

ABSTRACT
Mitochondria are associated with various radiation responses, including adaptive responses, mitophagy, the bystander effect, genomic instability, and apoptosis. We recently identified a unique radiation response in the mitochondria of human cells exposed to low-dose long-term fractionated radiation (FR). Such repeated radiation exposure inflicts chronic oxidative stresses on irradiated cells via the continuous release of mitochondrial reactive oxygen species (ROS) and decrease in cellular levels of the antioxidant glutathione. ROS-induced oxidative mitochondrial DNA (mtDNA) damage generates mutations upon DNA replication. Therefore, mtDNA mutation and dysfunction can be used as markers to assess the effects of low-dose radiation. In this study, we present an overview of the link between mitochondrial ROS and cell cycle perturbation associated with the genomic instability of low-dose irradiated cells. Excess mitochondrial ROS perturb AKT/cyclin D1 cell cycle signaling via oxidative inactivation of protein phosphatase 2A after low-dose long-term FR. The resulting abnormal nuclear accumulation of cyclin D1 induces genomic instability in low-dose irradiated cells.
KEYWORDS: cell cycle perturbation, damage response, genomic instability, low-dose radiation, mitochondria
Introduction
The main target of ionizing radiation (IR) is thought to be the nuclear DNA (nDNA) in the cell nucleus. Double strand breaks (DSBs) in nDNA generated by IR give rise to chromosomal aberrations. In response to genotoxic stress, mammalian cells activate cellular defense systems, including cell cycle checkpoints, apoptosis, and DNA repair mechanisms.1,2 Research has therefore focused on the effect of radiation on nDNA to elucidate DNA damage responses (DDR) in mammalian cells. Radiation also affects cell organelles, such as the plasma membrane, cytoskeleton, mitochondria, endoplasmic reticulum, Golgi apparatus, and lysosomes.3-6 Direct evidence for the effect of IR on the cytoplasmic structures has been obtained as bystander effects after cytoplasmic irradiation with α particles.7,8 Mitochondria contain their own DNA and can be directly damaged by IR. Although the whole mitochondrial genome, apart from the D-loop control region, consists of genes, only approximately 1% of nDNA encodes genes. Thus, mitochondrial DNA (mtDNA) mutations are more likely to cause functional loss than nDNA mutations. Hence, it is of special importance to maintain mtDNA integrity, particularly under oxidative stress conditions. Mitochondria, along with the nucleus, are therefore likely to be a major target of IR.6
Mitochondria regulate energy supply and are shown to be a source of endogenous reactive oxygen species (ROS) generation through oxidative phosphorylation.9 ROS function as a second messenger of intracellular signaling pathways for physiological processes10-12 by modifying the cysteine residues within redox-sensitive target proteins leading to reversible modification of enzymatic activity.13,14 However, ROS accumulation at high levels inflicts oxidative damage on cellular components, such as nucleic acids, proteins, and lipids, and inhibits cell proliferation.15 IR triggers genomic instability, a hallmark of cancer in irradiated cells as the late effect of IR.16-18 Changes in the pattern of DNA methylation are associated with cancerogenesis together with genetic mutations.19,20 Mitochondrial ROS induce genomic instability in irradiated cells.21-23 To control redox balance, mitochondria contain antioxidants, such as glutathione (GSH) and manganese superoxide dismutase (MnSOD), which scavenge ROS.24 KRIT1, a gene responsible for cerebral cavernous malformations regulates antioxidant pathway involving FoxO1 and MnSOD to maintain the homeostasis of intracellular ROS.25 Dismutation of superoxide anions in the mitochondria forms H2O2, either spontaneously or through the catalytic function of MnSOD, to maintain redox homeostasis. GSH peroxidases then further reduce H2O2 to water using GSH as a ROS receptor.
Mitochondria regulate apoptosis after high-dose IR in normal cells. Similarly, apoptosis was induced in radiosensitive ATM-deficient cells after low-dose long-term FR.26 In contrast, the same low-dose long-term FR activates mitochondrial function and causes chronic oxidative stresses due to elevated mitochondrial ROS in normal and complemented cells expressing ATM. So, there are different modes of mitochondrial radiation response according to intrinsic radiation sensitivity of the irradiated cells.26 This review is focused on our current understanding of a unique radiation response of mitochondria upon repeated low-dose IR. Mitochondrial ROS perturb cell cycle signaling and this is concomitant with genomic instability in human cells after low-dose long-term FR.
