–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Asfari M., Janjic D., Meda P., Li G., Halban P. A., and Wollheim C. B. (1992) Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology 130, 167–178 [DOI] [PubMed] [Google Scholar]
- 34. Ravassard P., Hazhouz Y., Pechberty S., Bricout-Neveu E., Armanet M., Czernichow P., and Scharfmann R. (2011) A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J. Clin. Invest. 121, 3589–3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eizirik D. L., and Mandrup-Poulsen T. (2001) A choice of death: the signal-transduction of immune-mediated beta cell apoptosis. Diabetologia 44, 2115–2133 [DOI] [PubMed] [Google Scholar]
- 36. Kutlu B., Cardozo A. K., Darville M. I., Kruhøffer M., Magnusson N., Ørntoft T., and Eizirik D. L. (2003) Discovery of gene networks regulating cytokine-induced dysfunction and apoptosis in insulin-producing INS-1 cells. Diabetes 52, 2701–2719 [DOI] [PubMed] [Google Scholar]
- 37. Ortis F., Cardozo A. K., Crispim D., Störling J., Mandrup-Poulsen T., and Eizirik D. L. (2006) Cytokine-induced proapoptotic gene expression in insulin-producing cells is related to rapid, sustained, and nonoscillatory nuclear factor-κB activation. Mol. Endocrinol. 20, 1867–1879 [DOI] [PubMed] [Google Scholar]
- 38. Eizirik D. L., Pipeleers D. G., Ling Z., Welsh N., Hellerström C., and Andersson A. (1994) Major species differences between humans and rodents in the susceptibility to pancreatic beta cell injury. Proc. Natl. Acad. Sci. U.S.A. 91, 9253–9256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eizirik D. L., Sandler S., Welsh N., Cetkovic-Cvrlje M., Nieman A., Geller D. A., Pipeleers D. G., Bendtzen K., and Hellerström C. (1994) Cytokines suppress human islet function irrespective of their effects on nitric oxide generation. J. Clin. Invest. 93, 1968–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lievens S., Vanderroost N., Defever D., Van der Heyden J., and Tavernier J. (2012) ArrayMAPPIT: a screening platform for human protein interactome analysis. Methods Mol. Biol. 812, 283–294 [DOI] [PubMed] [Google Scholar]
- 41. Eyckerman S., Verhee A., der Heyden J. V., Lemmens I., Ostade X. V., Vandekerckhove J., and Tavernier J. (2001) Design and application of a cytokine-receptor-based interaction trap. Nat. Cell Biol. 3, 1114–1119 [DOI] [PubMed] [Google Scholar]
- 42. Lievens S., Vanderroost N., Van der Heyden J., Gesellchen V., Vidal M., and Tavernier J. (2009) Array MAPPIT: high-throughput interactome analysis in mammalian cells. J. Proteome Res. 8, 877–886 [DOI] [PubMed] [Google Scholar]
- 43. Moore F., Cunha D. A., Mulder H., and Eizirik D. L. (2012) Use of RNA interference to investigate cytokine signal transduction in pancreatic beta cells. Methods Mol. Biol. 820, 179–194 [DOI] [PubMed] [Google Scholar]
- 44. Miani M., Barthson J., Colli M. L., Brozzi F., Cnop M., and Eizirik D. L. (2013) Endoplasmic reticulum stress sensitizes pancreatic beta cells to interleukin-1β-induced apoptosis via Bim/A1 imbalance. Cell Death Dis. 4, e701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cunha D. A., Hekerman P., Ladrière L., Bazarra-Castro A., Ortis F., Wakeham M. C., Moore F., Rasschaert J., Cardozo A. K., Bellomo E., Overbergh L., Mathieu C., Lupi R., Hai T., Herchuelz A., et al. (2008) Initiation and execution of lipotoxic ER stress in pancreatic beta cells. J. Cell Sci. 121, 2308–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hoorens A., Van de Casteele M., Klöppel G., and Pipeleers D. (1996) Glucose promotes survival of rat pancreatic beta cells by activating synthesis of proteins which suppress a constitutive apoptotic program. J. Clin. Invest. 98, 1568–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rasschaert J., Ladrière L., Urbain M., Dogusan Z., Katabua B., Sato S., Akira S., Gysemans C., Mathieu C., and Eizirik D. L. (2005) Toll-like receptor 3 and STAT-1 contribute to double-stranded RNA+ interferon-γ-induced apoptosis in primary pancreatic beta cells. J. Biol. Chem. 280, 33984–33991 [DOI] [PubMed] [Google Scholar]
