

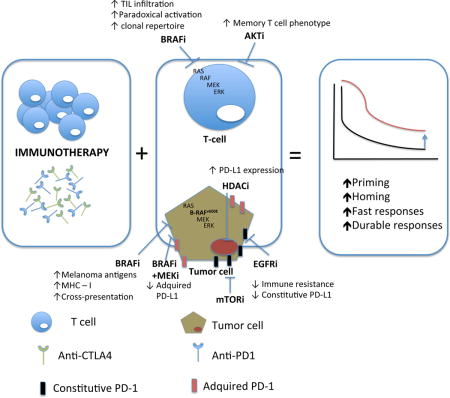
Graphical abstract
How can targeted therapy synergize immunotherapy?
1. Introduction
Recent advances in the understanding of anti-tumor immunity and tumor immune escape have facilitated the design of new immunotherapy agents for the treatment of cancer. The clinical development of these agents in clinical trials has resulted in long-term survival in a subset of patients with different cancer histologies and has opened the door to combinatorial therapies that could improve outcomes. In parallel, the discovery of oncogenic driver pathways in different tumor types and the development of inhibitory molecules targeting these pathways signified another major development in the treatment of metastatic cancer. Hence, there is considerable interest in testing the combination of both treatment modalities in ongoing early stage clinical trials.
The treatment of metastatic melanoma is at the center stage of this research effort. Prior to 2011, very limited treatments with demonstrated survival benefits were available. However, since the launch of ipilimumab in 2011, the US FDA has approved eight different single or combinations of agents, which has led to significant improvement in response rates and survival of patients with melanoma. These new agents are either targeted inhibitors of the mitogen-activated protein kinase (MAPK) oncogenic signaling pathway, or immune modulatory agents. This review will summarize currently available evidence and explain the rationale that supports the combination of immunotherapy and targeted therapy for the treatment of melanoma, and describe how this approach is being extended to patients with other histological types of cancer.
1.1. Immunotherapy
Studies have indicated the association of tumor T cell infiltrates with clinical benefit of immunotherapy in several tumor types(1–8). In addition, these immune infiltrates have been shown to include specific T cell clones that target somatic point mutations (also called neo-antigens (9)), as well as overexpressed cancer-testis antigens(10) or lineage-specific antigens(11–13). Rosenberg and colleagues at the National Cancer Institute (NCI) have been conducting clinical trials using ex vivo expanded autologous tumor-infiltrating lymphocytes (TILs) for adoptive cell transfer (14) (15). Thus far, the results have been reproducible and have demonstrated durable and relatively high complete remission (CR) rates(15). The newer generation of ACT, utilizing autologous T cells engineered to express chimeric antigen receptor (CAR) directed against CD19, has been highly successful in acute or chronic lymphoblastic leukemia and non-Hodgkin lymphomas(16, 17). However, less activity has been observed when engineered T cell receptors (TCR) were directed against solid tumor antigens, including melanoma antigen recognized by T cells 1 (MART1) and NY-ESO-1(18, 19).
The development of immune checkpoint inhibitors has been revolutionary in the field of cancer immunotherapy. Blockade of cytotoxic T lymphocyte antigen 4 (CTLA4) (20, 21) and programmed cell death protein 1 (PD-1) have demonstrated durable responses across different tumor types(22–24). In addition, the combination of these two checkpoint inhibitors has resulted in unprecedented high response rates in melanoma (nearly 60%), but has been associated with increased frequency of toxicities(25). Subsequently, pipelines of newer checkpoint inhibitors and other immunomodulatory agents are being developed. Most recently, FDA has approved intratumoral injection of talimogene laherparepvec (T-VEC), a genetically modified oncolytic virus, for the treatment of unresectable melanoma (26).
The success of these modern immunotherapy strategies has created great excitement in the cancer research field because it offers tumor specific response with durability due to the memory of effector cells. However, frequency of immunotherapy responses are relatively low in most cases, likely due to the tumor escape mechanisms that are different between individual patients and tumor types. Strategies to improve the response rate have been of high interest.
1.2. Targeted therapy
Small molecule inhibitors of driver mutation pathways, such as epidermal growth factor receptor (27) inhibitors for EGFR mutant lung cancer(28) or anaplastic lymphoma kinase (ALK) inhibitors for lung cancer patients who harbor the echinoderm microtubule-associated protein-like 4 (EML4)-ALK translocation(29), have been successfully developed for several cancer subtypes and can induce high response rates in tumors with underlying genetic alterations. Similarly, antibodies of human epidermal growth factor receptor 2 (HER2) have significantly improved survival in women with HER2 amplified breast cancer in both the adjuvant and metastatic settings(30). The identification of a prevalent driver mutation in BRAF has also led to the development of selective BRAF inhibitors and MEK inhibitors that shut down the MAPK pathway in melanomas(31–33). The initial response rates to targeted therapies have been high but the long-term effectiveness of these therapies has unfortunately been limited by the development of acquired resistance in the majority of patients (34–39).
2. The potential mechanisms of combined benefits of targeted therapy and immunotherapy
Targeted therapy can not only direct killing of tumor cells, but also have effects on the different components of the immune system, so called “immunesensitization”, suggesting a potentially synergistic benefit of combining targeted therapy and immunotherapy beyond the expected additive effect of two effective treatments (40).
2.1. Direct effects on tumor cells
The direct effects of BRAF and MEK inhibitors are achieved by the induction of cytotoxicity in melanoma cells through inhibition of the MAPK pathway, and subsequent cell death can create a more immunogenic environment in which tumor antigens can be cross-presented to T cells. Prior studies have demonstrated that decreased signaling through the MAPK pathway by BRAF and MEK inhibitors is correlated with increase in melanocyte differentiation antigens in both melanoma cell lines and clinical tumor samples from melanoma patients(41–43). Further, when resistance to BRAF inhibition occurs, it can be associated with loss of tumor antigen expression (41). BRAF inhibition has also been shown to cause the upregulation of major histocompatibility complex (MHC) class I molecules in tumor cells, which improves antigen presentation and recognition (44). In different preclinical models it has been described that both the apoptotic effect of inhibiting BRAF is being enhanced (45) or blocked (46) by cytokines produced by T cells such as TNF or IFN-γ. Several groups are actively studying to better characterize this interaction. These results have justified the clinical testing of a combinatorial approach between BRAF inhibition and ACT for BRAF-mutant metastatic melanoma.
