

Abstract
Background
Cardiac hypertrophy is the most potent cardiovascular risk factor after age, and relative mortality risk linked with cardiac hypertrophy is greater in women. Ischemic heart disease is the most common form of cardiovascular pathology for both men and women, yet significant differences in incidence and outcomes exist between the sexes. Cardiac hypertrophy and ischemia are frequently occurring dual pathologies. Whether the cellular (cardiomyocyte) mechanisms underlying myocardial damage differ in women and men remains to be determined. In this study, utilizing an in vitro experimental approach, our goal was to examine the proposition that responses of male/female cardiomyocytes to ischemic (and adrenergic) stress may be differentially modulated by the presence of pre-existing cardiac hypertrophy.
Methods
We used a novel normotensive custom-derived hypertrophic heart rat (HHR; vs control strain normal heart rat (NHR)). Cardiomyocyte morphologic and electromechanical functional studies were performed using microfluorimetric techniques involving simulated ischemia/reperfusion protocols.
Results
HHR females exhibited pronounced cardiac/cardiomyocyte enlargement, equivalent to males. Under basal conditions, a lower twitch amplitude in female myocytes was prominent in normal but not in hypertrophic myocytes. The cardiomyocyte Ca2+ responses to β-adrenergic challenge differed in hypertrophic male and female cardiomyocytes, with the accentuated response in males abrogated in females—even while contractile responses were similar. In simulated ischemia, a marked and selective elevation of end-ischemia Ca2+ in normal female myocytes was completely suppressed in hypertrophic female myocytes—even though all groups demonstrated similar shifts in myocyte contractile performance. After 30 min of simulated reperfusion, the Ca2+ desensitization characterizing the male response was distinctively absent in female cardiomyocytes.
Conclusions
Our data demonstrate that cardiac hypertrophy produces dramatically different basal and stress-induced pathophenotypes in female- and male-origin cardiomyocytes. The lower Ca2+ operational status characteristic of female (vs male) cardiomyocytes comprising normal hearts is not exhibited by myocytes of hypertrophic hearts. After ischemia/reperfusion, availability of activator Ca2+ is suppressed in female hypertrophic myocytes, whereas sensitivity to Ca2+ is blunted in male hypertrophic myocytes. These findings demonstrate that selective intervention strategies should be pursued to optimize post-ischemic electromechanical support for male and female hypertrophic hearts.
Electronic supplementary material
The online version of this article (doi:10.1186/s13293-016-0084-8) contains supplementary material, which is available to authorized users.
Keywords: Cardiomyocyte, Sex/gender, Cardiac hypertrophy, Ischemia/reperfusion, Stress response
Background
For both women and men, cardiovascular disease is the leading cause of death and disability [1, 2]. Cardiac hypertrophy is the most potent cardiovascular risk factor after age [3], and importantly, relative mortality risk linked with cardiac hypertrophy is greater in women [4]. There is a growing clinical recognition that myocardial cardiac stress responses, including neurohumoral activation, exhibit inherent sex difference [5].
Ischemic heart disease is the most common form of cardiovascular pathology for men and women [6]. Ischemic events occur earlier in men, and ischemia-related arrhythmia and sudden cardiac death incidence are higher in men. In contrast, while younger women have a lower risk of ischemic heart disease, in the event of a myocardial infarct, they experience higher mortality and rate of heart failure, even though reperfusion status is equivalent [7, 8]. Women with ischemic heart disease have higher rates of hospitalization than men [9]. The essential question of whether the mechanisms underlying ischemic heart disease in women differ from men remains unanswered [10]—and the cognate issue of whether ischemia coincident with hypertrophic comorbidity has differing gender aetiology and outcome has not been addressed.
