

Abstract
Earlier studies aimed at defining protective immunity induced by Mycobacterium bovis BCG immunization have largely focused on the induction of antituberculosis CD4+ and CD8+ T cell responses. Here we describe a vaccine consisting of a BCGΔmmaA4 deletion mutant formulated in dimethyl dioctadecyl-ammonium bromide (DDA) with d-(+)-trehalose 6,6′-dibehenate (TDB) (DDA/TDB) adjuvant (A4/Adj) that protected TCRδ−/− mice depleted of CD4+, CD8+, and NK1.1+ T cells against an aerosol challenge with M. tuberculosis. These mice were significantly protected relative to mice immunized with a nonadjuvanted BCGΔmmaA4 (BCG-A4) mutant and nonvaccinated controls at 2 months and 9 months postvaccination. In the absence of all T cells following treatment with anti-Thy1.2 antibody, the immunized mice lost the ability to control the infection. These results indicate that an unconventional T cell population was mediating protection in the absence of CD4+, CD8+, NK1.1+, and TCRγδ T cells and could exhibit memory. Focusing on CD4− CD8− double-negative (DN) T cells, we found that these cells accumulated in the lungs postchallenge significantly more in A4/Adj-immunized mice and induced significantly greater frequencies of pulmonary gamma interferon (IFN-γ)-producing cells than were seen in the nonvaccinated or nonadjuvanted BCG control groups. Moreover, pulmonary DN T cells from the A4/Adj group exhibited significantly higher IFN-γ integrated median fluorescence intensity (iMFI) values than were seen in the control groups. We also showed that enriched DN T cells from mice immunized with A4/Adj could control mycobacterial growth in vitro significantly better than naive whole-spleen cells. These results suggest that formulating BCG in DDA/TDB adjuvant confers superior protection in immunocompromised mice and likely involves the induction of long-lived memory DN T cells.
INTRODUCTION
Despite the widespread use of Mycobacterium bovis BCG vaccine and the availability of effective chemotherapy, tuberculosis (TB) remains an immense global public health challenge, with approximately 9 million new cases and 1.4 million deaths per year. Overall, an estimated 2 billion people are infected with M. tuberculosis worldwide (1, 2). These alarming statistics have made it obvious that current interventions are not controlling the epidemic. The reasons for the current TB problem are multifaceted and include the lack of an efficacious vaccine and the emergence of multidrug-resistant and extremely drug-resistant M. tuberculosis strains (1, 3). Importantly, the convergence of the HIV and TB epidemics has, without question, intensified the TB problem. Since HIV-infected individuals are considerably more susceptible to pathogens due to their immunocompromised state, coinfected individuals are 30 times more likely to develop active TB than those infected with M. tuberculosis only. In fact, TB causes 25% of all HIV-related deaths worldwide (2).
While BCG is one of the most widely used global vaccines, its impact on the current TB epidemic has clearly been inadequate. Randomized controlled clinical trials and retrospective case-control studies have shown that BCG immunization is effective in reducing cases of severe disseminated TB in children; however, the effectiveness of BCG in preventing pulmonary TB has been highly variable, ranging from 0% to 80% (4). Furthermore, protection is often not highly persistent, with substantial waning of BCG-induced protective responses generally seen during the first decade after immunization (5). Given the suboptimal efficacy in the context of the devastating TB epidemic, there is an urgent global health need to develop a new TB immunization strategy. Consequently, many TB researchers are developing strategies to amplify BCG-induced antituberculosis protective responses. A popular approach involves boosting with protein- or virus-vectored vaccines after a priming BCG immunization. Alternatively, a potentially simpler and less expensive strategy involves formulating BCG in a liposome-forming adjuvant. Lipid encapsulation of BCG has been shown to improve the immunogenicity and protective efficacy of BCG immunization in mice, guinea pigs, badgers, and cattle (6–10).
Our group recently demonstrated that formulation of a BCGΔmmaA4 (BCG-A4) mutant in DDA/TDB adjuvant (A4/Adj) increased the level and persistence of BCG-induced immune responses relative to those produced by conventional BCG and that the increased protection was associated with elevated CD4+ multifunctional T cell immune responses (11). In addition to the adjuvant, deletion of the mmaA4 gene may also enhance BCG-mediated immune responses. Dao and colleagues showed that deletion of the mmaA4 gene, which encodes a methyl transferase involved in mycolic acid synthesis, removed repression of interleukin-12 (IL-12) synthesis associated with M. tuberculosis infections (12). IL-12 has been shown to be a key molecule for polarizing Th1 differentiation, and both M. tuberculosis and BCG mmaA4 mutants were found to induce significantly elevated levels of IL-12 from infected macrophages.
We have consistently observed elevated protection with the A4/Adj formulation relative to that of wild-type BCG (wtBCG) formulated in DDA/TDB (11). For this reason, and given the unique property of this mutant strain to augment IL-12 production, we used the A4/Adj vaccine for the studies described here. To further evaluate the potential of the A4/Adj preparation, we tested the effectiveness of this vaccine preparation in immunocompromised mice and investigated the immune mechanisms that mediate antituberculosis protection in the context of immunodeficiency.
Given the acute susceptibility of HIV-infected individuals to TB, the development of efficacious TB vaccines for use in immunocompromised populations is a global public health priority. In an earlier study, we showed that immunization with a live attenuated M. tuberculosis strain (an RD1 panCD gene deletion mutant) protected CD4-deficient mice against an aerosol TB challenge and that this protection was associated with the activation of a population of TCRαβ+ CD4− CD8− double-negative (DN) T cells (13, 14). In the present study, we assessed whether mice lacking CD4+, CD8+, NK1.1+, and TCRγδ T cells were protected against a TB infection following immunization with a BCGΔmmaA4 mutant strain (BCG-A4) formulated with DDA/TDB adjuvant (A4/Adj). Here we show that significantly increased protection against a TB aerosol challenge was seen in severely immunocompromised mice immunized with the A4/Adj formulation. Interestingly, the enhanced protection detected in the mice vaccinated with A4/Adj was associated with an enhanced capacity to stimulate DN T cells.
MATERIALS AND METHODS
Animals.
C57BL/6 mice and breeding pairs of TCRδ−/− (B6.129P2-Tcrdtm1/Mom/J) mice were obtained from the Jackson Laboratories (Bar Harbor, ME). All mice used in this study were 6 to 8 weeks old and were maintained under appropriate conditions at the Center for Biologics Evaluation and Research, Bethesda, MD. This study was done in accordance with the guidelines for the care and use of laboratory animals specified by the National Institutes of Health. This protocol was approved by the Institutional Animal Care and Use Committee of the Center for Biologics Evaluation and Research under Animal Study Protocol 1993-09.
Immunizations.
The BCGΔmmaA4 mutant was derived from BCG Pasteur as previously described (12). The BCGΔmmaA4 strain was administered subcutaneously (s.c.) in phosphate-buffered saline (PBS) or adjuvant at 1 × 106 CFU per immunization in 0.2 ml for a total of three vaccinations 2 weeks apart. The adjuvant-containing formulation was prepared by mixing the mutant BCG with dimethyl dioctadecyl-ammonium bromide (DDA; Kodak, Rochester, NY) and d-(+)-trehalose 6,6′-dibehenate (TDB; Avanti Polar Lipids, Alabaster, AL). The formulation of the BCG–TDB/DDA adjuvant was performed as previously described (11).
In vivo cell depletions using monoclonal antibodies.
