

Abstract
Cellular membrane disruption induced by β-amyloid (Aβ) peptides has been considered one of the major pathological mechanisms for Alzheimer disease. Mechanistic studies of the membrane disruption process at a high-resolution level, on the other hand, are hindered by the co-existence of multiple possible pathways, even in simplified model systems such as the phospholipid liposome. Therefore, separation of these pathways is crucial to achieve an in-depth understanding of the Aβ-induced membrane disruption process. This study, which utilized a combination of multiple biophysical techniques, shows that the peptide-to-lipid (P:L) molar ratio is an important factor that regulates the selection of dominant membrane disruption pathways in the presence of 40-residue Aβ peptides in liposomes. Three distinct pathways (fibrillation with membrane content leakage, vesicle fusion, and lipid uptake through a temporarily stable ionic channel) become dominant in model liposome systems under specific conditions. These individual systems are characterized by both the initial states of Aβ peptides and the P:L molar ratio. Our results demonstrated the possibility to generate simplified Aβ-membrane model systems with a homogeneous membrane disruption pathway, which will benefit high-resolution mechanistic studies in the future. Fundamentally, the possibility of pathway selection controlled by P:L suggests that the driving forces for Aβ aggregation and Aβ-membrane interactions may be similar at the molecular level.
Keywords: Alzheimer disease, amyloid β, ion channel, liposome, membrane, NMR
Introduction
Membrane disruption induced by amyloid peptides has been considered a universal pathological mechanism for amyloid diseases (1–4). For Alzheimer disease, neuronal cellular membrane disruptions caused by β-amyloid (Aβ)3 peptides, including both 40- and 42-residue Aβ, have been reported within a broad range of systems, from synthetic phospholipid liposomes to the isolated plasma cellular membrane and living cells (5–17). However, mechanistic studies of the Aβ-induced membrane disruption process, particularly at the high-resolution level, are hindered by the co-existence of heterogeneous pathways even in the simplest model systems. For instance, Aβ fibrillation may occur on the membrane surface, which serves as a platform to enhance the local concentration of Aβ or to promote the formation of specific structures that facilitate fibrillar elongation (18). The oligomeric form of certain Aβ aggregates may produce ionic channels inside of the membrane bilayer, and the channels are reported to be selective to cations such as K+ and Ca2+ (19–21). In addition, it has been discovered recently that Aβ peptides (or oligomers) may also be fusogenic, i.e. inducing lipid mixing and vesicle fusion between liposomes (22, 23). Along individual pathways, the structural evolution of Aβ may be heterogeneous, i.e. with the co-existence of both on-pathway and off-pathway intermediates. It might be possible to distinguish these intermediate species using novel high-resolution techniques such as dynamic-nuclear-polarization-based solid-state NMR spectroscopy (24). However, the presence of multiple pathways will further increase complexity and make high-resolution studies infeasible. Therefore, it is important to generate individual Aβ-membrane systems with dominant pathways at least in the model phospholipid liposomes.
Studies in this work utilize model liposomes with a molar ratio between phosphatidylcholine (PC) and phosphatidylglycerol (PG) of ∼4:1, which mimics the ratio of neutral versus negatively charged phospholipids in neuronal cells (25). The presence of other lipid components and sterol molecules is not considered in this simplest liposome model. One experimentally adjusted parameter, the peptide-to-lipid molar ratio (P:L), is investigated. The P:L is inhomogeneous in human brains because the local Aβ concentration can be affected by many factors, such as the efficiency of the enzymatic cleavage of the amyloid precursor protein and the removal process of the eluted Aβ (14, 26). It has been shown that the quantities of Aβ peptides from the aged human brain cortex vary between 103∼104 pmol/g of wet brain tissue (27). Given the fact that the mass percentage of total lipids in the human brain is ∼2% (28), the estimated P:L ratio in the human brain may vary between 1:25,000 and 1:2500, which is far from most in vitro studies using phospholipid liposomes. However, the membrane distribution of Aβ is also heterogeneous, with more abundant Aβ concentrated in the sterol-enriched lipid raft domain (11, 12). For instance, it was reported previously that a volume fraction of 0.5% lipid raft region might contain ∼25% of the total 40-residue Aβ (29). Therefore, in these regions, the P:L ratio can be as high as 1:500–1:50, which is close to the P:L value used in typical biophysical and structural studies for Aβ-membrane interactions.
We have shown previously that, when monomeric Aβ was added into preformed PC/PG liposomes, the two evolution pathways, fibrillation and peptide-induced lipid mixing, were competing with each other when the P:L ratio changed from 1:30 to 1:120 (23). Specifically, the higher P:L ratio benefited fibril formation, and the lower ratio promoted lipid mixing. This result illustrated the importance of the P:L ratio in the generation of model systems for studying the Aβ-membrane interaction. In this work, we systematically investigate multiple membrane disruption pathways, including membrane content leakage, vesicle fusion, Aβ-lipid aggregation, and formation of ionic channels, and their correlations with the P:L ratio. Our results show that three of these pathways, fibrillation accompanied by membrane content leakage, Aβ-induced vesicle fusion, and the detachment of Aβ oligomers from membrane bilayers with lipid uptake, can be dominant within distinct model systems. The P:L ratio plays a crucial role in pathway selection. The results suggest that it is possible to study individual membrane disruption pathways at high-resolution levels in future works.
Experimental Procedures
Peptide Synthesis and Purification
All 40-residue Aβ peptides were synthesized manually using routine solid-phase peptide synthesis protocols and Fmoc chemistry. Isotope-labeled amino acids were purchased from Cambridge Isotope Inc., and the Fmoc protection groups were added on by the literature approach if necessary. Peptides were purified using reverse-phase HPLC (Agilent Inc.) and a C18 column (Zorbax, Agilent Inc.) and then lyophilized. The lyophilized powder was dissolved in 2% v/v NH3 and freeze-dried again to remove any preformed aggregation, according to a method described previously (32). The resulting peptides were stored at −20 °C until use.
Aβ-membrane Sample Preparation
“External addition” and “preincorporation” sample preparation protocols were described in detail previously (23, 30, 31). Briefly, the external addition samples were made by adding Aβ to preformed liposomes, which were prepared by resuspending dried lipid film in phosphate buffer (10 mm (pH 7.4) with 0.01% NaN3), followed by 10 cycles of freeze-thaw and 30× extrusion with the designed membrane sizes. The preincorporation samples were prepared from the Aβ/lipid film (co-dissolving of Aβ and lipids in hexafluoro-2-propanol and chloroform, respectively) through the same resuspension, freeze-thaw, and extrusion cycles. All samples that were utilized in this study had liposomes with PC and PG with a molar ratio of 4:1, extruded with 100-nm pore size membranes, and an Aβ concentration of 50 μm. Only the P:L ratio was varied for different samples. For the external addition samples, the peptide powder was freshly dissolved in 2% DMSO (v/v to the final volume of liposome) to ensure the initial monomeric form (32–35).
Analytical HPLC
The Aβ/liposome solutions from either external addition or preincorporation samples were centrifuged (Beckman Coulter Inc., 432,000 × g, 2 h, 4 °C) after the 4-h incubation period at 37 °C. The supernatant (∼1 ml for each sample) was collected, and 5% acetic acid (v/v) was added. The solution was then diluted by a factor of 2 using acetonitrile and sonicated for 3 min in a water bath sonicator to disassociate any oligomers. The supernatant was analyzed using an analytical C18 reversed-phase column (Zorbax, Agilent Inc.) and an acetonitrile-water linear solvent gradient (1–99% acetonitrile over 35 min). The concentration of Aβ was calculated by integrating the peptide elution peak and comparing it with the standard working curve from freshly dissolved Aβ peptides.