The importance of achieving a more detailed understanding of basic mechanisms of sex difference in pathophysiologic processes, which have particular relevance to cardiovascular disease and health demography, has been emphasized [11]. The value of sex-inclusive quality pre-clinical data to inform large-scale clinical studies has also been recently highlighted in the context of unexpected findings delivered by the RELAX trial—which sildenafil (a phosphodiesterase 5 inhibitor) did not show a beneficial effect in the treatment of heart failure with preserved ejection fraction [12]. In retrospect, it became apparent that this outcome might have been predicted if the rationale for the trial had not been reliant on data from male-only human and animal studies [13, 14].
Experimentally, important insights have been gained in relation to sex-specific differentials in the impacts of cardiac hypertrophy and of exposure to ischemic insult—yet still, knowledge is lacking [15, 16]. The extent of hypertrophic remodeling in female rodents in response to loading stress is generally attenuated, yet exogenous oestrogen and oestrogen receptor (oestrogen receptor α (ERα)) agonists have been shown to increase mortality in mice subjected to aortic constriction or to infarction [17–19]. Generally, female hearts are less susceptible to post-ischemic contractile dysfunction, findings which primarily derive from experiments involving animal models where function is not compromised by pre-existing pathologies [15, 20–22]. We have previously established that the intrinsic resistance of isolated female hearts to ischemic insult is abrogated in a setting of primary cardiac hypertrophy [23].
Most fundamentally, protection against hypertrophy and ischemia-driven arrhythmogenesis and contractile dysfunction is determined by cardiomyocyte Ca2+ regulatory status. Whilst limited sex-specific information is available at the myocyte level, in vitro findings suggest differential female and male contractile/Ca2+ handling characteristics in simulated ischemia and also sex differences in the responsiveness of myocytes to manipulation of adrenergic activation [24, 25]. Specifically, it is reported that during simulated ischemia, only female cardiomyocytes exhibit a reduction in Ca2+ transient amplitude (vs non-ischemic control) and that in reperfusion, female cardiomyocytes are hyper-contractile (vs males) and more likely to survive the ischemia/reperfusion challenge [24]. Sex differences in adrenergic responses apparently reflect reduced cyclic adenosine monophosphate (cAMP) levels in female myocytes (vs male), contributing to lower Ca2+ levels and internal Ca2+ store flux responses [25]. Importantly, no previous cardiomyocyte functional studies have interrogated the role of pre-existing hypertrophic pathology in determining the sex-specific responses to ischemic injury—a highly relevant clinical scenario of comorbidity [26, 27].
In this study, utilizing an in vitro experimental approach, our goal was to examine the proposition that responses of male and female cardiomyocytes to stress stimuli (β-adrenergic activation and simulated ischemia/reperfusion insult) may be differentially affected by the presence of pre-existing cardiac hypertrophy. Using a purpose-derived rat model of normotensive cardiac hypertrophy, the hypertrophic heart rat (HHR), a range of cardiomyocyte morphologic and electromechanical functional studies was performed using microfluorimetric techniques. We have previously reported premature mortality in the HHR (vs normal heart rat (NHR)), with ex vivo HHR exhibiting normal systolic function and an increased vulnerability to ischemia/reperfusion dysfunction/injury [23, 28]. The present study now demonstrates that cardiac hypertrophy produces dramatically different pathophenotypes in female- and male-origin cardiomyocytes exposed to ischemia. These findings suggest implications for sex-specific refinement of reperfusion therapeutic interventions.
Methods
Animals
Animals were obtained from established colonies of the HHR and the control NHR, maintained at the Biological Research Facility at the University of Melbourne, Australia, and derived as reported [29]. Briefly, these rats originated from the F2 progeny of a cross between the Fischer 344 and spontaneously hypertensive rat (SHR). Normotensive offspring were selected for either enlarged or normal heart size by echocardiography over successive generations to achieve genetic stability. Telemetry, tail-cuff plethysmography and direct intra-arterial recording methods have been used to determine the normotensive status of these animals [29]. Experiments were conducted and animals handled in the manner specified by the NHMRC/CSIRO/ACC Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (1997) and the EU Directive 2010/63/EU for animal experiments, with approval and oversight by the University of Melbourne Animal Ethics Committee.