Radiation responses of mitochondria
The radiation responses of mitochondria are well reviewed in other study6 and are summarized in Figure 1. IR increases mtDNA copy number in mammalian cells in vitro and in vivo.6 IR stimulates mitochondrial enzyme activity and gene expression to supply energy for radiation responses.6,27,28 Low-dose IR changes the dynamics of mitochondrial morphology and induces mitochondrial fusion to protect rat neurons.29 Mitochondrial fusion enables content mixing within a mitochondrial population including both damaged and healthy mitochondria to protect against mitochondrial dysfunction.30-32 On the other hand, mitophagy refers to the selective removal of damaged mitochondria by autophagy to control mitochondrial quality.33,34 The E3 ubiquitin ligase Parkin recognizes damaged mitochondria and promotes their clearance by mitophagy.35,36 Mitochondria are also associated with the non-targeted effects of radiation, including the adaptive response, the bystander effect, and genomic instability.21,22 Mitochondrial localization of MnSOD is required for radioprotection.37-39 IR at low doses induces NF-kappaB-mediated activation of MnSOD as a signaling regulator of cell survival pathways in low-dose IR-induced adaptive responses in mammalian cells.40,41 Constitutive active AKT is associated with induction of NF-kappaB signaling after chronic low-dose IR.41 Mitochondria-dependent NF-kappaB signaling pathways also implicates in the regulation of radiation-induced bystander.42 In contrast, IR at high doses results in the breakdown of the mitochondrial membrane potential, opening of the permeability transition pore (PTP), and release of cytochrome c for the induction of apoptosis in irradiated cells (Fig. 1, upper panel).43
Figure 1.

Radiation response of mitochondria The upper panel shows the difference in the radiation response of mitochondria according to the radiation dose after acute single radiation. The lower panel shows the mitochondrial radiation response to low-dose long-term fractionated radiation.
Although the health risks associated with low-dose radiation are currently under intensive investigation, the influences of low-dose long-term radiation remain unclear because of a lack of sufficient studies. We recently reported that low-dose long-term FR induces mitochondria-mediated oxidative stresses by increasing the generation of mitochondrial ROS in human cells (Fig. 1, lower panel).44 Thus, mitochondrial radiation responses change according to the radiation dose, duration of radiation exposure. The antioxidant GSH protects cells against oxygen toxicity mediated by mitochondrial ROS.44 However, GSH becomes exhausted after repeated low-dose IR. The accumulation of mitochondrial ROS owing to a GSH deficiency would activate oxidative stress responses and DDR over a prolonged period (Fig. 1, lower panel).
The role of ATM on radiation response of mitochondria
In ataxia-telangiectasia (AT), a disease characterized by high levels of radiosensitivity and neurodegeneration, ATM is mutated.45 ATM is a damage sensor kinase is essential for maintaining genome stability in response to various stresses. It has also been identified as a redox sensor and is activated by oxidization at a cysteine residue independent of DSBs under oxidative stress.46,47 Lower antioxidative capacity is reported in AT patients.48 Biogenesis of mitochondria in response to IR was investigated in radiosensitive human ATM-deficient cells compared with that in ATM-complemented cells. Consistent with the results in normal fibroblasts, low-dose long-term FR stimulated mitochondrial biogenesis with elevated ROS levels in ATM-complemented cells. In contrast, mitochondrial biogenesis was not triggered by the same radiation treatments in ATM-deficient cells.26 Thus, ATM is required for the radiation response of mitochondria. In response to DSB, ATM is shown to phosphorylate AMP-activated protein kinase (AMPK), which senses the cellular AMP/ATP ratio to induce mitochondrial biogenesis.49 ATM is associated with the induction of mitophagy in response to IR and oxidative stresses.50 ATM loss leads to defective mitophagy and increased frequency of abnormal mitochondria with decreased mitochondrial membrane potential.50 Severe mitochondrial damage, assessed from mitochondrial fragmentation, was induced by low-dose long-term FR in ATM-deficient cells, which showed highly radiosensitive phenotypes when subjected to low-dose long-term FR; cell death was through mitochondria-mediated apoptosis.26 Consequently, it became clear that the mitochondrial radiation response influences radiation sensitivity in human cells.
