

Abstract
The developmental regulation of globin gene expression has shaped research efforts to establish therapeutic modalities for individuals affected with sickle cell disease and β-thalassemia. Fetal hemoglobin has been shown to block sickle hemoglobin S polymerization to improve symptoms of sickle cell disease; moreover, fetal hemoglobin functions to replace inadequate hemoglobin A synthesis in β-thalassemia thus serving as an effective therapeutic target. In the perinatal period, fetal hemoglobin is synthesized at high levels followed by a decline to adult levels by one year of age. It is known that naturally occurring mutations in the γ-globin gene promoters and distant cis-acting transcription factors produce persistent fetal hemoglobin synthesis after birth to ameliorate clinical symptoms. Major repressor proteins that silence γ-globin during development have been targeted for gene therapy in β-hemoglobinopathies patients. In parallel effort, several classes of pharmacological agents that induce fetal hemoglobin expression through molecular and cell signaling mechanisms have been identified. Herein, we reviewed the progress made in the discovery of signaling molecules targeted by pharmacologic agents that enhance γ-globin expression and have the potential for future drug development to treat the β-hemoglobinopathies.
Keywords: Fetal hemoglobin, sickle cell disease, β-hemoglobinopathies, NRF2, p38 MAPK, cell signaling
Introduction
Sickle cell disease (SCD) and β-thalassemia are genetic disorders resulting in abnormal structure or decreased expression of the β-globin protein chain, respectively. SCD represents a significant global health problem, with more than 300,000 affected infants born each year, mainly in Africa. Fetal hemoglobin (HbF; α2γ2) is expressed in persons with SCD at variable levels and is known to alleviate the clinical severity of the disease.1 Although naturally occurring mutations in the β-locus or distant cis-acting factors contribute to HbF production during the adult developmental stage,2 many studies have been conducted to design pharmacologic HbF inducers to offer treatment options to the majority of individuals not carrying these mutations. To date, hydroxyurea is the only FDA-approved drug for the treatment of SCD; currently, the recommendation is to offer this agent to all children with sickle cell anemia after nine months of age regardless of clinical severity. Hydroxyurea is a potent HbF inducer in adults3,4 and children;5 however, it does not possess the ideal combination of efficacy, safety and ease of use. Data generated in the Cooperative Study of Sickle Cell Disease, verified that HbF levels above 8.6% significantly impacted mortality in SCD providing clinic proof of the protective effect of HbF.1 Thus, understanding mechanisms involved in γ-globin gene regulation is critical to designing additional therapeutic agents and a cure.
Two major mechanisms for γ-globin silencing during development have been established including competitive interaction of the γ- and β-globin genes with the upstream β-locus control region during the fetal to adult switch6,7 and gene-autonomous γ-globin silencing.8 The latter mechanism provides the basis for direct gene-based strategies to induce HbF after birth to cure individuals with β-hemoglobinopathies. Several transcription factors play a major role in γ-globin silencing during development including TR2/TR4, MYB, BCL11A, GATA1 and KLF1 (Figure 1). The direct repeat erythroid-definitive (DRED) complex binds a direct repeat element overlapping the distal CCAAT box in the ɛ- and γ-globin promoters.9 Point mutations in the direct repeat at base -117 produces elevated γ-globin expression after birth.10,11 Work by Cui et al. demonstrated that binding of the DRED complex composed of TR2/TR4 and the cofactors DNA methyltransferase 1 (DNMT1) and lysine-specific histone demethylase 1 (LSD1), directly represses γ-globin gene expression in definitive erythroid cells.12 DNMT1 methylates CpG dinucleotides to maintain stable DNA methylation and long-term gene silencing.13 By contrast, LSD1 removes the methyl group from mono- and dimethyl-histone H3 lysine 4 to decrease epigenetic marks associated with transcriptionally active genes.14 Drugs that directly inhibit DNMT1 and LSD1 serve as a foundation for the development of novel HbF inducers.
Figure 1.

Major transcription factors involved in developmental γ-globin silencing. Shown is a schematic of the β-globin locus including the locus control region (LCR) comprised on hypersensitive sites 1–5 (white boxes). The major repressor proteins involved in γ-globin regulation demonstrated by developmental studies in transgenic mice and humans are shown. KLF1 bind directly in the β-globin gene promoter and interacts mainly with hypersensitive site 3 to facilitate competitive β-globin gene expression during adult development (green arrows). KLF1 also activates BCL11A to promote γ-globin silencing through BCL11A binding in hypersensitive site 3 and downstream of Aγ-globin (red lines). Displayed are different BCL11A protein-partners involved in γ-globin repression. The non-steroidal nuclear receptors TR2 and TR4 along with major components of the DRED complex, DNMT1 and LSD1 bind the direct repeat elements in the ɛ-globin and γ-globin promoters.
LSD1: lysine-specific histone demethylase 1; DMNT1: DNA methyltransferase 1; BCL11A: B-cell CLL/lymphoma 11 A.
Insights into γ-globin regulation have been garnered from human genetic studies. Through quantitative trait locus analysis, Thein et al.15 identified the intergenic region between the HBS1L and MYB genes associated with inherited HbF levels. Later studies in newborns with trisomy 13 and high HbF levels led to the discovery of two microRNAs, miR-15 a and miR-16-1 mediating repression of MYB gene expression in adult CD34+ stem cell-derived erythroid progenitors.16 Disruption of MYB in mice produced increased ɛ-globin and γ-globin expression17 supporting a role of MYB in γ-gene silencing during development. Additional studies demonstrated the ability of MYB to activate KLF1 and TR2/TR4 in human erythroid cells18 as an indirect mechanism of γ-globin silencing. KLF1 is a master regulator of adult β-globin expression and many other aspects of normal erythropoiesis.19 In support of an inherited phenotype, Borg et al.20 identified a Maltese family with elevated HbF levels associated with a nonsense mutation in the KLF1 gene. Subsequently, other families have been identified with different KLF1 mutations that produce a haplo-insufficiency phenotype and elevated HbF21; experimental data determined that KLF1 represses γ-globin indirectly through B-cell CLL/lymphoma 11 A (BCL11A) activation.22
The discovery of BCL11A, a major repressor of γ-globin expression, has made a significant impact towards efforts to develop gene-based strategies to reactivate γ-globin expression during adult-stage development. During hematopoietic cell differentiation, BCL11A is down-regulated and is possibly involved in lymphoma pathogenesis since translocations associated with B-cell malignancies also deregulate BCL11A expression.23,24 Related to inherited HbF levels, BCL11A was first identified by human quantitative trait locus studies,25 and later confirmed to be a genetic modifier of γ-globin transcription by genome wide association studies in thalassemia26 and sickle cell patients.27 BCL11A interacts with GATA1, FOG1 and the NuRD repressor complex (Figure 1).28 Disruption of the Mi2β subunit of NuRD results in increased γ-globin gene expression in transgenic mice.29 Recently, it was shown that partial knockdown of Mi2β in primary erythroid cells results in significant γ-globin gene activation without affecting erythroid differentiation. Moreover, the effect of Mi2β on γ-globin gene silencing in part was due to down regulation of BCL11A and KLF1.
Using molecular analysis of human primary erythroid cultures, interactions between BCL11A and SOX6 across the β-locus were demonstrated as another mechanism of γ-globin silencing.30 However, significant concerns have been raised over the potential negative effects of knocking out BCL11A gene expression in hematopoietic stem cells and the risk for B-cell malignancies. Recent studies were conducted by Orkin and colleagues to characterize an enhancer in intron 1 of BCL11A, which can be target for erythroid-specific gene silencing.31 The guided knockout of this element in human stem cells using zinc finger nucleases and CRISPR-Cas approaches are underway for the development of gene therapy for β-hemoglobinopathies. Alternative strategies including drug-mediated modulation of major regulators of globin gene expression are also being explored. Macari et al.32 demonstrated inhibition of BCL11A and KLF1 along with activation of NRF2 signaling (discussed below) by the cholesterol reducing agent simvastatin, as a mechanism of γ-globin activation in primary erythroid cells. The development of additional therapeutic agents is desirable.
Modulation of epigenetic marks to activate HbF expression
There exists a large group of chromatin modifying proteins, which regulate epigenetic marks through modification of DNA and histone proteins which allows the interaction of transcription factors to consensus motifs to fine-tune gene expression during development. DNA methylation of the embryonic and fetal globin genes inversely correlates with the level of gene expression. The therapeutic activity of the DNMT1 inhibitors 5-azacytidine and decitabine produces demethylation at the γ-globin promoters and HbF induction.33,34 Decitabine is currently in clinical trials for the treatment of SCD (ClinicalTrials.gov Identifier: NCT01685515). Another chromatin modifying protein LSD1 demethylates mono- and dimethyl histone H3 lysine 4, an active histone mark. The FDA-approved antidepressant tranylcypromine, which is an LSD1 inhibitor, induces HbF synthesis in adult erythroid progenitors; combination therapy with decitabine produced a synergistic effect.35 Subsequent studies showed inhibition of erythroid maturation by tranylcypromine, which precludes further clinical development. However, another LDS1 inhibitor, RN-1, was recently shown to induce HbF in sickle cell mice with increased F-cells36 and decreased numbers of sickle red cells in the peripheral blood, suggesting RN-1 may also reduce disease pathology.
Another epigenetic mark targeted to achieve high-level HbF induction is histone acetylation, which is most commonly associated with active gene transcription. Of the large number of histone deacetylase inhibitors tested in tissue culture systems and demonstrated to induce HbF, subsequent human trials verified the ability of arginine butyrate to induce HbF to clinically relevant levels.37 Butyrate inhibits the function of several histone deacetylases producing hyperacetylation of mainly histones 3 and 4 and activation of the p38 mitogen activated protein kinase (MAPK) signaling pathway as a dual mechanism of γ-gene reactivation (discussed in detail below). However, further development of butyrate has been hindered by the requirement of intravenous administration. Major mediators of histone methylation (an active gene mark) are the protein arginine methyltransferases (PRMT). PRMT1 is associated with human γ-globin gene silencing through association with a repressor protein named friend of PRMT1.38 Knockdown of this repressor protein mediates increased γ-globin gene expression in cultured human primary erythroid cells.
Discovery of novel HbF inducers
Tissue culture systems are an invaluable tool for the development of novel HbF inducers; however, they have limitations in their predictive value with regard to response in people with β-hemoglobionopathies. Tissue culture provides an in vitro model that can be easily manipulated with different pharmacologic agents to determine the effects on globin gene expression; however, these observations may not accurately reflect developmental mechanisms involved in globin gene regulation. The most widely utilized cell lines namely K562 and mouse erythroleukemia cells serve as rapid screening tools, but the results obtained in these systems often are not reproduced in primary erythroid cultures or human trials.2,39,40 Our group characterized KU812 leukemia cells where significant γ-globin and low-level β-globin expression is observed at steady-state, as a system for chemical drug screens.41 Known HbF inducers and novel agents were identified in KU812 cells and later confirmed in primary erythroid cells.