In glioblastoma where PTEN loss is found in 60–80% of cases (47), a study showed that this loss of PTEN and subsequent up-regulation of the PI3K-AKT pathway resulted in increased constitutive expression of PD-L1 and is associated with immune resistance(48). This study also showed a decrease in the transcription of PD-L1 in cells treated with rapamycin, a mTOR inhibitor. Currently there is a clinical trial testing the efficacy of PI3K/mTOR inhibitors vs anti-PD-1 antibodies (NCT02430363) for patients with glioblastoma; however, testing a combination of these two agents would be of high interest. A similar justification has been extended to EGFR mutant non-small cell lung cancer (NSCLC) (approximately 10% of all NSCLC cases (49)), where this driver mutation has been shown to mediate immune escape through the upregulation of PD-1 and PD-L1(50). Treatment with EGFR inhibitors, on the other hand, has reduced PD-1 and PD-L1 expression (51). Based on this knowledge, clinical trials focusing on the combination of EGFR and PD-1 inhibitors have been initiated (NCT02039674). Both of these situations are examples of constitutive expression of PD-L1 as a result of an oncogenic event as opposed to adaptive immune expression. Blocking these driver mutations might downregulate the expression of PD-L1 at the tumor level. However, it remains unclear the consequence of inhibiting these mutations in tumors that lack infiltrating lymphocytes, which is more common in glioblastoma with PTEN loss and EGFR mutant NSCLC (52).
Epigenetic alterations are common in cancer cells, including global hypomethylation or hypermethylation of CpG islands in promoter regions. This hypermethylation can result in gene silencing of tumor suppressor genes, oncogenes, tumor associated antigens (TAA), antigen presentation machinery (APM) and co-stimulatory signaling (53, 54), leading to immune escape(55). Hypomethylating agents such as 5-azacitidine (AZA) have shown activity in up-regulating tumor associated antigens and increasing expression of HLA-A1 and other components of the antigen presenting machinery (APM) such as Transporter Associated with Antigen Processing 1 (TAP-1) (56) and -2(56–59). Another epigenetic process, histone acetylation, is a reversible mechanism enabling access of chromatin and transcription of genes (60). Histone deacetylase (HDAC), on the other hand, removes acetyl groups and suppresses the gene transcription process. HDAC inhibitors have been shown to restore expression of TAA and cancer-testis antigens such as NY-ESO-1 (61–63), enhance MHC class I and II expression(64, 65), and upregulate PD-L1 expression(66, 67). In preclinical models, Kim et al reported tumor eradication in 80% of mice bearing either CT26 colon cancer or 4T1 breast cancer, treated with combination of epigenetic-modulators (5-AZA plus entinostat, a HDAC inhibitor) and checkpoint inhibitors (anti-CTLA4 plus anti-PD1), but not checkpoint inhibitors alone, accomplished by depletion of myeloid derived suppressive cells (MDSC) (68).
2.2. Effects on effector T lymphocytes
Besides inhibiting oncogenic events in tumor cells, targeted therapy exerts important effects on normal cells that reply on the targeted pathway regulation, especially the cytotoxic T cells. For example, the selective inhibitors of BRAF V600 mutation could serve as activators of the MAPK pathway in cells with a wild-type BRAF genotype but a strong upstream signal, including keratinocytes or lymphocytes. This effect has been defined as paradoxical activation of the MAPK pathway (69) that results in the immune-sensitization effects of increased T cell function(70–72), and contributes to the skin toxicity of squamous cell carcinoma of BRAF inhibitors. Preclinical studies involving fully immunocompetent mice bearing BRAF-mutant melanoma tumors (as well as CDKN2A mutation and BRAF and MITF amplification) have shown that CD8 cells are critical to the benefits of BRAF inhibitors, as depletion of CD8 cells but not CD4 or NK cells in mice, can partially abrogate the anti-tumor effects of BRAF inhibition(70, 73). Furthermore, it was determined that despite the potential detrimental effects to the T effector cell function by directly inhibiting the MAPK pathway in cultured T cells in vitro (41, 74), addition of a MEK inhibitor in the in vivo setting could be synergistic to combined BRAF inhibition and immunotherapy, with maintained T cell function and a more immune permissive tumor microenvironment, and dampen the unwanted toxicity associated with the paradoxical activation of the MAPK pathway by BRAF inhibitors alone (75, 76). In addition, BRAF and MEK inhibitors can upregulate tumor PD-L1 expression and this has translated into a superior antitumor response when combining with PD-L1 antibodies (75, 76).
The number of TILs also appears to be affected by the inhibition of the MAPK pathway(73, 75, 76). A greater number of TILs has been described in tumor samples from patients treated by BRAF and/or MEK inhibitors, with increased TIL clonality in BRAF inhibitor treated tumors (42, 77, 78). Because increased tumoral or peritumoral infiltration of CD8 lymphocytes with high clonality has been shown to predict responses to PD-1 blockade(79), this clonal expansion towards a more specific repertoire induced by BRAF inhibition, along with the observed increase in PD-1 and PD-L1 soon after BRAF and/or MEK inhibition(42, 80), provide a sound rationale for this combination of targeted therapy and immunotherapy.
The combinatorial benefit of BRAF inhibitors and immunotherapy was demonstrated in another syngeneic melanoma model harboring oncogenic BRAF mutation (as well as CDKN2A −/−, PTEN −/−) (81), where blocking mutated BRAF increased CD8+ T cell infiltration, and the antitumor effects was enhanced when BRAF inhibitor was combined with PD-1 or PD-L1 inhibitors by enhanced T cell activity and improved survival of the treated mice.