Circular Dichroism Spectroscopy
All CD spectra were recorded on a Jasco J-810 spectrophotometer with a 0.1-cm path length quartz cuvette. Both external addition and preincorporation samples were incubated at 37 °C for the designated period of time. A 12-μl aliquot of sample solution was diluted with deionized water to a total volume of 300 μl and transferred into the CD cuvette. All spectra were recorded with a wavelength range of 190∼260 nm at a 1 nm/sec scanning rate, the temperature at 20 °C (controlled with a water bath), and a signal averaging over 40 scans. No spectral difference was observed between individual scans, indicating that the dilution prevented further structural evolution during CD measurements. Control spectra that contained only liposomes and were incubated under the same conditions as the corresponding Aβ-membrane samples were recorded and subtracted from the sample spectra. All spectra were processed using instrumental software.
Fluorescence Spectroscopy
All fluorescence experiments were performed using a PerkinElmer Life Sciences LS55 spectrometer with an external water bath for temperature control around 37 °C during the recording of kinetic traces. The detailed protocols for both ThT fluorescence and lipid mixing assays have been described in previous studies (23). The Aβ-membrane samples were prepared to measure the membrane content leakage as follows. For the external addition samples, the calcein-contained liposomes were prepared by co-dissolving lipids and calcein in chloroform (the final concentration of calcein in all samples was kept at 10 mm). This was followed by the formation of a lipid/calcein film under high-vacuum resuspension of the film in phosphate buffer (10 mm (pH 7.4) with 0.01% NaN3), extrusion through a 100-nm filter membrane, and dialysis against bulk phosphate buffer (1:1000 v/v, 2 × 24-hour dialysis at 4 °C) to remove any uncaptured calcein molecules. Monomeric Aβ peptides were freshly dissolved in 2% DMSO (v/v to the calcein-contained liposome solution) and added into liposomes. The mixture was transferred to a fluorescence cuvette and placed in the prewarmed sample holder, and the recording of the kinetic trace was started immediately. For the preincorporation samples, the Aβ peptides, lipids, and calcein were co-dissolved in organic solvents to form the film. This was followed by the same resuspension, extrusion, and dialysis process. The long dialysis period used in this protocol might have caused the negative results in the membrane content leakage measurements for the preincorporation samples.
Confocal Fluorescence Imaging
Fluorescence imaging measurements were performed on large unilamellar vesicles that were extruded using 1.0-μm filter membrane and giant unilamellar vesicles that were prepared using the fast evaporation method described previously (36). The large unilamellar vesicle samples, which were prepared using the preincorporation protocol, were utilized to identify the co-localization of lipids and Aβ peptides with 1% rhodamine B-labeled phosphatidylethanolamine (PE, molar percentage of total lipids) and rhodamine green-labeled 40-residue Aβ. The samples were prepared with P:L molar ratios of 1:30 and 1:120 and incubated at 37 °C for 48 h before testing. The giant unilamellar vesicle samples were prepared for the external addition condition only because the fast evaporation method did not efficiently incorporate Aβ peptides into vesicles. The giant unilamellar vesicle samples were utilized to monitor the changes in vesicle size upon addition of Aβ at both 1:30 and 1:120 P:L ratios.
The liposome suspension was gently vortexed before imaging. 400 μl of the liposome suspension was added to a glass-bottom confocal dish (MatTek Corp.). Images of the liposomes were captured at ambient temperature using a laser-scanning confocal microscope (Leica TCS SP2) with an HCX PL APO 63 × 1.4 oil immersion objective by ×4 digital zoom-in. Rhodamine green-labeled Aβ40 peptides were excited using a 488-nm argon laser, and rhodamine B-labeled PE was excited using a 543-nm HeNe laser. The laser power was kept at a minimum to allow sufficient signaling while avoiding unnecessary photobleaching. All images were processed with ImageJ software.
Black Lipid Membrane (BLM) Measurements of Ionic Channel Activities
To prepare the BLM, a 50-μl aliquot of 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine (in chloroform, 25 mg/ml) was dried under air flow to form a lipid film that was then redissolved in 75 μl of n-decane. The two-compartment electrophysiological bilayer chamber with a 200-μm aperture (Harvard Apparatus Inc., placed in a Faraday cage to reduce external vibration) was filled with 10 mm HEPES buffer (containing 150 mm KCl and 1 mm magnesium gluconate (pH 7.4)). A 10-μl aliquot of 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine/decane solution was deposited onto the aperture using a pipette to produce the BLM (which was monitored by measuring the resistance across the bilayer). All Aβ-liposomes used in the ionic channel measurements were prepared with the designed P:L ratio and a modified preincorporation protocol. 20 μl of 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine in chloroform was dried out to create a film. Then 75 μl of phosphate buffer was added to the film, and the solution was briefly sonicated in a bath sonicator. Then 11.7 μl of freshly prepared Aβ solution (2 mg/ml in deionized water) was added to the resuspended lipids. The mixture was freeze-thawed in liquid N2 and a room temperature water bath for 10 cycles to form vesicles with both lipids and Aβ. A transparent liposome solution was obtained using this approach. The solution was either used directly for the current trace recording or incubated for 48 h before testing. For the samples with chemical cross-linking, after the freeze-thaw cycles, 15.66 μl of glutaraldehyde (70% in water, Sigma-Aldrich) was added to cross-link the ion channels in the sample (19, 37). The sample was then spun on a rotator mixer for 20 min. Then 21.75 μl of Tris buffer (1 m, pH 7) was added to quench the cross-linking reaction, and the sample was set to incubate for 48 h at 37 °C.
To record an electronic current trace, the BLM was equilibrated for 5∼10 min after the starting point for baseline recording. Drops of 3 μl of 10 mm CaCl2 solution were added to both compartments of the chamber to facilitate the fusion of liposomes onto the BLM (38). The designed volume (20∼80 μl) of liposome solution was then added to one side of the compartments that was connected to the anode of an Ag/AgCl electrode with gentle mixing. Recording of the electronic current trace was continued for 30∼90 min (or until complete disruption of the BLM under certain circumstance).
Solid-state NMR Spectroscopy
All solid-state NMR measurements were performed on a 600 MHz Bruker NMR spectrometer installed with a 2.5-mm TriGamma magic angle spinning probe that was tuned to the 1H/31P/13C configuration. All NMR samples were incubated at 37 °C for the desired time periods before sample collection. NMR samples were collected by ultracentrifuging the Aβ/liposome solution (Beckmann Coulter Inc., TLX100.4 rotor, 100,000 rpm for 1 h at 4 °C) to form a gel-like pellet. For 31P static and T2 measurements, the wet pellet was packed into a 2.5-mm sample rotor with an Eppendorf centrifuge. For 13C-31P REDOR measurements, the wet pellet was lyophilized, packed into a rotor as dry powder, and rehydrated with deionized water.
The 31P static spectra were collected with the 1H-decoupled 31P direct excitation pulse sequence in which the acquisition was applied right after the 50-kHz, π/2 31P radiofrequency pulse. A continuous-wave 100-kHz 1H decoupling field was applied throughout the excitation and acquisition periods. The static spectra were processed with 100-Hz Gaussian line broadening. The 31P T2 measurements were performed with a Hahn echo pulse sequence with 50-kHz π/2 and π 31P pulses. Spectra with an array of delay times were recorded, and the normalized peak areas were plotted versus the delay period. The 13C,31P REDOR pulse sequence contains a 50-kHz 1H π/2 excitation pulse, a cross-polarization period (∼45-kHz 1H field and 40- to 65-kHz 13C field with a linear ramp over 1.5 ms), a series of rotor-synchronized π pulses with XY-8 phase cycling, and an acquisition period with a 100-kHz 1H decoupling field. The 13C transmitter was set to 165 ppm, which was close to the carbonyl carbon region of the spectrum. For each REDOR dephasing period, a pair of S0 and S1 spectra was collected without and with the 31P π pulses, respectively. The REDOR dephasing curve was obtained by plotting the normalized 13C signal attenuation (i.e. (S0-S1)/S0) as a function of dephasing time. All Spectra were processed with a 100-Hz Gaussian line broadening. For magic angle spinning spectroscopy, the spinning frequency was set to 10 kHz, and the temperature for all NMR measurements was controlled with a 270 K cooling N2 flow (corresponding to the ∼280 K sample temperature estimated from the 1H chemical shift of H2O). The REDOR curves were fit to a model 13C,31P two-spin system using a SIMPSON simulation package and the best fit internuclear distances were reported (39).
