

Abstract
Aims
The hedgehog (Hh) signalling pathway has been implicated in the pathogenesis and aggressiveness of prostate cancer through epithelial–mesenchymal interactions. The aim of this study was to elucidate the cell-type partitioned expression of the Hh pathway biomarkers in the non-neoplastic and tumour microenvironments and to correlate it with the grade and stage of prostate cancer.
Methods and results
Expression of the Hh pathway components (Shh, Smo, Ptch, Gli1) in the microenvironment of non-neoplastic peripheral zone (n = 119), hormone-naive primary prostate carcinoma (n = 141) and castrate-resistant bone marrow metastases (n = 53) was analysed using immunohistochemistry in tissue microarrays and bone marrow sections. Results showed that epithelial Shh, Smo and Ptch expression was up-regulated, whereas stromal Smo, Ptch, and Gli1 expression was down-regulated in prostate carcinomas compared to non-neoplastic peripheral zone tissue. Ptch expression was modulated further in high-grade and high-stage primary tumours and in bone marrow metastases. Hh signalling correlated with ki67 and vascular endothelial growth factor (VEGF) but not with CD31 expression.
Conclusion
Our results highlight the importance of Hh-mediated epithelial–mesenchymal interactions in the non-neoplastic prostate and imply that shifting the balance from paracrine towards autocrine signalling is important in the pathogenesis and progression of prostate carcinoma.
Keywords: hedgehog signalling, microenvironment, prostate carcinoma
Introduction
The hedgehog (Hh) signalling network is a highly conserved developmental pathway that orchestrates cell–cell interactions essential to organ formation.1 Of the three known mammalian Hh ligands, Sonic hedgehog (Shh) is the most abundantly expressed.2 After its secretion, Shh binds to the transmembrane protein receptor Patched (Ptch), releasing the transmembrane protein Smoothened (Smo) for downstream signalling.3 Downstream signalling is mediated by the Gli family of transcription factors. Three members of that family have been described in mammalian cells: Gli1, Gli2, and Gli3. Gli1, a target gene and transcriptional mediator of the Hh pathway, functions as an activator, whereas Gli2 can act as both an activator and a repressor, and Gli3 has weak activating and potent repressing functions.4 Gli1 and Ptch are target genes of Hh signalling, providing feedback regulatory loops, and are thus considered biomarkers of pathway activity.1
Hh signalling has been implicated in carcinogenesis.5-7 Germline mutations of the Ptch gene have been detected in patients with a predisposition for the development of basal cell carcinomas of the skin and central nervous system medulloblastomas.1,3 Further, aberrant Hh pathway activation has been identified in a variety of human malignancies, including glioblastoma, hematologic malignancies, and carcinomas of the breast, pancreas, and prostate.1
Hh signalling is fundamental to fetal development of the prostate8 and likely contributes to prostate regeneration.9 Interactions between epithelial cells and the surrounding stroma seem to drive Hh signalling activation in prostate development and neoplasia.10-14 Pathway activation has been reported to contribute to the aggressiveness of the disease and the development of the castrate-resistant phenotype of prostate carcinoma.15-16 Additionally, splice variants of the important Hh pathway components Ptch and Gli1 have been identified in prostate and other human cancer cell lines.17-19 However, only limited data exist on activity of the Hh pathway in non-metastatic hormone-naive prostate cancer.
We hypothesized that epithelial–mesenchymal interactions drive Hh pathway activation in the non-neoplastic and tumor microenvironments and that the expression levels of pathway components correlate with the grade and stage of prostate cancer. Thus, we investigated the expression of the biomarkers Shh, Ptch, Smo, and Gli1 in the microenvironments of 1) non-neoplastic peripheral zone (PZ) tissue, 2) primary hormone-naive prostate carcinomas, and 3) castrate-resistant bone marrow metastases. Since Hh signalling enhances tumor cell proliferation and has been associated with angiogenesis and vascular endothelial growth factor (VEGF) production,9,20 we also investigated whether Hh pathway activity correlates with the expression of biomarkers of proliferation (ki67) and angiogenesis (VEGF and CD31).
Our results highlight the importance of Hh-mediated epithelial–mesenchymal interactions in the non-neoplastic prostate and imply that shifting the balance from paracrine towards autocrine signalling, as reflected by upregulation of epithelial and downregulation of stromal Hh signalling, is important in the pathogenesis and progression of prostate carcinoma and thus provide support for therapeutic targeting of the Hh signalling pathway.
Materials and methods
Patients and Specimens
Our study included archived radical prostatectomy specimens (RPS) from 141 patients with hormone-naive primary prostate carcinoma and bone marrow biopsy specimens from 53 patients with castrate-resistant metastatic prostate carcinoma. All patients had given written informed consent at the time of their treatment for the use of their tissue. The protocol for our study was approved by the institutional review board of The University of Texas MD Anderson Cancer Center.
Western Blotting
To test the specificity of the antibodies used for immunohistohemistry, we performed Western blot analysis of the human neuroblastoma cell line IMR-32 that has been shown to have active Hh signalling. 21-22 Whole-cell lysate of IMR-32 was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). 40 μg of the cell lysate was separated on 4%–20% Tris-glycine polyacrylamide gels and transferred to nitrocellulose membranes [both from Novex (Invitrogen, Life Technologies), Carlsbad, CA, USA]. Membranes were then incubated overnight at 4° C with rabbit polyclonal primary antibodies (clone, dilution, company) against Shh (H-160, 1:200, Santa Cruz Biotechnology) and Ptch (1:100, Strategic Diagnostics, Inc., Newark, DE, USA); this was followed by incubation for one hour at room temperature with horseradish peroxidase– conjugated donkey antirabbit IgG (1:3000, Santa Cruz Biotechnology) and development by using enhanced chemiluminescence (Amersham, GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). To asses transfer efficiency and molecular weight of blotted protein, the Full Range Rainbow Molecular Weight Marker (Amersham, GE Healthcare Bio-Sciences Corp.) was used.