Mitochondria as target organelles for low-dose radiation
mtDNA is located at the inner mitochondrial membrane close to the sites of ROS production via the electron transport chain and suffers more oxidative damage than nDNA.51,52 Because mtDNA lacks histone protection and the efficient DNA repair system of nDNA, mtDNA damage due to IR is thought to be more extensive and persistent over time than nDNA damage. Mitochondria harbor base excision repair, but nucleotide excision repair is absent.53 DNA polymerase γ (Pol γ) is the enzyme for replication and repair of mtDNA. Although Pol γ exhibits high base substitution fidelity with exonucleolytic proofreading,54 it has low frameshift fidelity for repetitive sequences longer than 4 nucleotides.55 ROS-induced DNA lesions, such as abasic sites and single- and double-strand breaks, generate mutations upon DNA replication. mtDNA mutations induced by ROS-mediated oxidative modifications lead to progressive electron transport chain dysfunction and further increases in ROS production.56-58 IR permanently impairs mitochondria by induction of mtDNA mutations, leading to overproduction of mitochondrial ROS, which are implicated in many toxicities and disease processes as mediators of tissue injury.15 mtDNA mutations are frequently observed in various human cancers.59-62 Mutations in the nuclear-encoded mitochondrial gene have also been correlated with an increased cancer risk.63,64 mtDNA validation affects the radiotherapy and chemotherapy outcomes in cancer patients.63 Metabolic alterations associated with mitochondrial dysfunction increase tumorigenesis. Mitochondrial genomic instability is closely related to vascular disease, neurodegeneration, aging, and carcinogenesis.21,22,65,66 Therefore, mtDNA mutation and mitochondrial dysfunction can be used as markers to assess the effect of low-dose long-term FR. N-acetyl-cysteine, a glutathione precursor, increases the intracellular levels of GSH67 and suppresses the accumulation of mitochondrial ROS.44 Thus, increasing antioxidant capacity can prevent radiation toxicity induced by low-dose long-term FR.
Cyclin D1 as the molecular target associated with mitochondrial ROS-mediated genomic instability
Mitochondrial dysfunction can be communicated to the cell nucleus via mitochondrial ROS acting as signaling molecules. Elevated ROS generation stimulates stress-activated kinases and stress-signaling in cancer cells.68 Thus, ROS have multiple roles in tumor initiation, progression, and maintenance. We recently identified a target molecule associated with mitochondrial ROS-induced genomic instability in low-dose long-term FR cells.44 Figure 2 depicts a link between mitochondrial ROS and cell cycle perturbation in low-dose irradiated human cells. ROS damage to the molecules affects cell cycling, oxidizing PP2A on cysteine residues, and downregulating PP2A activity in long-term FR cells. This loss of PP2A activity can thus lead to a loss of negative feedback control of the AKT pathway,69 resulting in persistent AKT activity in cells after long-term FR. Consequently, constitutive AKT activation causes stabilization of nuclear cyclin D1 by inhibiting the nuclear export and subsequent GSK3β-mediated degradation of cyclin D1 (Fig. 2).70,71 Nuclear cyclin D1 accumulation was observed by low-dose long-term FR in most of PCNA-positive S-phase cells.70,71 Abnormal nuclear cyclin D1 accumulation during the S phase perturbs DNA replication including DNA re-replication and suppression of replication fork progression leading to DSBs.72,73 Perturbation of cyclin D1 expression is associated with cellular senescence and induction of genomic instability in irradiated cells.70,72,74,75 Aberrant cyclin D1 expression provides a driving force behind the development of tumorigenesis and is often detected in premalignant and malignant tissues. Collectively, cyclin D1 is thought to be the molecular target associated with mitochondrial ROS-mediated genomic instability.
Figure 2.