Cardiomyocyte isolation procedure
Ventricular myocytes were isolated from NHR and HHR rats, as described [30]. Briefly, animals were weighed, killed by decapitation under deep halothane anaesthesia, hearts were excised, trimmed, buffer washed and blotted prior to taking weight measurement by tare. Hearts were retrogradely perfused with bicarbonate-buffered Ca2+-free Krebs (in millimolars: 118 NaCl, 4.8 KCl, 1.2 KH2PO4, 1.2 MgCl2, 25 NaHCO3, 11 d-glucose) at 73 mmHg for 3 min at 37 °C. Cardiomyocytes were then perfused with type II collagenase (50 mg/ml, 295 U/mg; Worthington Biochemical Corporation, NJ, USA) to enable heart digestion. The heart was removed from the cannula, and the ventricular cardiomyocytes dispersed in Krebs/collagenase solution (in millimolars: 146.2 NaCl, 4.7 KCl, 0.4 NaH2PO4-H2O, 1.1 MgSO4-7H2O, 10 (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 11 d-glucose) supplemented with trypsin inhibitor (25 μg/ml; Sigma-Aldrich, MO, USA) to inhibit residual collagenase activity. For each heart, 100 rod-shaped and regularly striated cardiomyocytes were selected at random for length and width measurement at ×400 magnification using an inverted light microscope and calibrated eye piece as previously described [31]. Cardiomyocyte volume was derived from the product of cell length and width, multiplied by 7.59 × 10−3 pl/μm2 [32].
Cardiomyocyte Ca2+ handling and twitch analysis
Isolated myocyte Ca2+ levels and shortening performance were measured [30]. Cell suspensions were incubated with the Ca2+ fluorescent dye, fura2-AM (2.5 μM, 20 min, 25 °C; Invitrogen). Myocytes were superfused with 2 mM Ca2+-HEPES buffer (in millimolars: 146.2 NaCl, 4.7 KCl, 0.4 NaH2PO4H2O, 1.1 MgSO47H2O, 10 HEPES, 11 glucose, pH 7.4) and stimulated to establish steady state contractile performance at 4 Hz (5 min, 37 °C). Cardiomyocyte twitch properties were assessed by video-based edge detection (IonOptix, MA, USA). The indices used to describe twitch cycle were twitch amplitude normalized to diastolic cell length (twitch amplitude percentage) and maximal rate of shortening and lengthening (mrs and mrl, respectively; μm/s, normalized to diastolic cell length). Myocyte Ca2+ signals were measured by microfluorimetry (360:380 nm fluorescence ratio, 1000 Hz sampling; IonOptix, MA, USA) [30]. Fluorescence signals were corrected for background. The indices used to describe the Ca2+ transient were amplitude (F360:380) and the time constant of decay (tau, ms; time constant measured from curve fitted from 10 % below the peak level to baseline of transient). Cardiomyocyte twitch and Ca2+ fluorescent ratio properties were measured off-line using IonWizard (IonOptix) and were determined by average of ten steady state transients for each myocyte. Ca2+-shortening phase loops were constructed for individual activation cycles by plotting Ca2+ level vs myocyte shortening throughout the shortening and lengthening phases of the cycle. During the relaxation (i.e. lengthening) phase, the Ca2+ level at half (50 %) relaxation may be interpreted as an index of myofilament Ca2+ sensitivity [33].
Adrenergic challenge protocol
To assess the response of male and female NHR and HHR cardiomyocytes to an adrenergic challenge, myocytes stabilized for 5 min in 2 mM Ca2+-HEPES buffer were superfused with 10 nM isoproterenol for 5 min. Previous dose range and time-course studies confirmed this was an optimal time point to allow a maximal shortening and amplitude Ca2+ response to reach a new steady state [34].