Tissue Microarray Construction
Six tissue microarrays (TMAs) were constructed from the RPS. Hematoxylin and eosin–stained slides from formalin-fixed, paraffin-embedded blocks of the RPS were reviewed, and areas representative of the different patterns of the Gleason score of each tumour were selected for use in TMA construction. Each patient's case was represented by a median of six (range, 6–72) 0.6 mm–diameter cores from the tumour and a median of three (range, 0–6) cores from the adjacent non-neoplastic PZ tissue (available for 119 of the patients). In total, 2262 cores from the RPS of the 141 patients were included in the six TMAs.
Immunochistochemical Analyses
Serial 4-μm sections were cut from the TMAs and bone marrow biopsy specimens and subjected to immunohistochemical analysis by using a Dako autostainer (Dako North America, Inc., Carpinteria, CA, USA), as previously described.23 The following primary antibodies (including clone, dilution, and supplier) were used for staining the TMAs: Shh (H-160, 1:175, Santa Cruz Biotechnology), 24 Ptch (1:350, Strategic Diagnostics), Smo (1:80, Abcam, Cambridge, MA, USA), 25 Gli1 (1:300, Novus Biologicals, LLC, Littleton, CO, USA), ki67 (MIB-1, 1:50, Dako), VEGF (prediluted, Abcam), and CD31 (1:30, Dako). Antibodies against Shh, Ptch, and Gli1 were used for staining the bone marrow biopsy specimens. Sections were counterstained with hematoxylin.
The whole stained slide for each biomarker was scanned and viewed by using the Bliss imaging system with WebSlide Browser 4 (both from Bacus Laboratories, Inc., Lombard, IL, USA), and the images were automatically stored for later retrieval. Epithelial and stromal cells were evaluated separately. The percentage of positive-staining cells for each biomarker was determined for each TMA core and bone marrow specimen. Results for Shh, Smo, Ptch, Gli1 and VEGF expression were scored by using an 11-point scale: <1% positive-staining cells = 0; 1– 10% positive cells = 1; 11–20% = 2; 21–30% = 3; 31–40% = 4; 41–50% = 5; 51–60% = 6; 61–70% = 7; 71–80% = 8; 81–90% = 9; and 91–100% = 10. The percentage of cells expressing ki67 and the number of CD31-positive vessels per core were also determined.
Statistical Analyses
The patients' characteristics and the biomarker expression data were summarized by using descriptive statistics and exploratory data analysis. Categorical data were described with the use of contingency tables. Continuously scaled measures were summarized with descriptive statistical measures [e.g., mean with standard deviation (SD)]. Fisher's exact test was used to assess the association between categorical variables, and two-sample t testing was used to assess the mean difference of continuous variables, including biomarker expression between groups. Spearman's correlation on biomarker expression was calculated between biomarkers. To incorporate multiple observations (i.e., data from multiple cores) from an individual patient, mixed-effects models were fitted to allow estimates of both interpatient and intrapatient variability.
All reported P values are two-sided at a significance level of 0.05. To adjust for multiple comparisons, a Bonferroni correction, in which the significance level of each individual comparison is equal to 0.05 divided by the number of comparisons, was used.
Analyses were performed by using SAS for Windows (1999–2000, Release 8.1; SAS Institute, Inc., Cary, NC, USA) and S-PLUS 2000 (Insightful Corporation, Seattle, WA, USA) software.
Results
Patients' Characteristics
The mean age of the patients was 59 years (SD, 7; range, 41–74 years). For the purpose of this study, we considered patients with Gleason scores of 6 (3+3) and 7 (3+4) to have low-grade tumors. None of the 141 patients who had undergone prostatectomy had received therapy before surgery. The 53 patients with bone metastases had received long-term androgen-ablation therapy and chemotherapy and had castrate-resistant disease at the time of bone marrow biopsy. Table 1 summarizes the tumors' pathologic characteristics of the RPS.
Table 1. Pathological characteristics of the primary prostate adenocarcinomas (n = 141).
| Characteristic | n (%) |
|---|---|
| Gleason score | |
| 6 | 4 (2.8) |
| 7 (3+4) | 50 (35.5) |
| 7 (4+3) | 25 (17.7) |
| 8, 9 | 62 (43.9) |
| Pathological T (pT) stage | |
| T2 | 64 (45.4) |
| T3 | 77 (54.6) |
| Pathological N (pN) stage | |
| N0 | 120 (85.1) |
| N1 | 21 (14.9) |
Western Blotting
Two bands, corresponding to the precursor protein (45 kDa) and the 19-kDa N-terminal–secreted peptide of Shh, were recognized, as were the full-length protein and splice variants of Ptch (Figure 1).
Figure 1.
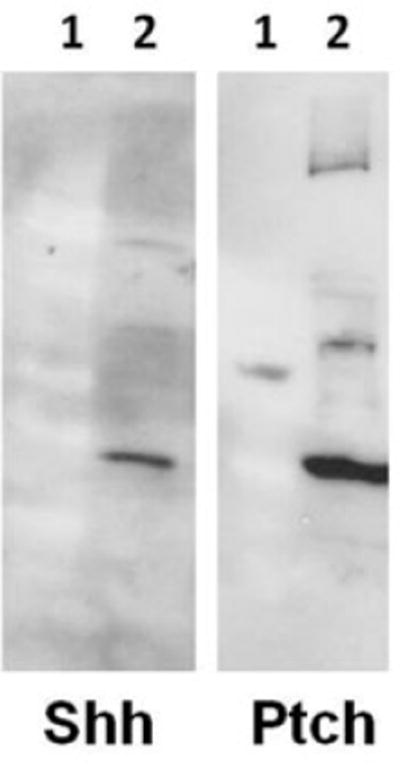
Western blot analysis of IMR-32 cell lysate using the anti-Shh and anti-Ptch antibodies (1 = molecular weight marker, 2 = IMR-32 cell lysate).