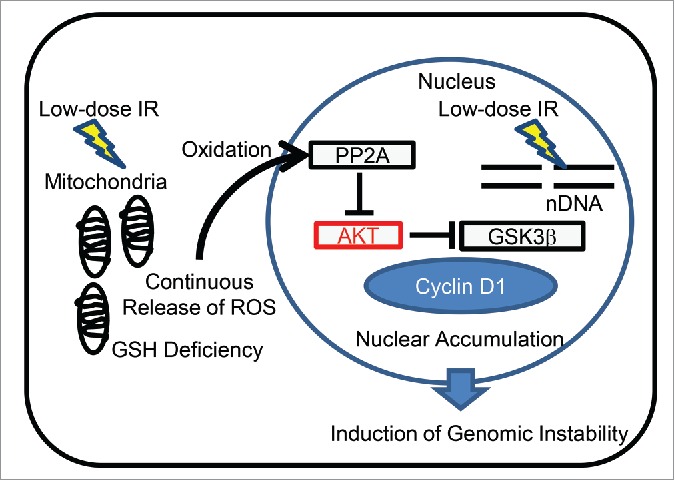
Nuclear retention of cyclin D1 by mitochondrial ROS. ROS are released from the mitochondria after low-dose long-term FR. Mitochondrial ROS inactivate PP2A, which in turn causes a loss of negative feedback control of the AKT pathway, leading to nuclear cyclin D1 accumulation. Perturbation of cyclin D1 expression causes genomic instability in irradiated cells.
Conclusion
This review assesses the role of the radiation response of mitochondria in radiation-induced genomic instability. Mitochondria ROS are primarily responsible for low-dose long-term radiation and affect AKT/cyclin D1 cell cycle signaling. Oxidative stress persists for prolonged periods after low-dose long-term FR. Oxidative damage will accumulate in mtDNA and result in mutagenesis, carcinogenesis, accelerated senescence, and cell death. Antioxidants may therefore be useful agents for radioprotection against mitochondrial damage induced by low-dose long-term FR.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was performed at the Joint Usage/Research Center (Radiation Biology Center), Kyoto University and at the Joint Usage/ Research Center (RIRBM), Hiroshima University.
Funding
Sources of support: This research was supported by a grant from the Japanese Ministry of Education and Science Houga (15K12220), Industrial Disease Clinical Research Grants from the Japanese Ministry of Health, Labor, and Welfare and in part by NIFS Collaborative Research Program (NIFS13KOBA028)..
References
- [1].Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature 2000; 408:433-9; PMID:11100718; http://dx.doi.org/ 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- [2].Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature 2012; 481:287-94; PMID:22258607; http://dx.doi.org/ 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- [3].Persson HL, Kurz T, Eaton JW, Brunk UT. Radiation-induced cell death: importance of lysosomal destabilization. Biochem J 2005; 389:877-84; PMID:15813701; http://dx.doi.org/ 10.1042/BJ20050271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Moretti L, Cha YI, Niermann KJ, Lu B. Switch between apoptosis and autophagy: radiation-induced endoplasmic reticulum stress? Cell Cycle 2007; 6:793-8; PMID:17377498; http://dx.doi.org/ 10.4161/cc.6.7.4036. [DOI] [PubMed] [Google Scholar]
- [5].Gabrys D, Greco O, Patel G, Prise KM, Tozer GM, Kanthou C. Radiation effects on the cytoskeleton of endothelial cells and endothelial monolayer permeability. Int J Radiat Oncol Biol Phys 2007; 69:1553-62; PMID:17920784; http://dx.doi.org/ 10.1016/j.ijrobp.2007.08.039. [DOI] [PubMed] [Google Scholar]
- [6].Kam WW, Banati RB. Effects of ionizing radiation on mitochondria. Free Radical Biol Med 2013; 65:607-19; http://dx.doi.org/ 10.1016/j.freeradbiomed.2013.07.024. [DOI] [PubMed] [Google Scholar]
- [7].Shao C, Folkard M, Michael BD, Prise KM. Targeted cytoplasmic irradiation induces bystander responses. Proc Natl Acad Sci U S A 2004; 101:13495-500; PMID:15345742; http://dx.doi.org/ 10.1073/pnas.0404930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Belyakov OV, Malcolmson AM, Folkard M, Prise KM, Michael BD. Direct evidence for a bystander effect of ionizing radiation in primary human fibroblasts. Br J Cancer 2001; 84:674-9; PMID:11237389; http://dx.doi.org/ 10.1054/bjoc.2000.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radical Biol Med 2009; 47:333-43; http://dx.doi.org/ 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- [10].Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell 2012; 48:158-67; PMID:23102266; http://dx.doi.org/ 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mandal S, Lindgren AG, Srivastava AS, Clark AT, Banerjee U. Mitochondrial function controls proliferation and early differentiation potential of embryonic stem cells. Stem Cells 2011; 29:486-95; PMID:21425411; http://dx.doi.org/ 10.1002/stem.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chiu J, Dawes IW. Redox control of cell proliferation. Trends Cell Biol 2012; 22:592-601; PMID:22951073; http://dx.doi.org/ 10.1016/j.tcb.2012.08.002. [DOI] [PubMed] [Google Scholar]