Results
Initial States of Aβ in Model Systems
We generated model systems using previously described external addition and preincorporation protocols with a range of P:L ratio from 1:30–1:120 (23, 30, 31). Peptides were pretreated using an approach reported in the literature to ensure the monomeric state (40). For the external addition protocol, we have shown that the thioflavin T (ThT) fluorescence emission was detected immediately after the addition of Aβ to liposomes for P:L values of 1:30, 1:60, and 1:90 but not for the P:L ratio of 1:120 (23). Additionally, the same concentration of free Aβ in aqueous buffer (i.e. 50 μm) does not show detectable ThT fluorescence within a similar incubation period (i.e. ∼6 h, data not shown), meaning that the rapid aggregation of Aβ is induced by peptide-membrane interactions. Analytical HPLC was performed to analyze the concentration of residual Aβ in the supernatant after the 4-h incubation period (Fig. 1). For the external addition samples, the percentage of residual Aβ in the supernatant decreases with the P:L ratio. At the lowest ratio (i.e. 1:120), ∼25% of Aβ remains in solution after the short incubation. The preincorporation samples, on the other hand, contains little residual Aβ (∼5%) in the supernatant, and the percentage seems to be independent of the P:L ratio. CD spectroscopy characterization of Aβ conformation in the external addition samples further confirmed the lack of abundant β strands (i.e. mostly random-coil, Fig. 2A) within the time period. Overall, for the external addition samples, the monomeric Aβ peptides seem to be absorbed by the liposome, and the initial nucleation process starts with membrane interactions in the first few hours of incubation. On the other hand, the CD spectra for preincorporation samples with different P:L ratios show a significantly higher population of β strand conformations within 4 h and increasing populations of β strands after 24 h (Fig. 2B). Therefore, high-order Aβ oligomers, but not fibrils (proved by transmission electron microscopy (TEM) in previous studies (31)), seem to be present as the dominant form of Aβ in the preincorporation samples. These oligomers may form during liposome preparation when a high concentration of Aβ peptides is suspended in aqueous buffer.
FIGURE 1.

Analytical HPLC of residual 40-residue Aβ in supernatants. A, HPLC chromatographs for the external addition and preincorporation samples with different P:L ratios. The Aβ eluted at ∼22 min. The dashed HPLC profile shown in the top left panel represents the blank without Aβ. The minor peak at ∼21 min was identified (using mass spectrometry) as the truncated Aβ impurity. B, plot of the residual Aβ (Ab) concentrations in the supernatant of external addition (sample index: 1, P:L = 1:30; 2, 1:60; 3, 1:90; 4, 1:120) and preincorporation (sample index: 5, 1:30; 6, 1:60; 7, 1:90; 8, 1:120) samples, calculated from the standard curve of freshly dissolved Aβ peptide. Error bars were determined from three independent runs. C, calibration curve from the freshly dissolved Aβ (with a concentration range from 1.5–30 μm) with a sample HPLC profile (30 μm) shown in the inset (x axis, elution time in minutes; y axis, intensity at 214 nm). Error bars were determined from three independent runs. a.u., arbitrary unit.
FIGURE 2.
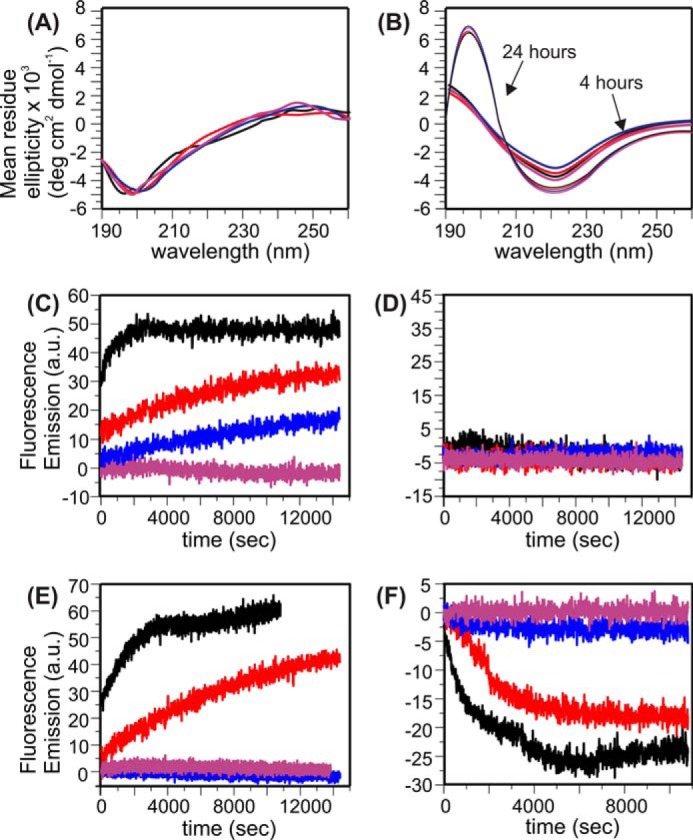
CD and fluorescence spectra for Aβ-liposome samples. Samples with different P:L ratios are color-coded in a uniform way for all panels (black, 1:30; red, 1:60; blue, 1:90; purple, 1:120). A and B, CD spectra for external addition (with 4-h incubation) (A) and preincorporation samples (with 4- and 24-h incubation) (B). Control spectra with liposomes in the absence of Aβ were subtracted from the corresponding Aβ-liposome samples. C and D, membrane content leakage traces from time-dependent calcein fluorescence emissions for external addition (C) and preincorporation (D) samples. All spectra were recorded with excitation and emission wavelengths at 485 and 520 nm, respectively. E, ThT fluorescence measurements on fibrillation kinetics for the preincorporation samples. Similar measurements for the external addition samples have been reported previously. Excitation and Emission wavelengths were set to 430 and 490 nm, respectively. F, lipid mixing assay on vesicle fusion for the preincorporation samples. Similar measurements for the external addition samples have been reported before. Excitation and Emission wavelengths were set to 480 and 585 nm, respectively. a.u., arbitrary unit.
Fibrillation and Membrane Content Leakage Are Associated, but They Compete with Vesicle Fusion
We measured the calcein fluorescence emission for both the external addition and preincorporation samples with various P:L ratios (Fig. 2, C and D). Calcein molecules were initially concentrated inside of the liposomes, and therefore the fluorescence signal was attenuated because of the self-quenching effect (18). The increment of the fluorescence emission indicates dilution of calcein and therefore leakage of the membrane bilayers. For the external addition samples, the calcein fluorescence emission increases at a higher P:L ratio, which follows the same trend as the ThT fluorescence but behaves oppositely to the trend of lipid mixing. Therefore, it is possible that the fibrillation process on the membrane surface induces local disruption of the phospholipid bilayers, which allows leakage of calcein molecules. At the highest P:L ratio (i.e. 1:30), the kinetics of membrane leakage is obviously faster than the build-up of ThT fluorescence emission and reaches plateau in less than 1 h. Because the CD results show that the peptide has mainly a random coil conformation within this short time period, the membrane content leakage is likely to be induced by the initial aggregation of Aβ, which does not involve the formation of a large hydrophobic core with β sheet-like structures. The absence of calcein fluorescence at a lower P:L ratio suggested that the vesicle fusion process did not induce membrane content leakage, which was consistent with the well known SNARE-mediated model through hemifusion and fusion pore formation (41).