Antibodies (Abs) specific for CD4 (clone GK1.5), CD8 (clone 2.43), NK1.1 (clone PK136), or Thy1.2 (clone 30-H12) molecules were used to deplete specific T cell populations (National Cell Culture Center, Minneapolis, MN). Antibodies were injected intraperitoneally at 0.5 mg per antibody at 4 and 2 days prior to the challenge and once per week thereafter. The efficacy of the antibody treatment was evaluated by bleeding the mice prior to the aerosol challenge and analyzing the peripheral blood mononuclear cells (PBMCs) by flow cytometry and by analysis of splenocytes at the time of sacrifice 1 month postchallenge by flow cytometry. Flow cytometry analysis of the PBMCs or splenocytes was done using antibodies specific for the Thy1.2 (rat anti-mouse Thy1.2 allophycocyanin [APC] Ab, clone 53-2.1), CD4 (rat anti-mouse CD4 Alexa Fluor 700 [AF-700] Ab, clone RM4-5), CD8 (rat anti-mouse CD8 peridinin chlorophyll protein complex [PerCP] Ab, clone 53-6.7), TCRγδ (hamster anti-mouse TCRγδ fluorescein isothiocyanate [FITC] Ab, clone GL3), NK1.1 (mouse anti-mouse NK1.1 phycoerythrin [PE] Ab, clone PK136), and B220 (rat anti-mouse/human NK1.1 Pacific blue [PB] Ab, clone RA3-6B2) molecules. The analysis was performed using an LSRII flow cytometer (Becton Dickinson, Sparks, MD) and FlowJo software (Tree Star Inc., Ashland, OR). The PB anti-B220 antibody was purchased from Biolegend (San Diego, CA). All other antibodies were purchased from BD Biosciences (San Jose, CA). Flow analysis revealed that treatment with antibodies consistently reduced the target T cell population by >90% at up to 1 month postchallenge.
Evaluation of vaccine-induced protective immunity in a murine model of pulmonary TB.
Unless otherwise stated, 2 months following the final immunization, the mice (5 mice per group) were infected with M. tuberculosis Erdman by aerosol at a concentration known to deliver about 300 CFU into the lungs over a 30-min exposure in a Middlebrook chamber (Glas Col, Terre Haute, IN) (15). At each time point, the lungs and spleens were homogenized separately in PBS with 0.05% Tween 80 using a Seward Stomacher 80 blender (Tekmar, Cincinnati, OH). The homogenates were serially diluted in PBS–0.05% Tween 80 and plated on Middlebrook 7H11 agar (Difco) plates containing 10% oleic acid-albumin-dextrose-catalase (OADC) enrichment (Becton Dickinson, Sparks, MD) medium, 10 μg/ml ampicillin, 50 μg/ml cycloheximide, and 2 μg/ml 2-thiophenecarboxylic acid hydride (TCH) (Sigma, St. Louis, MO). The concentration of TCH added to the agar plates inhibits BCG growth while permitting M. tuberculosis growth. Plates were incubated at 37°C for 2 to 3 weeks before counting was performed to determine the number of mycobacterial CFU per organ.
Quantitation of pulmonary DN T cells postchallenge.
The frequency of pulmonary DN T cells from challenged mice was quantified by flow cytometry to determine the percentage of DN T cells in the lungs and the frequency of the cells secreting gamma interferon (IFN-γ). Prior to being challenged, naive mice or mice immunized with BCG-A4 with or without adjuvant were depleted of CD4+, CD8+, and NK1.1+ T cells. Cells were isolated from the lungs 10 days following an aerosol challenge by disrupting the lung tissue with razor blades and incubating the lung homogenates with type I collagenase (Invitrogen, Carlsbad, CA) (0.7 mg/ml) for 1 h. The homogenates were then passed through 70-μm-pore-size cell strainers to removed tissue clumps. The resulting single-cell suspension was then treated with 5 ml ACK lysing buffer (Lanza, Walkersville, MD) for 1 min to deplete the cell suspension of erythrocytes. After washing of the cells with PBS, near-infrared (near-IR) live-dead stain (Invitrogen) (10 μl of a 1:100 dilution) was added to the cells and the mixture was incubated 30 min in a volume of 50 μl to allow gating on viable cells. The cells were then washed with PBS–2% fetal bovine serum (PBS-FBS). Subsequently, antibody against CD16/CD32 (FcγIII/II receptor; clone 2.4G2) (Fc block) was added in a volume of 50 μl and the mixture was incubated at 4°C for 15 min. To allow gating on DN T cells, anti-B220 (PB), anti-Thy1.2 (rat anti-mouse Thy1.2 Alexa Fluor 700; clone 53-2.1) (BD Biosciences), and biotinylated anti-TCRβ (rat anti-mouse TCRβ; clone H57-597) (BD Biosciences) antibody molecules were added to the cells at 0.1 to 0.4 μg per tube and incubated for 30 min at 4°C. Biotinylated anti-TCRβ was detected by incubating the cells with streptavidin-conjugated Qdot605 fluorochrome (Invitrogen). The cells were then washed twice with PBS and then fixed for 30 min at 4°C with 2% paraformaldehyde–PBS. After being fixed, the cells were pelleted, washed twice with PBS-FBS, and stored at 4°C. Fixed cells were washed twice with perm-wash buffer (1% FBS–0.01 M HEPES–0.1% saponin–PBS) followed by intracellular staining using rat anti-mouse PE–IFN-γ antibody (clone XMG1.2) (BD Biosciences) at 0.2 μg. The cells were incubated at 4°C for 30 min and washed twice with perm-wash buffer and then twice with PBS-FBS. The cells were analyzed using an LSRII flow cytometer (Becton Dickinson) and FlowJo software (Tree Star Inc., Ashland, OR). About 100,000 events per sample were acquired and then, using FlowJo software, gated on live, single-cell Thy1.2+ lymphocytes to determine the frequency of DN T cells in the lungs postchallenge and the frequency of DN T cells staining positive for IFN-γ.
iMFI assessments.
The median fluorescence intensity (MFI) for IFN-γ from pulmonary DN T cells at 10 days postchallenge was evaluated in the different vaccine groups using FlowJo software, and the value determined represents the middle number of the range of values representing the distributions of DN T cells secreting IFN-γ. The integrated MFI (iMFI) was calculated by multiplying the percentage of cell frequency by the MFI values. The data are presented as means of the individual iMFI assessments from the lungs of 3 mice.
DN T cell enrichment.
For enrichment of double-negative (DN) T cells, splenocyte single-cell suspensions were obtained from wild-type (WT) C57BL/6 mice that had been treated with antibodies to deplete CD4+, CD8+, and NK1.1+ T cells. The spleen cells were isolated by homogenizing the spleens in DMEM containing 10% bovine calf serum (cDMEM)–10% FBS with a 3-ml syringe barrel and passing the homogenate through a 0.7-μm-pore-size cell strainer. After centrifugation, the pellet was resuspended in ACK buffer (Lanza), incubated for 1 min to remove erythrocytes, and then washed with PBS containing 2 mM EDTA and 0.5% FBS. After being counted using a hemacytometer, the T cells were enriched using a Dynal mouse T cell negative isolation kit (Invitrogen) according to the manufacturer's instructions. The antibody mix used with the kit to deplete non-T cells contains antibodies against mouse CD45R (B220), CD11b, Ter-119, and CD16/32 molecules. The enriched DN T cells were analyzed by flow cytometry to determine the purity of the DN T cell preparation.
MGIA.