- [13].Finkel T. Signal transduction by reactive oxygen species. J Cell Biol 2011; 194:7-15; PMID:21746850; http://dx.doi.org/ 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012; 24:981-90; PMID:22286106; http://dx.doi.org/ 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kehrer JP, Klotz LO. Free radicals and related reactive species as mediators of tissue injury and disease: implications for Health. Critical Rev Toxicol 2015; 45:765-98; http://dx.doi.org/ 10.3109/10408444.2015.1074159. [DOI] [PubMed] [Google Scholar]
- [16].Little JB, Nagasawa H, Pfenning T, Vetrovs H. Radiation-induced genomic instability: delayed mutagenic and cytogenetic effects of X rays and α particles. Radiat Res 1997; 148:299-307; PMID:9339945; http://dx.doi.org/ 10.2307/3579514. [DOI] [PubMed] [Google Scholar]
- [17].Streffer C. Strong association between cancer and genomic instability. Radiat Environ Biophys 2010; 49:125-31; PMID:20033424; http://dx.doi.org/ 10.1007/s00411-009-0258-4. [DOI] [PubMed] [Google Scholar]
- [18].Kadhim M, Salomaa S, Wright E, Hildebrandt G, Belyakov OV, Prise KM, Little MP. Non-targeted effects of ionising radiation–implications for low dose risk. Mutation Res 2013; 752:84-98; PMID:23262375; http://dx.doi.org/ 10.1016/j.mrrev.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Murnane JP. Role of induced genetic instability in the mutagenic effects of chemicals and radiation. Mutation Res 1996; 367:11-23; PMID:8596542; http://dx.doi.org/ 10.1016/S0165-1218(96)90017-8. [DOI] [PubMed] [Google Scholar]
- [20].Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer 2013; 13:497-510; PMID:23760024; http://dx.doi.org/ 10.1038/nrc3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kim GJ, Chandrasekaran K, Morgan WF. Mitochondrial dysfunction, persistently elevated levels of reactive oxygen species and radiation-induced genomic instability: a review. Mutagenesis 2006; 21:361-7; PMID:17065161; http://dx.doi.org/ 10.1093/mutage/gel048. [DOI] [PubMed] [Google Scholar]
- [22].Kim GJ, Fiskum GM, Morgan WF. A role for mitochondrial dysfunction in perpetuating radiation-induced genomic instability. Cancer Res 2006; 66:10377-83; PMID:17079457; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Clutton SM, Townsend KM, Walker C, Ansell JD, Wright EG. Radiation-induced genomic instability and persisting oxidative stress in primary bone marrow cultures. Carcinogenesis 1996; 17:1633-9; PMID:8761419; http://dx.doi.org/ 10.1093/carcin/17.8.1633. [DOI] [PubMed] [Google Scholar]
- [24].Kohen R, Nyska A. Oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicologic pathol 2002; 30:620-50; http://dx.doi.org/ 10.1080/01926230290166724. [DOI] [PubMed] [Google Scholar]
- [25].Goitre L, Balzac F, Degani S, Degan P, Marchi S, Pinton P, Retta SF. KRIT1 regulates the homeostasis of intracellular reactive oxygen species. PloS One 2010; 5:e11786; PMID:20668652; http://dx.doi.org/ 10.1371/journal.pone.0011786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shimura T, Kobayashi J, Komatsu K, Kunugita N. Severe mitochondrial damage associated with low-dose radiation sensitivity in ATM- and NBS1-deficient cells. Cell Cycle 2016; http://dx.doi.org/ 10.1080/15384101.2016.1156276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yu J, Wang Q, Chen N, Sun Y, Wang X, Wu L, Chen S, Yuan H, Xu A, Wang J. Mitochondrial transcription factor A regulated ionizing radiation-induced mitochondrial biogenesis in human lung adenocarcinoma A549 cells. J Radiat Res 2013; 54:998-1004; PMID:23645454; http://dx.doi.org/ 10.1093/jrr/rrt046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kulkarni R, Marples B, Balasubramaniam M, Thomas RA, Tucker JD. Mitochondrial gene expression changes in normal and mitochondrial mutant cells after exposure to ionizing radiation. Radiat Res 2010; 173:635-44; PMID:20426663; http://dx.doi.org/ 10.1667/RR1737.1. [DOI] [PubMed] [Google Scholar]