For the preincorporation samples, calcein fluorescence emissions were not observed for any P:L ratio. This result is surprising because we have proposed previously (and will prove with later evidence) that membranes are seriously disrupted by the elution of Aβ oligomers under this condition (30, 31). However, it is worth noting that the calcein-contained liposomes used in this fluorescence assay were separated from the uncaptured calcein through slow dialysis (see “Experimental Procedures”). It is likely that membrane disruption had already occurred during the dialysis process and the system had been in equilibrium before the calcein fluorescence assay. ThT fluorescence (Fig. 2E) and lipid mixing measurements (Fig. 2F) were performed on preincorporation samples to further study the evolution of Aβ oligomers. At high P:L ratios (i.e. 1:30 and 1:60), increments in the ThT fluorescence are detected, meaning that further Aβ aggregation occurs during the incubation. Given the fact that there had already been β strand-enriched structures at the beginning of incubation, it is likely that the size of the β sheet core increased. On the other hand, the lipid-mixing assay resulted in a decreased fluorescence signal, which means that the distances between fluorophores and quenching chromophores are shortened (see “Experimental Procedures”). This is opposite to the expected fluorescence enhancement in lipid mixing, suggesting that there is no vesicle fusion along the evolution pathway of the preincorporation samples. At low P:L ratios (i.e. 1:90 and 1:120), neither ThT fluorescence nor lipid mixing activity was detected.
Membrane-associated Aβ Oligomers Form Peptide-Lipid Complexes Rapidly at a High P:L Ratio
We have previously proposed the formation of Aβ-lipid complexes in preincorporation samples with a P:L of ∼1:30 (30). Experimentally, spherical oligomer species were observed by TEM with 4-h incubation (31), and solid-state NMR spectroscopy identified the parallel β sheet core and residue-specific lipid contact within the oligomers (30). To further investigate the fates of membrane-associated Aβ oligomers at different P:L ratios, we perform solid-state NMR 31P static spectroscopy (Fig. 3A) and relaxation measurements (Fig. 3B). The data show that the isotropic 31P peak intensity increases significantly during the 4-h incubation period for the samples with high P:L ratios (i.e. 1:30 and 1:60), but samples with lower ratios have less change. Particularly the phospholipid bilayers seem to be unaffected at a P:L of ∼1:120 within 4 h. The large isotropic 31P peak intensities for samples with a high P:L ratio decrease after 24-h incubation, but the isotropic peak intensities for low P:L ratio samples seem to slightly increase within the same incubation time. This suggests that, along with the formation of spherical oligomer species (within the first few hours based on previous TEM measurements (31)), at least a fraction of lipid molecules experience fast tumbling, which is different from the typical uniaxial rotation and wobbling motion of lipids in integrated bilayers. For samples with a high P:L ratio, this lipid motion was restricted after a long incubation period. In addition, the spin-spin relaxation (T2) time constants of 31P for samples with 1:30 and 1:60 P:L ratios decrease within the same period of incubation (i.e. ∼4 h) but increase again to their initial values after 24 h. For lower P:L ratios such as 1:90 and 1:120, no obvious change in T2 is detected. Because the T2 constant is mainly influenced by low-frequency motion and therefore reflects the overall rigidity of membrane bilayers (42, 43), the result again suggests that, at a high P:L ratio, membranes are likely to be disrupted within a few hours.
FIGURE 3.

31P solid-state NMR measurements for preincorporation Aβ-liposome samples. A, static 31P NMR spectra collected at 1- (black), 4- (red), and 24-h (blue) incubation times for samples with different P:L ratios. Isotropic peaks were observed at ∼5 ppm for the sample with P:L ratios of 1:30 and 1:60 but less significantly for samples with P:L ratios of 1:90 and 1:120. B, plots of 31P T2 relaxation curves versus incubation time (1, 4, and 24 h). A faster relaxation rate was observed for 1:30 and 1:60 samples at a 4-h incubation period. All experimental data sets fit reasonably well to single exponential decay curves.
Our previous solid-state NMR measurements suggested residue-specific contacts between Aβ and lipids in the preincorporation sample with a P:L ratio of 1:30 (30). For instance, the residues Asp-23 and Leu-34 are detected to be in close proximity to the lipid phosphate headgroups. We performed confocal fluorescence imaging measurements to further study the locations of Aβ and lipids in the external addition and preincorporation samples with different P:L ratios. All samples were prepared with a 48-h incubation period and therefore reflected the equilibrium states of the corresponding Aβ-membrane system. Fig. 4A shows that, in the systems with fluorophore-labeled lipids, the external addition samples, at both 1:30 and 1:120 P:L ratios, contain only liposomes with mostly spherical morphology. Therefore, the externally added Aβ did not induce disruption of the overall integrity of membranes at either a higher or lower P:L ratio. However, at the lower P:L ratio, there are a number of cases where fusion between individual vesicles is observed, resulting in non-spherical species. Detailed analysis of the liposome size suggests that the distribution of vesicle size in the external addition sample with a 1:120 P:L ratio is broader and that there are larger populations of vesicles with greater sizes compared with the 1:30 sample (as well as the control liposomes without peptides), indicating the presence of vesicle fusion. For the preincorporation samples, confocal fluorescence imaging experiments were performed on the double fluorophore-labeled system where the rhodamine B-labeled (red) lipids and rhodamine green-labeled Aβ were utilized. We observed co-localization of lipids and peptides within the amorphous aggregates (Fig. 4B) for P:L ratios 1:30 and 1:120. For the 1:30 preincorporation sample, there seems to be a larger population of amorphous aggregates and few spherical vesicles. For the 1:120 sample, the aggregates are smaller in size and less abundant, whereas the spherical vesicles are greater in population. At the 1:120 ratio, the spherical vesicles contain only lipids (red) but no peptide (green). The co-localization of lipids and peptides in aggregates illustrates the final fate of Aβ oligomers in preincorporation samples, i.e. they eventually disassociated from the bilayer by phospholipid uptake and therefore caused membrane disruption. This process is likely to happen at all P:L ratios that have been tested in this work. However, a higher P:L ratio accelerates the process.
FIGURE 4.

Confocal fluorescence imaging for Aβ-liposome samples. A, representative confocal imaging for rhodamine B-labeled (red channel only) liposomes in the external addition giant unilamellar vesicle samples and the distribution of vesicle sizes after 48-h incubation. The histograms for the control (without peptide) and the sample with 1:30 P:L ratio and 1:120 P:L ratio are shown in the top left, center left, and bottom left panels, respectively. The arrows highlighted non-spherical species that indicate fusion between individual vesicles. B, representative confocal imaging for rhodamine B-labeled (red channel) liposomes and rhodamine green-labeled (green channel) 40-residue Aβ peptides in the preincorporation large unilamellar vesicle samples with a P:L ratio of 1:30 (left panels) and 1:120 (right panels). The white dotted contours highlight the morphologies of aggregates, and the arrows highlight spherical vesicles only composed of lipids (lack of green channel signal at the same locations). Scale bars = 5 μm.