Over the last two decades, numerous primary liquid culture systems have been established to generate hematopoietic progenitors from peripheral blood mononuclear and CD34+ stem cells.42,43 Primary erythroid cells provide a valuable model for identifying transcription factors and epigenetic targets for modulating γ-globin transcription. Yet, there is not a one-to-one correlation between drug-mediated HbF induction in primary cultures and clinical response in β-hemoglobinopathy patients. One caveat for studies using primary erythroid cells is that γ-globin gene expression should be measured after considerable erythroid differentiation has occurred, i.e. when γ-globin and β-globin expression levels approach those observed in adult erythroid cells. Drugs that interfere with erythroid differentiation might result in significant increases in γ-globin expression in this model but may have deleterious effects on erythropoiesis. Variation in erythroid maturation must be taken into consideration when assessing the therapeutic potential of different agents.
Over the last four decades, more than 70 pharmacologic compounds have been shown to induce HbF synthesis mainly in vitro in tissue culture systems with limited preclinical animal studies and clinical trials. While major transcription factors involved in developmentally regulated hemoglobin switching have been identified through human and murine genetic studies (described above), by contrast most drug therapies reactivate HbF through modulation of DNA-binding proteins and/or epigenetic modifications in adult erythroid cells. These agents include 5-azacytidine,44 decitabine,45,46 hydroxyurea,47 butyrate,48,49 short-chain fatty acid derivatives,50,51 simvastatin,32 pomalidimide,52 and fumaric acid esters53 among others. HbF induction is accomplished by diverse mechanisms such as inhibition of DNA methyl transferases and histone deacetylases, DNA damage, and immune-modulation.2,54 Recently, expanded insights into γ-globin regulation have been gained through the investigation of cell signaling pathways activated by pharmacologic agents, which is the focus of the remainder of this review.
Cell signaling mechanisms involved in erythroid differentiation
Commitment to the erythroid lineage and terminal differentiation requires activation of multiple signaling pathways mediated by interaction between erythropoietin (EPO) and the erythropoietin receptor (EPO-R). The latter is activated by EPO-induced dimerization55,56 and subsequent activation of signal transduction through Janus kinase 2 and signal transducer and activator 5 A (STAT5A) and STAT5B57; subsequent activation of MAPK and phosphatidylinositol 3-kinase also occurs.58 MAPK signaling involves three pathways including SAPK/JNK, ERK1/2 and p38 MAPK.59 ERK1/2 plays a role in proliferation of early erythroid precursors and terminal maturation,60 while SAPK/JNK and p38 MAPK are involved in erythroid differentiation.61,62 During normal erythropoiesis in the adult bone marrow, hemoglobin synthesis is controlled by erythroid-specific and ubiquitous transcription factors2; for example, ATF-2 is phosphorylated and activated by p38 MAPK to alter target gene transcription.63 The study of cell signaling molecules involved in γ-globin activation during erythropoiesis or in response to various pharmacologic agents has shed light on mechanisms of HbF induction thus providing therapeutic targets for the development of novel clinical treatments (Table 1). A full discussion of the complex network of proteins required to achieve normal erythropoiesis is beyond the scope of this review. However, these observations have provided a conceptual model for experimental approaches to ascertain the role of cell signaling effector molecules in γ-globin gene reactivation during adult development.
Table 1.
Major cell signaling pathways involved in drug-mediated HbF induction
| Drug | Class of drug | Cell signaling pathway | References |
|---|---|---|---|
| Erythropoietin | Growth factor | JAK/STAT, MAPK | Remy et al.,56 Wojchowski et al.,57 Seger and Krebs59 |
| Thalidomide | Immune modulator | p38 MAPK, cAMP | Aerbajinai et al.88 |
| Hydroxyurea | Ribonucleotide reductase inhibitor | p38 MAPK, cAMP, Cgmp | Park et al.,75 Lou et al.,77 Ikuta et al.,93 Tang et al.96 |
| Butyrate | HDAC inhibitor | p38, cGMP, cAMP | Witt et al.,71 Pace et al.,78 Sangerman et al.81 |
| Trichostatin A | HDAC inhibitor | p38 MAPK | Pace et al.78 |
| Scriptaid | HDAC inhibitor | p38 MAPK | Johnson et al.72 |
| Apicidin | HDAC inhibitor | p38 MAPK | Witt et al.73 |
| 5-Azacytidine | DNMT inhibitor | cAMP | Keefer et al.98 |
cAMP: cyclic adenosine monophosphate; cGMP: cyclic guanosine monophosphate; DNMT: DNA methyltransferase; HDAC: histone deacetylase; JAK/STAT: Janus kinase/signal transducers and activators of transcription; MAPK: mitogen-activated protein kinases; SCFAD: short-chain fatty acid derivative.
JAK/STAT signaling pathway
Erythropoietin was one of the first agents to be tested as an HbF inducer in baboons;64 however, its efficacy was not demonstrated in human trials.65 The STAT transcription factors are the most common downstream signaling molecules activated by EPO. There are six major STAT protein family members including STAT-1 through STAT-6, which exist as latent cytoplasmic proteins66 that can be activated by pharmacologic agents. Studies by Boosalis et al. demonstrated that short-chain fatty acid derivatives induce prolonged expression of the growth-promoting genes MYB and MYC.67 Furthermore, sodium butyrate and dimethyl butyrate mediate STAT-5 activation in the absence of histone hyperacetylation contributing to cellular proliferation and HbF induction by these agents.
Two STAT3 variants, STAT-3α and STAT-3β, have identical cognate DNA-binding motifs, but the latter acts as a dominant-negative isoform.68 Work from our laboratory (Figure 2) demonstrated that STAT3β silences γ-globin expression by means of a consensus-binding site at nucleotide +9 in the γ-globin 5′-untranslated region.69 In subsequent studies, we observed in vivo binding of STAT3 and GATA-1 to this region by chromatin immunoprecipitation assay in K562 and mouse erythroleukemia cells.70 DNA–protein interaction studies confirmed that a STAT3/GATA-1 complex is assembled in this region. STAT3β enforced expression in K562 cells reduced γ-globin expression which was reversed by GATA-1 over-expression. These data provide evidence that GATA-1 can reverse STAT3-mediated γ-globin silencing in erythroid cells.
Figure 2.

Transcription factors mediating γ-globin gene regulation via cell signaling pathways. Shown are the cells signaling pathways and downstream transcription factors demonstrated to bind the γ-globin gene promoter after drug treatments in erythroid cells.
ARE: antioxidant response element; ATF-2: activating transcription factor-2; CREB1: cAMP response element binding protein; Epo: erythropoietin; Gγ-globin cAMP response element; Jak: Janus kinase; MAPK: mitogen activated protein kinase; NRF2: Nuclear factor (erythroid-derived 2)-like 2; ROS: reactive oxygen species; STAT3: signal transducer and activator of transcription 3
γ-globin activation via p38 MAPK
A role for p38 MAPK cell signaling in HbF induction (Table 1) has been demonstrated for histone deacetylase inhibitors such as butyrate, trichostatin A, scriptaid, and apicidin71–73; by contrast, ERK MAPK is a negative regulator of γ-globin expression.71 Studies from our laboratory showed that butyrate and trichostastin A stimulated p38 MAPK phosphorylation via a H2O2-dependent mechanism, supporting a role for reactive oxygen species in HbF induction.74 Studies to define the mechanisms by which hydroxyurea induces HbF demonstrated its ability to active p38 MAPK signaling75 and act as a nitric oxide (NO) donor.76 We later demonstrated increased levels of nitrites and nitrates in K562 cells after hydroxurea treatment, which was reversed by the NO synthase inhibitor NG-monomethyl-L-arginine77 confirming the complexity of how hydroxyurea functions in erythroid progenitors.
Studies using K562 stable cell lines78 and primary erythroid progenitors79 have clarified molecular mechanisms for γ-globin gene regulation. Enforced p38 MAPK expression in K562 stable lines increased γ-globin transcription supporting transactivation by downstream DNA-binding proteins. Several transcription factors acting as downstream effectors of p38 MAPK such as ATF-1-4, CREB, and CREM80–82 are candidate regulators of γ-globin. We confirmed that butyrate and trichostatin A mediate CREB1 and ATF-2 phosphorylation and binding to an upstream cyclic AMP response element (CRE) located at -1222 in the Gγ-globin promoter (G-CRE)81 (Figure 2). We then completed studies to determine if p38 MAPK signaling is required for steady-state γ-globin expression independent of drug induction. We observed γ-globin silencing after p38 or CREB1 siRNA knockdown.83 Subsequent chromatin immunoprecipitation assays verified that CREB1 and its binding partner cAMP response element-binding protein (CBP) co-localize at the G-CRE region. These data support the role of p38 MAPK/CREB1 signaling in Gγ-globin gene transcription under steady-state conditions.
CREB1 is known to have a wide array of binding partners including CBP, ATF-2, cJun,81,82 KLF4,84 and MYB85 among others. To determine whether a multiprotein complex bind to the G-CRE, we conducted studies using erythroid progenitors generated from normal adult bone marrow CD34+ stem cells. Through co-immunoprecipitation studies, we confirmed protein–protein interactions between cJun/ATF-2 and CREB1/ATF-2 but not between CREB1 and cJun. Promoter pull-down assay confirmed co-localization of cJun, CREB1, and ATF-2 on the G-CRE.86 Subsequent studies were conducted using a chromatographically purified G-CRE/ATF2 protein complex and mass spectrometry analysis.87 We identified the major binding partners of ATF-2 including CREB1, cJun, Brg1, and histone deacetylase 2 among others. Immunoprecipitation assays verified interaction of these proteins with ATF2 in human CD34+ cells undergoing erythroid differentiation, which was correlated with γ-globin expression. We are currently characterizing the changes in chromatin domains and epigenetic marks in the G-CRE region as it related to γ-globin expression during hemoglobin switching.