- [29].Chien L, Chen WK, Liu ST, Chang CR, Kao MC, Chen KW, Chiu SC, Hsu ML, Hsiang IC, Chen YJ, et al.. Low-dose ionizing radiation induces mitochondrial fusion and increases expression of mitochondrial complexes I and III in hippocampal neurons. Oncotarget 2015; 6:30628-39; PMID:26415228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007; 130:548-62; PMID:17693261; http://dx.doi.org/ 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- [31].Chen H, Chan DC. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum Mol Genetics 2009; 18:R169-76; http://dx.doi.org/ 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ono T, Isobe K, Nakada K, Hayashi JI. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genetics 2001; 28:272-5; PMID:11431699; http://dx.doi.org/ 10.1038/90116. [DOI] [PubMed] [Google Scholar]
- [33].Tolkovsky AM. Mitophagy. Biochim Et Biophysica Acta 2009; 1793:1508-15; http://dx.doi.org/ 10.1016/j.bbamcr.2009.03.002. [DOI] [PubMed] [Google Scholar]
- [34].Zhang J. Autophagy and Mitophagy in Cellular Damage Control. Redox Biol 2013; 1:19-23; PMID:23946931; http://dx.doi.org/ 10.1016/j.redox.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A 2003; 100:4078-83; PMID:12642658; http://dx.doi.org/ 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183:795-803; PMID:19029340; http://dx.doi.org/ 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Epperly MW, Gretton JE, Sikora CA, Jefferson M, Bernarding M, Nie S, Greenberger JS. Mitochondrial localization of superoxide dismutase is required for decreasing radiation-induced cellular damage. Radiat Res 2003; 160:568-78; PMID:14565825; http://dx.doi.org/ 10.1667/RR3081. [DOI] [PubMed] [Google Scholar]
- [38].Hosoki A, Yonekura S, Zhao QL, Wei ZL, Takasaki I, Tabuchi Y, Wang LL, Hasuike S, Nomura T, Tachibana A, et al.. Mitochondria-targeted superoxide dismutase (SOD2) regulates radiation resistance and radiation stress response in HeLa cells. J Radiat Res 2012; 53:58-71; PMID:22302046; http://dx.doi.org/ 10.1269/jrr.11034. [DOI] [PubMed] [Google Scholar]
- [39].Murley JS, Kataoka Y, Cao D, Li JJ, Oberley LW, Grdina DJ. Delayed radioprotection by NFkappaB-mediated induction of Sod2 (MnSOD) in SA-NH tumor cells after exposure to clinically used thiol-containing drugs. Radiat Res 2004; 162:536-46; PMID: 15624308; http://dx.doi.org/ 10.1667/RR3256. [DOI] [PubMed] [Google Scholar]
- [40].Eldridge A, Fan M, Woloschak G, Grdina DJ, Chromy BA, Li JJ. Manganese superoxide dismutase interacts with a large scale of cellular and mitochondrial proteins in low-dose radiation-induced adaptive radioprotection. Free Radical Biol Med 2012; 53:1838-47; http://dx.doi.org/ 10.1016/j.freeradbiomed.2012.08.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Park HS, Seong KM, Kim JY, Kim CS, Yang KH, Jin YW, Nam SY. Chronic low-dose radiation inhibits the cells death by cytotoxic high-dose radiation increasing the level of AKT and acinus proteins via NF-kappaB activation. Int J Radiat Biol 2013; 89:371-7; PMID:23205493; http://dx.doi.org/ 10.3109/09553002.2013.754560. [DOI] [PubMed] [Google Scholar]
- [42].Zhou H, Ivanov VN, Lien YC, Davidson M, Hei TK. Mitochondrial function and nuclear factor-kappaB-mediated signaling in radiation-induced bystander effects. Cancer Res 2008; 68:2233-40; PMID:18381429; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wang X. The expanding role of mitochondria in apoptosis. Genes Dev 2001; 15:2922-33; PMID:11711427. [PubMed] [Google Scholar]