Simulated ischemia/reperfusion protocol
To assess the response of male and female NHR and HHR cardiomyocytes to an ischemia and reperfusion challenge, cardiomyocytes were superfused with a modified HEPES buffer (in millimolars: 136 NaCl, 8 KCl, 0.4 NaH2PO4-H2O, 1.1 MgSO4-7H2O, 2.0 CaCl2, 10 HEPES, 10 lactate, pH 6.8, 100 % N2 gas saturation) for 20 min to simulate conditions associated with ischemia, including hypoxia, hypercapnia, acidosis, substrate deprivation and lactate accumulation [30, 35, 36]. At the end of the 20-min superfusion with the simulated ischemia solution, the perfusate was switched back to the basal 2-mM Ca2+-HEPES buffer for a further 30 min. Cardiomyocytes were electrically stimulated (4 Hz) throughout the ischemia/reperfusion protocol.
Statistics
Data are presented as mean ± SEM. Differences between groups were assessed by two-way ANOVA and with repeated measures as appropriate. Myocyte survival curves were assessed by log-rank Mantel-Cox test. Data were considered significant at p < 0.05. All statistical calculations were performed using SPSS V.22.0 (IBM Corp, NY, USA). Statistical annotation used throughout the study to designate significance level: #, strain effect; *, sex effect; †, sex-strain interaction. Post hoc analysis was performed on data in which a sex-strain interaction was evident, with significant differences in post hoc analysis represented in the figures (§, p < 0.05, male vs female of same strain) directly above the relevant figure bar or data point.
Results
Cardiac and cardiomyocyte hypertrophy in male and female HHR
Female body and heart weights were significantly lower than strain and age-matched NHR and HHR values (Table 1). Heart weight and cardiomyocyte dimensions were smaller in female NHR (Table 1 and Fig. 1, respectively), yet cardiac weight index (CWI; g/kg) was greater compared with male NHR. In HHR, cardiac and cardiomyocyte hypertrophy was most pronounced in female with augmented CWI associated with more substantial myocyte enlargement in width (and consequently volume) dimension (Table 1 and Fig. 1, †p < 0.05; Fig. 1b–d). These data confirm a robust hypertrophic phenotype in female and male HHR.
Table 1.
Age and body/heart weights of male and female NHR and HHR
| Parameter | NHR | HHR | ||
|---|---|---|---|---|
| Male | Female | Male | Female | |
| Age (weeks) | 14.6 ± 0.7 (15) | 13.3 ± 0.3 (13) | 14.8 ± 1.0 (14) | 13.7 ± 0.6 (13) |
| Body weight (g) | 352 ± 7.9 (15) | 180 ± 5.5# (13) | 299 ± 10.2## (14) | 171 ± 4.7## (13) |
| Heart weight (mg) | 1410 ± 28.5 (15) | 882 ± 27.7# (13) | 1514 ± 85.8 (14) | 1202 ± 69.2#,## (13) |
| CWI (mg/g) | 4.02 ± 0.1 (15) | 4.91 ± 0.1# (13) | 5.12 ± 0.3## (14) | 7.04 ± 0.4#, ## (13) |
#sex p < 0.05; ##strain p < 0.05, mean ± SEM, n = hearts or animals in brackets for each group
Fig. 1.