Biomarker Expression
Shh was expressed in the cytoplasm and primarily localized in epithelial cells (Figure 2A–2C). Smo was expressed in both the membrane and the cytoplasm of epithelial and stromal cells (Figure 2D–2F). Consistent with its localization in cytoplasmic vesicular structures 26, Ptch was expressed in the cytoplasm of both epithelial and stromal cells (Figure 2G–2I). Gli1 was expressed in the nuclei and faintly in the cytoplasm of the cells (Figure 2J–2L); since Gli1 is a transcription factor and its cytoplasmic expression was faint and inconsistent, only its nuclear expression was analyzed further.
Figure 2.
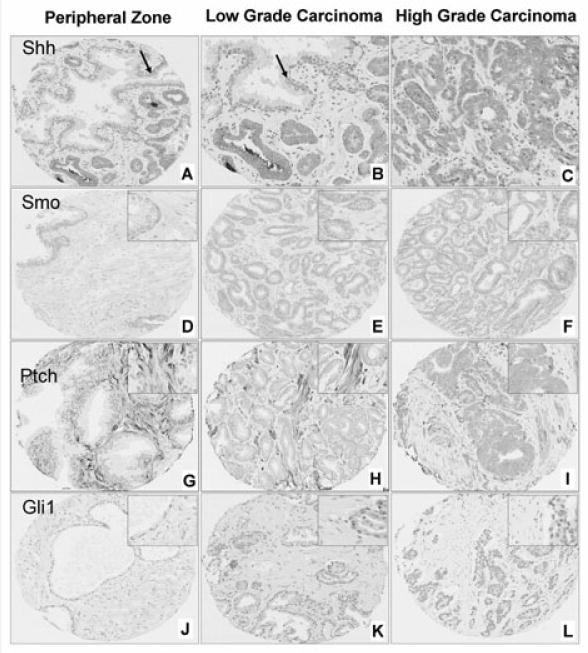
Representative images of Shh (A–C), Smo (D–F), Ptch (G–I) and Gli1 (J–L) expression in non-neoplastic peripheral zone (D, G, J), low-grade prostate carcinoma [Gleason scores 6 and 7 (3+4)] (A, B, E, H, K) and high-grade carcinoma [Gleason scores 7 (4+3), 8 and 9] (C, F, I, L). Arrows in A and B depict the non-neoplastic epithelium. Note that epithelial Shh (A–C), Smo (D–F) and Ptch (G–I) expression is increased and stromal expression is decreased in tumours compared to non-neoplastic tissue. Ptch expression is increased in epithelial cells and decreased in stromal cells of high-grade compared to low-grade tumours (H, I). Stromal expression of Gli1 (J–L) is down-regulated in carcinomas compared to peripheral zone tissue.
The following subsections, as well as Tables 2–5 and Figures 2 and 3, provide further details about the expression of the seven biomarkers studied.
Table 2. Mean expression levels of the biomarkers in non-neoplastic peripheral zone tissue, primary prostate carcinomas and bone marrow metastases.
| Location | Biomarker | Non-neoplastic | Primary tumour | Bone Marrow metastases | SD between patients | SD within patients | P-value (non-neoplastic versus tumour) | P-value (primary versus metastatic) |
|---|---|---|---|---|---|---|---|---|
| Epithelium | Shh* | 2.8 | 7.9 | 8.2 | 1.7 | 2.1 | <0.001 | 0.511 |
| SMO* | 7.9 | 9.1 | 1.3 | 1.80 | <0.001 | |||
| Ptch* | 1.6 | 2.9 | 5.2 | 2.2 | 2 | <0.001 | <0.001 | |
| Gli* | 7.4 | 7 | 7.3 | 2.3 | 2 | 0.04 | 0.644 | |
| ki67** | 0.3 | 3.4 | 2.6 | 3.3 | <0.001 | |||
| VEGF* | 3.5 | 7.5 | 2.1 | 2.2 | <0.001 | |||
|
| ||||||||
| Stroma | Shh* | 1.5 | 1.2 | 2 | 0.8 | 0.7 | 0.04 | 0.04 |
| SMO* | 6.7 | 4.1 | 1.8 | 1.9 | <0.001 | |||
| Ptch* | 8.4 | 7.2 | 4.7 | 1.5 | 1.5 | <0.001 | <0.001 | |
| Glil* | 5.8 | 4.2 | 4.9 | 2 | 2 | <0.001 | 0.04 | |
| VEGF* | 3.5 | 2.4 | 1.3 | 1.4 | <0.001 | |||
| CD31*** | 16 | 21 | 6 | 8 | <0.001 | |||
11-point scale system.
Percentage of positive cells.
CD31 -positive vessels per core.
SD: standard deviation; VEGF: vascular endothelial growth factor.