The mycobacterial growth inhibition assay (MGIA) was done as previously described (16). Briefly, bone marrow cells were collected from the tibias and femurs of C57BL/6 mice and cultured for 7 days in wells of 24-well plates in growth media consisting of DMEM supplemented with 10% FBS, 10 mM HEPES, 2.0 mM l-glutamine, 0.1 mM minimal essential medium (MEM) nonessential amino acids, and 10% L929 cell conditioned medium. After 7 days, the bone marrow-derived macrophages (BMMΦ) were incubated with BCG (multiplicity of infection [MOI] = 1:1,000) 2 h before the DN T cells or splenocytes from nonvaccinated or vaccinated mice were added to the wells. Following a 7-day incubation period, the wells were rinsed with PBS and the macrophages were lysed with water containing 0.1% (wt/vol) saponin. The lysates were serially diluted, and aliquots were plated on 7H11-plus-OADC plates to quantify bacterial CFU. After the plates were incubated for 2 to 3 weeks, the colonies were counted and the reduction in CFU associated with the immune cell cocultures relative to nonvaccinated spleen cell cocultures was determined.
Statistical analysis.
The Graph Pad Prism 5 program was used to analyze the data for these experiments (Graphpad Software Inc., La Jolla, CA). The protection data, the cell flow cytometry results, the growth inhibition data, and the MFI data were evaluated using two-tailed, unpaired t test analysis for comparison of a single experimental group to the nonvaccinated control group, and one-way analysis of variance (ANOVA) was used for multigroup comparisons.
RESULTS
Vaccinated mice lacking CD4+, CD8+, NK1.1+, and TCR γδ+ T cells were protected against an aerosol TB challenge.
We previously found that both wtBCG and an M. tuberculosis mutant strain protected mice in the absence of both CD4+ and CD8+ T cells (14). In the present study, we examined whether a BCG mutant formulated in DDA/TDB adjuvant would augment protection in immunocompromised mice as has been demonstrated in immunocompetent mice (11). We chose the BCG-A4 mutant for this study since we have found that the mutant formulated in DDA/TDB protects consistently better than adjuvanted wtBCG. In initial experiments, WT C57BL/6 mice were immunized with the A4/Adj preparation and were then treated with antibodies to deplete either CD4+ T cells alone or both CD4+ and CD8+ T cells. As seen in Fig. 1, the immunocompetent, vaccinated mice were significantly protected against the TB infection in both the lungs (Fig. 1A) (−0.85 log10 reduction relative to naive mice) and the spleen (Fig. 1B) (−1.7 log10) at 1 month postchallenge. Furthermore, a significant protective response was also seen in CD4+ T cell-depleted, vaccinated mice compared to similarly treated nonvaccinated mice (−1.4 log10 and −1.8 log10 reductions in the lungs and spleens, respectively). Finally, reduced bacterial burdens at 1 month postchallenge were detected in the lungs (−1.85 log10) and spleens (−2.50 log10) of vaccinated relative to nonvaccinated mice depleted of both CD4+ and CD8+ T cells. Interestingly, the bacterial CFU values in the lungs of the CD4+ and CD4/CD8+ T cell-depleted mice were nearly identical, suggesting that the protection mediated by the A4/Adj vaccine in the absence of CD4+ T cells was not CD8+ T cell dependent.
FIG 1.

Mutant BCGΔmmaA4 plus DDA/TDB protects mice in the absence of CD4+ and CD8+ T cells. WT C57BL/6 mice were challenged with M. tuberculosis 2 months after vaccination with the BCGΔmmaA4 mutant (BCG-A4) or with BCG-A4 plus DDA/TDB adjuvant (A4/Adj). Mice were treated with antibodies for depleting CD4+ or CD8+ T cells 4 and 2 days before and once per week after challenge. The mean and standard deviation (SD) of the bacterial CFU were determined in the lungs (A) and spleens (B) 1 month postchallenge from one experiment. n = 4 to 5 mice per group. *, statistical significance relative to nonvaccinated (Naive) mice (P < 0.05). ^, statistical significance relative to nonvaccinated mice treated with anti-CD4 monoclonal Abs (MAbs) (nonvaccinated + α-CD4 MAbs) (P < 0.05). #, statistical significance relative to nonvaccinated + anti-CD4 (α-CD4)/CD8 MAbs (P < 0.05).
To determine whether BCG-A4 protects in the absence of other specific T cells and whether the adjuvant augments protection, experiments were conducted using TCRδ−/− mice. Initially, we observed that there was no difference in protection or the level of M. tuberculosis infection between WT C57BL/6 and TCRδ−/− mice. In other experiments, TCRδ−/− mice were vaccinated with BCG-A4 either alone or formulated with DDA/TDB and then treated with antibodies to deplete CD4+, CD8+, and NK1.1+ T cells (+Ab Combo) before being challenged with M. tuberculosis Erdman 2 months after the last vaccination. The data shown in Fig. 2 are representative of results of two independent experiments. At 1 month postchallenge, mice vaccinated with both BCG formulations were protected significantly better than nonvaccinated, TCRδ−/− mice in both the lungs (Fig. 2A) and spleens (Fig. 2B). Immunization with A4/Adj induced significantly improved protection relative to nonadjuvanted BCG-A4 in both non-Ab-treated TCRδ−/− mice (1.8 versus 1.2 log10 CFU reduction, respectively, relative to untreated nonvaccinated mice) and Ab-treated TCRδ−/− mice (2.3 versus 1.5 log10 CFU reduction, respectively, relative to Ab-treated, nonvaccinated mice). Similar results were also seen in mouse spleens for both vaccine formulations. Significant splenic protection was detected in immunocompetent mice relative to nonvaccinated TCRδ−/− controls after A4/Adj immunization (1.4 log10 CFU reduction) and vaccination with BCG-A4 alone (1.0 log10 CFU reduction) in non-Ab-treated mice. Splenic protective immune responses evoked by BCG-A4 alone and A4/Adj immunization were also significantly elevated relative to the levels seen with nonvaccinated immunodeficient mice (1.3 and 1.76 log10 CFU reduction relative to Ab Combo-treated nonvaccinated mice, respectively). Although reduced in the spleens of the A4/Adj-immunized mice, the splenic bacterial burdens were not significantly different from those observed in the nonadjuvanted BCG-A4 spleens. Thus, we observed that vaccination with the BCG-A4 and the A4/Adj formulation resulted in a significant reduction in pulmonary and splenic CFU relative to nonimmunized controls in both immunocompetent and severely immunocompromised mice and that A4/Adj vaccination resulted in a significant reduction in pulmonary CFU levels relative to those seen with BCG-A4 alone in the presence or absence of conventional T cells. It should be noted that mice treated with the adjuvant alone were not protected in either the lungs or the spleens in the absence of conventional T cells and were modestly protected only in the lungs of immunocompetent mice (0.36 log10 CFU reduction relative to nonvaccinated mice) (not shown). Remarkably, these data show that none of the conventional T cell populations that were depleted likely mediated the observed protection in wild-type C57BL/6 or TCRδ−/− mice in the absence of CD4+ T cells.
FIG 2.

Formulating the BCGΔmmaA4 mutant in adjuvant enhanced protection in mice lacking CD4+, CD8+, NK1.1+, or TCRγδ T cells. TCRδ−/− mice were challenged with M. tuberculosis by aerosol 2 months after vaccination. Mice were treated with antibodies (Ab Combo) for depleting CD4+, CD8+, and NK1.1+ T cells 4 and 2 days before and once per week after challenge. The mean and SD of the bacterial CFU were determined in the lungs (A) and spleens (B) 1 month postchallenge. Data shown are from an experiment representative of 2 experiments. n = 4 to 5 mice per group. *, significance relative to nonvaccinated mice (P < 0.05). ^, significance relative to nonvaccinated + Ab Combo (P < 0.05). #, significance relative to nonadjuvanted BCG-A4 (P < 0.05). ∞, significance relative to nonadjuvanted BCG-A4 + Ab Combo (P < 0.05).