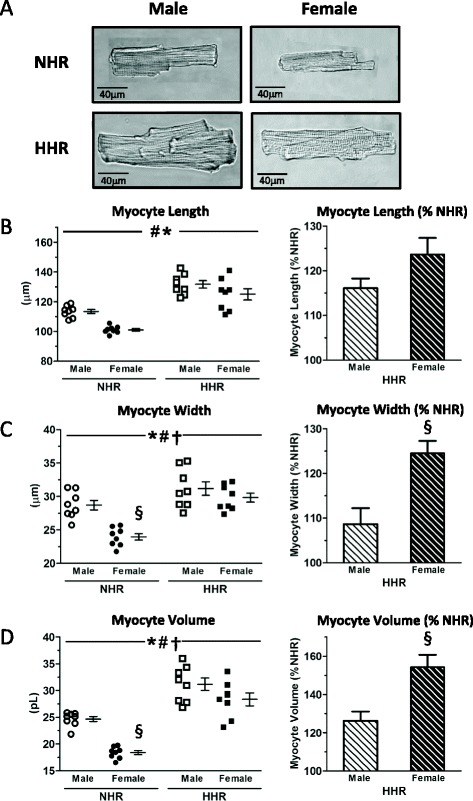
Cardiomyocyte hypertrophy in male and female HHR. a Representative images of cardiomyocytes from male and female NHR and HHR hearts. b–d Augmented CWI associated with more substantial myocyte enlargement in width (and consequently volume) dimension in female vs male HHR (*sex p < 0.05; #strain p < 0.05; †sex-strain interaction; §post hoc male vs female p < 0.05, mean ± SEM. Each data point mean of n = 50 cells from each of eight hearts/group, total 400 cells/gp)
Cardiomyocyte basal performance—sex differences suppressed with hypertrophy
Basal NHR cardiomyocyte contractile performance was lower in female cardiomyocytes compared with males (Fig. 2a–d, Additional file 1: Table S1 *p < 0.05). This lower female contractility was marked in normal female myocytes (approximately 50 % lower of the percentage shortening values vs male NHR), though time to peak shortening was particularly slow in female HHR cardiomyocytes (Additional file 1: Table S1). Lower contractility in normal female myocytes was associated with a trend for lower Ca2+ transient amplitude and systolic Ca2+ (Fig. 2e, f, Additional file 1: Table S1). No sex difference was observed in NHR cardiomyocyte diastolic Ca2+ levels or the time constant of Ca2+ transient decay (Fig. 2g, h). Lower contractility was not apparent in female HHR cardiomyocytes relative to male (Fig. 2b). As for NHR, overall Ca2+ levels did not differ between male and female HHR myocytes (Fig. 2e–h). These data show that basal contractile performance in non-hypertrophic male and female cardiomyocytes is fundamentally different, but this sex differential is lost when myocytes exhibit pathological hypertrophy.
Fig. 2.
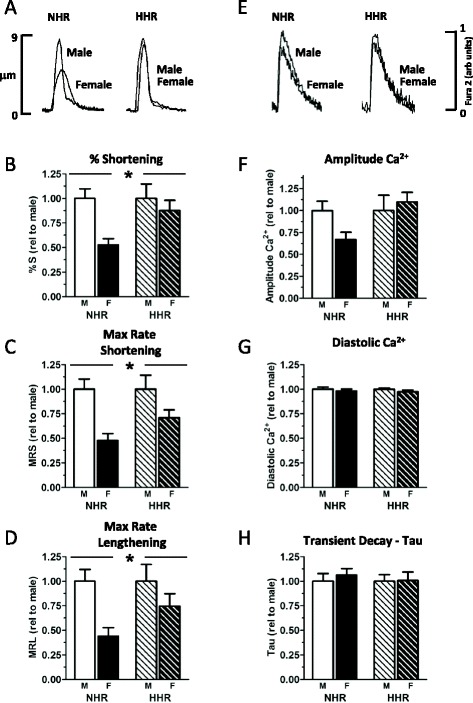
Basal sex differences in cardiomyocyte performance suppressed with hypertrophy. a Representative records of myocyte shortening in male and female NHR and HHR cardiomyocytes. b–d Basal twitch amplitude (% shortening), maximum rate of shortening/lengthening are lower in female cardiomyocytes compared with males—an effect that is accentuated in NHR compared with HHR. e No sex difference was observed in Ca2+ transients, though a trend for lower Ca2+ transient amplitude was evident in female NHR cardiomyocytes (f). No sex difference was observed in cardiomyocyte diastolic Ca2+ levels or the time constant of Ca2+ transient decay (g, h). (*sex p < 0.05; p < 0.05; mean ± SEM; n = 8–13 cells from four to five hearts/group)
Augmented HHR Ca2+ activator response to β-adrenoceptor stimulation in male cardiomyocytes absent in female cardiomyocytes