Residue-specific Peptide-to-Lipid Contact in the Aβ-Lipid Complex
We have previously determined residue-specific contacts between lipid phosphate headgroups (i.e. 31P) and residues Asp-23 and Leu-34 within the Aβ-lipid aggregates in the preincorporation sample with a P:L ratio of 1:30 using solid-state NMR rotational echo double-resonance (REDOR) spectroscopy (31). To perform more quantitative measurements, we synthesized four doubly labeled Aβ sequences. Within each sequence, one glycine and one non-glycine amino acid (i.e. Ala, Val, or Leu) were 13C isotope-labeled at the carbonyl carbons. This design utilized the distinct 13C′ (carbonyl carbon) chemical shifts between glycine and other residues. Based on the fluorescence imaging results on the same sample, peptides were only located in the aggregates. Therefore, the REDOR distances reflect the 13C,31P proximities within the aggregates. Fig. 5, A–D, plots the representative REDOR spectra with 17.8- and 23.8-ms mixing periods. The experimental REDOR buildup curves and the fitting of the experimental data to a theoretical 13C,31P two-spin system are shown in Fig. 5, E–H. Certain residues within the Aβ sequence have closer contacts (and therefore faster buildup curves) to lipid 31P than the others, indicating that there are specific Aβ-lipid binding sites within the complex and that the amorphous Aβ-lipid aggregates are composed of ordered Aβ-lipid complexes at the molecular level. This is consistent with the fact that the morphologies of aggregates observed in red (i.e. lipids) and green (i.e. peptides) channels in confocal imaging are similar. The residues Gly-25, Gly-29, Gly-33, and Val-36 showed relatively stronger 31P contacts and therefore shorter internuclear distances, whereas residues Gly-9, Val-12, Leu-17, and Ala-21 were farther away from 31P. Quantitative REDOR distance fitting showed that two residues, Gly-25 (with the best fit 13C,31P distance of 6.3 ± 0.2 Å) and Val-36 (with the best fit 13C,31P distance of 6.0 ± 0.2 Å), had particularly close contacts to 31P, which was consistent with our previous observations for residues Asp-23 and Leu-34. Fittings for residues Gly-33 (7.0 ± 0.1 Å) and Gly-29 (7.3 ± 0.1 Å) showed longer distances. It is worth noting that the fittings for Gly-25 and Val-36 were less perfect compared with Gly-33 and Gly-29 (with larger χ2min values). It is possible that multiple lipid molecules are located closer to these residues so their REDOR data were more significantly deviated from an ideal two-spin system. We have also shown previously that Aβ peptides were likely to adopt a parallel in-register β sheet core structure in the Aβ-lipid complex, which was similar to the Aβ fibrils (31). This means residues Gly-25 and Val-36 are located far away from each other within one Aβ molecule. Therefore, it is reasonable to conclude that there is more than one possible binding site on the Aβ sequence for the lipid molecule.
FIGURE 5.

A–D, 13C,31P REDOR measurements on the preincorporation sample with a P:L ratio of 1:30. For each labeled sequence, pairs of representative S0/S1 REDOR spectra at 17.8 and 23.8 ms dephasing times. Each spectrum shows a doublet where the right-side peak represents Gly C' and the left-side peak is from the C' of the second type of amino acid (i.e. Val/Leu/Ala). E–H, the experimental REDOR dephasing (i.e. ΔS = (S0-S1)/S0) was calculated by integrating over a 1-ppm range around the corresponding S0 and S1 peaks. Shown is fitting of the experimental ΔS versus REDOR dephasing time to a single 13C,31P spin pair using SIMPSON. For each set of experimental data, the best fit 13C,31P dipolar coupling frequency (fopt) and the standard deviation (χ2min) are shown. The best fit 13C,31P distances (r) were calculated using the relationship r = (12,250/fopt)1/3.
Membrane-associated Aβ Oligomers Form Temporarily Stable Ionic Channels at a Low P:L Ratio
Although the preincorporation samples from Aβ-lipid complexes and aggregates at both P:L ratios 1:30 and 1:120, they behave differently within the first few hours of incubation according to our fluorescence and 31P NMR measurements. It has been suggested that Aβ peptides form an ionic channel in the model liposomes that were prepared using similar preincorporation protocols and a P:L ratio around 1:120 (19, 21, 37). We prepared fresh preincorporation samples with this low P:L ratio and fused the Aβ-oligomer-contained vesicles to the BLM deposited on a 200-μm aperture within an electrophysiological chamber. Without the addition of Aβ-contained liposomes, the BLM is sufficiently stable. Electronic current induced by the K+ cation across the BLM bilayer, which indicates the opening and closing activities of ionic channels, was observed (Fig. 6A). Meanwhile, resistance across the planar BLM decreases and the noise level of the current trace increases continuously, suggesting the destabilization of membrane bilayer. Interestingly, when the same sample (the preincorporation sample with a P:L ratio of 1:120) was incubated at physiological temperature for 48 h, no obvious ionic channel activity was recorded. The planar BLM, however, still seems to be destabilized (Fig. 6B), as indicated by the resistance decreasing over time. In addition, a slow time scale oscillation of the electronic current trace was recorded, which might suggest membrane leakage through nonspecific interactions other than ion channels. When samples were freshly prepared with the P:L ratio of 1:30, the BLM seemed to be unaffected and was stable for a long time (Fig. 6C). Fluorescence imaging and previous TEM results showed that, at this high P:L ratio, the Aβ oligomers elute from the membrane bilayer, form a peptide-lipid complex rapidly, and form larger aggregates after the 48-h incubation period (30). These aggregates are likely to sediment in the electrophysiological chamber and will not fuse to the BLM bilayer. In addition, the chemically cross-linked Aβ-liposome system with a P:L ratio of 1:120 showed ionic channel activity after 48-h incubation (Fig. 6D). These findings suggest that the Aβ ionic channels could form within a short period time at relatively a low P:L ratio but may not be stable over time. Chemical cross-linking may help stabilize the ionic channel structures (19). Similar temporary cation-selected membrane pore formation for the membrane-associated 40-residue Aβ has been reported in a previous article where the monomeric peptide was added to preformed liposomes (40). The peptide concentration (∼10 μm) utilized in that work was approximately five times lower than our Aβ concentration, and the P:L ratio (1:500∼1:1000) was also much lower. For this system, the authors proposed a two-step membrane disruption process that involved the formation of a membrane pore first, followed by the detergent-like lipid disruption. In fact, this seems to be similar to our current observation for the preincorporation sample with a P:L ratio of 1:120, which represents membrane interactions of Aβ oligomers. Although monomeric Aβ was added into preformed liposomes in the literature, it was shown that the peptide was probably in the nucleation step while interacting with membranes. Therefore, the initial Aβ oligomers were probably stabilized by excessive amount of lipids. This is different from our external addition samples, where the Aβ concentration was higher but there were less surrounding lipids. In our case, the Aβ peptides are more likely to have peptide-peptide interactions to form fibrils.
FIGURE 6.

Electronic current traces for the preincorporation samples. Each panel contains a 1-min current trace that is ∼10 min after the data collection and a 3-s expanded region that highlights the signal. A, freshly prepared sample with a P:L ratio of 1:120. B, the same sample as in A but after 48-h incubation at 37 °C. C, cross-linked sample with a P:L ratio of 1:120 and after 48-h incubation at 37 °C. D, freshly prepared sample with a P:L ratio of 1:30.
Discussion
Three Distinct Aβ Evolution Pathways in the Presence of the Membrane
Three distinct membrane-associated Aβ evolution pathways are identified based on this as well as previous studies, and each individual pathway seems to result in membrane disruption differently. Elution of Aβ occurs after amyloid precursor protein cleavage (44). Whether the peptide aggregates before interacting with the membrane depends on the local Aβ concentration, which can be affected by a variety of factors, such as the efficiency of Aβ elution from the membrane and the clearance of existing Aβ aggregates. In addition, the critical concentration of aggregation for Aβ peptides may also vary in a broad range, from below 0.1 μm to above 10 μm, as determined previously using a number of techniques (45–48). Therefore, the interactions between Aβ and the membrane may start from a highly heterogeneous system that involves the co-existence of Aβ monomers and small and large oligomers. In this work, the external addition model systems contain largely unstructured Aβ at the initial stage of incubation (Fig. 2A), and the peptides are slowly absorbed by membrane. It is likely that it represents membrane interactions of Aβ monomers or small oligomers. On the other hand, the preincorporation model contains β strand-enriched Aβ constructs (Fig. 2B) that are initially bound to liposomes. Therefore, it mimics the interaction between large Aβ oligomers and membranes.