More recently, inhibition of Pi3K/Akt/mTOR pathway was shown to induce the expansion of TILs promoting a memory T cell phenotype (82), which provides a rationale for enhancing the persistence of transferred tumor specific T cells in the adoptive cell transfer (82) immunotherapy approach. Therefore, a phase I trial investigating the persistence of adoptively transferred TILs cultured with an AKT inhibitor in patients with metastatic melanoma is currently ongoing by Steven Rosenberg’s groups at NCI (NCT02489266).
A newer class of drugs targeting focal adhesion kinase (83) has attracted interest as inhibition of FAK is associated with decrease of pluripotent cells that are considered cancer stem/progenitor cells (CSCs). This was initially described in breast cancer where ablation of FAK reduced the pool of CSCs in primary tumors of FAK-targeted mice and impaired their self-renewal and migration in vitro. In addition, CSCs isolated from FAK-targeted mice have compromised tumorigenicity and impaired maintenance in vivo (84). Recently FAK inhibitor has been showed to reduce CSCs and delay tumor growth following cisplatin plus pemetrexed treatment in a patient-derived xenograft model of malignant mesothelioma and other tumor types (85, 86). Interestingly, FAK inhibitors have been recently reported to help proliferation of CD8 cytotoxic T cells and inhibit tumor associated macrophages (87), and combination of FAK inhibitor (VS-4718) and anti-PD-1 agent extended survival of mice bearing colon cancer tumors, providing the rationale for translation into clinical trials.
2.3. Effects on the tumor microenvironment
In melanoma, besides the direct effects on cytotoxic T cells, BRAF and MEK inhibition has also demonstrated immunomodulatory effects in the melanoma tumor microenvironment. The work referenced above combining BRAF and MEK inhibition with immunotherapy (75), showed that the triple combination therapy resulted in increased melanosomal antigen and MHC expression and global immune-related gene up-regulation. Single-agent dabrafenib increased tumor-associated macrophages and T regulatory cells (Tregs) in tumors, which decreased with the addition of trametinib. MEK inhibitors have been shown to decrease immunosuppressive cytokines, including IL-1, IL-6, and IL-8, IL-10 as well as decrease angiogenic factors such as vascular endothelial growth factor (VEGF) (88). Inhibition of BRAF reduced expression of IL-1 in cell lines and tumor biopsies, and because the immune inhibitory activity of tumor-associated fibroblasts (89) is enhanced by IL-1, treatment with BRAF inhibition can potentially decrease the number of tumor-associated TAF in the stroma, as suggested by Khalili et al (90). When a xenograft model of BRAF-mutated human melanoma cell line transduced with gp100 and H-2D was used to assess melanocyte differentiation antigen-independent enhancement of immune responses by BRAF inhibitor, it was found that administration of vemurafenib significantly increased the tumor infiltration and function of adoptively transferred gp100-specific pmel-1 T cells in vivo (91), primarily mediated by the ability of vemurafenib to inhibit melanoma tumor cell production of VEGF. Analysis of human melanoma biopsies showed down-regulated VEGF before and during BRAF inhibitor treatment.
Imatinib, a selective inhibitor of the c-kit receptor tyrosine kinase that has shown great success in treating chronic myelogenous leukemia (CML) and gastrointestinal stromal tumors (92) was found to be associated with activation of CD8 T cells and reduction of Tregs, through reduction of indoleamine 2,3-dioxygenase (IDO) in the tumor microenvironment (93). Sunitinib, a multi-targeted receptor tyrosine kinase (RTK) inhibitor against platelet-derived growth factor receptors (PDGF-Rs), vascular endothelial growth factor receptors (VEGFRs) as well as c-kit and approved by the FDA for the treatment of renal cell carcinoma (RCC) and imatinib-resistant gastrointestinal stromal tumor (92), has been shown to decrease the amount of immune-suppressive MDSCs and Tregs (94) in the tumor microenvironment.
3. Clinical trials testing the combination of immunotherapy and targeted therapy
Based on the rationale detailed above, the concept of combining targeted therapy and immunotherapy are being tested in clinical trials. The first reported combination therapy with ipilimumab and vemurafenib had been terminated early due to grade 3 hepatotoxicities (73). The etiology of this hepatotoxicity was unclear, but could be related to the paradoxical activation of the MAPK pathway in BRAF wild type cells. However, a separate trial (NCT01767454) involving ipilimumab and dabrafenib did not encounter hepatotoxicity, suggesting a drug-specific process. Interestingly, a second treatment arm that examined the triple combination of dabrafenib, trametinib and ipilimumab had to be discontinued because of two out of seven serious adverse events involving colon perforations (95). Currently at least three clinical trials have been currently ongoing to test the combination of BRAF plus MEK inhibitors with PD1/L1 blockade and have shown encouraging results.
These unexpected toxicities highlighted the complexity of the translational clinical scenario, and a pressing need for judicious evaluation both in the clinic and in a wide-range of potentially clinically predictive animal models to better guide the clinical adaptability of these two promising modalities. For that reason, development of more genetically relevant animal models that can closely resemble the human circumstance across all tumor histologies is warranted. The current tumor models, which are frequently derived from genetically engineered mice (GEM) with constitutively active oncogenic signaling, fail to recapitulate the antigenic complexity derived from skin UV damage in human melanoma. New models are being developed and hopefully they will succeed in better translating the biology behind each tumor-type. On the other hand, the biology and immune response in mouse models does not correlate well with activity in the corresponding human cancer, exemplified by the success of anti-PD1 therapies in human cancers in contrast to the lack of response to these antibodies in majority of tumor models. Therefore, if strong rationale stands, testing judiciously in the clinical setting should be warranted, even wirh lack of activities in the preclinical setting.