Another class of drugs that mediate immunomodulatory effects, namely thalidomide and pomalidimide were shown to mediate HbF induction; pomalidomide is a structural analog of thalidomide. Thalidomide induces γ-globin expression in a dose-dependent manner but had no effect on β-globin expression.88 Furthermore, thalidomide treatment increased reactive oxygen species and p38 MAPK signaling along with histone H4 hyperacetylation suggesting different mechanisms of HbF induction by this agent. Using erythroid progenitors derived from human CD34+ stem cells isolated from normal and sickle cell donors, it was shown that pomalidomide is a potent HbF inducer but delays erythroid maturation.89 Subsequent studies in the preclinical sickle cell transgenic mice confirmed HbF induction at levels similar to hydroxyurea.52 Whether pomalidimide actives p38 MAPK signaling, as a mechanism of HbF induction was not studied; however, it is an inhibitor of tumor necrosis factor-α, Interleukin-6 and Interleukin-1β.90
NO and cyclic guanosine monophosphate signaling pathway
NO is a second messenger signaling molecule generated from L-arginine by the action of NO synthase.91 In the vasculature, NO reacts with iron in the active site of the enzyme guanylyl cyclase to stimulate cyclic guanosine monophosphate (cGMP) signaling.92 Elevated soluble guanylyl cyclase levels have been observed in erythroid progenitors expressing a γ-globin program compared with those expressing β-globin.93 Moreover, NO/cGMP signaling activates several transcription factors such as c-Fos and c-Jun,92 known to regulate γ-globin expression through binding at hypersensitive site 2 in the β-locus control region. In addition, NO increases Sp-1 binding to the CACCC box and may activate γ-globin by this mechanism.94
Of the known HbF inducers, hydroxyurea, hemin, and butyrate have been shown to induce cGMP signaling to increase γ-globin expression in K562 cells and human erythroid progenitors.93,95 Hydroxyurea is metabolized to NO to activate cGMP signaling. Secretion-associated and RAS-related (SAR) is a small guanosine triphosphate-binding protein known to be involved in protein trafficking from the endoplasmic reticulum to the Golgi complex. Tang et al. demonstrated that SAR and γ-globin gene mRNAs are induced after hydroxyurea treatment.96 Additional studies showed that stable expression of SAR in K562 cells increased γ-globin mRNA, while inhibiting ERK cell signaling consistent with a previously demonstrated negative role for ERK in γ-globin gene regulation.71
Later studies by the Ikuta lab focused on the ability of cGMP to stimulate cAMP-dependent pathways as a mechanism of γ-globin activation.97 Ikuta’s work suggested that the cAMP-dependent pathway plays a negative role in γ-globin regulation in K562 cells. By contrast, another group demonstrated cAMP production is required for HbF induction by hydroxyurea, butyrate, and 5-azacytidine in human CD34+ erythroid progenitors, while perturbation of cGMP production had only minimal effects.98 Therefore, the exact role of cAMP and cGMP in γ-globin regulation in erythroid cells requires additional studies.
In clinical trials, it was observed that hydroxyurea increases circulating NO levels in sickle cell patients;99 therefore, we quantified the level of nitrate and nitrite generated by this agent in erythroid progenitors in the presence of three NO synthase inhibitors.77 Hydroxyurea increased nitrates and nitrites levels and γ-globin transcription in this system. Furthermore, pretreatment with cGMP pathway inhibitors reversed γ-gene activation by hydroxyurea demonstrating direct activation of NO and cGMP cell signaling as a mechanism of HbF induction.
Oxidative stress and antioxidant response
When cells are exposed to high-level environmental oxidants such as ionizing and UV irradiation or elevated reactive oxygen species, they undergo oxidative stress.100 To counter these extrinsic and intrinsic attacks, cells invoke defense mechanisms through basal and inducible regulation of phase II cytoprotective enzymes including heme oxygenase 1, superoxide dismutase, catalase, NADPH: quinone oxidoreductase 1 (NQO1), and glutathione S-transferase among others.101 These enzymes share a common consensus sequence TGACnnnGC encoded in the 5′ upstream region of gene promoters known as the antioxidant response element (ARE).102–104 Later studies demonstrated the ubiquitously expressed transcription factor NRF2 binds the ARE as a heterodimer with the small Maf proteins (MafF, MafG or MafK) in response to oxidative stress.105 These data support NRF2 as a key regulator the cytoprotective enzymes through the ARE.106 NRF2 was initially identified as a protein bound to tandem NFE2/AP1 repeats in the locus control region of the β-globin locus in K562 cells.107 Subsequent knockout studies in mice determined that NRF2 was not essential for murine erythropoiesis.108 However, recent publications have brought attention to NRF2 related to its role in γ-globin activation and HbF induction in response to several NRF2 chemical inducers.32,109
NRF2/Keap1 repressor complex and NRF2 activation
The human NRF2 protein consists of 605 amino acid residues, which are organized in seven NRF2-ECH homology (Neh) domains (Figure 3a). The Neh2 domain interacts with the adapter repressor protein Kelch-like ECH-associated protein1 (Keap1)110 enriched with lysine residues that are targets for ubiquitination by ubiquitin ligase E3.111,112 The Neh 4 and 5 domains are bound by the transcription activators CBP and p300 when NRF2 interacts with different AREs to activate target gene transcription.113 Neh1 consists of a CNC-basic leucine zipper region, which dimerizes with small Maf proteins and serves as the NRF2 DNA-binding domain.114
Figure 3.

Schematic structure of NRF2 and Keap1 proteins. (a) Different domains in NRF2 including the Keap1 binding site (Neh2) and region of heterodimerization with small Maf proteins and DNA-binding domain (Neh1). Neh4 and Neh5 are domains for binding the co-activators CBP/p300. Neh6 domain is the binding domain to βTrCP, another ubiquitin ligase E3. (b) The regions in Keap1 required for binding to the E3 ligase Cul3/Rbx1 complex which ubiquitinates NRF2 and direct proteasome degradation. Also displayed is the NF2 binding domain including BTB , and DGR. The red circles represent residues reported to be phosphorylated by protein kinases. The triangles represent cysteine residues. The green triangles indicate reactive cysteine residues that function in oxidative stress sensing in the cell. Many antioxidant reagents target cysteine residue 151 which is labeled separately.
NRF2: Nuclear factor (erythroid-derived 2)-like 2; IVR: intervening region; Keap1: Kelch-like ECH-associated protein1; DGR: double glycine repeats; Neh: NRF2-ECH homology
The human Keap1 protein is composed of 625 amino acid residues (Figure 3b). It has three major domains, Broad complex, Tramtrack and Bric a brac (BTB), intervening region (IVR) and six Kelch/double glycine repeats (DGR) repeats. Keap1 forms homodimers and associates with the Cul3-dependent ubiquitin ligase E3 at the BTB domain.115 The DGR and the C-terminal regions associate with NRF2 under non-oxidative stress conditions.116 Keap1 protein has 27 cysteine residues, which mediates its role as a sensor for redox conditions in cells.
Mechanisms by which NRF2 is stabilized away from the Keap1-Cul3 inhibitory complex under oxidative condition have been studied extensively.101,117,118 In general, in the non-oxidative state, newly translated NRF2 is captured in the cytoplasm by Keap1 dimers through the DLG and ETGE motifs.119 The Keap1 dimer also interacts with the N-terminus of Cul3 to mediate NRF2 ubiquitination by a Cul3-dependent E3 ubiquitin ligase and rapid degradation through proteasome-dependent mechanisms.115 By contrast, when cellular oxidative insults or antioxidant drug treatment occur, reactive cysteine residues in Keap1 are oxidized.120 Mutagenesis studies identified three cysteine residues in Keap1 (Green arrows in Figure 3b) that alter NRF2 function under oxidative stress conditions.121,122 This reaction results in a conformational change in the structure of Keap1, dissociation from Cul3 and stabilization of NRF2. Subsequently, NRF2 is phosphorylated and translocated to the nucleus where it forms heterodimer with small Maf proteins and bind to AREs of many genes encoding cytoprotective enzymes.123
Recently, another ubiquitin E3 ligase, β-transducin repeats-containing protein (βTrCP) was shown to associate with NRF2 at the Neh6 domain after GSK3β mediated NRF2 phosphorylation at two serine residues (Ser335 and Ser338).124,125 Inhibition of GSK3β activity causes NRF2 activation by dephosphorylation and increased heme oxygenase 1 expression in Keap1−/− mouse embryonic stem cells126 suggesting the βTrCP-GSK3-NRF2 axis is another NRF2 regulation system independent of Keap1 regulation. Interestingly, transcriptome profiles demonstrated that Keap1 is highly expressed in CD34+ and CD71+ hematopoietic cells compared to other cell types whereas βTrCP is only highly expressed in CD71+ erythroid cells.127 Other mechanisms of NRF2 regulation include activation by MAPK signaling in diverse cell types.118 Studies using inhibitors of p38 MAPK (SB203580), ERK (PD98059), and JNK (SP600125) signaling, showed attenuation of the ability of d-aminolevulinic acid/sodium ferrous citrate treatment to mediate NRF2 induction of heme oxygenase 1 expression in cardiomyocytes.128 These studies demonstrate that MAPK signaling is involved in NRF2 activation and subsequent induction of cytoprotective enzymes.
HbF induction by NRF2 activators
The activators of NRF2 are categorized into nine chemical classes based upon differences in their capability to induce the NQO1 gene: (1) oxidizable diphenols and quione; (2) Michael reaction acceptor (olefins or acetylenes conjugated to electron-withdrawing groups; (3) isothiocyanates; (4) hydroperoxides; (5) trivalent arsenic derivatives; (6) divalent heavy-metal cations (lead and cadmium); (7) vicinal dithiols; (8) 1,2-dithiole-3-thiones; and (9) carotenoids and other conjugated polyenes.129–131 These molecules are chemically active and able to covalently modify sulfhydryl groups by alkylation, oxidation or reduction. The presence of ortho-hydroxyl group on the aromatic rings of the inducers enhances rapid reactivity with sulfhydryl compounds to raise inducer potency.131 By contrast, Keap1 serves as a sensor for the presence of NRF2 activators through its high content of cysteine residues. Many of these compounds have clinical utility for a wide range of diseases through NRF2 activation and induction of cytoprotective genes.132–135
Recently, NRF2 inducers have attracted attention in the globin field due to their ability to activate γ-globin transcription and HbF production in human erythroid cells (Table 2). Macari and Lowrey109 reported that the known NRF2 inducers tert-butylhydroquione (tBHQ), curcumin and 2-dithiole-3-thione, induce γ-globin and NQO1 transcription in K562 cells; moreover, increased NRF2 binding to the ARE region was observed in the promoters of these genes. In later studies, Lowrey and colleagues showed combining tBHQ and simvastatin (an FDA-approved drug used for cardiovascular disease), induced γ-globin and NQO1 transcription in an additive manner;32 simvastatin also induced HbF production in human primary erythroid progenitors (Table 2). To gain insights into molecular mechanisms, they observed that combined tBHQ and simvastatin treatment reduced the expression of two γ-globin repressors, KLF1 and BCL11A, suggesting a dual mechanism of γ-globin regulation. These agents could potentially be developed as γ-globin inducers to define a new class of therapeutic drugs for treating SCD and β-thalassemia.
Table 2.