When monomeric or small oligomeric Aβ is exposed to phospholipid liposomes with a relatively high P:L ratio, the peptide forms amyloid fibrils (Fig. 7A). The whole process under physiological temperature and pH value takes several days, and it was previously traced by both TEM and ThT fluorescence (30). The ThT fluorescence was characterized by an initial slow enhancement, indicating the formation of a nucleus on the membrane surface, followed by a rapid increase in fibril elongation. Fig. 4A shows that there is no lipid involved in Aβ fibrils because no filament-like morphology was observed in the red channel. During the fibrillation process (i.e. 48 h after the initial incubation), the liposome is largely intact. However, the very beginning stage of nucleation is accompanied by membrane content leakage. Based on previous solid-state NMR 31P relaxation measurements, the overall membrane rigidity was reduced during the time period of membrane leakage, and this membrane disruption was less significant in the absence of negatively charged lipids or in the presence of cholesterol (31).
FIGURE 7.

A, proposed membrane interaction pathways under different conditions of initial Aβ oligomeric states and P:L molar ratio. Fibrillation and vesicle fusion are induced by membrane interactions of monomeric or small oligomeric Aβ peptides. Membrane fragmentation and Aβ-lipid aggregates are induced by membrane interactions of preformed large Aβ oligomers, with the possibility of forming ionic channels. B, schematic of binding between Aβ and lipids in the Aβ-lipid aggregates based on solid-state NMR measurements. A possible binding model between Aβ and phospholipids, derived from solid-state NMR 13C,31P REDOR experiments. The best fit distances for four specific residues, Gly-25, Gly-29, Gly-33, and Val-36, are shown with dashed circles, which indicate the possible binding sites of phospholipids. At least two binding sites are required to fulfill the experimental results. The schematic is drawn in scale.
When the P:L ratio is relatively low, monomeric or small oligomeric Aβ will not form fibrils but induce lipid mixing and vesicle fusion (Fig. 7A). This was observed previously by a fluorescence lipid mixing assay and confirmed by the increment of vesicle size in confocal imaging (23). The peptide does not exist in the form of large β sheet-like oligomers at the fusion site because an increase of ThT fluorescence was not detected. We have shown previously that the same segments in the Aβ sequence were involved in both the initial fibrillation (i.e. peptide-peptide interaction) and vesicle fusion (i.e. peptide-lipid interaction) and therefore induced the competition between these two evolution pathways (23).
The interaction of preformed large Aβ oligomers with membrane bilayers takes a distinct pathway (Fig. 7A) because it does not lead to vesicle fusion. Meanwhile, it will not form long filament-like amyloid fibrils even after having been incubated for a long time. Previous TEM images have shown that the oligomers will finally form amorphous aggregates of spherical species and short and curvy protofilaments (30). We further show here that the aggregates are composed of both Aβ and lipids and that there is little Aβ remaining in the liposomes. However, the detailed elution process of Aβ oligomers from the membrane may depend on the P:L ratio, which defines the amount of lipids available to stabilize the oligomers. At a high P:L ratio (e.g. 1:30), the elution process occurs rapidly and results in the formation of a stable Aβ-lipid complex. This process is accompanied by further increments of oligomer size (increase of ThT fluorescence) and an overall decrease in membrane rigidity (decrease of the 31P T2 constant). At this stage, we obtained the following structural features for the Aβ-lipid complex (31): previous chemical shift measurements on individual residues showed that Phe-19, Ala-21, Asp-23, Ala-30, and Leu-34 were in β strand conformation and that Ser-26, Lys-28, and Val-40 were in coil conformation. Residues Val-18, Ala-21, Ala-30, and Met-35 were in parallel-in-register β sheet conformation. The Aβ oligomer core had a similar structure as the Aβ fibril because close contacts were detected within residue pairs Asp-23/Lys-28 and Phe-19/Leu-34. Qualitatively, there were close contacts between lipid 31P and residues Asp-23 and Leu-34. Quantitatively, the 13Cs of Gly-25 (6.5 ± 0.2 Å), Gly-29 (7.3 ± 0.1 Å), Gly-33 (7.0 ± 0.1 Å), and Val-36 (6.2 ± 0.2 Å) are closer to 31P, and the N-terminal residues Gly-9 (> 10.0 Å), Val-12 (> 10.0 Å), Leu-17 (> 10.0 Å), and Ala-21 (> 10.0 Å) are farther away. A sketched model of binding between Aβ and lipid molecules that satisfies all these constraints is shown in Fig. 7B. Interestingly, the proposed structural model of Aβ oligomers in the ionic channel by molecular dynamics simulation showed similar features in terms of the Aβ core and possible lipid contacts (21, 49–51). At a low P:L ratio (e.g. 1:120), we observed the formation of K+ ionic channels with freshly prepared preincorporation samples, and the channel activity was stabilized with chemical cross-linking that stabilizes the membrane-contained oligomers. Therefore, it is possible that the channel activity disappears upon incubation because the Αβ oligomers eventually aggregate to form an Aβ-lipid complex. However, we do not rule out the possibility that the ionic channel may be stabilized with a higher molar ratio of lipids or changes in lipid composition.
It is worth noting that the three distinct pathways shown in Fig. 7 are predominant in particular model systems. The membrane-incorporated large Aβ oligomers do not form mature fibrils or induce membrane content leakage or lipid fusion. Meanwhile, monomeric or small oligomeric Aβ peptides do not seem to form an Aβ-lipid complex or other aggregates that involve co-localization of both peptides and lipids. In addition, the Aβ fibrils and the hydrophobic core of the Aβ-lipid complex are structurally different at a high-resolution level. Therefore, our model systems can be utilized to simplify the heterogeneous structural evolution of membrane-associated Aβ peptides. Along each pathway, liposomes are disrupted through distinct mechanisms, and, therefore, the high-resolution peptide-lipid interactions that cause the disruptions are expected to be different. Simplified model systems can minimize the complexity and facilitate future high-resolution studies along each pathway.
Implications for the Cellular Toxicity of Aβ
Different Aβ constructs seem to possess different levels of cellular toxicity that are usually evaluated by cell viability assays upon incubation with these species. Typically, it is considered that oligomers have a higher toxicity level compared with either monomers or fibrils (1, 51–54). In addition, different high-resolution fibrillar structures might also possess variable levels of toxicity (32, 33, 55, 56). Cellular membrane disruption has been considered as a major mechanism for the neurotoxicity induced by amyloid peptides, including Aβ. Our results have the following implications. Cellular membranes can be disrupted through a variety of pathways that involve interactions between lipids and different Aβ species. This means one needs to be cautious when evaluating the relative cellular toxicity in two types of Aβ species. For instance, different Aβ sequences (i.e. wild-type versus mutants) may have distinct critical concentrations of aggregation (57). Therefore, they might be in different oligomer states when the same concentration is used, and this might affect the dominant membrane disruption pathways, as we have shown. Additionally, the P:L ratio is also critical for pathway selection. Therefore, to evaluate the relative cellular toxicity between the two Aβ species, it is important to quantify the lipid components in cells (2). Our results show that cellular membrane disruption is a dynamic process that is accompanied by changes in Aβ. For example, the membrane content leakage occurs simultaneously with the nucleation process of Aβ fibrillation in external addition samples. For preincorporation samples, the changes in membrane overall rigidity is observed together with the increment of ThT fluorescence, which is an indication of the growth of the Aβ hydrophobic core. Therefore, cellular toxicity might be related to the dynamic properties of an Aβ species. It has been reported previously that the cellular toxicity of mature Aβ fibrils increases with ultrasound sonication (33, 56). This can be explained by the fact that the fibrillar ends are more dynamic for the short pieces (i.e. typically ∼50–100 nm) than in long filaments. It is possible that the shrinkage and/or elongation processes induce cellular membrane disruption and therefore possesses a high cellular toxicity (3). Formation of ionic channels has been proposed as a universal mechanism for the cellular toxicity of amyloid peptides because the channel disrupts Ca2+ homeostasis across cellular membranes. We show that transient ion channel activities can be detected for the membrane-associated Aβ oligomers at relatively low P:L ratios. Using the same preincorporation protocols, aggregations of the Aβ-lipid complex form rapidly at a higher P:L ratio. Lowing the ratio seems to postpone the aggregation process. It is possible that the ionic channel shares similar structural features with the Aβ-lipid complex as an intermediate species along the aggregation process. In fact, we have shown, using NMR, that a number of structural results agreed with the computational model of ionic channels. In the future, it may be possible to stabilize the intermediate ionic channel constructs by chemical cross-linking and to perform NMR studies to characterize their high-resolution structures.