Table 5. Spearman's correlation between the Shh, Ptch and Gli1 in bone marrow metastases.
| Bone marrow metastases | Epithelium | Stroma | |||||
|---|---|---|---|---|---|---|---|
| Ptch | Gli1 | Shh | Ptch | Gli1 | |||
| Epithelium | Shh | Correlation coefficient | 0.405 | 0.281 | 0.475 | 0.111 | 0.247 |
| Sig. (two-tailed) | 0.003 | 0.041 | 0.000 | 0.443 | 0.087 | ||
| Ptch | Correlation coefficient | 0.355 | 0.388 | 0.462 | 0.301 | ||
| Sig. (two-tailed) | 0.009 | 0.005 | 0.001 | 0.036 | |||
| Gli1 | Correlation coefficient | 0.160 | 0.370 | 0.484 | |||
| Sig. (two-tailed) | 0.263 | 0.008 | 0.000 | ||||
| Stroma | Shh | Correlation coefficient | 0.443 | 0.425 | |||
| Sig. (two-tailed) | 0.001 | 0.002 | |||||
| Ptch | Correlation coefficient | 0.502 | |||||
| Sig. (two-tailed) | 0.000 | ||||||
Figure 3.
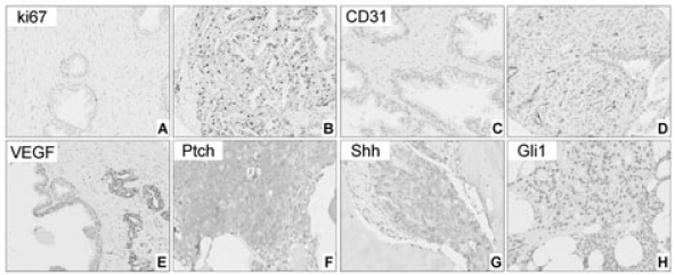
A–E, Expression of ki67, CD31 and vascular endothelial growth factor (VEGF) in non-neoplastic peripheral zone (PZ) and primary prostate carcinoma. Expression of Ki67 is lower in non-neoplastic PZ (A) compared to prostate carcinoma (B). Number of CD31-positive vessels increases from non-neoplastic prostate (C) to prostate carcinoma (D). Low expression levels of VEGF in adjacent non-neoplastic epithelium compared to the malignant cells (E). F–H, Expression of Ptch (F), Shh (G) and Gli1 (H) in bone marrow metastases. Note that Ptch expression in epithelial cells of a metastatic tumour is higher than that in primary carcinomas in Figure 2H and 2I.
Non-neoplastic prostate PZ tissue
Differences in the cellular localization of Shh and Ptch expression were noted in the microenvironment of PZ tissue: Shh and Ptch were primarily expressed in epithelial and stromal cells, respectively (Figure 2A, 2G). Smo and Gli1, however, were expressed in both epithelial and stromal cells (Figure 2D, 2J).
Spearman's correlation coefficient testing revealed that epithelial Shh expression correlated with epithelial Ptch (P < 0.001, r = 0.4) and epithelial VEGF (P < 0.001, r = 0.589) expression. A correlation between epithelial and stromal Gli1 expression was noted (P < 0.001, r = 0.523). In addition, the cell-proliferation rate strongly correlated with epithelial Gli1 expression (P < 0.001, r = 0.507), and microvessel density (as evaluated by CD31 positivity) correlated with stromal Gli1 expression (P < 0.001, r = 0.523).
Primary prostate carcinoma
As Table 2 and Figure 2 illustrate, in the primary prostate carcinomas, Shh was expressed primarily in epithelial cells, whereas the other components of the pathway were expressed in both epithelial and stromal cells. Statistical analysis revealed that Shh expression was higher in tumour epithelial cells than it was in the non-neoplastic epithelium (P < 0.001). Expression of Smo was higher in the tumor epithelial cells than it was in the non-neoplastic epithelium, but lower in the tumor stroma than it was in the non-neoplastic stroma (P < 0.001 for both comparisons). Concordantly, epithelial expression of Ptch was higher in the primary tumor than it was in the non-neoplastic tissue, but its stromal expression was lower in the tumor than it was in the non-neoplastic PZ tissue (P < 0.001). Nuclear Gli1 expression was lower in tumor stromal cells than it was in the non-neoplastic PZ stromal cells (P < 0.001), whereas its epithelial expression did not differ between the tumor and non-neoplastic tissues.
Considering the mean values for the expression of Shh, Ptch, Smo, and Gli1 in the non-neoplastic PZ tissue as cutoff values (Table 2), we found that the epithelial expression of all four biomarkers was upregulated in 42 tumors and that the expression of three biomarkers was upregulated in another 42 tumors; that is, the expression of at least three of the Hh pathway components was upregulated in the epithelial cells in 84 of the 141 (60%) primary tumors. In 10 (7%) of the primary tumours, only one biomarker's expression was upregulated in the epithelial cells, and in one (0.7%) tumor, the expression of no biomarkers was upregulated. In the stromal cells of the 141 tumors, the expression of all four biomarkers was downregulated in 62 (44%), and that of three biomarkers was downregulated in 36 (26%) tumors. In the stromal cells of only 10 tumors (7%), the expression of all four biomarkers was upregulated.
The results of testing for any correlation between the expression of the Hh pathway biomarkers and Gleason score and pathologic disease stage are shown in Tables 3 and 4, respectively. No difference was noted in the expression levels of Shh, Smo, and Gli1 in relation to Gleason score (Figure 2B, 2C, 2E, 2F, 2K, 2L) and pT stage. However, expression of Ptch in the epithelial cells of tumours with Gleason scores 6 and 7 (3+4) was marginally lower than it was in tumors with Gleason score 7 (4+3) (P = 0.017) and significantly lower than it was in tumours with Gleason scores 8 and 9 (P < 0.001) (Figure 2H, 2I). A significant difference was also noted in the expression level of Ptch in epithelial cells between pT2- and pT3- stage tumours (mean expression values of 1.4 and 3.1, respectively; P < 0.001). In contrast, Ptch expression in the stroma was lower in tumours with Gleason scores 8 and 9 and in pT3- stage tumours than it was in low-grade tumours and in p T2- stage tumours, respectively (P < 0.001). No significant differences were found in the expression of the Hh pathway biomarkers in relation to pN disease stage (data not shown).