Summary of drugs that induce HbF through the Keap1/NRF2 pathway
| Drug | Structure | Effect | References |
|---|---|---|---|
| tert-Butylhydroquinone (tBHQ) |  |
Induces NRF2 activation and binding to γ-globin promoter; Increase γ-globin expression and HbF level in K562 and primary erythroid progenitors | Macari and Lowrey109 |
| Simvastatin |  |
Increase γ-globin expression and HbF level in K562 and primary erythroid progenitors by NRF2 activation and inhibits β-globin transcription by suppression KLF1 and BCL11A | Macari et al.32 |
| MLN9708 (proteasome inhibitor) |  |
Inhibits proteasome mediated ubiquitination and degradation of NRF2; induces ROS generation and antioxidant response in B-CFU cells from SCD patient; induces NRF2 nuclear localization; Increases HbF level in K562 cells | Kunze et al.142 |
| Dimethylfumarate (DMF) | 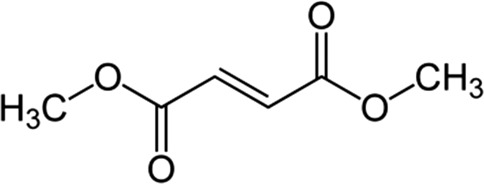 |
Increase γ-globin expression and HbF level in primary erythroid progenitors; induces NRF2 to activate γ-globin promoter | Promsote et al.53 |
NRF2: NRF2: Nuclear factor (erythroid-derived 2)-like 2; SCD: sickle cell disease; HbF: fetal hemoglobin.
Another group demonstrated a curcumin derivative, bisdemethoxy curcumin, induces γ-globin expression in primary erythroid progenitors.136 Although it is unclear whether NRF2 signaling is involved in this induction, another report showed that this agent induces heme oxygenase 1 transcription through NRF2 nuclear localization in a mouse B-cell line.137 Other drugs that target NRF2 have been investigated as HbF inducers; for example, sulforaphane is a potent NRF2 activator that is produced from its precursor, glucoraphanin.138 Young broccoli sprouts and cauliflower are enriched with glucoraphanin, from which sulforaphane is released by myrosinase when the plant is physically damaged.139 Recently, a clinical trial was initiated to test the ability of sulforaphane in broccoli sprout homogenates to induce HbF expression in human primary erythroid progenitors from healthy and sickle cell donors (ClinicalTrials.gov Identifier: NCT01715480). Triterpenoids are another group of NRF2 inducers140 with potential use in sickle cell patients, since they experience inflammation and oxidative stress. These agents have anti-inflammatory and antioxidative stress properties, especially bardoxolone methyl and 2-cyano-3,12-dioxooleana-1,9-dien-28-imidazolide. Additional research is required to demonstrate, if this class of drugs induces HbF in β-hemoglobinopathies.
Among the compounds discussed, tBHQ and sulforaphane preferably target cysteine residue 151 in the BTB domain of Keap1 (Figure 2), whereas the other agents do not target this cysteine.122 By contrast, it is known that NRF2 activation can occur by mechanisms independent of Keap1 inactivation. For example, in a recent report, the ability of the proteasome inhibitor MLN9708 to stabilize and enhance NRF2 nuclear localization was demonstrated.141 Studies in erythroid progenitors from sickle cell patients showed HbF induction by MLN9708, which was reversed by siRNA-mediated NRF2 silencing. These data suggest other regulatory steps of the NRF2/Keap1/Cul3-Rbx1 pathway can be targeted to achieve HbF induction in erythroid cells.
HbF induction by dimethyl fumarate
The fumaric acid esters are known NRF2 activators in different cell types such as cerebral endothelial cells and astrocytes,142 and lymphocytes and monocytes.143 These compounds have pleiotropic actions in a broad spectrum of tissues, and they have high tolerability and oral bioavailability. Pharmacokinetic studies indicate following oral intake, dimethyl fumarate (DMF) is rapidly hydrolyzed and converted into monomethyl fumate (MMF), which mediates a covalent modification at cysteine residue 151 of Keap1 to inactivate the repressor protein followed by NRF2 stabilization and target gene activation.144 Recent FDA approval of Tecfidera (DMF, Biogen Idec) for use in adults with multiple sclerosis makes this agent attractive for rapid extrapolation to clinical trials in SCD and β-thalassemia. A phase 3 trial to test the safety and efficacy of Tecfidera in pediatrics was initiated in Europe (ClinicalTrials.gov Identifier: NCT02283853).
Recent studies from our laboratory confirmed the ability of DMF and MMF to induce γ-globin transcription and HbF expression to levels comparable to hydroxyurea in primary erythroid cells.53 In our published works, DMF treatment for 48 h produced four-fold γ-globin induction and a concomitant 26-fold increase in HbF-positive cells by flow cytometry analysis with anti-γ-FITC antibody. Western blot confirmed an increase in HbF protein by DMF and its active metabolite MMF comparable to hydroxyurea. Later studies were conducted to address the question whether DMF induces HbF via NRF2 activation. To test this mechanism, we generated human primary erythroid cells in liquid culture from adult bone marrow CD34+ stem cells. As shown in Figure 4(a), after DMF treatment the level of NRF2 transcription increased 2.7-fold with a simultaneous increase in NRF2 protein by Western blot analysis (Figure 4b); by contrast, hydroxyurea did not increase NRF2 levels.
Figure 4.

DMF induces NRF2 expression in human erythroid progenitors. Primary erythroid cells were generated in liquid culture established with adult bone marrow CD34+ stem cells as previously published.53 On day 8, cells were treated with the various drugs for 48 h and then harvested for RNA isolation. NRF2 expression levels were measured by reverse transcription-quantitative PCR (a) and protein was isolated for western blot analysis (b). *p < 0.05 Primary erythroid cells were treated with siRNA targeted to NRF2 (siNRF2) using Dharmafect reagent (Thermo Fisher Scientific, Pittsburg, PA) on day 8 in culture. After 48 h RNA was harvested to measure NRF2 expression by reverse transcription-quantitative PCR (c). In the same sample, the effect of siNRF2 treatment on the ability of DMF to induce γ-globin was investigated. Pretreatment with siNRF2 inhibited DMF-mediated γ-globin activation (d). **p < 0.01.
NRF2: Nuclear factor (erythroid-derived 2)-like 2; DMF: dimethylfumarate
Subsequent studies were conducted in primary erythroid cells to test the effects of NRF2 gene silencing using siRNA treatment in the presence and absence of DMF; siNRF2 treatment alone produced 60% target gene silencing compared to scrambled siRNA controls (Figure 4(c)). Combination treatment with siNRF2 inhibited the ability of DMF to activate γ-globin transcription demonstrated by 50% γ-globin silencing, below steady-state levels (Figure 4(d)). These studies support a role of NRF2 signaling in DMF-mediated γ-globin activation; however, whether this agent will be useful for the treatment of β-hemoglobinopathies required animal studies and clinical trials.
Based on these finding we propose the model shown in Figure 5 for γ-globin activation by DMF which is known to target cysteine residue 151 to deactivate the repressor protein Keap1.144 As a result, NRF2 is stabilized and translocated to the nucleus to activate the ARE located in the γ-globin promoters. A similar mechanism for HbF induction has been established for simvastatin.32 We also have experimental data that DMF stimulates reactive oxygen species in K562 cells, which might activate p38 MAPK signaling as a second mechanism of γ-globin activation by this agent (B. P. unpublished). Additional studies are underway in our lab to test the ability of DMF to induce HbF in vivo using the preclinical β-YAC mouse model carrying the entire 248 kb β-locus.145 This transgenic line has a γ-globin promoter inducible by 5-azacytidine, butyrate146 and tranylcypromine, a lysine-specific demethylase inhibitor.147 Our data support the possible re-purposing of Tecfidera (DMF) as a stand-alone therapy or combined with hydroxyurea for the treatment of SCD or β-thalassemia.
Figure 5.

Model of drug-mediated γ-globin activation via NRF2 stabilization. Under basal conditions, NRF2 is sequestered in the cytoplasm by Keap1 dimers, ubiquitinated by the Cul3-Rbx1 ubiquitin ligase E3 complex, and targeted for proteasome-dependent degradation (left side). Treatment with tBHQ, simvastatin and DMF result in chemical modification of the repressor protein Keap1 at cysteine residue 151 which changes the conformation of Keap1 and facilitates NRF2 stabilization and nuclear translocation (right side). Subsequent heterodimerization with small Maf proteins mediates binding of NRF2 to the γ-globin promoter ARE (antioxidant response element) located at -100 base pairs upstream of the cap site.
NRF2: Nuclear factor (erythroid-derived 2)-like 2; Keap1: Kelch-like ECH-associated protein1; DMF: dimethylfumarate; tBHQ: tert-butylhydroquinone; ARE: antioxidant response element
Future perspectives
Strategies to induce HbF expression include the use of pharmacologic agents that alter epigenetic marks or cell signaling pathways which allow DNA-binding proteins to transactivate gene expression. Alternate strategies include inhibition of major repressor proteins that silence γ-globin transcription during development. After decades of investigations only one drug, hydroxyurea, is FDA approved for the treatment of adults with SCD.4 However, hydroxyurea is not effective in all sickle cell patients and it is less effective in β-thalassemia.148 These observations provide a rational for continued research to develop safe and useful alternative drug therapies. Changes in chromatin structure mediated by histone deacetylase and DNA methyltransferase inhibitors is a valuable approach to reactivate γ-globin but there remains challenges in translating tissue culture findings into clinical efficacy. For example, butyrate must be given intravenously49 and decitabine requires stabilization with tetrahydrouridine to achieve oral bioavailability in sickle cell patients.149
As cell signaling mechanisms of gene regulation have emerged, additional approaches have been pursued to reactivate γ-globin transcription. It is known that stage-specific transcription factors2 activated by multiple cellular signaling pathways are required to achieve normal hemoglobin switching and erythroid differentiation. Interestingly, treatment with erythropoietin increases red cell production and HbF in culture150 but was not effective in clinical trials in sickle cell patients.151 By contrast, a wide variety of drugs stimulate p38 MAPK signaling including hydroxyurea,75 butyrate,71,79,81 and thalidomide88 thus distinguishing several agents with clinical potential. Despite progress made in identifying novel agents using tissue culture systems, there remains limitation on the number of preclinical sickle cell mouse and baboons models to test drug efficacy to for the translation of new agents in to clinical trials. Realizing the cost and length of time required to bring drugs from bench to bedside, the NIH partnered with pharmaceutical companies to support efforts to re-purpose FDA-approved drugs. This provides opportunity to test agents with different mechanisms of action to develop combination drug regimens for treating β-hemoglobinopathies similar to that used for cancer and other diseases.