- [44].Shimura T, Sasatani M, Kamiya K, Kawai H, Inaba Y, Kunugita N. Mitochondrial reactive oxygen species perturb AKT/cyclin D1 cell cycle signaling via oxidative inactivation of PP2A in low-dose irradiated human fibroblasts. Oncotarget 2016; 7(3):3559-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].McKinnon PJ. ATM and ataxia telangiectasia. EMBO Rep 2004; 5:772-6; PMID:15289825; http://dx.doi.org/ 10.1038/sj.embor.7400210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science 2010; 330:517-21; PMID:20966255; http://dx.doi.org/ 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- [47].Guo Z, Deshpande R, Paull TT. ATM activation in the presence of oxidative stress. Cell Cycle 2010; 9:4805-11; PMID:21150274; http://dx.doi.org/ 10.4161/cc.9.24.14323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Reichenbach J, Schubert R, Schwan C, Muller K, Bohles HJ, Zielen S. Anti-oxidative capacity in patients with ataxia telangiectasia. Clin Exp Immunol 1999; 117:535-9; PMID:10469059; http://dx.doi.org/ 10.1046/j.1365-2249.1999.01000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fu X, Wan S, Lyu YL, Liu LF, Qi H. Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PloS One 2008; 3:e2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Valentin-Vega YA, Maclean KH, Tait-Mulder J, Milasta S, Steeves M, Dorsey FC, Cleveland JL, Green DR, Kastan MB. Mitochondrial dysfunction in ataxia-telangiectasia. Blood 2012; 119:1490-500; PMID:22144182; http://dx.doi.org/ 10.1182/blood-2011-08-373639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci U S A 1988; 85:6465-7; PMID:3413108; http://dx.doi.org/ 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A 1997; 94:514-9; PMID:9012815; http://dx.doi.org/ 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kazak L, Reyes A, Holt IJ. Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol 2012; 13:659-71; PMID:22992591; http://dx.doi.org/ 10.1038/nrm3439. [DOI] [PubMed] [Google Scholar]
- [54].Longley MJ, Nguyen D, Kunkel TA, Copeland WC. The fidelity of human DNA polymerase gamma with and without exonucleolytic proofreading and the p55 accessory subunit. J Biol Chem 2001; 276:38555-62; PMID:11504725; http://dx.doi.org/ 10.1074/jbc.M105230200. [DOI] [PubMed] [Google Scholar]
- [55].Copeland WC, Longley MJ. DNA polymerase gamma in mitochondrial DNA replication and repair. Scientific World J 2003; 3:34-44; http://dx.doi.org/ 10.1100/tsw.2003.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 2014; 94:909-50; PMID:24987008; http://dx.doi.org/ 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Et Biophysica Acta 2006; 1757:509-17; http://dx.doi.org/ 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]
- [58].Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 2000; 192:1001-14; PMID:11015441; http://dx.doi.org/ 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, Markowitz SD, Trush MA, Kinzler KW, Vogelstein B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genetics 1998; 20:291-3; PMID:9806551; http://dx.doi.org/ 10.1038/3108. [DOI] [PubMed] [Google Scholar]
- [60].Fliss MS, Usadel H, Caballero OL, Wu L, Buta MR, Eleff SM, Jen J, Sidransky D. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Sci 2000; 287:2017-9; http://dx.doi.org/ 10.1126/science.287.5460.2017. [DOI] [PubMed] [Google Scholar]
- [61].Liu VW, Shi HH, Cheung AN, Chiu PM, Leung TW, Nagley P, Wong LC, Ngan HY. High incidence of somatic mitochondrial DNA mutations in human ovarian carcinomas. Cancer Res 2001; 61:5998-6001; PMID:11507041. [PubMed] [Google Scholar]