Tables 1 to 5 summarize ongoing clinical trials involving the combination of targeted therapy and immune checkpoint blockade. The number of clinical trials has increased exponentially in the last few years, and the involved tumor types have become more diverse and are no longer limited to the traditionally “immunotherapy-sensitive” melanoma or kidney cancers (Table 1). Some studies are assessing increased tumor cell killing and antigen expression/presentation that could enhance T cell activation, whereas other studies are attempting to overcome the immune suppressive environment within the tumors. These tables also included clinical trials exploring the combination of targeted therapy with ACT, majority of which involve combination with TIL therapy conducted by NCI. The rationale behind the combination of targeted therapy and immunotherapy is strongly supported by the above mentioned preclinical data, however, translation into a clinical setting will require carefully selection of the targets in a case by case setting, and optimization of the schedule and sequence of the involved drugs.
Table 1.
Clinical trials involving the combination of tyrosine kinase inhibitors (TKIs) and immunotherapy.
| Immune checkpoint blockade +TKIs | |||
|---|---|---|---|
| Clinical trial | Condition | Phase | Intervention |
| NCT01738139 | Advanced tumors | Phase I | Ipilimumab+Imatinib Mesylate |
| NCT02133742 | CCmRC | Phase I | Pembrolizumab+Axitinib |
| NCT02420912 | CLL | Phase II | Nivolumab + Ibrutinib |
| NCT02011945 | CML | Phase I | Nivolumab+Dasatinib |
| NCT02329847 | Hematologic | Phase I/II | Nivolumab + Ibrutinib |
| NCT02446457 | Lymphoma | Phase II | Pembrolizumab+Rituximab |
| NCT01656642 | Melanoma | PhaseI | PD-L1inhibitor+vemurafenib PD-L1inhibitor+vemurafenib+cobimetinib |
| NCT01659151 | Melanoma | Phase II | ACT with TIL+High Dose IL-2+Lymphodepletion+Vemurafenib |
| NCT01940809 | Melanoma | PhaseI | Ipilimumab Ipilimumab+trametinib Nivolumab+ipilimumab Ipilimumab+dabrafenib Ipilimumab+dabrafenib+trametinib Nivolumab+ipilimumab+trametinib Nivolumab+ipilimumab+Dabrafenib Nivolumab+Ipilimumab+Dabrafenib+Trametinib |
| NCT02027961 | Melanoma | PhaseI/II | PD-L1inhibitor+Trametinib PD-L1 inhibitor+ Trametinib+Dabrafenib |
| NCT02489266 | Melanoma | Phase I | Lymphodepletion+AKTi-treated TIL+IL-2 |
| NCT02354690 | Melanoma | Phase I/II | Vemurafenib –> TIL+lymphodepletion+IL2 |
| NCT02357732 | Melanoma | Phase I | Nivolumab+Dabrafenib Nivolumab+Trametinib Nivolumab+Dabrafenib+Trametinib |
| NCT02400385 | Melanoma | Phase II | Sunitinib+Nivolumab |
| NCT02130466 | Melanoma | Phase I/II | Dabrafenib+Trametinib Pembrolizumab+Dabrafenib Pembrlizumab+Trametinib Pembrolizumab+Dabrafenib+Trametinib |
| NCT01454102 | NSCLC | Phase I | (19 arms at different dose combination) Nivolumab + Gemcitabine + Cisplatin Nivolumab + Pemetrexed + Cisplatin Nivolumab + Paclitaxel + Carboplatin Nivolumab + Bevacizumab maintenance Nivolumab + Erlotinib Nivolumab Nivolumab + Ipilimumab Nivolumab Nivolumab + Ipilimumab |
| NCT02323126 | NSCLC | Phase II | Nivolumab + EGF816 (EGFRinhibitor) Nivolumab + INC280 (cMET inhibitor) |
| NCT02039674 | NSCLC | Phase I/II | Pembrolizumab+Paclitaxel+Carboplatin Pembrolizumab+Paclitaxel+Carboplatin+Bevacizumab Pembrolizumab+Pemetrexed+Carboplatin Pembrolizumab+Ipilimumab Pembro+Erlotinib Pembrolizumab+Gefitinib Carboplatin+Pemetrexed+/− Pembrolizumab) Pembrolizumab + ipilimumab |
| NCT01998126 | NSCLC | Phase I | Ipilimumab+Erlotinib Ipilimumab+Crizotinib |
| NCT02364609 | NSCLC | Phase I | Pembrolizumab+Afatinib |
| NCT02448303 | NSCLC | Phase II | Pembrolizumab Pembrolizumab+ACP-196 (BTKi) |
| NCT02511184 | NSCLC | Phase I | Pembrolizumab+Crizotinib |
| NCT01767454 | Solid tumors | Phase I | Ipilimumab+Dabrafenib Ipilimumab+Dabrafenib+Trametinib |
| NCT02423343 | Solid tumors | Phase I/II | Nivolumab+Galunisertib |
TKI = tyrosine kinase inhibitors; CLL = chronic lymphocytic leukemia; NSCLC = Non-small cell lung cancer; CCmRC = Clear cell metastatic renal cancer; CML = chronic myelogenous leukemia; IL-2 – interleukin-2; TIL – tumor infiltrating lymphocytes;
Table 5.
Clinical trials involving the combination of CSC inhibitors and immunotherapy.
| Immune Checkpoint blockade + CSC inhibitor or others | |||
|---|---|---|---|
| Clinical trial | Condition | Phase | Intervention |
| NCT02467361 | Solid tumors | Phase I/II | Ipilimumab+BBI608 Nivolumab+BBI608 Pembrolizumab+BBI608 |
| NCT02546531 | Solid tumors | Phase I | Pembrolizumab+Defactinib+Gemcitabine |
CSC = Cancer stem cell;
Monoclonal antibodies targeting the human epidermal growth factor receptor (HER) family members can enhance dendritic cell mediated T cell priming and antibody dependent cellular cytotoxicity (ADCC), therefore is another potential candidate to combine with PD-1 checkpoint inhibitors (96, 97). Table 2 summarized ongoing clinical trials designed to investigate such combinations involving HER2 inhibitors such as trastuzumab (NCT02318901) and HER1 (27) inhibitor cetuximab (NCT02105636, NCT02252042).