- 48. Chen M. C., Proost P., Gysemans C., Mathieu C., and Eizirik D. L. (2001) Monocyte chemoattractant protein-1 is expressed in pancreatic islets from prediabetic NOD mice and in interleukin-1β-exposed human and rat islet cells. Diabetologia 44, 325–332 [DOI] [PubMed] [Google Scholar]
- 49. Overbergh L., Valckx D., Waer M., and Mathieu C. (1999) Quantification of murine cytokine mRNAs using real time quantitative reverse transcriptase PCR. Cytokine 11, 305–312 [DOI] [PubMed] [Google Scholar]
- 50. Cardozo A. K., Kruhøffer M., Leeman R., Orntoft T., and Eizirik D. L. (2001) Identification of novel cytokine-induced genes in pancreatic beta cells by high-density oligonucleotide arrays. Diabetes 50, 909–920 [DOI] [PubMed] [Google Scholar]
- 51. Tirasophon W., Lee K., Callaghan B., Welihinda A., and Kaufman R. J. (2000) The endoribonuclease activity of mammalian IRE1 autoregulates its mRNA and is required for the unfolded protein response. Genes Dev. 14, 2725–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Moore F., Naamane N., Colli M. L., Bouckenooghe T., Ortis F., Gurzov E. N., Igoillo-Esteve M., Mathieu C., Bontempi G., Thykjaer T., Ørntoft T. F., and Eizirik D. L. (2011) STAT1 is a master regulator of pancreatic beta cell apoptosis and islet inflammation. J. Biol. Chem. 286, 929–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ortis F., Naamane N., Flamez D., Ladrière L., Moore F., Cunha D. A., Colli M. L., Thykjaer T., Thorsen K., Orntoft T. F., and Eizirik D. L. (2010) Cytokines interleukin-1β and tumor necrosis factor-α regulate different transcriptional and alternative splicing networks in primary beta cells. Diabetes 59, 358–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ammendrup A., Maillard A., Nielsen K., Aabenhus Andersen N., Serup P., Dragsbaek Madsen O., Mandrup-Poulsen T., and Bonny C. (2000) The c-Jun amino-terminal kinase pathway is preferentially activated by interleukin-1 and controls apoptosis in differentiating pancreatic beta cells. Diabetes 49, 1468–1476 [DOI] [PubMed] [Google Scholar]
- 55. Bonny C., Oberson A., Steinmann M., Schorderet D. F., Nicod P., and Waeber G. (2000) IB1 reduces cytokine-induced apoptosis of insulin-secreting cells. J. Biol. Chem. 275, 16466–16472 [DOI] [PubMed] [Google Scholar]
- 56. Bonny C., Oberson A., Negri S., Sauser C., and Schorderet D. F. (2001) Cell-permeable peptide inhibitors of JNK: novel blockers of beta cell death. Diabetes 50, 77–82 [DOI] [PubMed] [Google Scholar]
- 57. Marroquí L., Santin I., Dos Santos R. S., Marselli L., Marchetti P., and Eizirik D. L. (2014) BACH2, a candidate risk gene for type 1 diabetes, regulates apoptosis in pancreatic beta cells via JNK1 modulation and crosstalk with the candidate gene PTPN2. Diabetes 63, 2516–2527 [DOI] [PubMed] [Google Scholar]
- 58. Gurzov E. N., Ortis F., Cunha D. A., Gosset G., Li M., Cardozo A. K., and Eizirik D. L. (2009) Signaling by IL-1β+IFN-γ and ER stress converge on DP5/Hrk activation: a novel mechanism for pancreatic beta cell apoptosis. Cell Death Differ. 16, 1539–1550 [DOI] [PubMed] [Google Scholar]
- 59. Miani M., Colli M. L., Ladrière L., Cnop M., and Eizirik D. L. (2012) Mild endoplasmic reticulum stress augments the proinflammatory effect of IL-1β in pancreatic rat beta cells via the IRE1α/XBP1s pathway. Endocrinology 153, 3017–3028 [DOI] [PubMed] [Google Scholar]
- 60. Ron D., and Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 [DOI] [PubMed] [Google Scholar]
- 61. Gong P., Canaan A., Wang B., Leventhal J., Snyder A., Nair V., Cohen C. D., Kretzler M., D'Agati V., Weissman S., and Ross M. J. (2010) The ubiquitin-like protein FAT10 mediates NF-κB activation. J. Am. Soc. Nephrol. 21, 316–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ortis F., Pirot P., Naamane N., Kreins A. Y., Rasschaert J., Moore F., Théâtre E., Verhaeghe C., Magnusson N. E., Chariot A., Orntoft T. F., and Eizirik D. L. (2008) Induction of nuclear factor-κB and its downstream genes by TNF-α and IL-1β has a pro-apoptotic role in pancreatic beta cells. Diabetologia 51, 1213–1225 [DOI] [PubMed] [Google Scholar]