- [62].Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene 2006; 25:4663-74; PMID:16892080; http://dx.doi.org/ 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- [63].van Gisbergen MW, Voets AM, Starmans MH, de Coo IF, Yadak R, Hoffmann RF, Boutros PC, Smeets HJ, Dubois L, Lambin P. How do changes in the mtDNA and mitochondrial dysfunction influence cancer and cancer therapy? Challenges, opportunities and models. Mutation Res Rev Mutat Res 2015; 764:16-30; http://dx.doi.org/ 10.1016/j.mrrev.2015.01.001. [DOI] [PubMed] [Google Scholar]
- [64].Chandra D, Singh KK. Genetic insights into OXPHOS defect and its role in cancer. Biochim Et Biophysica Acta 2011; 1807:620-5; http://dx.doi.org/ 10.1016/j.bbabio.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yu E, Mercer J, Bennett M. Mitochondria in vascular disease. Cardio Vascular Res 2012; 95:173-82; http://dx.doi.org/ 10.1093/cvr/cvs111. [DOI] [PubMed] [Google Scholar]
- [66].Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, et al.. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005; 309:481-4; PMID:16020738; http://dx.doi.org/ 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- [67].Staal FJ, Roederer M, Herzenberg LA, Herzenberg LA. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc Natl Acad Sci U S A 1990; 87:9943-7; PMID:2263644; http://dx.doi.org/ 10.1073/pnas.87.24.9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Benhar M, Engelberg D, Levitzki A. ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep 2002; 3:420-5; PMID:11991946; http://dx.doi.org/ 10.1093/embo-reports/kvf094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Liu W, Akhand AA, Takeda K, Kawamoto Y, Itoigawa M, Kato M, Suzuki H, Ishikawa N, Nakashima I. Protein phosphatase 2A-linked and -unlinked caspase-dependent pathways for downregulation of Akt kinase triggered by 4-hydroxynonenal. Cell Death Differentiation 2003; 10:772-81; PMID:12815460; http://dx.doi.org/ 10.1038/sj.cdd.4401238. [DOI] [PubMed] [Google Scholar]
- [70].Shimura T, Hamada N, Sasatani M, Kamiya K, Kunugita N. Nuclear accumulation of cyclin D1 following long-term fractionated exposures to low-dose ionizing radiation in normal human diploid cells. Cell Cycle 2014; 13:1248-55; PMID:24583467; http://dx.doi.org/ 10.4161/cc.28139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Shimura T, Kakuda S, Ochiai Y, Nakagawa H, Kuwahara Y, Takai Y, Kobayashi J, Komatsu K, Fukumoto M. Acquired radioresistance of human tumor cells by DNA-PK/AKT/GSK3beta-mediated cyclin D1 overexpression. Oncogene 2010; 29:4826-37; PMID:20562919; http://dx.doi.org/ 10.1038/onc.2010.238. [DOI] [PubMed] [Google Scholar]
- [72].Shimura T, Ochiai Y, Noma N, Oikawa T, Sano Y, Fukumoto M. Cyclin D1 overexpression perturbs DNA replication and induces replication-associated DNA double-strand breaks in acquired radioresistant cells. Cell Cycle 2013; 12:773-82; PMID:23388457; http://dx.doi.org/ 10.4161/cc.23719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Aggarwal P, Lessie MD, Lin DI, Pontano L, Gladden AB, Nuskey B, Goradia A, Wasik MA, Klein-Szanto AJ, Rustgi AK, et al.. Nuclear accumulation of cyclin D1 during S phase inhibits Cul4-dependent Cdt1 proteolysis and triggers p53-dependent DNA rereplication. Genes Dev 2007; 21:2908-22; PMID:18006686; http://dx.doi.org/ 10.1101/gad.1586007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Leontieva OV, Lenzo F, Demidenko ZN, Blagosklonny MV. Hyper-mitogenic drive coexists with mitotic incompetence in senescent cells. Cell Cycle 2012; 11:4642-9; PMID:23187803; http://dx.doi.org/ 10.4161/cc.22937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Dulic V, Drullinger LF, Lees E, Reed SI, Stein GH. Altered regulation of G1 cyclins in senescent human diploid fibroblasts: accumulation of inactive cyclin E-Cdk2 and cyclin D1-Cdk2 complexes. Proc Natl Acad Sci U S A 1993; 90:11034-8; PMID:8248208; http://dx.doi.org/ 10.1073/pnas.90.23.11034. [DOI] [PMC free article] [PubMed] [Google Scholar]