β-Adrenoceptor agonist challenge in vitro elicited a substantial increase in cardiomyocyte contractility (Fig. 3a, Additional file 2: Table S2). The magnitude of this response was similar in male and female cardiomyocytes, but more pronounced in the presence of hypertrophy (Fig. 3b–d, M NHR 178.9 ± 16.1 %, F NHR 213.5 ± 32.1 %, M HHR 282.9 ± 36.3 %, F NHR 300.7 ± 25.2 %, #p < 0.05). Sex differences in the Ca2+ response to β-adrenoceptor stimulation were evident. The extent of isoproterenol-induced increase in Ca2+ transient amplitude was selectively blunted in female HHR myocytes compared with males (Fig. 3e, †p < 0.05). This sex-specific response was not observed in non-hypertrophic (NHR) myocytes. Diastolic Ca2+ levels were stable with isoproterenol challenge in all treatment groups (Fig. 3f). A more rapid transient decay (decrease in tau) was evident in hypertrophic male and female cardiomyocytes, consistent with a trend for increased maximum rate of lengthening (Fig. 3d, g). These data demonstrate that the Ca2+ response to β-adrenergic stimulation is different in male and female hypertrophic cardiomyocytes—the accentuated response in HHR male myocytes is not achieved in female myocytes.
Fig. 3.
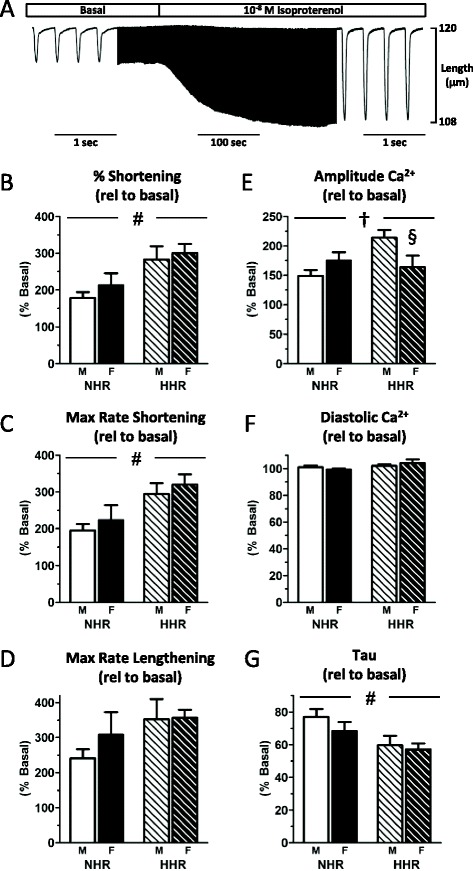
β-Adrenoceptor stimulation Ca2+response blunted in hypertrophic female HHR cardiomyocytes. a Representative cardiomyocyte shortening trace (central portion time compressed) showing isoproterenol (10 nM) exposure-elicited response of rapid and sustained increase in cell twitch. b–d The extent of increase in shortening and maximum rate of shortening was similar in male and female cardiomyocytes, but more pronounced in the presence of pathological hypertrophy. Rate of lengthening was unchanged. e The extent of isoproterenol-induced increase in Ca2+ transient amplitude was selectively blunted in female HHR myocytes. f Diastolic Ca2+ levels were stable with isoproterenol challenge in all treatment groups, whilst tau (g) was significantly faster in HHR of both sexes (#strain p < 0.05; †sex-strain interaction; §post hoc male vs female p < 0.05, mean ± SEM, n = 9–11 cells from four to five hearts/group)
Selective Ca2+ elevation in female myocytes at end ischemia abrogated with hypertrophy
Myocyte contractile performance and Ca2+ levels were tracked throughout the time-course of a simulated ischemia/reperfusion challenge (sample time-compressed record, Fig. 4a, Additional file 2: Table S2). At the commencement of ischemia, all cell groups demonstrated an abrupt decrease in twitch amplitude and a small increase in resting length. This reduction in twitch amplitude reached a plateau within the first 5 min of simulated ischemia (Fig. 4b), with no significant difference between groups in the extent of twitch amplitude reduction at end ischemia (Fig. 4c). Ca2+ transient responses to simulated ischemia were very different in male and female cardiomyocytes (Fig. 4d, †p < 0.05). In contrast to males, female NHR cardiomyocytes exhibited a substantial increase in amplitude Ca2+ at the end of ischemia (relative to pre-ischemic basal; M NHR 104.8 ± 13.3 %, F NHR 186.2 ± 35.9 %, *p < 0.05), which was not associated with a concomitant increase in shortening (Fig. 4e). This marked and selective elevation of end-ischemia Ca2+ in female NHR was