Table 2.
Clinical trials involving the combination of target-specific monoclonal antibodies and immunotherapy.
| Immune Checkpoint blockade + Monoclonal antibodies | |||
|---|---|---|---|
| Clinical trial | Condition | Phase | Intervention |
| NCT00182650 | HL | Phase I | IL-2+rituximab+lymphodepletion+therapeutic autologous lymphocytes |
| NCT02318901 | Solid tumors | Phase I/II | Pembrolizumab+Trastuzumab Pembrolizumab+T-DM1 Pembrolizumab+Cetuximab |
SSC = Squamous cell carcinoma; T-DM1 – trastuzumab-DM1; IL-2 – interleukine-2
Similar to the melanoma setting, both checkpoint inhibitors (immunotherapy) and anti-angiogenic agents (targeted therapy) are successful in the treatment of renal cell carcinoma, therefore there is great interest in combining these two classes of agents for better disease control and potential synergy in RCC. Preliminary results from trials involving the combination of nivolumab with sunitinib or pazopanib revealed response rates as high as 50%, but increased toxicity, especially hepatotoxicity was observed(98) (NCT02014636, NCT 01472081). Similarly, a phase I trial combining CTLA-4 monoclonal antibody tremelimumab with sunitinib resulted in unexpected renal toxicity(99). Several other trials are subsequently open to evaluate alternative anti-angiogenic drugs (Table 3).
Table 3.
Clinical trials involving the combination of anti-angiogenic agents and immunotherapy.
| Immune Checkpoint blockade + anti-angiogenic agents | |||
|---|---|---|---|
| Clinical trial | Condition | Phase | Intervention |
| NCT02348008 | CCmRC | Phase I/II | Pembrolizumab+Bevacizumab |
| NCT02014636 | CCmRC | Phase I | Pazopanib Pembrolizumab Pembrolizumab+Pazopanib |
| NCT01472081 | CCmRC | Phase I | Nivolumab+Ipilimumab Nivolumab+Pazopanib Nivolumab+ Sunitinib |
| NCT02337491 | Glioblastoma | Phase II | Pembrolizumab Pembrolizumab+Bevecizumab |
| NCT02501096 | Solid tumors | Phase I/II | Pembrolizumab+Lenvatinib |
CCmRC = clear cell metastatic renal cancer;
As described earlier, the role of epigenetic modulation to improve the tumor immune microenvironment using HDAC inhibitors is also being investigated. Table 4 summarizes the current clinical trials involving HDAC inhibitors and immune checkpoint blockade. A phase I trial testing priming with azacytidine (a hypomethylation agent) plus entinostat (a HDAC inhibitor) prior to nivolumab in advanced NSCLC, has induced responses in six patients (100). Three of these patients experienced durable responses and two had stable diseases for 9 months (101). Based on these results, a phase II trial (NCT01928576) is similarly designed and currently recruiting. Previous clinical data combining another HDAC inhibitor vorinostat and tamoxifen has provided 19% response rate in hormone therapy resistant breast cancer (102). More recently, the combination of vorinostat, tamoxifen and PD-1 inhibition is being investigated (NCT02395627) with both concurrent and sequential schedules.
Table 4.
Clinical trials involving the combination with epigenetic modulators and immunotherapy.
| Immune Checkpoint blockade + Epigenetic modulator | |||
|---|---|---|---|
| Clinical trial | Condition | Phase | Intervention |
| NCT02395627 | Breast | Phase II | Pembrolizumab+Tamoxifen+Vorinostat |
| NCT02538510 | Head&neck | Phase I/II | Pembrolizumab+Vorinostat |
| NCT02437136 | NSCLC/Melanoma | Phase I/II | Pembrolizumab+Entinostat |
| NCT01928576 | NSCLC | Phase II | Azacitidine –> Nivolumab Azacitidine+Entinostat –> Nivolumab |
NSCLC = non-small cell lung cancer;
Finally, Table 5 summarizes trials that combine drugs that target cancer stem cells (CSC) with immune checkpoint blockade, including a FAK inhibitor defactinib in combination with pembrolizumab and gemcitabine for advanced pancreatic cancer patients (NCT02546531), and a cancer stem cell inhibitor BBI608 combined with ipilimumab or nivolumab or pembrolizumab (at the investigator’s discretion) in advanced solid tumors (NCT02467361).
4. Conclusions
The advance in cancer immunotherapy has resulted in a paradigm shift in the management of patients across several tumor types, including the traditionally non-immunotherapy responsive histologies, with the promise of long-term disease control. The immediate challenge facing the field is how to improve the response towards majority of patients and tumor types. The readily available targeted therapies that are already approved in many tumor types with an association with high response rate for the indicated patient population provide an attractive combination strategy. The potential synergy of targeted therapy and immunotherapy has been shown in both preclinical models and patient derived samples. However, critical questions have to be answered before translation of this approach into clinical applications. Particular concepts need to be explored and confirmed in relevant animal models and optimized in clinical trials, and toxicity needs to be evaluated. In all of the ongoing trials, tumor biopsies and translational studies need to be incorporated into the study design.
Highlights.
Targeted therapy has the potential to enhance immunotherapy by inducing immune effects in tumor cells, modulating T cell homing and function, as well as the tumor immune microenvironment.
The potential benefits of combined targeted therapy and immunotherapy are not limited to tyrosine kinase inhibitors (TKIs) or melanoma, but extend to other molecular targets and tumors histologies.