In this review, we discussed several compounds which stimulate NRF2 signaling to reactivate γ-globin transcription, holding promise for development as HbF inducers. For example tBHQ, simvastatin and DMF activate NRF2 signaling to facilitate HbF synthesis in human primary erythroid cells. Other small molecules that target the Keap1/NRF2 axis have been identified such as supercurcumin, hemin152 and bardoxolone methyl that might be targeted for therapeutic development. Of interest to our group is the FDA-approved drug Tecfidera (DMF) which we established as an HbF inducer in human primary erythroid cells.53 Recently, Belcher et al. demonstrated that DMF stimulated expression of heme oxingenase 1, ferritin and other antioxidant proteins in NY1DD transgenic sickle mice.153 Furthermore, blood cells isolated from DMF-treated sickle mice showed significantly decreased adhesion to heme- and TNF-activated human umbilical vein endothelial cells. They concluded that DMF may be an effective agent to prevent sickle pain episodes. Tecfidera holds promise for the treatment of SCD; however, additional preclinical data are needed to move this agent to clinical trials.
Gene therapy for the β-hemoglobinopathies involves several approaches either the addition of a normal γ- or β-globin gene using lentivirus-based vectors, correction of β-globin mutations or silencing of major γ-globin repressor proteins. The addition of a normal β-globin gene has been used to cure a few β-thalassemia patients;154 work continues to extend this approach to sickle cell patients. To identify repressors of γ-globin expression, human genetic and developmental murine studies established TR2/TR4, BCL11A, and KLF1 as potential targets for gene therapy. Naturally occurring mutations in KLF1 provide nucleotides to target for mutagenesis to create a haplosufficiency state and high HbF levels in mature erythrocytes. An alternate approach was made possible with the discovery of the erythroid-specific enhancer in BCL11A,31 which address concerns about total knockout of this factor and risks for carcinogenesis. Rapid advances in selective DNA mutation using CRISPR-Cas techniques provide the technology to achieve erythroid-specific BCL11A knockout to inhibit γ-globin silencing during development. Once these techniques are proven safe in human stem cells then gene therapy for β-hemoglobinopathies by this approach will be possible.
Other molecular molecules are being pursued as potential γ-globin inducers include miRNAs that target the 3′untranslated region of mRNA to mediate gene silencing. Recently, it was shown that miR-144 targets α-globin to prevent its precipitation in red blood cells,155 and it is associated with severe anemia in sickle cell patients. Interestingly, miR-144 is a negative regulator of NRF2156 a known γ-globin transactivator. Many miRNA molecules are targeted for clinical development, thus treatment with antagomirs to repress miR-144 might hold potential as a therapeutic option to induce HbF in β-hemoglobinopathy disorders.
Acknowledgements
This work was supported by grant HL69234 from the National Heart, Lung, and Blood Institute to Dr. Betty Pace. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authors’ contributions
BSP designed and supervised all experimental studies and writing and editing of the review article. LL conducted a literature search and contributed to writing the review article and previously published experimental data. LHM conducted studies to generate data related to mechanism of γ-globin induction by dimethyl fumarate and contributed to writing and editing the review article. BL conducted experiments to generate data related to dimethyl fumarate experiments and contributed to writing and editing the review article.
References
- 1.Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Life expectancy and risk factors for early death. N Engl J Med 1994; 330: 1639–44. [DOI] [PubMed] [Google Scholar]
- 2.Stamatoyannopoulos G, Grosveld F. Hemoglobin switching. In: Stamatoyannopoulos G, Majerus PW, Perlmutter RM, Varmus H (eds) The molecular basis of blood disease (Vol. 3). Philadelphia: Saunders, 2001.
- 3.Platt OS, Orkin SH, Dover G, Beardsley GP, Miller B, Nathan DG. Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J Clin Invest 1984; 74: 652–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, McMahon RP, Bonds DR. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the multicenter study of hydroxyurea in sickle cell anemia. N Engl J Med 1995; 33: 1317–22. [DOI] [PubMed] [Google Scholar]
- 5.Wang WC, Ware RE, Miller ST, Iyer RV, Casella JF, Minniti CP, Rana S, Thornburg CD, Rogers ZR, Kalpatthi RV, Barredo JC, Brown RC, Sarnaik SA, Howard TH, Wynn LW, Kutlar A, Armstrong FD, Files BA, Goldsmith JC, Waclawiw MA, Huang X, Thompson BW. BABY HUG investigators. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 2011; 377: 1663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carter D, Chakalova L, Osborne CS, Dai YF, Fraser P. Long-range chromatin regulatory interactions in vivo. Nat Genet 2002; 32: 623–6. [DOI] [PubMed] [Google Scholar]
- 7.Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W. Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol Cell 2002; 10: 1453–65. [DOI] [PubMed] [Google Scholar]
- 8.Raich N, Enver T, Nakamoto B, Josephson B, Papayannopoulou T, Stamatoyannopoulos G. Autonomous developmental control of human embryonic globin gene switching in transgenic mice. Science 1990; 250: 1147–9. [DOI] [PubMed] [Google Scholar]
- 9.Tanabe O, Katsuoka F, Campbell AD, Song W, Yamamoto M, Tanimoto K, Engel JD. An embryonic/fetal beta-type globin gene repressor contains a nuclear receptor TR2/TR4 heterodimer. EMBO J 2002; 21: 3434–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gelinas R, Endlich B, Pfeiffer C, Yagi M, Stamatoyannopoulos G. G to A substitution in the distal CCAAT box of the Aγ-globin gene in Greek hereditary persistence of fetal haemoglobin. Nature 1985; 313: 323–5. [DOI] [PubMed] [Google Scholar]
- 11.Fucharoen S, Shimizu K, Fukumaki Y. A novel C-T transition within the distal CCAAT motif of the Gγ-globin gene in the Japanese HPFH: implication of factor binding in elevated fetal globin expression. Nucleic Acids Res 1990; 18: 5245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cui S, Kolodziej KE, Obara N, Amaral-Psarris A, Demmers J, Shi L, Engel JD, Grosveld F, Strouboulis J, Tanabe O. Nuclear receptors TR2 and TR4 recruit multiple epigenetic transcriptional corepressors that associate specifically with the embryonic beta-type globin promoters in differentiated adult erythroid cells. Mol Cell Biol 2011; 31: 3298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lei H, Oh SP, Okano M, Jüttermann R, Goss KA, Jaenisch R, Li E. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development 1996; 122: 3195–205. [DOI] [PubMed] [Google Scholar]
- 14.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004; 119: 941–53. [DOI] [PubMed] [Google Scholar]
- 15.Thein SL, Menzel S, Peng X, Best S, Jiang J, Close J, Silver N, Gerovasilli A, Ping C, Yamaguchi M, Wahlberg K, Ulug P, Spector TD, Garner C, Matsuda F, Farrall M, Lathrop M. Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc Natl Acad Sci USA 2007; 104: 11346–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sankaran VG, Menne TF, Šćepanović D, Vergilio JA, Ji P, Kim J, Thiru P, Orkin SH, Lander ES, Lodish HF., Sankaran VG, Menne TF, Šćepanović D, Vergilio JA, Ji P, Kim J, Thiru P, Orkin SH, Lander ES, Lodish HF. MicroRNA-15a and -16-1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proc Natl Acad Sci USA 2011; 108: 1519–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki M, Yamazaki H, Mukai HY, Motohashi H, Shi L, Tanabe O, Engel JD, Yamamoto M, Suzuki M, Yamazaki H, Mukai HY, Motohashi H, Shi L, Tanabe O, Engel JD, Yamamoto M. Disruption of the Hbs1l-Myb locus causes hereditary persistence of fetal hemoglobin in a mouse model. Mol Cell Biol 2013; 33: 1687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bianchi E, Zini R, Salati S, Tenedini E, Norfo R, Tagliafico E, Manfredini R, Ferrari S. c-myb supports erythropoiesis through the transactivation of KLF1 and LMO2 expression. Blood 2010; 116: e99–e110. [DOI] [PubMed] [Google Scholar]
- 19.Siatecka M, Bieker JJ. The multifunctional role of EKLF/KLF1 during erythropoiesis. Blood 2011; 118: 2044–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borg J, Papadopoulos P, Georgitsi M, Gutiérrez L, Grech G, Fanis P, Phylactides M, Verkerk AJ, van der Spek PJ, Scerri CA, Cassar W, Galdies R, van Ijcken W, Ozgür Z, Gillemans N, Hou J, Bugeja M, Grosveld FG, von Lindern M, Felice AE, Patrinos GP, Philipsen S. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet 2010; 42: 801–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gallienne AE, Dréau HM, Schuh A, Old JM, Henderson S. Ten novel mutations in the erythroid transcription factor KLF1 gene associated with increased fetal hemoglobin levels in adults. Haematologica 2012; 97: 340–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou D, Liu K, Sun CW, Pawlik KM, Townes TM. KLF1 regulates BCL11A expression and gamma- to beta-globin gene switching. Nat Genet 2010; 42: 742–4. [DOI] [PubMed] [Google Scholar]
- 23.Khaled WT, Choon Lee S, Stingl J, Chen X, Raza Ali H, Rueda OM, Hadi F, Wang J, Yu Y, Chin SF, Stratton M, Futreal A, Jenkins NA, Aparicio S, Copeland NG8Watson CJ, Caldas C, Liu P. BCL11A is a triple-negative breast cancer gene with critical functions in stem and progenitor cells. Nat Commun 2015; 6: 5987–5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dyer MJ. The pathogenetic role of oncogenes deregulated by chromosomal translocation in B-cell malignancies. Int J Hematol 2003; 77: 315–20. [DOI] [PubMed] [Google Scholar]
- 25.Menzel S, Garner C, Gut I, Matsuda F, Yamaguchi M, Heath S, Foglio M, Zelenika D, Boland A, Rooks H, Best S, Spector TD, Farrall M, Lathrop M, Thein SL. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet 2007; 39: 1197–9. [DOI] [PubMed] [Google Scholar]