To determine if T cells were absolutely required for the protective immunity elicited by the A4/Adj vaccine, TCRδ−/− mice were depleted of all T cells using anti-Thy1.2 antibody along with the antibodies included in the combination. Flow cytometry analysis of the PBMCs prior to challenge showed that ≥90% of T cells had been depleted in the treated mice. As shown in Fig. 3, the level of M. tuberculosis infection in both naive and A4/Adj-immunized mice increased to over 8.0 log10 CFU in the lungs and over 7.0 log10 in the spleens at 1 month following an aerosol infection, indicating that nonimmunized and vaccinated mice had lost the ability to control the infection in the absence of all T cells. In contrast, CFU levels in untreated nonvaccinated mice did not exceed 6.5 log10 in the lungs and 5.3 log10 in the spleens. As expected, CFU values were significantly lower in A4/Adj-vaccinated mice (untreated), with a 1.6 log10 CFU reduction in both the lungs and the spleens, than in nonvaccinated mice. Thus, the cumulative experimental evidence strongly suggested that protection in the absence of CD4+ T cells in mice immunized with A4/Adj involves an unconventional T cell population.
FIG 3.

A4/Adj-induced protection is T cell dependent. TCRδ−/− mice were challenged with M. tuberculosis by aerosol 2 months postvaccination. Mice were treated with antibodies against CD4, CD8, and NK1.1 (Ab Combo) and anti-Thy1.2 Ab for depleting all T cells 4 and 2 days before and once per week after challenge. The mean and SD of the bacterial CFU were determined in the lungs (A) and spleens (B) 1 month postchallenge from one experiment. *, significance relative to nonvaccinated mice (P < 0.05). ^, significance relative to nonvaccinated + Ab Combo (P < 0.05). n = 4 to 5 mice per group.
It should be emphasized that the protection seen in mice treated with the antibody combination was not due to cellular repopulation at 1 month postchallenge. To demonstrate that the antibody combination treatment regimen efficiently removed the target cell populations for up to 1 month postchallenge, splenocytes from treated mice were obtained at the time of sacrifice and stained with antibodies against the CD4, CD8, and NK1.1 T cell molecules. Representative flow cytometry results are shown in Fig. 4 for splenocytes from immunocompetent (non-Ab-treated) mice (Fig. 4A) and mice treated with the Ab combination (Fig. 4B). These flow cytometry results show that the target cell populations remained depleted 1 month postchallenge, with the DN T cells comprising >95% of the T cell population in the Ab-treated mice at the time of sacrifice.
FIG 4.

Representative flow cytometry data showing the depletion efficiency of the antibody combination. TCRδ−/− mice were treated with the antibody combination (anti-CD4, anti-CD8, and anti-NK1.1) on two separate days 1 day apart before receiving an aerosol challenge with M. tuberculosis the same week as the antibody treatment. Following the challenge, the mice were treated with the antibody combination once per week for a total of 4 weeks, after which the spleen cells were isolated and stained with antibodies to identify the target cell populations by flow cytometry. The upper panels (A) show the frequency of target cells from a mouse not treated with the antibody combination, while the lower panels (B) show the frequency of target cells after treatment with the antibody combination.
Long-term protection seen in vaccinated, immunocompromised mice.
The ability to generate persistent memory cells is an essential property of any efficacious vaccine. To determine whether the cells mediating the protective responses in the A4/Adj-vaccinated, Ab combination-treated mice induced durable memory responses, TCRδ−/− mice were vaccinated with the A4/Adj preparation either 2 or 9 months before the depletion of CD4+, CD8+, and NK1.1+ T cells and then subsequently challenged with M. tuberculosis by aerosol. As seen in Fig. 5, TCRδ−/− mice that were immunized with the A4/Adj formulation either 2 or 9 months prior to being challenged were protected equally, and this observation was consistent in both immunocompetent mice and mice depleted of conventional T cells. Immunocompetent (non-Ab-treated) mice challenged 2 months (1.6 log10 CFU reduction in the mouse lungs) or 9 months (1.2 log10 CFU reduction in the mouse lungs) postvaccination were similarly protected. Importantly, there were no significant differences in pulmonary protection between vaccinated, Ab-treated mice challenged 9 months postvaccination (1.8 log10 CFU reduction) and those challenged 2 months postvaccination (2.0 log10 CFU reduction) and bacterial burdens in the lungs were nearly equivalent. Furthermore, no difference in the levels of protection in the spleen was observed over this 9-month time period. At 9 months postvaccination, the splenic protection was comparable to that seen at the 2-month time point in both untreated mice (1.65 versus 1.9 log10 CFU reduction, respectively, relative to untreated nonvaccinated mice) and Ab-treated mice (1.3 versus 1.4 log10 CFU reduction relative to Ab-treated naive mice). Since there was no significant decline in protection during the 9-month postvaccination study period, these results suggest that a population of protective, unconventional memory T cells is generated in response to vaccination with adjuvanted BCG-A4.
FIG 5.

A4/Adj immunization generated long-lived memory cells that do not express TCRγδ, CD4, CD8, or NK1.1. TCRδ−/− mice were challenged with M. tuberculosis by aerosol 2 months or 9 months (9 M) postvaccination (PV). Mice were treated with antibodies (Ab Combo) for depleting CD4+, CD8+, and NK1.1+ T cells 4 and 2 days before and once per week after challenge. The mean and SD of the bacterial CFU in the lungs (A) and spleens (B) were determined 1 month postchallenge from one experiment. n = 4 to 5 mice per group. *, significance relative to nonvaccinated mice (P < 0.05). ^, significance relative to nonvaccinated + Ab Combo (P < 0.05).
Flow cytometry analysis of vaccine-induced DN T cells.
Since protection was found to be T cell dependent but could be demonstrated in the absence of conventional T cells, we focused on double-negative (CD4− CD8−) (DN) T cells in subsequent experiments. Using flow cytometry, we examined the frequency of DN T cells in the lungs of mice depleted of CD4+, CD8+, and NK1.1+ T cells 10 days post-aerosol infection with M. tuberculosis (Fig. 6A) and the frequency of DN T cells producing IFN-γ (Fig. 6B). This time point was chosen because we have previously shown that vaccine-induced antituberculosis immune responses are greatest 10 to 14 days post-aerosol infection (relative to those seen with nonvaccinated controls) in this murine M. tuberculosis challenge model (17). At 10 days postchallenge, the frequency of DN T cells in the lungs of A4/Adj-immunized mice (6.7% ± 2%) was significantly greater than that seen with nonvaccinated mice (3.0% ± 0.5%), while the frequency of these cells in the lungs of mice vaccinated with BCG-A4 alone (3.6% ± 2%) was elevated but not significantly different from that seen with nonimmunized mice. Mice vaccinated with both formulations exhibited significantly elevated frequencies of pulmonary DN T cells producing IFN-γ relative to frequencies in nonvaccinated mice (Fig. 6B). Interestingly, the frequency of IFN-γ-producing DN T cells in the A4/Adj group (2.75% ± 0.2%) was significantly elevated relative to the frequency of DN T cells recovered from mice vaccinated with nonadjuvanted BCG-A4 (1.87% ± 0.32%).
FIG 6.