Many clinical trials are currently underway, but optimization of the dosing regimen and schedule is needed to confirm benefits and avoid toxicity associated with these combinations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Clemente CG, Mihm MC, Jr, Bufalino R, Zurrida S, Collini P, Cascinelli N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 1996;77(7):1303–10. doi: 10.1002/(SICI)1097-0142(19960401)77:7<1303::AID-CNCR12>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 2.Vesalainen S, Lipponen P, Talja M, Syrjanen K. Histological grade, perineural infiltration, tumour-infiltrating lymphocytes and apoptosis as determinants of long-term prognosis in prostatic adenocarcinoma. European journal of cancer. 1994;30A(12):1797–803. doi: 10.1016/0959-8049(94)e0159-2. [DOI] [PubMed] [Google Scholar]
- 3.Leffers N, Gooden MJ, de Jong RA, Hoogeboom BN, ten Hoor KA, Hollema H, et al. Prognostic significance of tumor-infiltrating T-lymphocytes in primary and metastatic lesions of advanced stage ovarian cancer. Cancer immunology, immunotherapy: CII. 2009;58(3):449–59. doi: 10.1007/s00262-008-0583-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A. 2007;104(9):3360–5. doi: 10.1073/pnas.0611533104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–4. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 6.Gooden MJ, de Bock GH, Leffers N, Daemen T, Nijman HW. The prognostic influence of tumour-infiltrating lymphocytes in cancer: a systematic review with meta-analysis. Br J Cancer. 2011;105(1):93–103. doi: 10.1038/bjc.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schumacher K, Haensch W, Roefzaad C, Schlag PM. Prognostic significance of activated CD8(+) T cell infiltrations within esophageal carcinomas. Cancer Res. 2001;61(10):3932–6. [PubMed] [Google Scholar]
- 8.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. The New England journal of medicine. 2003;348(3):203–13. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 9.Lu YC, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res. 2014;20(13):3401–10. doi: 10.1158/1078-0432.CCR-14-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kessler JH, Beekman NJ, Bres-Vloemans SA, Verdijk P, van Veelen PA, Kloosterman-Joosten AM, et al. Efficient identification of novel HLA-A(*)0201-presented cytotoxic T lymphocyte epitopes in the widely expressed tumor antigen PRAME by proteasome-mediated digestion analysis. J Exp Med. 2001;193(1):73–88. doi: 10.1084/jem.193.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brichard V, Van Pel A, Wolfel T, Wolfel C, De Plaen E, Lethe B, et al. The tyrosinase gene codes for an antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med. 1993;178(2):489–95. doi: 10.1084/jem.178.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bakker AB, Schreurs MW, de Boer AJ, Kawakami Y, Rosenberg SA, Adema GJ, et al. Melanocyte lineage-specific antigen gp100 is recognized by melanoma-derived tumor-infiltrating lymphocytes. J Exp Med. 1994;179(3):1005–9. doi: 10.1084/jem.179.3.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawakami Y, Eliyahu S, Sakaguchi K, Robbins PF, Rivoltini L, Yannelli JR, et al. Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2-restricted tumor infiltrating lymphocytes. J Exp Med. 1994;180(1):347–52. doi: 10.1084/jem.180.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McNeil S, Shinde V, Andrew M, Hatchette T, Leblanc J, Ambrose A, et al. Interim estimates of 2013/14 influenza clinical severity and vaccine effectiveness in the prevention of laboratory-confirmed influenza-related hospitalisation, Canada, February 2014. Euro surveillance: bulletin Europeen sur les maladies transmissibles = European communicable disease bulletin. 2014;19(9) doi: 10.2807/1560-7917.es2014.19.9.20729. [DOI] [PubMed] [Google Scholar]
- 15.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17(13):4550–7. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116(20):4099–102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chodon T, Comin-Anduix B, Chmielowski B, Koya RC, Wu Z, Auerbach M, et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20(9):2457–65. doi: 10.1158/1078-0432.CCR-13-3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–26. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 22.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. The New England journal of medicine. 2015;372(21):2018–28. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 25.Larkin J, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. The New England journal of medicine. 2015;373(13):1270–1. doi: 10.1056/NEJMc1509660. [DOI] [PubMed] [Google Scholar]
- 26.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol. 2015;33(25):2780–8. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 27.Sfaxi F, Scamuffa N, Lalou C, Ma J, Metrakos P, Siegfried G, et al. Repression of liver colorectal metastasis by the serpin Spn4A a naturally occurring inhibitor of the constitutive secretory proprotein convertases. Oncotarget. 2014;5(12):4195–210. doi: 10.18632/oncotarget.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, et al. Erlotinib in lung cancer – molecular and clinical predictors of outcome. N Engl J Med. 2005;353(2):133–44. doi: 10.1056/NEJMoa050736. [DOI] [PubMed] [Google Scholar]
- 29.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–94. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 30.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 31.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC, et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer cell. 2015;27(2):240–56. doi: 10.1016/j.ccell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–7. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell. 2015;162(6):1271–85. doi: 10.1016/j.cell.2015.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi H, Hong A, Kong X, Koya RC, Song C, Moriceau G, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer discovery. 2014;4(1):69–79. doi: 10.1158/2159-8290.CD-13-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer discovery. 2014;4(1):80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ribas A, Wolchok JD. Combining cancer immunotherapy and targeted therapy. Curr Opin Immunol. 2013;25(2):291–6. doi: 10.1016/j.coi.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer research. 2010;70(13):5213–9. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 42.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19(5):1225–31. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kono M, Dunn IS, Durda PJ, Butera D, Rose LB, Haggerty TJ, et al. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Mol Cancer Res. 2006;4(10):779–92. doi: 10.1158/1541-7786.MCR-06-0077. [DOI] [PubMed] [Google Scholar]