- 26.Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, Usala G, Busonero F, Maschio A, Albai G, Piras MG, Sestu N, Lai S, Dei M, Mulas A, Crisponi L, Naitza S, Asunis I, Deiana M, Nagaraja R, Perseu L, Satta S, Cipollina MD, Sollaino C, Moi P, Hirschhorn JN, Orkin SH, Abecasis GR, Schlessinger D, Cao A. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci USA 2008; 105: 1620–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Solovieff N, Milton JN, Hartley SW, Sherva R, Sebastiani P, Dworkis DA, Klings ES, Farrer LA, Garrett ME, Ashley-Koch A, Telen MJ, Fucharoen S, Ha SY, Li CK, Chui DH, Baldwin CT, Steinberg MH. Fetal hemoglobin in sickle cell anemia: genome-wide association studies suggest a regulatory region in the 5' olfactory receptor gene cluster. Blood 2010; 115: 1815–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu J, Sankaran VG, Ni M, Menne TF, Puram RV, Kim W, Orkin SH. Transcriptional silencing of γ-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev 2010; 24: 783–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Costa FC, Fedosyuk H, Chazelle AM, Neades RY, Peterson KR. Mi2beta is required for gamma-globin gene silencing: temporal assembly of a GATA1-FOG1-Mi2 repressor complex in beta-YAC transgenic mice. PLoS Genet 2012; 8: e1003155–e1003155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yi Z, Cohen-Barak O, Hagiwara N, Kingsley PD, Fuchs DA, Erickson DT, Epner EM, Palis J, Brilliant MH. Sox6 directly silences epsilon globin expression in definitive erythropoiesis. PLoS Genet 2006; 2: e14–e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, Shao Z, Canver MC, Smith EC, Pinello L, Sabo PJ, Vierstra J, Voit RA, Yuan GC, Porteus MH, Stamatoyannopoulos JA, Lettre G, Orkin SH. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 2013; 342: 253–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Macari ER, Schaeffer EK, West RJ, Lowrey CH. Simvastatin and t-butylhydroquinone suppress KLF1 and BCL11A gene expression and additively increase fetal hemoglobin in primary human erythroid cell. Blood 2013; 121: 830–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeSimone J, Heller P, Hall L, Zwiers D. 5-Azacytidine stimulates fetal hemoglobin synthesis in anemic baboons. Proc Natl Acad Sci USA 1982; 79: 4428–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruiz MA, Rivers A, Ibanez V, Vaitkus K, Mahmud N, DeSimone J, Lavelle D. Hydroxymethylcytosine and demethylation of the γ-globin gene promoter during erythroid differentiation. Epigenetics 2015; 10: 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi L, Cui S, Engel JD, Tanabe O. Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nat Med 2013; 19: 291–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cui S, Lim KC, Shi L, Lee M, Jearawiriyapaisarn N, Myers G, Campbell A, Harro D, Iwase S, Trievel RC, Rivers A, DeSimone J, Lavelle D, Saunthararajah Y, Engel JD. The LSD1 inhibitor RN-1 induces fetal hemoglobin synthesis and reduces disease pathology in sickle cell mice. Blood 2015; pii: blood-2015-02–626259. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Atweh GF, Sutton M, Nassif I, Boosalis V, Dover GJ, Wallenstein S, et al. Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease. Blood 1999; 93: 1790–7. [PMC free article] [PubMed] [Google Scholar]
- 38.van Dijk TB, Gillemans N, Pourfarzad F, van Lom K, von Lindern M, Grosveld F, Philipsen S. Fetal globin expression is regulated by friend of PRMT1. Blood 2010; 116: 4349–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klein E, Ben-Bassat H, Neumann H, Ralph P, Zeuthen J, Polliack A, Vánky F. Properties of the K562 cell line, derived from a patient with chronic myeloid leukemia. Int J Cancer 1976; 18: 421–31. [DOI] [PubMed] [Google Scholar]
- 40.Ross J, Gielen J, Packman S, Ikawa Y, Leder P. Globin gene expression in cultured erythroleukemic cells. J Mol Biol 1974; 87: 697–714. [DOI] [PubMed] [Google Scholar]
- 41.Zein S, Lou RF, Sivanand S, Ramakrishnan V, Mackie A, Li W, Pace BS. KU812 Cell Line: model for identifying fetal hemoglobin inducing drugs. Expl Biol Med 2010; 235: 1385–94. [DOI] [PubMed] [Google Scholar]
- 42.Fibach E, Rachmilewitz EA. The two-step liquid culture: a novel procedure for studying maturation of human normal and pathological erythroid precursors. Stem Cells 1993; 11(Suppl 1): 36–41. [DOI] [PubMed] [Google Scholar]
- 43.Tsiftsoglou AS, Vizirianakis IS, Strouboulis J. Erythropoiesis: model systems, molecular regulators, and developmental programs. IUBMB Life 2009; 61: 800–30. [DOI] [PubMed] [Google Scholar]
- 44.Charache S, Dover G, Smith K, Talbot CC, Jr, Moyer M, Boyer S. Treatment of sickle cell anemia with 5-azacytidine results in increased fetal hemoglobin production and is associated with nonrandom hypomethylation of DNA around the gamma-delta-beta-globin gene complex. Proc Natl Acad Sci USA 1983; 80: 4842–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saunthararajah Y, Hillery CA, Lavelle D, Molokie R, Dorn L, Bressler L, Gavazova S, Chen YH, Hoffman R, DeSimone J. Effects of 5-aza-2'-deoxycytidine on fetal hemoglobin levels, red cell adhesion, and hematopoietic differentiation in patients with sickle cell disease. Blood 2003; 102: 3865–70. [DOI] [PubMed] [Google Scholar]
- 46.Lavelle D, Chin J, Vaitkus K, Redkar S, Phiasivongsa P, Tang C, Will R, Hankewych M, Roxas B, Singh M, Saunthararajah Y, Desimone J. Oral decitabine reactivates expression of the methylated gamma-globin gene in Papio anubis. Am J Hematol 2007; 82: 981–5. [DOI] [PubMed] [Google Scholar]
- 47.Charache S, Dover GJ, Moyer MA, Moore JW. Hydroxyurea-induced augmentation of fetal hemoglobin production in patients with sickle cell anemia. Blood 1987; 69: 109–16. [PubMed] [Google Scholar]
- 48.Perrine SP, Ginder GD, Faller DV, Dover GH, Ikuta T, Witkowska HE, Cai SP, Vichinsky EP, Olivieri NF. A short-term trial of butyrate to stimulate fetal-globin-gene expression in the beta-globin disorders. N Engl J Med 1993; 328: 81–6. [DOI] [PubMed] [Google Scholar]
- 49.Weinberg RS, Ji X, Sutton M, Perrine S, Galperin Y, Li Q, Liebhaber SA, Stamatoyannopoulos G, Atweh GF. Butyrate increases the efficiency of translation of gamma-globin mRNA. Blood 2005; 105: 1807–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pace BS, White GL, Dover GJ, Boosalis MS, Faller DV, Perrine SP. Short chain fatty acid derivatives induce fetal globin expression and erythropoiesis in vivo. Blood 2002; 100: 4640–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fucharoen S, Inati A, Siritanaratku N, Thein SL, Wargin WC, Koussa S, Taher A, Chaneim N, Boosalis M, Berenson R, Perrine SP. A randomized phase I/II trial of HQK-1001, an oral fetal globin gene inducer, in β-thalassaemia intermedia and HbE/β-thalassaemia. Br J Haematol 2013; 161: 587–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meiler SE, Wade M, Kutlar F, Yerigenahally SD, Xue Y, Moutouh-de Parseval LA, Corral LG, Swerdlow PS, Kutlar A. Pomalidomide augments fetal hemoglobin production without the myelosuppressive effects of hydroxyurea in transgenic sickle cell mice. Blood 2011; 118: 1109–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Promsote W, Makala L, Li B, Smith SB, Singh N, Ganapathy V, Pace BS, Martin PM. Monomethylfumarate induces γ-globin expression and fetal hemoglobin production in cultured human retinal pigment epithelial (RPE) and erythroid cells, and in intact retina. Invest Ophthalmol Vis Sci 2014; 55: 5382–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perrine SP, Pace BS, Faller DV. Targeted fetal hemoglobin induction for treatment of beta hemoglobinopathies. Hematol Oncol Clin North Am 2014; 28: 233–48. [DOI] [PubMed] [Google Scholar]
- 55.Watowich SS, Wu H, Socolovsky M, Klingmuller U, Constantinescu SN, Lodish HF. Cytokine receptor signal transduction and the control of hematopoietic cell development. Annu Rev Cell Dev Biol 1996; 12: 91–128. [DOI] [PubMed] [Google Scholar]
- 56.Remy I, Wilson IA, Michnick SW. Erythropoietin receptor activation by a ligand-induced conformation change. Science 1999; 283: 990–3. [DOI] [PubMed] [Google Scholar]
- 57.Wojchowski DM, Gregory RC, Miller CP, Pandit AK, Pircher TJ. Signal transduction in the erythropoietin receptor system. Exp Cell Res 1999; 253: 143–56. [DOI] [PubMed] [Google Scholar]
- 58.Munugalavadla V, Kapur R. Role of c-Kit and erythropoietin receptor in erythropoiesis. Crit Rev Oncol Hematol 2005; 54: 63–75. [DOI] [PubMed] [Google Scholar]
- 59.Seger R, Krebs EG. The MAPK signaling cascade. FASEB J 1995; 9: 726–35. [PubMed] [Google Scholar]
- 60.Arcasoy MO, Jiang X. Co-operative signalling mechanisms required for erythroid precursor expansion in response to erythropoietin and stem cell factor. Br J Haematol 2005; 130: 121–9. [DOI] [PubMed] [Google Scholar]
- 61.Bose C, Udupa KB. Erythropoietin enhancement of rat pancreatic tumor cell proliferation requires the activation of ERK and JNK signals. Am J Physiol Cell Physiol 2008; 295: C394–405. [DOI] [PubMed] [Google Scholar]
- 62.Dalmas DA, Tierney LA, Zhang C, Narayanan PK, Boyce RW, Schwartz LW, et al. Effects of p38 MAP kinase inhibitors on the differentiation and maturation of erythroid progenitors. Toxicol Pathol 2008; 36: 958–71. [DOI] [PubMed] [Google Scholar]
- 63.Baan B, van der Zon GC, Maassen JA, Ouwens DM. The nuclear appearance of ERK1/2 and p38 determines the sequential induction of ATF2-Thr71 and ATF2-Thr69 phosphorylation by serum in JNK-deficient cells. Mol Cell Endocrinol 2009; 311: 94–100. [DOI] [PubMed] [Google Scholar]
- 64.Al-Khatti A, Veith RW, Papayannopoulou T, Fritsch EF, Goldwasser E, Stamatoyannopoulos G. Stimulation of fetal hemoglobin synthesis by erythropoietin in baboons. N Engl J Med 1987; 317: 415–20. [DOI] [PubMed] [Google Scholar]
- 65.Nagel RL, Vichinsky E, Shah M, Johnson R, Spadacino E, Fabry ME, Mangahas L, Abel R, Stamatoyannopoulos G. F-reticulocyte response in sickle cell anemia treated with recombinant human erythropoietin: a double-blind study. Blood 1993; 81: 9–14. [PubMed] [Google Scholar]