Author Contributions
W. Q. conceived and coordinated the study and wrote the paper. D. A. D. designed, performed, and analyzed the experiments shown in Figs. 1 and 2. K. D. performed and analyzed the experiments shown in Figs. 3 and 6. C. G. designed and helped with the experiments shown in Fig. 6. K. D., D. X., and H. D. designed, performed, and analyzed the experiments shown in Fig. 4. D. A. D., H. K., and Q. C. performed and analyzed the experiments shown in Fig. 5. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
The NMR facility at Binghamton University was supported by the National Science Foundation Major Research Instrumentation Program (NSF 0922815). We acknowledge support from Dr. Ming An for using the rotatory evaporator for sample preparation and the mass spectrometry facility from the University of Illinois.
This work was supported by the startup package from SUNY Research Foundation and the Department of Chemistry of Binghamton University (to W. Q.) and by National Science Foundation Grant 1515028 (to C. G.). The authors declare that they have no conflicts of interest with the contents of this article.
- Aβ
- amyloid β
- PC
- phosphatidylcholine
- PG
- phosphatidylglycerol
- P:L
- peptide:lipid
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- ThT
- thioflavin T
- PE
- phosphatidylethanolamine
- BLM
- black lipid membrane
- REDOR
- rotational echo double-resonance
- TEM
- transmission electron microscopy
- T2
- spin-spin relaxation time.
References
- 1. Chiti F., and Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 2. Chiang P. K., Lam M. A., and Luo Y. (2008) The many faces of amyloid β in Alzheimer's disease. Curr. Mol. Med. 8, 580–584 [DOI] [PubMed] [Google Scholar]
- 3. Eckert G. P., Wood W. G., and Müller W. E. (2010) Lipid membranes and β-amyloid: a harmful connection. Curr. Protein Pept. Sci. 11, 319–325 [DOI] [PubMed] [Google Scholar]
- 4. Butterfield S. M., and Lashuel H. A. (2010) Amyloidogenic protein-membrane interactions: mechanistic insight from model systems. Angew. Chem. Int. Ed. Engl. 49, 5628–5654 [DOI] [PubMed] [Google Scholar]
- 5. Mason R. P., Estermyer J. D., Kelly J. F., and Mason P. E. (1996) Alzheimer's disease amyloid β peptide 25–35 is localized in the membrane hydrophobic core: x-ray diffraction analysis. Biochem. Biophys. Res. Comm. 222, 78–82 [DOI] [PubMed] [Google Scholar]
- 6. Mason R. P., Jacob R. F., Walter M. F., Mason P. E., Avdulov N. A., Chochina S. V., Igbavboa U., and Wood W. G. (1999) Distribution and fluidizing action of soluble and aggregated amyloid β-peptide in rat synaptic plasma membranes. J. Biol. Chem. 274, 18801–18807 [DOI] [PubMed] [Google Scholar]
- 7. Fletcher T. G., Keire D. A. (1997) The interaction of β-amyloid protein fragment (12–28) with lipid environments. Protein Sci. 6, 666–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McLaurin J., and Chakrabartty A. (1997) Characterization of the interactions of Alzheimer's β-amyloid peptides with phospholipid membranes. Eur. J. Biochem. 245, 355–363 [DOI] [PubMed] [Google Scholar]
- 9. Widenbrant M. J., Rajadas J., Sutardja C., and Fuller G. G. (2006) Lipid-induced β-amyloid peptide assemblage fragmentation. Biophys. J. 91, 4071–4080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Terzi E., Hölzemann G., and Seelig J. (1997) Interaction of Alzheimer β-amyloid peptide(1–40) with lipid membranes. Biochemistry 36, 14845–14852 [DOI] [PubMed] [Google Scholar]
- 11. Oshima N., Morishima-Kawashima M., Yamaguchi H., Yoshimura M., Sugihara S., Khan K., Games D., Schenk D., Ihara Y. (2001) Accumulation of amyloid β-protein in the low-density membrane domain accurately reflects the extent of β-amyloid deposition in the brain. Am. J. Pathol. 158, 2209–2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hayashi H., Mizuno T., Michikawa M., Haass C., Yanagisawa K. (2000) Amyloid precursor protein in unique cholesterol-rich microdomains different from caveolae-like domains. Biochim. Biophys. Acta 1483, 81–90 [DOI] [PubMed] [Google Scholar]
- 13. Müller W. E., Koch S., Eckert A., Hartmann H., and Scheuer K. (1995) β-Amyloid peptide decreases membrane fluidity. Brain. Res. 674, 133–136 [DOI] [PubMed] [Google Scholar]
- 14. Gibson Wood W., Eckert G. P., Igbavboa U., and Müller W. E. (2003) Amyloid β-protein interactions with membranes and cholesterol: causes or casualties of Alzheimer's disease. Biochim. Biophys. Acta 1610, 281–290 [DOI] [PubMed] [Google Scholar]
- 15. Yip C. M., Darabie A. A., and McLaurin J. (2002) Aβ42-peptide assembly on lipid bilayers. J. Mol. Biol. 318, 97–107 [DOI] [PubMed] [Google Scholar]
- 16. Yip C. M., and McLaurin J. (2001) Amyloid-β peptide assembly: a critical step in fibrillogenesis and membrane disruption. Biophys. J. 80, 1359–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Peters I., Igbavboa U., Schütt T., Haidari S., Hartig U., Rosello X., Böttner S., Copanaki E., Deller T., Kögel D., Wood W. G., Müller W. E., and Eckert G. P. (2009) The interaction of β-amyloid protein with cellular membrane stimulates its own production. Biochim. Biophys. Acta 1788, 964–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Engel M. F., Khemtémourian L., Kleijer C. C., Meeldijk H. J., Jacobs J., Verkleij A. J., de Kruijff B., Killian J. A., and Höppener J. W. (2008) Membrane damage by human islet amyloid polypeptide through fibril growth at the membranes. Proc. Natl. Acad. Sci. 105, 6033–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Quist A., Doudevski I., Lin H., Azimova R., Ng D., Frangione B., Kagan B., Ghiso J., and Lal R. (2005) Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. 102, 10427–10432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kawahara M., Arispe N., Kuroda Y., and Rojas E. (1997) Alzheimer's disease amyloid β-protein forms Zn2+-sensitive, cation-selective channels across excised membrane patches from hypothalamic neurons. Biophys. J. 73, 67–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lal R., Lin H., and Quist A. P. (2007) Amyloid β ion channel: 3D structure and relevance to amyloid channel paradigm. Biochim. Biophys. Acta 1768, 1966–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vestergaard M. C., Morita M., Hamada T., and Takagi M. (2013) Membrane fusion and vesicular transformation induced by Alzheimer's amyloid β. Biochim. Biophys. Acta 1828, 1314–1321 [DOI] [PubMed] [Google Scholar]
- 23. Akinlolu R. D., Nam M., and Qiang W. (2015) Competition between fibrillation and induction of vesicle fusion for the membrane-associated 40-residue β amyloid peptides. Biochemistry 54, 3416–3419 [DOI] [PubMed] [Google Scholar]
- 24. Potapov A., Yau W. M., Ghirlando R., Thurber K. R., and Tycko R. (2015) Successive stages of amyloid-β self-assembly characterized by solid-state nuclear magnetic resonance with dynamic nuclear polarization. J. Am. Chem. Soc. 137, 8294–8307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Calderon R. O., Attema B., and DeVries G. H. (1995) Lipid composition of neuronal cell bodies and neurites from cultured dorsal root ganglia. J. Neurochem. 64, 424–429 [DOI] [PubMed] [Google Scholar]