- 44.Sapkota B, Hill CE, Pollack BP. Vemurafenib enhances MHC induction in BRAF homozygous melanoma cells. Oncoimmunology. 2013;2(1):e22890. doi: 10.4161/onci.22890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Acquavella N, Clever D, Yu Z, Roelke-Parker M, Palmer DC, Xi L, et al. Type I cytokines synergize with oncogene inhibition to induce tumor growth arrest. Cancer Immunol Res. 2015;3(1):37–47. doi: 10.1158/2326-6066.CIR-14-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gray-Schopfer VC, Karasarides M, Hayward R, Marais R. Tumor necrosis factor-alpha blocks apoptosis in melanoma cells when BRAF signaling is inhibited. Cancer research. 2007;67(1):122–9. doi: 10.1158/0008-5472.CAN-06-1880. [DOI] [PubMed] [Google Scholar]
- 47.Srividya MR, Thota B, Shailaja BC, Arivazhagan A, Thennarasu K, Chandramouli BA, et al. Homozygous 10q23/PTEN deletion and its impact on outcome in glioblastoma: a prospective translational study on a uniformly treated cohort of adult patients. Neuropathology. 2011;31(4):376–83. doi: 10.1111/j.1440-1789.2010.01178.x. [DOI] [PubMed] [Google Scholar]
- 48.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nature medicine. 2007;13(1):84–8. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 49.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 50.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer discovery. 2013;3(12):1355–63. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Azuma K, Ota K, Kawahara A, Hattori S, Iwama E, Harada T, et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2014;25(10):1935–40. doi: 10.1093/annonc/mdu242. [DOI] [PubMed] [Google Scholar]
- 52.Rutledge WC, Kong J, Gao J, Gutman DA, Cooper LA, Appin C, et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin Cancer Res. 2013;19(18):4951–60. doi: 10.1158/1078-0432.CCR-13-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wachowska M, Muchowicz A, Golab J. Targeting Epigenetic Processes in Photodynamic Therapy-Induced Anticancer Immunity. Front Oncol. 2015;5:176. doi: 10.3389/fonc.2015.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447(7143):433–40. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 55.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 56.Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014;5(3):587–98. doi: 10.18632/oncotarget.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weber J, Salgaller M, Samid D, Johnson B, Herlyn M, Lassam N, et al. Expression of the MAGE-1 tumor antigen is up-regulated by the demethylating agent 5-aza-2′-deoxycytidine. Cancer Res. 1994;54(7):1766–71. [PubMed] [Google Scholar]
- 58.Coral S, Sigalotti L, Altomonte M, Engelsberg A, Colizzi F, Cattarossi I, et al. 5-aza-2′-deoxycytidine-induced expression of functional cancer testis antigens in human renal cell carcinoma: immunotherapeutic implications. Clin Cancer Res. 2002;8(8):2690–5. [PubMed] [Google Scholar]
- 59.Sigalotti L, Fratta E, Coral S, Maio M. Epigenetic drugs as immunomodulators for combination therapies in solid tumors. Pharmacol Ther. 2014;142(3):339–50. doi: 10.1016/j.pharmthera.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 60.Wanczyk M, Roszczenko K, Marcinkiewicz K, Bojarczuk K, Kowara M, Winiarska M. HDACi–going through the mechanisms. Front Biosci (Landmark Ed) 2011;16:340–59. doi: 10.2741/3691. [DOI] [PubMed] [Google Scholar]
- 61.Van den Eynde BJ, Boon T. Tumor antigens recognized by T lymphocytes. Int J Clin Lab Res. 1997;27(2):81–6. doi: 10.1007/BF02912440. [DOI] [PubMed] [Google Scholar]
- 62.Guo ZS, Hong JA, Irvine KR, Chen GA, Spiess PJ, Liu Y, et al. De novo induction of a cancer/testis antigen by 5-aza-2′-deoxycytidine augments adoptive immunotherapy in a murine tumor model. Cancer Res. 2006;66(2):1105–13. doi: 10.1158/0008-5472.CAN-05-3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roulois D, Blanquart C, Panterne C, Gueugnon F, Gregoire M, Fonteneau JF. Downregulation of MUC1 expression and its recognition by CD8(+) T cells on the surface of malignant pleural mesothelioma cells treated with HDACi. Eur J Immunol. 2012;42(3):783–9. doi: 10.1002/eji.201141800. [DOI] [PubMed] [Google Scholar]
- 64.Woods DM, Woan K, Cheng F, Wang H, Perez-Villarroel P, Lee C, et al. The antimelanoma activity of the histone deacetylase inhibitor panobinostat (LBH589) is mediated by direct tumor cytotoxicity and increased tumor immunogenicity. Melanoma Res. 2013;23(5):341–8. doi: 10.1097/CMR.0b013e328364c0ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gregorie CJ, Wiesen JL, Magner WJ, Lin AW, Tomasi TB. Restoration of immune response gene induction in trophoblast tumor cells associated with cellular senescence. J Reprod Immunol. 2009;81(1):25–33. doi: 10.1016/j.jri.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014;28(6):1280–8. doi: 10.1038/leu.2013.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Woods DM, Sodre AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC Inhibition Upregulates PD-1 Ligands in Melanoma and Augments Immunotherapy with PD-1 Blockade. Cancer Immunol Res. 2015 doi: 10.1158/2326-6066.CIR-15-0077-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. 2014;111(32):11774–9. doi: 10.1073/pnas.1410626111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. The New England journal of medicine. 2012;366(3):207–15. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res. 2012;72(16):3928–37. doi: 10.1158/0008-5472.CAN-11-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Comin-Anduix B, Chodon T, Sazegar H, Matsunaga D, Mock S, Jalil J, et al. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clin Cancer Res. 2010;16(24):6040–8. doi: 10.1158/1078-0432.CCR-10-1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hong DS, Vence L, Falchook G, Radvanyi LG, Liu C, Goodman V, et al. BRAF(V600) inhibitor GSK2118436 targeted inhibition of mutant BRAF in cancer patients does not impair overall immune competency. Clin Cancer Res. 2012;18(8):2326–35. doi: 10.1158/1078-0432.CCR-11-2515. [DOI] [PubMed] [Google Scholar]