DDA/TDB adjuvant enhanced the frequency of DN T cells in the lungs and of pulmonary DN T cells secreting IFN-γ post-aerosol M. tuberculosis infection. Mice were treated with antibodies to deplete CD4+, CD8+, and NK1.1+ T cells before receiving an aerosol challenge with M. tuberculosis Erdman. Lung cells from nonvaccinated mice or mice vaccinated with the BCGΔmmaA4 mutant (BCG-A4) with or without formulation with DDA/TDB adjuvant (Adj) were isolated 10 days postchallenge. The frequency of DN T cells recovered from the lung (A) or secretion of IFN-γ (B) was measured by flow cytometry. Means and SD were determined from 3 mice per group from one experiment. *, significant differences relative to nonvaccinated mice (P < 0.05). #, significant differences relative to BCG-A4 results (P < 0.05).
To further assess the impact of immunization with the A4/Adj vaccine, we determined the differences between nonvaccinated mice and mice vaccinated with either BCG-A4 or A4/Adj with respect to integrated median fluorescent intensity (iMFI) values. The IFN-γ iMFI is a metric for evaluating the total response of an IFN-γ-producing population and is calculated by multiplying the frequency of cytokine expressing cells by the MFI (11). As shown in Fig. 7, the IFN-γ iMFI values for both nonadjuvanted BCG-A4 (1,790 ± 409) and A4/Adj (2,729 ± 350) mice were significantly elevated relative to those seen with nonvaccinated mice (597 ± 208). Moreover, the iMFI for A4/Adj was also significantly elevated relative to that determined for BCG-A4 alone. These data indicate that the DDA/TDB adjuvant not only enhanced the frequency of DN T cells in the lungs early after an aerosol infection with M. tuberculosis but also enhanced BCG's capacity to induce antigen-specific pulmonary T cells and also improved the ability of the BCGΔmmaA4 strain to evoke increased IFN-γ responses from these DN T cells.
FIG 7.
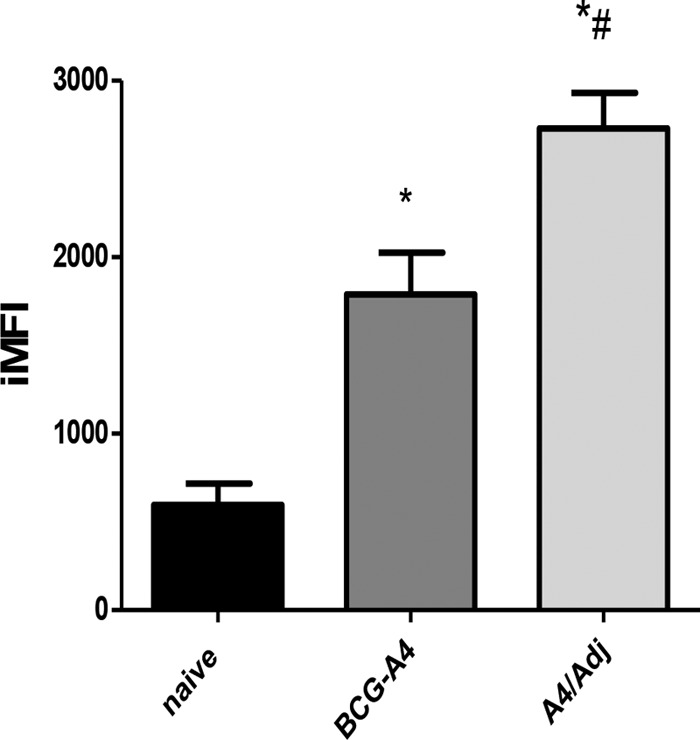
DDA/TDB adjuvant significantly increased BCG-A4-mediated IFN-γ secretion from pulmonary DN T cells. Mice were treated with antibodies to deplete CD4+, CD8+, and NK1.1+ T cells before receiving an aerosol challenge with M. tuberculosis Erdman. Lung cells were isolated from nonvaccinated mice or mice vaccinated with the BCGΔmmaA4 mutant (BCG-A4) or A4/Adj 10 days postchallenge. The frequency and MFI values of pulmonary DN T cells secreting IFN-γ were measured by flow cytometry. The iMFI is the integrated median fluorescence intensity, which is calculated by multiplying the frequency of cells producing IFN-γ by the MFI value. Means and SD were calculated from 3 mice per group from one experiment. *, significant differences relative to nonvaccinated mice (P < 0.05). #, significant differences relative to BCG-A4-immunized mice (P < 0.05).
Vaccine-induced unconventional T cells restrict in vitro mycobacterial growth.
To determine whether DN T cells could mediate inhibition of mycobacterial growth, an in vitro coculture assay was utilized. This growth inhibition assay has been used extensively to investigate the cellular immunity associated with mycobacterial and Francisella infections. Initially, enriched DN T cell populations recovered from mice vaccinated with A4/Adj and treated with the Ab Combo were cocultured with BCG-infected bone marrow-derived macrophages (BMMΦ). Inhibition of mycobacterial growth was measured by quantifying the CFU in the DN T cell coculture following a 7-day incubation period. As controls, whole splenocyte preparations from nonvaccinated or A4/Adj mice were overlaid onto BCG-infected BMMΦ. As shown in Fig. 8, splenocytes or DN T cells from A4/Adj-immunized mice showed mycobacterial CFU levels that were significantly reduced by 0.5 or 0.45 log10 CFU, respectively, relative to splenocytes from nonimmunized mice. Importantly, the level of growth inhibition detected in the A4/Adj-induced DN T cell coculture (−0.45 log10 CFU) was similar to the results seen in assays using BCG immune splenocytes.
FIG 8.
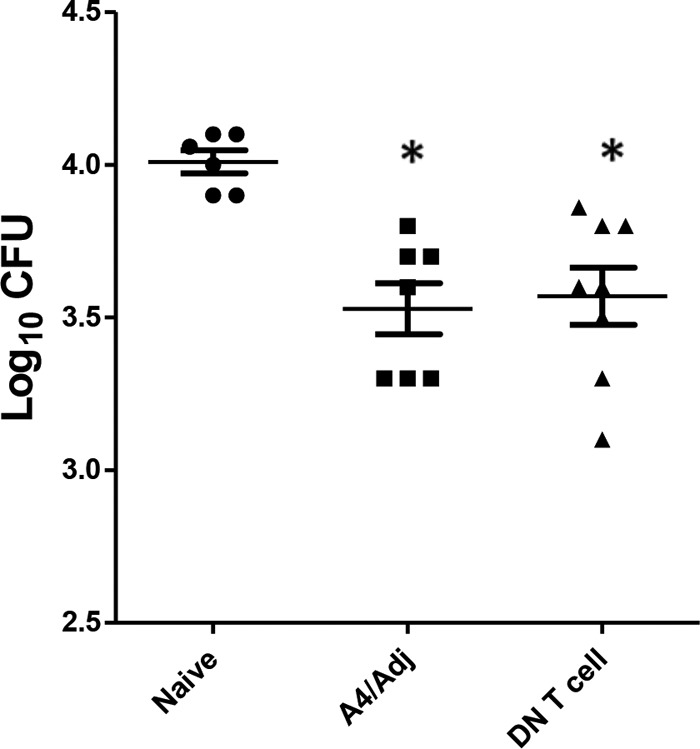
DN T cells significantly inhibited mycobacterial growth in vitro relative to nonvaccinated splenocytes. DN T cells enriched from the spleens of mice vaccinated with A4/Adj or splenocytes from nonvaccinated or immunized mice were cocultured with BCG-infected bone marrow-derived macrophages for 7 days, after which the cells were lysed and plated onto mycobacterial growth media. Mycobacterial CFU levels were quantified after a 2- to 3-week incubation. Means and SD are from three independent experiments. *, significant differences relative to nonvaccinated mice (P < 0.05).