- 26. Bokvist M., Lindström F., Watts A., and Gröbner G. (2004) Two types of Alzheimer's β-amyloid (1–40) peptide membrane interactions: aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J. Mol. Biol. 335, 1039–1049 [DOI] [PubMed] [Google Scholar]
- 27. Funato H., Yoshimura M., Kusui K., Tamaoka A., Ishikawa K., Ohkoshi N., Namekata K., Okeda R., and Ihara Y. (1998) Quantitation of amyloid β-protein (A β) in the cortex during aging and in Alzheimer's disease. Am. J. Pathol. 152, 1633–1640 [PMC free article] [PubMed] [Google Scholar]
- 28. O'Brien J. S., and Sampson E. L. (1965) Lipid composition of the normal human brain: grey matter, white matter and myelin. J. Lipid Res. 6, 537–544 [PubMed] [Google Scholar]
- 29. Kawarabayashi T., Shoji M., Younkin L. H., Wen-Lang L., Dickson D. W., Murakami T., Matsubara E., Abe K., Ashe K. H., and Younkin S. G. (2004) Dimeric amyloid β protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated Tau accumulation in the Tg2576 mouse model of Alzheimer's disease. J. Neurosci. 24, 3801–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Qiang W., Yau W. M., and Schulte J. (2015) Fibrillation of β amyloid peptides in the presence of phospholipid bilayers and the consequent membrane disruption. Biochim. Biophys. Acta 1848, 266–276 [DOI] [PubMed] [Google Scholar]
- 31. Qiang W., Akinlolu R. D., Nam M., and Shu N. (2014) Structural evolution and membrane interaction of the 40-residue β amyloid peptides: differences in the initial proximity between peptides and the membrane bilayer studied by solid-state nuclear magnetic resonance spectroscopy. Biochemistry 53, 7503–7514 [DOI] [PubMed] [Google Scholar]
- 32. Lu J. X., Qiang W., Yau W. M., Schwieters C. D., Meredith S. C., and Tycko R. (2013) Molecular structure of β-amyloid fibrils in Alzheimer's disease brain tissue. Cell 154, 1257–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qiang W., Yau W. M., Luo Y., Mattson M. P., and Tycko R. (2012) Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 109, 4443–4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qiang W., Yau W. M., and Tycko R. (2011) Structural evolution of Iowa mutant β-amyloid fibrils from polymorphic to homogeneous states under repeated seeded growth. J. Am. Chem. Soc. 133, 4018–4029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Paravastu A. K., Leapman R. D., Yau W. M., and Tycko R. (2008) Molecular structural basis for polymorphism in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 18349–18354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moscho A., Orwar O., Chiu D. T., Modi B. P., and Zare R. N. (1996) Rapid preparation of giant unilamellar vesicles. Proc. Natl. Acad. Sci. 93, 11443–11447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin H., Bhatia R., and Lal R. (2001) Amyloid β protein forms ion channels: implications for Alzheimer's disease pathophysiology. FASEB J. 15, 2433–2444 [DOI] [PubMed] [Google Scholar]
- 38. Ostroumova O. S., Schagina L. V., Mosevitsky M. I., and Zakharov V. V. (2011) Ion channel activity of brain abundant protein BASP1 in planar lipid bilayers. FEBS J. 278, 461–469 [DOI] [PubMed] [Google Scholar]
- 39. Bak M., Rasmussen J. T., and Nielsen N. C. (2000) SIMPSON: A general simulation program for solid-state NMR spectroscopy. J. Magn. Reson. 147, 296–330 [DOI] [PubMed] [Google Scholar]
- 40. Sciacca M. F., Kotler S. A., Brender J. R., Chen J., Lee D. K., and Ramamoorthy A. (2012) Two-step mechanism of membrane disruption by Aβ through membrane fragmentation and pore formation. Biophys. J. 103, 702–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen Y. A., and Scheller R. H. (2001) SNARE-mediated membrane fusion. Nat. Rev. Mol. Cell Biol. 2, 98–106 [DOI] [PubMed] [Google Scholar]
- 42. Drechsler A., Anderluh G., Norton R. S., and Separovic F. (2010) Solid-state NMR study of membrane interactions of the pore-forming cytolysin, equinatoxin II. Biochim. Biophys. Acta 1798, 244–251 [DOI] [PubMed] [Google Scholar]
- 43. Wang T., Cady S. D., and Hong M. (2012) NMR determination of protein partitioning into membrane domains with different curvatures and application to the influenza M2 peptide. Biophys. J. 102, 787–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang Y. W., Thompson R., Zhang H., and Xu H. (2011) APP processing in Alzheimer's disease. Mol. Brain 4:3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Qiang W., Kelley K., and Tycko R. (2013) Polymorph-specific kinetics and thermodynamics of β-amyloid fibril growth. J. Am. Chem. Soc. 135, 6860–6871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Komatsu H., Feingold-Link E., Sharp K. A., Rastogi T., and Axelsen P. H. (2010) Intrinsic linear heterogeneity of amyloid β protein fibrils revealed by higher resolution mass-per-length determinations. J. Biol. Chem. 285, 41843–41851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sengupta P., Garai K., Sahoo B., Shi Y., Callaway D. J., and Maiti S. (2003) The amyloid β peptide (Aβ(1–40)) is thermodynamically soluble at physiological concentrations. Biochemistry 42, 10506–10513 [DOI] [PubMed] [Google Scholar]
- 48. Hasegawa K., Ono K., Yamada M., and Naiki H. (2002) Kinetic modeling and determination of reaction constants of Alzheimer's β-amyloid fibril extension and dissociation using surface plasmon resonance. Biochemistry 41, 13489–13498 [DOI] [PubMed] [Google Scholar]
- 49. Connelly L., Jang H., Arce F. T., Ramachandran S., Kagan B. L., Nussinov R., and Lal R. (2012) Effects of point substitutions on the structure of toxic Alzheimer's β-amyloid channels: atomic force microscopy and molecular dynamics simulations. Biochemistry 51, 3031–3038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Capone R., Jang H., Kotler S. A., Connelly L., Teran Arce F., Ramachandran S., Kagan B. L., Nussinov R., and Lal R. (2012) All-D-enantiomer of β-amyloid peptide forms ion channels in lipid bilayers. J. Chem. Theory Comput. 8, 1143–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yu X., Wang Q., Pan Q., Zhou F., and Zheng J. (2013) Molecular interactions of Alzheimer amyloid-β oligomers with neutral and negatively charged lipid bilayers. Phys. Chem. Chem. Phys. 15, 8878–8889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Benilova I., Karran E., and De Strooper B. (2012) The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357 [DOI] [PubMed] [Google Scholar]
- 53. Ahmed M., Davis J., Aucoin D., Sato T., Ahuja S., Aimoto S., Elliott J. I., Van Nostrand W. E., and Smith S. O. (2010) Structural conversion of neurotoxic amyloid-β(1–42) oligomers to fibrils. Nat. Struct. Mol. Biol. 17, 561–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 55. Tycko R. (2011) Solid-state NMR studies of amyloid fibril structure. Annu. Rev. Phys. Chem. 62, 279–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Petkova A. T., Leapman R. D., Guo Z., Yau W. M., Mattson M. P., and Tycko R. (2005) Self-propagating, molecular-level polymorphism in Alzheimer's β-amyloid fibrils. Science 307, 262–265 [DOI] [PubMed] [Google Scholar]
- 57. Tycko R., Sciarretta K. L., Orgel J. P., and Meredith S. C. (2009) Evidence for novel β-sheet structures in Iowa mutant β-amyloid fibrils. Biochemistry 48, 6072–6084 [DOI] [PMC free article] [PubMed] [Google Scholar]