- 73.Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. The Journal of clinical investigation. 2013;123(3):1371–81. doi: 10.1172/JCI66236. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 74.Vella LJ, Pasam A, Dimopoulos N, Andrews M, Knights A, Puaux AL, et al. MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells. Cancer Immunol Res. 2014;2(4):351–60. doi: 10.1158/2326-6066.CIR-13-0181. [DOI] [PubMed] [Google Scholar]
- 75.Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci Transl Med. 2015;7(279):279ra41. doi: 10.1126/scitranslmed.aaa4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Homet Moreno B, Mok S, Comin-Anduix B, hu-Lieskovan S, Ribas A. Combined treatment with Dabrafenib and Trametinib with Immune-stimulating Antibodies for BRAF mutant melanoma. Oncoimmunology. 2015 doi: 10.1080/2162402X.2015.1052212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Long GV, Wilmott JS, Haydu LE, Tembe V, Sharma R, Rizos H, et al. Effects of BRAF inhibitors on human melanoma tissue before treatment, early during treatment, and on progression. Pigment Cell Melanoma Res. 2013;26(4):499–508. doi: 10.1111/pcmr.12098. [DOI] [PubMed] [Google Scholar]
- 78.Cooper ZA, Frederick DT, Juneja VR, Sullivan RJ, Lawrence DP, Piris A, et al. BRAF inhibition is associated with increased clonality in tumor-infiltrating lymphocytes. Oncoimmunology. 2013;2(10):e26615. doi: 10.4161/onci.26615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5(200):200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cooper ZA, Juneja VR, Sage PT, Frederick DT, Piris A, Mitra D, et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol Res. 2014;2(7):643–54. doi: 10.1158/2326-6066.CIR-13-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res. 2015;75(2):296–305. doi: 10.1158/0008-5472.CAN-14-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vaziri K, Roland JC, Robinson LL, Reines HD, Fakhry SM. Extreme anemia in an injured Jehovah’s Witness: a test of our understanding of the physiology of severe anemia and the threshold for blood transfusion. The Journal of trauma. 2009;67(1):E11–3. doi: 10.1097/TA.0b013e318047bfc8. [DOI] [PubMed] [Google Scholar]
- 84.Luo M, Fan H, Nagy T, Wei H, Wang C, Liu S, et al. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. 2009;69(2):466–74. doi: 10.1158/0008-5472.CAN-08-3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pachter JA, Kolev VN, Schunselaar L, Shapiro IM, Bueno R, Baas P, et al. FAK inhibitor VS-6063 (defactinib) targets mesothelioma cancer stem cells, which are enriched by standard of care chemotherapy. American Association for Cancer Research, Poster. 2015 [Google Scholar]
- 86.Stokes JB, Adair SJ, Slack-Davis JK, Walters DM, Tilghman RW, Hershey ED, et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther. 2011;10(11):2135–45. doi: 10.1158/1535-7163.MCT-11-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ring J, Li Y, Shapiro IM, Wang Y, Weaver DT, Pachter JA. FAK/PYK2 inhibitors defatinib and VS-4718 enhance immune checkpoint inhibitor efficacy. SITC poste 2015. 2015 [Google Scholar]
- 88.Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203(7):1651–6. doi: 10.1084/jem.20051848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. The New England journal of medicine. 2015;373(1):23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Khalili JS, Liu S, Rodriguez-Cruz TG, Whittington M, Wardell S, Liu C, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res. 2012;18(19):5329–40. doi: 10.1158/1078-0432.CCR-12-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res. 2013;19(2):393–403. doi: 10.1158/1078-0432.CCR-12-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nicholson AM, Ledgister S, Williams T, Robinson S, Gayle P, Lindo T, et al. Distribution of nosocomial organisms and their resistance patterns in the intensive care unit of the University Hospital of the West Indies, Kingston, Jamaica. The West Indian medical journal. 2009;58(2):142–8. [PubMed] [Google Scholar]
- 93.Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17(9):1094–100. doi: 10.1038/nm.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69(6):2514–22. doi: 10.1158/0008-5472.CAN-08-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Minor DR, Puzanov I, Callahan MK, Hug BA, Hoos A. Severe gastrointestinal toxicity with administration of trametinib in combination with dabrafenib and ipilimumab. Pigment Cell Melanoma Res. 2015;28(5):611–2. doi: 10.1111/pcmr.12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Collins DM, O’Donovan N, McGowan PM, O’Sullivan F, Duffy MJ, Crown J. Trastuzumab induces antibody-dependent cell-mediated cytotoxicity (ADCC) in HER-2-non-amplified breast cancer cell lines. Ann Oncol. 2012;23(7):1788–95. doi: 10.1093/annonc/mdr484. [DOI] [PubMed] [Google Scholar]
- 97.Ming Lim C, Stephenson R, Salazar A, Ferris R. TLR3 agonists improve the immunostimulatory potential of cetuximab against EGFR+head and neck cancer cells. Oncoimmunology. 2013 doi: 10.4161/onci.24677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Amin A. Nivolumab (anti-PD-1; BMS-936558, ONO-4538) in combination with sunitinib or pazopanib in patients (pts) with metastatic renal cell carcinoma (mRCC) ASCO 2014. 2014 [Google Scholar]
- 99.Rini BI, Stein M, Shannon P, Eddy S, Tyler A, Stephenson JJ, Jr, et al. Phase 1 dose-escalation trial of tremelimumab plus sunitinib in patients with metastatic renal cell carcinoma. Cancer. 2011;117(4):758–67. doi: 10.1002/cncr.25639. [DOI] [PubMed] [Google Scholar]
- 100.Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1(7):598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, et al. Alterations of immune response of Non-Small Cell Lung Cancer with Azacytidine. Oncotarget. 2013;4(11):2067–79. doi: 10.18632/oncotarget.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer. 2011;104(12):1828–35. doi: 10.1038/bjc.2011.156. [DOI] [PMC free article] [PubMed] [Google Scholar]