DISCUSSION
To model TB vaccine protection in the context of severe immunodeficiency, we examined the protective efficacy of a BCGΔmmaA4 mutant with or without formulation in DDA/TDB adjuvant in immunocompromised mice. This mutant strain has been shown to enhance IL-12 secretion from infected macrophages (12). Furthermore, we previously showed that formulating this BCG mutant with DDA/TDB adjuvant improved protection against an M. tuberculosis pulmonary infection relative to that with nonadjuvanted BCG in immunocompetent mice (11). Our previous studies suggested that the enhanced protection of the A4/Adj vaccine may partially result from increased targeting of the vaccine to relevant lymph nodes and the subsequent amplified antigen-specific multifunctional CD4+ T cell responses. In this study, vaccination with the adjuvanted BCG mutant significantly protected both immunocompetent mice and TCRδ−/− mice depleted of CD4+, CD8+, and NK1.1+ T cells against an aerosol challenge with M. tuberculosis relative to results for nonvaccinated controls. Furthermore, we showed that immunization with the A4/Adj vaccine formulation elicited significantly increased antituberculosis protective responses relative to that with a nonadjuvanted BCG-A4 preparation in these severely immunocompromised mice. The protective immune responses persisted 9 months after vaccination, indicating that the A4/Adj formulation induced long-lived memory cells. The results of these cell depletion vaccination challenge experiments suggest that the A4/Adj formulation induced DN T cells that appear to be involved in protection in the absence of conventional T cells. In an earlier study, we demonstrated that a live attenuated TB strain also protected mice in the absence of conventional T cells by induction of DN T cells (14).
To gain a clearer understanding of the role of DN T cells in vaccine-mediated protection, we examined the frequency of these cells in the lungs of mice postchallenge. We found that the frequency of these cells in the lungs was significantly elevated in mice immunized with A4/Adj relative to their frequency in nonvaccinated mice 10 days after an aerosol challenge, with a significantly greater number of DN T cells secreting IFN-γ in the adjuvanted vaccine group than in nonvaccinated and BCG-A4-immunized mice. Not only were there significantly higher frequencies of cytokine-producing pulmonary DN T cells, we observed significantly greater iMFI values associated with this T cell population from the A4/Adj vaccine group than from the nonvaccinated mice and the nonadjuvanted BCG-A4 group. Importantly, we also found that DN T cells from A4/Adj-immunized mice significantly restricted intramacrophage mycobacterial growth in vitro relative to splenocytes from nonvaccinated mice. These findings suggest that the DN T cells were important for protecting the vaccinated, immunocompromised mice and that the adjuvant not only led to an enrichment of these cells in the lungs but also enhanced their ability to produce IFN-γ after an aerosol challenge with M. tuberculosis.
DN T cells represent an interesting mixed population of Th1-like T cells (1% to 3%) in mice and humans that have been difficult to isolate and study because of an absence of specific phenotypic markers. It has been proposed that DN T cells originate in the thymus by escaping negative selection and, following migration to the periphery, expand in number after encountering antigen. Several potential roles for DN T cells in host immunity have been proposed. For example, DN T cells can be involved in the development of tolerance to transplantation and may play a homeostatic role in autoimmune disease (18). Although CD4+ and CD8+ T cells are considered the primary mediators of protective immunity against microbial pathogens, recent studies have indicated that DN T cells can contribute to the control of pathogen growth. We previously demonstrated that DN T cells could potently and specifically inhibit the proliferation of F. tularensis and M. tuberculosis in in vitro cultures and promote the survival of mice infected with these organisms in vivo (19). Furthermore, Stenger et al. showed that a human CD1-restricted DN T cell line lysed TB-infected macrophages and that the results were fatty acid synthase (FAS) dependent (20), and Pinheiro et al. observed high frequencies of DN T cells expressing activation markers (HLA-DR and CD69) in the PBMCs of TB-infected patients (21). In a study conducted by Zufferey and others, DN T cells from PBMCs isolated from infants and children 10 weeks post-BCG vaccination were examined (22). After stimulation with BCG, CD4+ and DN T cells were found to be the major IFN-γ-producing cells, and the iMFI values for the DN T cells was higher than those observed for CD4+ T cells. Additionally, Mou et al. have shown that major histocompatibility complex (MHC) class II-restricted DN T cells contribute to optimal immunity against Leishmania major in mice (23). For simian immunodeficiency virus (SIV) infections, Sundaravaradan and colleagues reported that multifunctional DN T cells mediated a T helper cell function during SIV infection of mangabeys; these animals remained healthy despite high viremia and CD4+ T cell depletion (24). While the role of DN T cells in protecting humans against microbial pathogens is less clear, DN T cell populations have been shown to expand during staphylococcal and mycobacterial infections (21, 25). The percentage of DN T cells has also been shown to be increased during HIV disease, especially in people with low numbers of CD4+ T cells and in HIV patients with M. avium infections (26–28). Given their potential role in contributing to the host immune response to intracellular pathogens, vaccines that amplify DN T cell responses (such as the A4/Adj preparation) may be beneficial for controlling and protecting against intracellular infections, particularly in the context of T cell immunodeficiency.
The immune mechanisms associated with the induction of DN T cells by the A4/Adj vaccine are currently being investigated. Earlier studies had clearly shown that the DDA/TDB mixture induces potent Th1 cellular responses and augments BCG-mediated Th1 immune responses (9, 11). More specifically, it has been suggested that DDA cationic liposomes can create depot-like effects where antigen is slowly released over time and may act as carriers to deliver antigens to antigen-presenting cells (29). The TDB component, a synthetic analog of the mycobacterial cord factor, is an immunostimulant which can enhance T cell differentiation and increase migration of monocytes to the injection site and draining to lymph nodes, suggesting that the adjuvant enhances both cell-mediated and innate immune responses (29). Thus, it is likely that innate immune responses contribute to the enhanced protection associated with the adjuvant. It is of interest that, in humans, mycobacterial lipid extracts can stimulate CD1-restricted DN T cells (30).
An increasing number of studies have shown memory-like T and B cell-independent cross-protection between infections, indicating a heterologous memory mechanism for the innate immune system. This process of trained immunity has been seen when innate immune cells such as monocytes, macrophages, and NK cells provide protection against infections independently of the presence of lymphocytes (31). Recently, trained immunity was suggested as a mechanism of early clearance of TB infection by innate immunity (32); therefore, the innate immune response may play a larger role in protection against TB infection than has been previously appreciated. It has been recognized that BCG immunization decreases overall childhood mortality from non-TB infections, animal studies have shown that BCG immunization conferred partial protection against Babesia microti, Plasmodium berghei, and Toxoplasma gondii, and immunization of SCID mice with BCG was demonstrated to protect against Candida albicans (32, 33). Although our findings showed that the majority of protection seen in immunodeficient mice was mediated by T cells, we did observe a significant reduction in CFU in (Thy1.2 antibody-treated) A4/Adj-immunized mice depleted of all T cells relative to depleted naive controls (0.7 log reduction), suggesting that an innate immune component was likely contributing to protection in those severely immunocompromised mice.