Table 3. Differential expression of the biomarkers in relation to Gleason score (GS).
| Location | Biomarker | Gleason score (GS) | SD between patients | SD within patients | P-value (GS2 versus GS1) | P-value (GS2 versus GS3) | P-value (GS3 versus GS1) | ||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| GS1 [6 and 7 (3+4)] | GS2 [7 (4+3)] | GS3 (8 and 9) | |||||||
| Epithelium | Shh* | 8.6 | 7.5 | 7.6 | 2.1 | 1.8 | 0.044 | 0.746 | 0.04 |
| SMO* | 9.3 | 8.4 | 9.5 | 1.3 | 1.4 | 0.05 | 0.04 | 0.558 | |
| Ptch* | 1.3 | 2.6 | 3.2 | 2.2 | 1.8 | 0.017 | 0.337 | <0.001 | |
| Gli* | 6.7 | 7.5 | 7.2 | 2.8 | 1.5 | 0.243 | 0.658 | 0.344 | |
| ki67** | 1.1 | 1 | 4.6 | 6.8 | 3 | 0.973 | 0.306 | 0.368 | |
| VEGF* | 8.1 | 7.1 | 7.2 | 2.4 | 2 | 0.117 | 0.87 | ||
|
| |||||||||
| Stroma | Shh* | 1.2 | 1 | 0.9 | 0.5 | 0.6 | 0.279 | 0.587 | 0.04 |
| SMO* | 4.5 | 4.4 | 3.9 | 2.1 | 1.7 | 0.794 | 0.379 | 0.154 | |
| Ptch* | 8.3 | 7.8 | 7.2 | 1.2 | 1.5 | 0.102 | 0.068 | <0.001 | |
| Gli* | 4 | 4.4 | 3.9 | 2 | 1.9 | 0.537 | 0.337 | 0.685 | |
| VEGF* | 2.7 | 2.4 | 1.8 | 1.1 | 1.2 | 0.416 | 0.039 | <0.001 | |
| CD31*** | 18 | 25 | 21 | 7.3 | 7.4 | <0.001 | 0.054 | 0.04 | |
11-point scale system.
Percentage of positive cells.
CD31-positive vessels per core.
SD: standard deviation; VEGF: vascular endothelial growth factor.
Table 4. Differential expression of the biomarkers in relation to pT stage.
| Location | Biomarker | pT2 | pT3 | SD between patients | SD within patients | P-value |
|---|---|---|---|---|---|---|
| Epithelium | Shh* | 8.3 | 7.7 | 2.2 | 1.9 | 0.159 |
| SMO* | 9.4 | 9 | 1.3 | 1.4 | 0.233 | |
| Ptch* | 1.4 | 3.1 | 2.2 | 1.8 | <0.001 | |
| Gli1* | 6.5 | 7.4 | 2.8 | 1.6 | 0.066 | |
| ki67** | 1.1 | 5 | 6.5 | 3 | 0.152 | |
| VEGF* | 7.7 | 7.3 | 2.4 | 2 | 0.33 | |
|
| ||||||
| Stroma | Shh* | 1.1 | 0.9 | 0.5 | 0.6 | 0.059 |
| SMO* | 4.3 | 4 | 2 | 1.7 | 0.453 | |
| Ptch* | 8.2 | 7.3 | 1.2 | 1.5 | <0.001 | |
| Gli1* | 4.2 | 3.9 | 2 | 1.9 | 0.53 | |
| VEGF* | 2.6 | 2 | 1.2 | 1.2 | 0.003 | |
| CD31*** | 19 | 22 | 7 | 7 | 0.012 | |
11-point scale system.
Percentage of positive cells.
CD31-positive vessels per core.
SD: standard deviation; VEGF: vascular endothelial growth factor.
Figure 3 (A–E) illustrates the expression of the biomarkers ki67, VEGF, and CD31 in non-neoplastic prostate PZ tissue and primary prostate carcinomas. As expected, the epithelial-cell proliferation rate (as determined by ki67 expression) and microvessel density (as determined by the number of CD31-positive vessels) were higher in the tumours than they were in the non-neoplastic PZ tissue (P < 0.001). Microvessel density was lower in low-grade than in higher-grade tumuors (P < 0.001). VEGF expression was higher in the epithelial cells and lower in the stromal cells of primary prostate carcinomas than it was in the corresponding cell compartments of the non-neoplastic PZ tissues (P < 0.001). In addition, stromal expression of VEGF decreased with increasing disease grade (i.e., Gleason score) (P < 0.001) and pT stage (P = 0.003). No significant differences were found in the expression of the three biomarkers in relation to pN disease stage (data not shown).
Spearman's correlation coefficient testing revealed coordinated expression of Hh pathway components, as shown by significant correlations between epithelial Shh and epithelial Ptch (P < 0.001, r = 0.347), epithelial Smo (P < 0.001, r = 0.454) and stromal Smo (P < 0.001, r = 0.308) expression. A correlation between epithelial and stromal expression was noted for Gli1 (P < 0.001, r = 0.523) and Shh (P < 0.001, r = 0.398). Also, epithelial expression of Ptch correlated with epithelial (P < 0.001, r = 0.466) and stromal (P < 0.001, r = 0.316) Gli1 expression. Cancers with relatively higher proliferation rates also displayed relatively higher Ptch and Gli1 expression levels, as shown by correlations between ki67 and epithelial Ptch (P < 0.001, r = 0.496) and Gli1 (P < 0.001, r = 0.315) expression. Microvessel density, as determined by counts of CD31-positive–stained vessels, did not correlate with the expression of any of the biomarkers, but epithelial VEGF expression correlated strongly with epithelial Shh (P < 0.001, r = 0.786) and epithelial Smo expression (P < 0.001, r = 0.361). Additionally, stromal expression of VEGF correlated with stromal Shh (P < 0.001, r = 0.395) and Ptch expression (P < 0.001, r = 0.376).