totally abrogated with hypertrophy (HHR, Fig. 4e, M HHR 73.1 ± 9.5, F HHR 66.8 ± 10.9 %). Despite a significantly lower Ca2+ signal in hypertrophic cardiomyocytes (Fig. 4e, #p < 0.05), the shortening responses between male and female HHR were not different (Fig. 4c, p = ns). A sex-strain interaction was evident for diastolic Ca2+ during simulated ischemia (Fig. 4f, g). An increase in diastolic Ca2+ in male HHR cardiomyocytes was not evident in female HHR, which remained at near-basal levels of diastolic Ca2+ (Fig. 4g). This contrasted with NHR myocytes, with a trend for greater diastolic Ca2+ at the end of simulated ischemia compared with males.
Fig. 4.
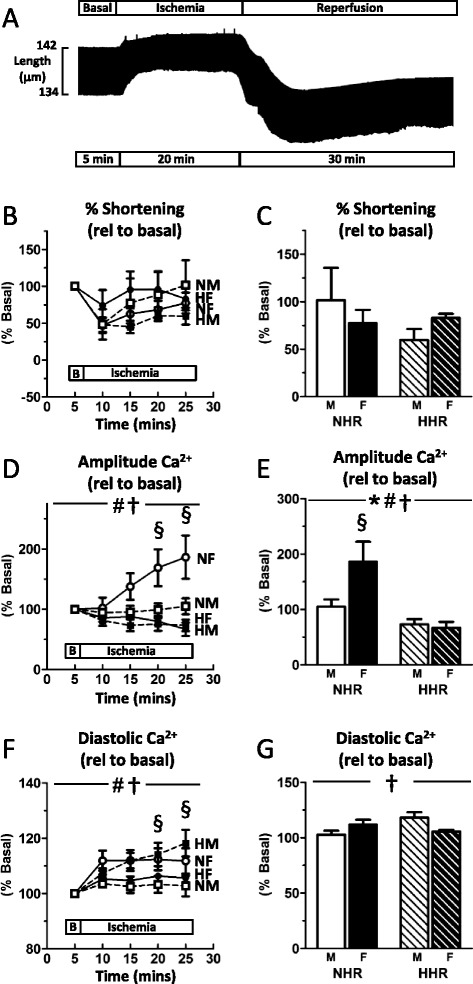
Selective Ca2+ elevation in female myocytes at end ischemia is abrogated with hypertrophy. a Representative cardiomyocyte shortening record shows modulated diastolic length and twitch amplitude in simulated ischemia and reperfusion (time compressed). b All groups show a similar pattern of shortening response to simulated ischemia. c No significant difference between groups was detected in the extent of change in twitch reduction at end ischemia. d Ca2+ transient responses during the time-course of simulated ischemia. e Female NHR cardiomyocytes exhibited a substantial increase in amplitude Ca2+ at the end of ischemia which was not evident in female hypertrophic myocytes. f Diastolic Ca2+ increased throughout ischemia in male HHR cardiomyoctes. g At end ischemia, increased diastolic Ca2+ in male HHR was absent in female HHR myocytes. (*sex p < 0.05; #strain p < 0.05; †sex-strain interaction; §post hoc male vs female p < 0.05; mean ± SEM; n = 7–14 cells from 6 to 11 hearts/group)
Myofilament Ca2+ desensitization in reperfused hypertrophic male myocytes is absent in females
During the simulated reperfusion phase of the protocol, myocytes of all groups most typically exhibited a rapid decrease in resting length (i.e. a diastolic contracture response) with decrement in twitch amplitude, followed by gradual return to a new steady state in the extent of myocyte shortening, albeit diastolic relaxation state was only partially regained (Fig. 4a). Myocyte attrition (acute necrosis/oncosis) was observed throughout both the ischemic and reperfusions phases of the protocol (Fig. 5a). In males, there was a trend for hypertrophic myocytes (HHR) to be more resistant to cell death during the simulated ischemia/reperfusion challenge compared with normal myocytes (NHR), though this did not reach significance (Fig. 5a). A strain trend was not apparent in female myocytes. All groups showed full recovery from ischemic diastolic contracture, with resting cardiomyocyte length lower at end reperfusion compared with the pre-ischemic basal level (Fig. 5b). The extent of this reduction was greatest in male NHR cardiomyocytes and significantly greater than in female NHR cardiomyocytes. This sex difference was not apparent when cardiomyocytes