Studies in several animal models have shown that formulating the BCG vaccine in adjuvant enhances its capacity to induce antituberculosis protective immunity (6–11). Although the biological reasons for the enhanced immunogenicity of lipid-encapsulated BCG have not been fully defined, recent reports have suggested possible mechanisms for the increased vaccine-induced immunity. Formulating BCG in adjuvant seems to increase targeting of the vaccine to relevant lymph nodes (29). We recently showed that elevated T cell responses were detected within the lymph nodes of mice vaccinated with adjuvanted BCG relative to those vaccinated with nonadjuvanted BCG (34). Also, formulating BCG with lipid-containing adjuvants increases splenic antigen-specific multifunctional CD4+ T cell responses, including the central memory cells producing tumor necrosis factor alpha (TNF-α) and IL-2 (11). This finding is important because, although BCG is effective at inducing effector memory cells, it is relatively ineffective at evoking the more persistent faster-acting central memory cells (35).
In addition to formulation of BCG in adjuvant, different strategies to improve BCG have been explored in preclinical studies and many are now being investigated in the clinic. These include constructing recombinant BCG strains overexpressing M. tuberculosis antigens or lysins to allow BCG to escape phagocytic endosomes (36). Once in the cytoplasm, induction of apoptosis is followed by cross-presentation of mycobacterial antigens, allowing more-efficient priming of T cells. Synthetic long peptides, which have been shown to induce antitumor immunity, were also shown to protect against TB in mice after both prophylactic and therapeutic immunizations and augmented BCG-induced protection (37). Others found that a recombinant BCG (rBCG) strain overexpressing a TLR2 binding protein, PPE57, was able to protect better than wild-type BCG following an intravenous (i.v.) challenge with M. tuberculosis (38). Another potential strategy to augment induction of T cells by vaccination was described by Pere et al. (39). Those authors showed that vaccine-mediated protection against tumors in mice was augmented when regulatory T (TReg) cells were suppressed by inclusion of a CCR4 antagonist with the vaccine. It would be interesting to explore this strategy in the context of immunization against intracellular pathogens and, in particular, to determine if BCG protective immune responses may be more easily boosted in the absence of TReg cells.
In this study, we extended earlier findings about the immunogenicity of BCG/adjuvant formulations by demonstrating that an adjuvanted BCG vaccine can stimulate antituberculosis responses from DN T cells. Our data suggest that the vaccine-induced protection we observed in severely immunocompromised mice against an aerogenic TB challenge likely involved the activation of DN T cells. These data indicate that future TB vaccines should be designed to stimulate DN and other unconventional T cells in addition to CD4+ and CD8+ T cells. Overall, the validation of the antituberculosis protective capacity of unconventional T cells from this study should impact the future development of TB vaccines, especially those vaccines designed for use in immunocompromised individuals.
Funding Statement
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Pitt JM, Blankley S, McShane H, O'Garra A. 2013. Vaccination against tuberculosis: how can we better BCG? Microb Pathog 58:2–16. doi: 10.1016/j.micpath.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. 2015. Tuberculosis fact sheet no. 104. World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs104/en. [Google Scholar]
- 3.Shenoi S, Friedland G. 2009. Extensively drug-resistant tuberculosis: a new face to an old pathogen. Annu Rev Med 60:307–320. doi: 10.1146/annurev.med.60.053107.103955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodrigues LC, Diwan VK, Wheeler JG. 1993. Protective effect of BCG against tuberculous meningitis and miliary tuberculosis: a meta-analysis. Int J Epidemiol 22:1154–1158. doi: 10.1093/ije/22.6.1154. [DOI] [PubMed] [Google Scholar]
- 5.Fine PE. 2001. BCG: the challenge continues. Scand J Infect Dis 33:243–245. doi: 10.1080/003655401300077144. [DOI] [PubMed] [Google Scholar]
- 6.Aldwell FE, Brandt L, Fitzpatrick C, Orme IM. 2005. Mice fed lipid-encapsulated Mycobacterium bovis BCG are protected against aerosol challenge with Mycobacterium tuberculosis. Infect Immun 73:1903–1905. doi: 10.1128/IAI.73.3.1903-1905.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buddle BM, Aldwell FE, Skinner MA, de Lisle GW, Denis M, Vordermeier HM, Hewinson RG, Wedlock DN. 2005. Effect of oral vaccination of cattle with lipid-formulated BCG on immune responses and protection against bovine tuberculosis. Vaccine 23:3581–3589. doi: 10.1016/j.vaccine.2005.01.150. [DOI] [PubMed] [Google Scholar]
- 8.Aldwell FE, Cross ML, Fitzpatrick CE, Lambeth MR, de Lisle GW, Buddle BM. 2006. Oral delivery of lipid-encapsulated Mycobacterium bovis BCG extends survival of the bacillus in vivo and induces a long-term protective immune response against tuberculosis. Vaccine 24:2071–2078. doi: 10.1016/j.vaccine.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 9.Dietrich J, Billeskov R, Doherty TM, Andersen P. 2007. Synergistic effect of bacillus calmette Guerin and a tuberculosis subunit vaccine in cationic liposomes: increased immunogenicity and protection. J Immunol 178:3721–3730. doi: 10.4049/jimmunol.178.6.3721. [DOI] [PubMed] [Google Scholar]
- 10.Clark S, Cross ML, Nadian A, Vipond J, Court P, William A, Hewinson RG, Aldwell FE, Chambers MA. 2008. Oral vaccination of guinea pigs with Mycobacterium bovis bacillus Calmette-Guerin vaccine in a lipid matrix protects against aerosol infection with virulent M. bovis. Infect Immun 76:3771–3776. doi: 10.1128/IAI.00052-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Derrick SC, Dao D, Yang A, Kolibab K, Jacobs WR, Morris SL. 2012. Formulation of a mmaA4 gene deletion mutant of Mycobacterium bovis BCG in cationic liposomes significantly enhances protection against tuberculosis. PLoS One 7:e32959. doi: 10.1371/journal.pone.0032959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dao DN, Sweeney K, Hsu T, Gurcha SS, Nascimento IP, Roshevsky D, Besra GS, Chan J, Porcelli SA, Jacobs WR. 2008. Mycolic acid modification by the mmaA4 gene of M. tuberculosis modulates IL-12 production. PLoS Pathog 4:e1000081. doi: 10.1371/journal.ppat.1000081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sambandamurthy VK, Derrick SC, Hsu T, Chen B, Larsen MH, Jalapathy KV, Chen M, Kim J, Porcelli SA, Chan J, Morris SL, Jacobs WR Jr. 2006. Mycobacterium tuberculosis DeltaRD1 DeltapanCD: a safe and limited replicating mutant strain that protects immunocompetent and immunocompromised mice against experimental tuberculosis. Vaccine 24:6309–6320. doi: 10.1016/j.vaccine.2006.05.097. [DOI] [PubMed] [Google Scholar]