Castrate-resistant bone marrow metastases
Epithelial Ptch expression was higher in castrate-resistant bone marrow metastases than it was in the primary prostate carcinomas (P < 0.001). In contrast, stromal expression of Ptch was lower in the bone marrow metastases than it was in the primary tumours (P < 0.001) (Figure 3F; compare with Figure 2H, 2I). Shh and Gli1 expression levels did not differ significantly between the primary tumours and the bone marrow metastases (Figure 3G, 3H). Finally, as shown in Table 5, significant correlations were found between the pathway components in the bone marrow metastases.
Discussion
In vivo and correlative tissue-based data have highlighted the involvement of the Hh signalling pathway in prostate carcinogenesis.9,12,15,16,27 We hypothesized that epithelial–mesenchymal interactions drive Hh pathway activation in the non-neoplastic and tumour microenvironments and that the expression levels of pathway components correlate with the grade and stage of prostate carcinoma. To the best of our knowledge, ours is the first study to identify partitioned modulation of Hh signalling in clinical specimens of primary prostate carcinoma and bone marrow metastases. This information might be helpful for determining the optimal timing for administering Hh pathway inhibitors in the treatment of men with prostate cancer.
The few reported localization studies of the Hh pathway components in human specimens have included a limited number of specimens. Ptch has been reported to be expressed in epithelial cells27 and results regarding Gli1 localization have been inconclusive.12,27 Our study which included a larger number of specimens, revealed expression of pathway components in both epithelial and stromal cells in the tumour microenvironment. Consistent with previous findings, 9,12,15,27 epithelial expression of Shh, Smo, and Ptch was higher in primary carcinomas than non-neoplastic PZ tissue. Epithelial Ptch was further upregulated in high-grade and high-stage tumours and in castrate-resistant bone marrow metastases.
In contrast to our findings in epithelial cells, stromal Ptch, Gli1, and Smo expression were downregulated in primary carcinomas relative to non-neoplastic PZ tissue. Moreover stromal Ptch expression was lower in high grade and stage than it was in low-grade and stage tumors, and was also further downregulated in metastatic lesions. Such a shift in the balance of signalling from paracrine towards autocrine is likely not unique to prostate cancer as it has been reported during small cell lung carcinogenesis.28 The stromal Hh signalling decrease observed with increased disease aggressiveness is also consistent with the emergence of an epitheliocentric phenotype during prostate cancer progression.29
A possible limitation of our study is that the non-neoplastic PZ tissues were obtained from morphologically benign areas of the tumour-containing specimens. As molecular alterations reportedly occur in benign-looking tissues from prostates harboring tumors,30,31 investigation of Hh signalling in normal PZ tissue is warranted for conclusive statements regarding normal prostate. Nonetheless, our findings clearly show that Hh signalling in morphologically malignant tissue is upregulated relative to that in benign-looking PZ tissue, which supports the concept that activation of Hh pathway signalling is important in the phenotypic development of primary prostate carcinoma.
Our findings of steady levels of Gli1 expression at the protein level in primary and metastatic prostate carcinoma need to be interpreted in the light of the reported upregulation of Gli1 mRNA levels in metastatic samples compared to that in primary tumors.9 Several explanations may account for this discordance. It has been shown that mRNA levels do not always correspond to protein levels. 32 The low number of patients examined in the later study might also explain the discrepancy. Additionally, in our study, Gli1, although a transcriptional target of Hh pathway activation, did not follow Ptch expression level modulations in primary prostate carcinomas and bone marrow metastases. This is not surprising, given the complexity of the Hh signalling network. 33 -38 Cytoplasmic Gli1, although faint and inconsistent, could also contribute to total protein levels.39 Additionally, other Gli family members, especially Gli2, may be more important for pathway activation in humans. 40,41 The complexity of Gli1 regulation is further highlighted by the lack of correlation between Shh and Gli1 in our study. Conversely, Shh correlated with Ptch and Smo expression. Limited Gli1 antibody specificity could also account for this finding. It is noteworthy that Western blot experiments demonstrated a high degree of specificity of the anti-Shh antibody and adequate specificity of the anti-Ptch antibody, validating our immunohistochemical results.
Expression of ki67 correlated with biomarkers of epithelial Hh pathway activation (Gli1 and Ptch) and VEGF expression correlated with both epithelial and stromal Hh signalling. Previous studies have linked Hh signalling with prostate cancer cell proliferation9,27 and angiogenesis.20 Our correlative findings provide support of an association of Hh signalling with proliferation and VEGF production in the clinical setting and rationalize Hh targeting in prostate carcinoma as a therapeutic strategy.
In summary, we present a detailed analysis of the expression of the Hh pathway in non-neoplastic PZ tissue and prostate carcinoma. Shh was predominantly expressed in the epithelial cells, whereas downstream effectors were present in both the epithelium and the stroma, highlighting the importance of Hh-mediated epithelial–mesenchymal interactions. Upregulation of epithelial and downregulation of stromal Hh signalling in malignant compared to non-neoplastic tissue and in high grade and stage compared to low grade and stage tumors imply that shifting the balance towards autocrine signalling may be implicated in the pathogenesis and progression of prostate carcinoma. Our findings add to the general concept that Hh signalling is central to prostate carcinoma microenvironment. More broadly, the findings are in line with the view that prostate carcinoma progresses from a paracrine to an autocrine state over time and with disease evolution. These data provide additional support for Hh pathway inhibition in prostate cancer therapeutics. Further study of the mechanistic roles of autocrine and paracrine Hh signalling in prostate carcinoma microenvironment is warranted in order to better define the role of Hh inhibition as a therapeutic strategy.