- 66.Darnell JE., Jr STATs and gene regulation. Science 1997; 277: 1630–5. [DOI] [PubMed] [Google Scholar]
- 67.Boosalis MS, Bandyopadhyay R, Bresnick EH, Pace BS, Van DeMark K, Zhang B, Faller DV, Perrine SP. Short-chain fatty acid derivatives stimulate cell proliferation and induce STAT-5 activation. Blood 2001; 97: 3259–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schaefer TS, Sanders LK, Nathans D. Cooperative transcriptional activity of Jun and Stat3 beta, a short form of Stat3. Proc Natl Acad Sci USA 1995; 92: 9097–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Foley H, Ofori-Acquah S, Baliga BS, Pace BS. STAT3 mediates globin repression by interleukin-6 in K562 cells. J Biol Chem 2002; 77: 16211–9. [DOI] [PubMed] [Google Scholar]
- 70.Yao X, Kodeboyina S, Liu L, Dzandu J, Sangerman J, Ofori-Acquah SF, Pace BS. Role of STAT3 and GATA-1 interactions in gamma-globin gene expression. Exp Hematol 2009; 7: 889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Witt O, Sand K, Pekrun A. Butyrate-induced erythroid differentiation of human K562 leukemia cells involves inhibition of ERK and activation of p38 MAP kinase pathways. Blood 2000; 95: 2391–6. [PubMed] [Google Scholar]
- 72.Johnson J, Hunter R, McElveen R, Qian XH, Baliga BS, Pace BS. Fetal hemoglobin induction by the histone deacetylase inhibitor, scriptaid. Cell Mol Biol 2005; 51: 229–38. [PubMed] [Google Scholar]
- 73.Witt O, Monkemeyer S, Rönndahl G, Erdlenbruch B, Reinhardt D, Kanbach K, et al. Induction of fetal hemoglobin expression by the histone deacetylase inhibitor apicidin. Blood 2003; 101: 2001–7. [DOI] [PubMed] [Google Scholar]
- 74.Hsiao CH, Li W, Lou TF, Baliga BS, Pace BS. Fetal hemoglobin induction by histone deacetylase inhibitors involves generation of reactive oxygen species. Exp Hematol 2006; 34: 264–73. [DOI] [PubMed] [Google Scholar]
- 75.Park JI, Choi HS, Jeong JS, Han JY, Kim IH. Involvement of p38 kinase in hydroxyurea-induced differentiation of K562 cells. Cell Growth Differ 2001; 12: 481–6. [PubMed] [Google Scholar]
- 76.King SB. Arole for nitric oxide in hydroxyurea-mediated fetal hemoglobin induction. J Clin Invest 2003; 111: 171–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lou TF, Singh M, Mackie A, Li W, Pace BS. Hydroxyurea generates nitric oxide in human erythroid cells: mechanisms for gamma-globin gene activation. Exp Biol Med 2009; 234: 1374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pace BS, Qian XH, Sangerman J, Ofori-Acquah SF, Baliga BS, Han J, Critz SD. p38 MAP kinase activation mediates gamma-globin gene induction in erythroid progenitors. Exp Hematol 2003; 31: 1089–96. [DOI] [PubMed] [Google Scholar]
- 79.Uddin S, Ah-Kang J, Ulaszek J, Mahmud D, Wickrema A. Differentiation stage-specific activation of p38 mitogen activated protein kinase isoforms in primary human erythroid cells. Proc Natl Acad Sci USA 2004; 101: 147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Foulkes NS, Laoide BM, Schlotter F, Sassone-Corsi P. Transcriptional antagonist cAMP-responsive element modulator (CREM) down-regulates c-fos cAMP-induced expression. Proc Natl Acad Sci USA 1991; 88: 5448–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sangerman J, Lee MS, Yao X, Oteng E, Hsiao CH, Li W, Zein S, Ofori-Acquah SF, Pace BS. Mechanism for fetal hemoglobin induction by histone deacetylase inhibitors involves gamma-globin activation by CREB1 and ATF-2. Blood 2006; 108: 3590–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Benbrook DM, Jone NC. Heterodimer formation between CREB and JUN proteins. Oncogene 1990; 5: 295–302. [PubMed] [Google Scholar]
- 83.Ramakrishnan V, Pace BS. Regulation of γ-globin gene expression involves signaling through the p38 MAPK/CREB1 pathway. Blood Cells Mol Dis 2001; 47: 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kalra IS, Alam MM, Choudhary PK, Pace BS. Krüppel-like Factor 4 activates HBG gene expression in primary erythroid cells. Br J Haematol 2001; 154: 248–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sampurno S, Bijenhof A, Cheasley D, Xu H, Robine S, Hilton D, Alexander WS, Pereira L, Mantamadiotis T, Malaterre J, Ramsay RG., Sampurno S1, Bijenhof A, Cheasley D, Xu H, Robine S, Hilton D, Alexander WS, Pereira L, Mantamadiotis T, Malaterre J, Ramsay RG. The Myb-p300-CREB axis modulates intestine homeostasis, radiosensitivity and tumorigenesis. Cell Death Dis 2013; 4: e605–e605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kodeboyina S, Balamurugan P, Liu L, Pace BS. cJun modulates Ggamma-globin gene expression via an upstream cAMP response element. Blood Cells Mol Dis 2010; 44: 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu L, Karmakar S, Dhar R, Mahajan M, Choudhury A, Weissman S, Pace BS. Regulation of Gγ-globin gene by ATF2 and its associated proteins through the cAMP-response element. PLoS One 2013; 8: e78253–e78253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Aerbajinai W, Zhu J, Gao Z, Chin K, Rodgers GP. Thalidomide induces gamma-globin gene expression through increased reactive oxygen species-mediated p38 MAPK signaling and histone H4 acetylation in adult erythropoiesis. Blood 2007; 110: 2864–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moutouh-de Parseval LA, Verhelle D, Glezer E, Jensen-Pergakes K, Ferguson GD, Corral LG, Morris CL, Muller G, Brady H, Chan K. Pomalidomide and lenalidomide regulate erythropoiesis and fetal hemoglobin production in human CD34+ cells. J Clin Invest 2008; 118: 248–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Corral LG, Haslett PA, Muller GW, Chen R, Wong LM, Ocampo CJ, Patterson RT, Stirling DI, Kaplan G. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha. J Immunol 1999; 163: 380–6. [PubMed] [Google Scholar]
- 91.Gladwin MT, Crawford JH, Patel RP. The biochemistry of nitric oxide, nitrite, and hemoglobin: role in blood flow regulation. Free Radic Biol Med 2004; 36: 707–17. [DOI] [PubMed] [Google Scholar]
- 92.Haby C, Lisovoski F, Aunis D, Zwiller J. Stimulation of the cyclic GMP pathway by NO induces expression of the immediate early genes c-fos and junB in PC12 cells. J Neurochem 1994; 62: 496–501. [DOI] [PubMed] [Google Scholar]
- 93.Ikuta T, Ausenda S, Cappellini MD. Mechanism for fetal globin gene expression: role of the soluble guanylate cyclase-cGMP-dependent protein kinase pathway. Proc Natl Acad Sci USA 2001; 98: 1847–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fischer KD, Haese A, Nowock J. Cooperation of GATA-1 and Sp1 can result in synergistic transcriptional activation or interference. J Biol Chem 1993; 268: 23915–23. [PubMed] [Google Scholar]
- 95.Haynes J, Baliga BS, Obiako B, Pace BS. Zileuton induces hemoglobin F synthesis in erythroid progenitors: role of 12-lipoxygenase and nitric oxide. Blood 2004; 103: 3945–50. [DOI] [PubMed] [Google Scholar]
- 96.Tang DC, Zhu J, Liu W, Chin K, Sun J, Chen L, Hanover JA, Rodgers GP. The hydroxyurea-induced small GTP-binding protein SAR modulates gammaglobin gene expression in human erythroid cells. Blood 2005; 106: 3256–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Inoue A, Kuroyanagi Y, Terui K, Moi P, Ikuta T. Negative regulation of gamma-globin gene expression by cyclic AMP-dependent pathway in erythroid cells. Exp Hematol 2004; 32: 244–53. [DOI] [PubMed] [Google Scholar]
- 98.Keefer JR, Schneidereith TA, Mays A, Purvis SH, Dover GJ, Smith KD. Role of cyclic nucleotides in fetal hemoglobin induction in cultured CD34+ cells. Exp Hematol 2006; 34: 1151–61. [DOI] [PubMed] [Google Scholar]
- 99.Gladwin MT, Shelhamer JH, Ognibene FP, Pease-Fye ME, Nichols JS, Link B, Patel DB, Jankowski MA, Pannell LK, Schechter AN, Rodgers GP. Nitric oxide donor properties of hydroxyurea in patients with sickle cell disease. Br J Haematol 2002; 116: 436–44. [DOI] [PubMed] [Google Scholar]
- 100.Klaunig JE, Kamendulis LM, Hocevar BA. Oxidative stress and oxidative damage in carcinogenesis. Toxicol Pathol 2010; 38: 96–109. [DOI] [PubMed] [Google Scholar]
- 101.Niture SK, Khatri R, Jaiswal AK. Regulation of Nrf2-an update. Free Radic Biol Med 2014; 66: 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li Y, Jaiswal AK. Regulation of human NAD(P)H:quinone oxidoreductase gene. Role of AP1 binding site contained within human antioxidant response element. J Biol Chem 1992; 267: 15097–104. [PubMed] [Google Scholar]
- 103.Wasserman WW, Fahl WE. Functional antioxidant responsive elements. Proc Natl Acad Sci USA 1997; 94: 5361–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Prestera T, Holtzclaw WD, Zhang Y, Talalay P. Chemical and molecular regulation of enzymes that detoxify carcinogens. Proc Natl Acad Sci USA 1993; 90: 2965–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Motohashi H, Katsuoka F, Engel JD, Yamamoto M. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc Natl Acad Sci USA 2004; 101: 6379–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yamazaki H, Tanji K, Wakabayashi K, Matsuura S, Itoh K. Role of the Keap1/Nrf2 pathway in neurodegenerative diseases. Pathol Int 2015; 65: 210–9. [DOI] [PubMed] [Google Scholar]
- 107.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci USA 1994; 91: 9926–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci USA 1996; 93: 13943–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Macari ER, Lowrey CH. Induction of human fetal hemoglobin via the NRF2 antioxidant response signaling pathway. Blood 2011; 117: 5987–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999; 13: 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 2004; 24: 7130–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol 2004; 24: 10941–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001; 6: 857–68. [DOI] [PubMed] [Google Scholar]
- 114.Hirotsu Y, Katsuoka F, Funayama R, Nagashima T, Nishida Y, Nakayama K, Engel JD, Yamamoto M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res 2012; 40: 10228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J Biol Chem 2006; 281: 24756–68. [DOI] [PubMed] [Google Scholar]