- 14.Derrick SC, Evering TH, Sambandamurthy VK, Jalapathy KV, Hsu T, Chen B, Chen M, Russell RG, Junqueira-Kipnis AP, Orme IM, Porcelli SA, Jacobs WR Jr, Morris SL. 2007. Characterization of the protective T-cell response generated in CD4-deficient mice by a live attenuated Mycobacterium tuberculosis vaccine. Immunology 120:192–206. doi: 10.1111/j.1365-2567.2006.02491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Derrick SC, Yabe IM, Yang A, Morris SL. 2011. Vaccine-induced anti-tuberculosis protective immunity in mice correlates with the magnitude and quality of multifunctional CD4 T cells. Vaccine 29:2902–2909. doi: 10.1016/j.vaccine.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 16.Kolibab K, Parra M, Yang AL, Perera LP, Derrick SC, Morris SL. 2009. A practical in vitro growth inhibition assay for the evaluation of TB vaccines. Vaccine 28:317–322. doi: 10.1016/j.vaccine.2009.10.047. [DOI] [PubMed] [Google Scholar]
- 17.Lim J, Derrick SC, Kolibab K, Yang AL, Porcelli S, Jacobs WR, Morris SL. 2009. Early pulmonary cytokine and chemokine responses in mice immunized with three different vaccines against Mycobacterium tuberculosis determined by PCR array. Clin Vaccine Immunol 16:122–126. doi: 10.1128/CVI.00359-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hillhouse EE, Delisle JS, Lesage S. 2013. Immunoregulatory CD4(-)CD8(-) T cells as a potential therapeutic tool for transplantation, autoimmunity, and cancer. Front Immunol 4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cowley SC, Hamilton E, Frelinger JA, Su J, Forman J, Elkin KL. 2005. CD4− CD8− T cells control intracellular bacterial infections both in vitro and in vivo. J Exp Med 202:309–319. doi: 10.1084/jem.20050569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stenger S, Mazzaccaro RJ, Uyemura K, Cho S, Barnes PF, Rosat J-P, Sette A, Brenner MB, Porcelli SA, Bloom BR, Modlin RL. 1997. Differential effects of cytolytic T cell subsets on intracellular infection. Science 276:1684–1687. doi: 10.1126/science.276.5319.1684. [DOI] [PubMed] [Google Scholar]
- 21.Pinheiro MB, Antonelli LR, Sathler-Avelar R, Vitelli-Avelar DM, Spindola-de-Miranda S. 2012. CD4-CD8-αβ and γδ T cells display inflammatory and regulatory potentials during human tuberculosis. PLoS One 7:e50923. doi: 10.1371/journal.pone.0050923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zufferey C, Germano S, Dutta B, Ritz N, Curtis N. 2013. The contribution of non-conventional T cells and NK cells in the mycobacterial-specific IFNγ response in Bacille Calmette-Guerin (BCG)-immunized infants. PLoS One 8:e77334. doi: 10.1371/journal.pone.0077334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mou Z, Liu D, Okwor I, Jia P, Orihara K, Uzonna JE. 2014. MHC class II restricted innate-like double negative T cells contribute to optimal primary and secondary immunity to Leishmania major. PLoS Pathog 10:e1004396. doi: 10.1371/journal.ppat.1004396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sundaravaradan V, Seleem R, Micci L, Gasper MA, Ortiz AM, Else J, Silvestri G, Paiardini M, Aitchison JD, Sodora DL. 2013. Multifunctional double-negative T cells in sooty mangabeys mediate T-helper functions irrespective of SIV infection. PLoS Pathog 9:e1003441. doi: 10.1371/journal.ppat.1003441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carulli G, Lagomarsini G, Azzara A, Testi R, Riccioni R, Petrini M. 2004. Expansion of TcRalphabeta+CD3+CD4-CD8- (CD4/CD8 double-negative) T lymphocytes in a case of staphylococcal toxic shock syndrome. Acta Haematol 111:163–167. doi: 10.1159/000076526. [DOI] [PubMed] [Google Scholar]
- 26.Mathiot ND, Keueger R, French MA, Price P. 2001. Percentage of CD3+CD4-CD8-γδTCR- T cells is increased by HIV disease. AIDS Res Hum Retroviruses 17:977–980. doi: 10.1089/088922201750290096. [DOI] [PubMed] [Google Scholar]
- 27.Margolick JB, Carey V, Munoz A, Polk BF, Giorgi JV, Gauer KD, Kaslow R, Rinaldo C. 1989. Development of antibodies to HIV-1 is associated with an increase in circulating CD3+CD4-CD8- lymphocytes. Clin Immunol Immunopathol 51:348–361. doi: 10.1016/0090-1229(89)90033-0. [DOI] [PubMed] [Google Scholar]
- 28.Moreau J-F, Taupin J-L, Dupon M, Carron J-C, Ragnaud J-M, Marimoutou C, Bernard N, Constans J, Texier-Maugein J, Barbeau P, Journot V, Dabis F, Bonneville M, Pellegrin JL. 1996. Increases in CD3+CD4-CD8- T lymphocytes in AIDS patients with disseminated Mycobacterium avium-intracellulare complex infection. J Infect Dis 174:969–976. doi: 10.1093/infdis/174.5.969. [DOI] [PubMed] [Google Scholar]
- 29.Henriksen-Lacey M, Christensen D, Bramwell VW, Lindenstrom T, Agger EM, Andersen P, Perrie Y. 2010. Liposomal cationic charge and antigen adsorption are important properties for the efficient deposition of antigen at the injection site and ability of the vaccine to induce a CMI response. J Control Release 145:102–108. doi: 10.1016/j.jconrel.2010.03.027. [DOI] [PubMed] [Google Scholar]
- 30.Moody DB, Porcelli SA. 2003. Intracellular pathways of CD1 antigen presentation. Nat Rev Immunol 3:11–22. doi: 10.1038/nri979. [DOI] [PubMed] [Google Scholar]
- 31.Blok BA, Arts RJW, van Crevel R, Benn CS, Netea MG. 2015. Trained innate immunity as underlying mechanism for the long-term, nonspecific effects of vaccines. J Leukoc Biol 98:347–356. doi: 10.1189/jlb.5RI0315-096R. [DOI] [PubMed] [Google Scholar]
- 32.Lerm M, Netea MG. 24 November 2015. Trained immunity: a new avenue for tuberculosis vaccine development. J Intern Med doi: 10.1111/joim.12449. [DOI] [PubMed] [Google Scholar]
- 33.Parra M, Liu X, Derrick SC, Yang A, Tian J, Kolibab K, Kumar S, Morris SL. 2013. Molecular analysis of non-specific protection against murine malaria induced by BCG vaccination. PLoS One 8:e66115. doi: 10.1371/journal.pone.0066115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Derrick SC, Yang A, Parra M, Kolibab K, Morris SL. 2015. Effect of cationic liposomes on BCG trafficking and vaccine-induced immune responses following a subcutaneous immunization in mice. Vaccine 33:126–132. doi: 10.1016/j.vaccine.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 35.Orme IM. 2010. The Achilles heel of BCG. Tuberculosis 90:329–332. doi: 10.1016/j.tube.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 36.Orme IM. 2015. Tuberculosis vaccine types and timings. Clin Vaccine Immunol 22:249–257. doi: 10.1128/CVI.00718-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coppola M, van den Eeden SJ, Wilson L, Franken KL, Ottenhoff TH, Geluk A. 2015. Synthetic long peptide derived from Mycobacterium tuberculosis latency antigen Rv1733c protects against tuberculosis. Clin Vaccine Immunol 22:1060–1069. doi: 10.1128/CVI.00271-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu Y, Yang E, Huang Q, Ni W, Kong C, Liu G, Li G, Su H, Wang H. 2015. PPE57 induces activation of macrophages and drives Th1-type immune responses through TLR2. J Mol Med 93:645–662. doi: 10.1007/s00109-014-1243-1. [DOI] [PubMed] [Google Scholar]
- 39.Pere H, Montier Y, Bayry J, Quintin-Colonna F, Merillon N, Dransart E, Badoual C, Gey A, Ravel P, Marcheteau E, Batteux F, Sandoval F, Adotevi O, Chiu C, Garcia S, Tanchot C, Lone Y-C, Ferreira LC, Nelson BH, Hanahan D, Fridman WH, Johannes L, Tartour E. 2011. A CCR4 antagonist combined with vaccines induces antigen-specific CD8+ T cells and tumor immunity against self antigens. Blood 118:4853–4862. doi: 10.1182/blood-2011-01-329656. [DOI] [PubMed] [Google Scholar]