Acknowledgments
This study was supported in part by a Young Investigator Award to Dr Efstathiou, Prostate Cancer Foundation and in part by the National Institutes of Health through MD Anderson's Cancer Center Support Grant, CA016672.
The authors would like to thank Karen F. Phillips, ELS, from the Department of Genitourinary Medical Oncology, MD Anderson, for her editing of the manuscript.
Abbreviations
- Hh
hedgehog
- Ptch
Patched
- Shh
Sonic hedgehog
- PZ
peripheral zone
- RPS
radical prostatectomy specimens
- Smo
Smoothened
- TMA
tissue microarray
- VEGF
vascular endothelial growth factor
References
- 1.Teglund S, Toftgård R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta. 2010;1805:181–208. doi: 10.1016/j.bbcan.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Azoulay S, Terry S, Chimingqi M, et al. Comparative expression of Hedgehog ligands at different stages of prostate carcinoma progression. J Pathol. 2008;216:460–470. doi: 10.1002/path.2427. [DOI] [PubMed] [Google Scholar]
- 3.Cohen MM., Jr The hedgehog signaling network. Am J Med Genet A. 2003;123A:5–28. doi: 10.1002/ajmg.a.20495. [DOI] [PubMed] [Google Scholar]
- 4.Stecca B, Ruiz I, Altaba A. Context-dependent regulation of the GLI code in cancer by HEDGEHOG and non-HEDGEHOG signals. J Mol Cell Biol. 2010;2:84–95. doi: 10.1093/jmcb/mjp052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marker PC. Does prostate cancer co-opt the developmental program? Differentiation. 2008;76:736–744. doi: 10.1111/j.1432-0436.2008.00297.x. [DOI] [PubMed] [Google Scholar]
- 6.Yang L, Xie G, Fan Q, Xie J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene. 2010;29:469–481. doi: 10.1038/onc.2009.392. [DOI] [PubMed] [Google Scholar]
- 7.Shaw A, Bushman W. Hedgehog signaling in the prostate. J Urol. 2007;177:832–838. doi: 10.1016/j.juro.2006.10.061. [DOI] [PubMed] [Google Scholar]
- 8.Prins GS, Putz O. Molecular signaling pathways that regulate prostate gland development. Differentiation. 2008;76:641–659. doi: 10.1111/j.1432-0436.2008.00277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karhadkar SS, Bova GS, Abdallah N, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707–712. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 10.Lamm ML, Catbagan WS, Laciak RJ, et al. Sonic hedgehog activates mesenchymal Gli1 expression during prostate ductal bud formation. Dev Biol. 2002;49:349–366. doi: 10.1006/dbio.2002.0774. [DOI] [PubMed] [Google Scholar]
- 11.Wang BE, Shou J, Ross S, Koeppen H, De Sauvage FJ, Gao WQ. Inhibition of epithelial ductal branching in the prostate by sonic hedgehog is indirectly mediated by stromal cells. J Biol Chem. 2003;278:18506–18513. doi: 10.1074/jbc.M300968200. [DOI] [PubMed] [Google Scholar]
- 12.Fan L, Pepicelli CV, Dibble CC, et al. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology. 2004;145:3961–3970. doi: 10.1210/en.2004-0079. [DOI] [PubMed] [Google Scholar]
- 13.Shaw A, Gipp J, Bushman W. The Sonic Hedgehog pathway stimulates prostate tumor growth by paracrine signaling and recapitulates embryonic gene expression in tumor myofibroblasts. Oncogene. 2009;28:4480–4490. doi: 10.1038/onc.2009.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zunich SM, Douglas T, Valdovinos M, et al. Paracrine sonic hedgehog signaling by prostate cancer cells induces osteoblast differentiation. Mol Cancer. 2009;8:12. doi: 10.1186/1476-4598-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheng T, Li C, Zhang X, Chi S, et al. Activation of the hedgehog pathway in advanced prostate cancer. Mol Cancer. 2004;3:29. doi: 10.1186/1476-4598-3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaw C, Price AM, Ktori E, et al. Hedgehog signaling in androgen independent prostate cancer. Eur Urol. 2008;54:1333–1343. doi: 10.1016/j.eururo.2008.01.070. [DOI] [PubMed] [Google Scholar]
- 17.Lo HW, Zhu H, Cao X, Aldrich A, Ali-Osman F. A novel splice variant of GLI1 that promotes glioblastoma cell migration and invasion. Cancer Res. 2009;69:6790–6798. doi: 10.1158/0008-5472.CAN-09-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimokawa T, Tostar U, Lauth M, et al. Novel human glioma-associated oncogene 1 (GLI1) splice variants reveal distinct mechanisms in the terminal transduction of the hedgehog signal. J Biol Chem. 2008;283:14345–14354. doi: 10.1074/jbc.M800299200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagao K, Togawa N, Fujii K, et al. Detecting tissue-specific alternative splicing and disease-associated aberrant splicing of the PTCH gene with exon junction microarrays. Hum Mol Genet. 2005;14:3379–3388. doi: 10.1093/hmg/ddi369. [DOI] [PubMed] [Google Scholar]
- 20.Yamazaki M, Nakamura K, Mizukami Y, et al. Sonic hedgehog derived from human pancreatic cancer cells augments angiogenic function of endothelial progenitor cells. Cancer Sci. 2008;99:1131–1138. doi: 10.1111/j.1349-7006.2008.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dahmane N, Sánchez P, Gitton Y, et al. The Sonic Hedgehog-Gli pathway regulates dorsal brain growth and tumorigenesis. Development. 2001;128:5201–5212. doi: 10.1242/dev.128.24.5201. [DOI] [PubMed] [Google Scholar]