- 116.Ogura T, Tong KI, Mio K, Maruyama Y, Kurokawa H, Sato C, Yamamoto M. Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc Natl Acad Sci USA 2010; 107: 2842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol 2013; 1: 45–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bhakkiyalakshmi E, Sireesh D, Rajaguru P, Paulmurugan R, Ramkumar KM. The emerging role of redox-sensitive Nrf2-Keap1 pathway in diabetes. Pharmacol Res 2015; 91: 104–14. [DOI] [PubMed] [Google Scholar]
- 119.Tong KI, Padmanabhan B, Kobayashi A, Shang C, Hirotsu Y, Yokoyama S, Yamamoto M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol Cell Biol 2007; 27: 7511–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Holland R, Hawkins AE, Eggler AL, Mesecar AD, Fabris D, Fishbein JC. Prospective type 1 and type 2 disulfides of Keap1 protein. Chem Res Toxicol 2008; 21: 2051–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Uruno A, Motohashi H. The Keap1-Nrf2 system as an in vivo sensor for electrophiles. Nitric Oxide 2011; 25: 153–60. [DOI] [PubMed] [Google Scholar]
- 122.Takaya K, Suzuki T, Motohashi H, Onodera K, Satomi S, Kensler TW, Yamamoto M. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic Biol Med 2012; 53: 817–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Marini MG, Chan K, Casula L, Kan YW, Cao A, Moi P. hMAF, a small human transcription factor that heterodimerizes specifically with Nrf1 and Nrf2. J Biol Chem 1997; 272: 16490–97. [DOI] [PubMed] [Google Scholar]
- 124.Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013; 32: 3765–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, Tobón-Velasco JC, Devijver H, García-Mayoral MF, Van Leuven F, Hayes JD, Bertho G, Cuadrado A. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/β-TrCP axis. Mol Cell Biol 2012; 2: 3486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rojo AI, Medina-Campos ON, Rada P, Zúñiga-Toalá A, López-Gazcón A, Espada S. Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a Keap1-independent regulation of Nrf2 stability: role of glycogen synthase kinase-3. Free Radic Biol. Med 2011; 52: 473–87. [DOI] [PubMed] [Google Scholar]
- 127.Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA 2004; 101: 6062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhao M, Guo H, Chen J, Wang J, Huang H, Zheng S, Hei M, Li J, Huang S, Li J, Ma X, Chen Y, Zhao L, Zhuang J, Zhu P. 5-Aminolevulinic acid combined with sodium ferrous citrate ameliorates H2O2-induced cardiomyocyte hypertrophy via activating the MAPK/Nrf2/HO-1 pathway. Am J Physiol Cell Physiol 2015; 308: C665–72. [DOI] [PubMed] [Google Scholar]
- 129.Talalay P, Fahey JW, Holtzclaw WD, Prestera T, Zhang Y. Chemoprotection against cancer by phase 2 enzyme induction. Toxicol Lett 1995; 82–83: 173–9. [DOI] [PubMed] [Google Scholar]
- 130.Dinkova-Kostova AT, Talalay P. Persuasive evidence that quinone reductase type 1 (DT diaphorase) protects cells against the toxicity of electrophiles and reactive forms of oxygen. Free Radic Biol Med 2000; 29: 231–40. [DOI] [PubMed] [Google Scholar]
- 131.Dinkova-Kostova AT, Massiah MA, Bozak RE, Hicks RJ, Talalay P. Potency of Michael reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulfhydryl groups. Proc Natl Acad Sci USA 2001; 98: 3404–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kweider N, Wruck C, Rath W. New insights into the pathogenesis of preeclampsia – the role of nrf2 activators and their potential therapeutic impact. Geburtshilfe Frauenheilkd 2013; 73: 1236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yang JH, Shin BY, Han JY, Kim MG, Wi JE, Kim YW, Cho IJ, Kim SC, Shin SM, Ki SH. Isorhamnetin protects against oxidative stress by activating Nrf2 and inducing the expression of its target genes. Toxicol Appl Pharmacol 2014; 274: 293–301. [DOI] [PubMed] [Google Scholar]
- 134.Palsamy P, Subramanian S. Resveratrol protects diabetic kidney by attenuating hyperglycemia-mediated oxidative stress and renal inflammatory cytokines via Nrf2-Keap1 signaling. Biochim Biophys Acta 2011; 1812: 719–31. [DOI] [PubMed] [Google Scholar]
- 135.Cui W, Bai Y, Luo P, Miao L, Cai L. Preventive and therapeutic effects of MG132 by activating Nrf2-ARE signaling pathway on oxidative stress-induced cardiovascular and renal injury. Oxid Med Cell Longev 2013; 2013: 306073–306073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chaneiam N, Changtam C, Mungkongdee T, Suthatvoravut U, Winichagoon P, Vadolas J, Suksamrarn A, Fucharoen S, Svasti S. A reduced curcuminoid analog as a novel inducer of fetal hemoglobin. Ann Hematol 2013; 92: 379–86. [DOI] [PubMed] [Google Scholar]
- 137.Pugazhenthi S, Akhov L, Selvaraj G, Wang M, Alam J. Regulation of heme oxygenase-1 expression by demethoxy curcuminoids through Nrf2 by a PI3-kinase/Akt-mediated pathway in mouse beta-cells. Am J Physiol Endocrinol Metab 2007; 293: E645–55. [DOI] [PubMed] [Google Scholar]
- 138.Perocco P, Bronzetti G, Canistro D, Valgimigli L, Sapone A, Affatato A, Pedulli GF, Pozzetti L, Broccoli M, Iori R, Barillari J, Sblendorio V, Legator MS, Paolini M, Abdel-Rahman SZ. Glucoraphanin, the bioprecursor of the widely extolled chemopreventive agent sulforaphane found in broccoli, induces phase-I xenobiotic metabolizing enzymes and increases free radical generation in rat liver. Mutat Res 2006; 595: 125–36. [DOI] [PubMed] [Google Scholar]
- 139.Thangstad OP, Winge P, Husebye H, Bones A. The myrosinase (thioglucoside glucohydrolase) gene family in Brassicaceae. Plant Mol Biol 1993; 23: 511–24. [DOI] [PubMed] [Google Scholar]
- 140.Owusu-Ansah A, Choi SH, Petrosiute A, Letterio JJ, Huang AY. Triterpenoid inducers of Nrf2 signaling as potential therapeutic agents in sickle cell disease: a review. Front Med 2015; 9: 46–56. [DOI] [PubMed] [Google Scholar]
- 141.Pullarkat V, Meng Z, Tahara SM, Johnson CS, Kalra VK. Proteasome inhibition induces both antioxidant and hb f responses in sickle cell disease via the nrf2 pathway. Hemoglobin 2014; 38: 188–95. [DOI] [PubMed] [Google Scholar]
- 142.Kunze R, Urrutia A, Hoffmann A, Liu H, Helluy X, Pham M, Reischl S, Korff T, Marti HH. Dimethyl fumarate attenuates cerebral edema formation by protecting the blood-brain barrier integrity. Exp Neurol 2015; pii: S0014–4886, 00052–7. [DOI] [PubMed] [Google Scholar]
- 143.Gill AJ, Kolson DL. Dimethyl fumarate modulation of immune and antioxidant responses: application to HIV therapy. Crit Rev Immunol 2013; 33: 307–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P, Zeng W, Hronowsky X, Buko A, Chollate S, Ellrichmann G, Brück W, Dawson K, Goelz S, Wiese S, Scannevin RH, Lukashev M, Gold R. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011; 134: 678–92. [DOI] [PubMed] [Google Scholar]
- 145.Peterson KR, Clegg CH, Huxley C, Josephson BM, Haugen HS, Furukawa T, et al. Transgenic mice containing a 248-kb yeast artificial chromosome carrying the human beta-globin locus display proper developmental control of human globin genes. Proc Natl Acad Sci USA 1993; 90: 7593–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pace B, Li Q, Peterson K, Stamatoyannopoulos G. Alpha-Amino butyric acid fails to reactivate the totally silenced gamma gene of the beta locus YAC. Blood 1994; 84: 4344–53. [PubMed] [Google Scholar]
- 147.Shi L, Cui S, Engel JD, Tanabe O. Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nat Med 2013; 19: 291–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kosaryan M, Zafari M, Alipur A, Hedayatizadeh-Omran A. The effect and side effect of hydroxyurea therapy on patients with β-thalassemia: a systematic review to December 2012. Hemoglobin 2014; 38: 262–71. [DOI] [PubMed] [Google Scholar]
- 149.Lavelle D, Vaitkus K, Ling Y, Ruiz MA, Mahfouz R, Ng KP, Negrotto S, Smith N, Terse P, Engelke KJ, Covey J, Chan KK, Desimone J, Saunthararajah Y. Effects of tetrahydrouridine on pharmacokinetics and pharmacodynamics of oral decitabine. Blood 2012; 119: 1240–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fibach E, Schechter AN, Noguchi CT, Rodgers GP. Reducing erythropoietin in cultures of human erythroid precursors elevates the proportion of fetal haemoglobin. Br J Haematol 1994; 88: 39–45. [DOI] [PubMed] [Google Scholar]
- 151.Rodgers GP, Dover GJ, Uyesaka N, Noguchi CT, Schechter AN, Nienhuis AW. Augmentation by erythropoietin of the fetal-hemoglobin response to hydroxyurea in sickle cell disease. N Engl J Med 1993; 328: 73–80. [DOI] [PubMed] [Google Scholar]
- 152.Foresti R, Bains SK, Pitchumony TS, de Castro Brás LE, Drago F, Dubois-Randé JL, Bucolo C, Motterlini R. Small molecule activators of the Nrf2-HO-1 antioxidant axis modulate heme metabolism and inflammation in BV2 microglia cells. Pharmacol Res 2013; 76: 132–48. [DOI] [PubMed] [Google Scholar]
- 153.Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, Smith A, Nath KA, Hebbel RP, Vercellotti GM. Dimethyl fumarate induces cytoprotection and inhibits inflammation and vaso-occlusion in transgenic sickle mice. Blood 2014; 124: 219–219. [Google Scholar]
- 154.Tubsuwan A, Abed S, Deichmann A, Kardel MD, Bartholomä C, Cheung A, Negre O, Kadri Z, Fucharoen S, von Kalle C, Payen E, Chrétien S, Schmidt M, Eaves CJ, Leboulch P, Maouche-Chrétien L. Parallel assessment of globin lentiviral transfer in induced pluripotent stem cells and adult hematopoietic stem cells derived from the same transplanted β-thalassemia patient. Stem Cells 2013; 31: 1785–94. [DOI] [PubMed] [Google Scholar]
- 155.Fu YF, Du TT, Dong M, Zhu KY, Jing CB, Zhang Y, Wang L, Fan HB, Chen Y, Jin Y, Yue GP, Chen SJ, Chen Z, Huang QH, Jing Q, Deng M, Liu TX. Mir-144 selectively regulates embryonic α-hemoglobin synthesis during primitive erythropoiesis. Blood 2009; 113: 1340–9. [DOI] [PubMed] [Google Scholar]
- 156.Sangokoya C, Telen MJ, Chi JT. MicroRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood 2010; 116: 4338–48. [DOI] [PMC free article] [PubMed] [Google Scholar]