- 22.Mao L, Xia YP, Zhou YN, et al. A critical role of Sonic Hedgehog signaling in maintaining the tumorigenicity of neuroblastoma cells. Cancer Sci. 2009;100:1848–1855. doi: 10.1111/j.1349-7006.2009.01262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Efstathiou E, Troncoso P, Wen S, et al. Initial modulation of the tumor microenvironment accounts for thalidomide activity in prostate cancer. Clin Cancer Res. 2007;13:1224–1231. doi: 10.1158/1078-0432.CCR-06-1938. [DOI] [PubMed] [Google Scholar]
- 24.Nakashima H, Nakamura M, Yamaguchi H, et al. Nuclear factor-kappaB contributes to hedgehog signaling pathway activation through sonic hedgehog induction in pancreatic cancer. Cancer Res. 2006;66:7041–7049. doi: 10.1158/0008-5472.CAN-05-4588. [DOI] [PubMed] [Google Scholar]
- 25.Wehrens XH, Mies B, Gimona M, Ramaekers FC, Van Eys GJ, Small JV. Localization of smoothelin in avian smooth muscle and identification of a vascular-specific isoform. FEBS Lett. 1997;405:315–320. doi: 10.1016/s0014-5793(97)00207-x. [DOI] [PubMed] [Google Scholar]
- 26.Uchikawa H, Toyoda M, Nagao K, et al. Brain- and heart-specific Patched-1 containing exon 12b is a dominant negative isoform and is expressed in medulloblastomas. Biochem Biophys Res Commun. 2006;349:277–283. doi: 10.1016/j.bbrc.2006.08.046. [DOI] [PubMed] [Google Scholar]
- 27.Sanchez P, Hernández AM, Stecca B, et al. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc Natl Acad Sci USA. 2004;101:12561–12566. doi: 10.1073/pnas.0404956101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watkins DN, Berman DM, Baylin SB. Hedgehog signaling: progenitor phenotype in small-cell lung cancer. Cell Cycle. 2003;2:196–198. [PubMed] [Google Scholar]
- 29.Efstathiou E, Logothetis CJ. A new therapy paradigm for prostate cancer founded on clinical observations. Clin Cancer Res. 2010;16:1100–1107. doi: 10.1158/1078-0432.CCR-09-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chandran UR, Dhir R, Ma C, Michalopoulos G, Becich MJ, Gilbertson JR. Differences in gene expression in prostate cancer, normal appearing prostate tissue adjacent to cancer and prostate tissue from cancer free organ donors. BMC Cancer. 2005;5:45. doi: 10.1186/1471-2407-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parr RL, Dakubo GD, Crandall KA, et al. Somatic mitochondrial DNA mutations in prostate cancer and normal appearing adjacent glands in comparison to age-matched prostate samples without malignant histology. J Mol Diagn. 2006;8:312–319. doi: 10.2353/jmoldx.2006.050112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim KH, Kim JM, Choi YL, et al. Expression of sonic hedgehog signaling molecules in normal, hyperplastic and carcinomatous endometrium. Pathol Int. 2009;59:279–287. doi: 10.1111/j.1440-1827.2009.02366.x. [DOI] [PubMed] [Google Scholar]
- 33.Chang H, Li Q, Moraes RC, Lewis MT, Hamel PA. Activation of Erk by sonic hedgehog independent of canonical hedgehog signaling. Int J Biochem Cell Biol. 2010;42:1462–1471. doi: 10.1016/j.biocel.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Riobó NA, Lu K, Ai X, Haines GM, Emerson CP., Jr Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc Natl Acad Sci USA. 2006;103:4505–4510. doi: 10.1073/pnas.0504337103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seto M, Ohta M, Asaoka Y, et al. Regulation of the hedgehog signaling by the mitogen-activated protein kinase cascade in gastric cancer. Mol Carcinog. 2009;48:703–712. doi: 10.1002/mc.20516. [DOI] [PubMed] [Google Scholar]
- 36.Riobo NA, Haines GM, Emerson CP., Jr Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 2006;66:839–845. doi: 10.1158/0008-5472.CAN-05-2539. [DOI] [PubMed] [Google Scholar]
- 37.Dennler S, André J, Alexaki I, et al. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007;67:6981–6986. doi: 10.1158/0008-5472.CAN-07-0491. [DOI] [PubMed] [Google Scholar]
- 38.Palaniswamy R, Teglund S, Lauth M, Zaphiropoulos PG, Shimokawa T. Genetic variations regulate alternative splicing in the 5′ untranslated regions of the mouse glioma-associated oncogene 1, Gli1. BMC Mol Biol. 2010;11:32. doi: 10.1186/1471-2199-11-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kogerman P, Grimm T, Kogerman L, et al. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. 1999;13:12–19. doi: 10.1038/13031. [DOI] [PubMed] [Google Scholar]
- 40.Doles J, Cook C, Shi X, Valosky J, Lipinski R, Bushman W. Functional compensation in Hedgehog signaling during mouse prostate development. Dev Biol. 2006;295:13–25. doi: 10.1016/j.ydbio.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 41.Thiyagarajan S, Bhatia N, Reagan-Shaw S, et al. Role of GLI2 transcription factor in growth and tumorigenicity of prostate cells. Cancer Res. 2007;67:10642–10646. doi: 10.1158/0008-5472.CAN-07-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]