were hypertrophic. During the first 5 min of reperfusion, spontaneous contractions were evident in male NHR (one of eight cardiomyocytes) and HHR (four of nine cardiomyocytes) cardiomyocytes, contrasting with a lack of spontaneous contractions in both female NHR and HHR cardiomyocytes, At the end of the 30-min simulated reperfusion challenge, surviving male and female cardiomyocytes from normal and hypertrophic hearts exhibited very different recovery parameters (Fig. 5c, †p < 0.05). Male HHR cardiomyocytes demonstrated equivalent shortening to male NHR controls, yet amplitude Ca2+ was significantly increased. This directly contrasted with female HHR cardiomyocytes, where shortening was significantly increased compared with female NHR controls, yet amplitude Ca2+ was not different. Phase loop analyses were performed to assess individual myocyte Ca2+-shortening relationships (Fig. 5d). Male, but not female, HHR cardiomyocytes exhibited a large decrease in myofilament Ca2+ sensitivity, with an approximate two- to threefold increase in Ca2+ levels at 50 % myocyte relaxation compared with male NHR and female NHR and HHR cardiomyocytes (Fig. 5e, †p < 0.05). These data demonstrate that, in contrast to hypertrophic male myocytes, myofilament Ca2+ responsiveness in hypertrophic female cardiomyocytes was not characterized by ‘desensitization’. Thus, a major sex-specific stress response phenotype difference emerges between female and male hypertrophic cardiomyocytes when subjected to simulated ischemia and reperfusion challenge.
Fig. 5.

Myofilament Ca2+ desensitization in hypertrophic male myocytes at the end of reperfusion is absent in females. a Cardiomyocyte survival plots throughout simulated ischemia and reperfusion. b Resting myocyte length decreased to a greater extent in male NHR cardiomyocytes compared with females, with this sex difference absent in the HHR. c At end reperfusion, male HHR cardiomyocytes maintain contractility yet exhibit significantly increased Ca2+. Female HHR myocytes show augmented contractility with substantially lower transient Ca2+. d Phase loop analysis shows male, but not female, HHR cardiomyocytes exhibit a modified Ca2+-shortening relationship. e Male HHR cardiomyocytes exhibit significantly lower level of myofilament Ca2+ sensitivity, with an approximate two to threefold increase in Ca2+ levels at 50 % myocyte relaxation. (*sex p < 0.05; #strain p < 0.05; †sex-strain interaction; §post hoc male vs female p < 0.05; mean ± SEM; n = 5–8 cells from four to six hearts/group)
Discussion
This investigation provides new evidence that male and female hearts with an underlying pathological hypertrophy respond differently to cellular stresses. Using the novel HHR model (a model of normotensive intrinsic hypertrophy derived through a multigenerational program of selective breeding), our findings document pronounced cardiac and cardiomyocyte enlargement in females—including cardiomyocyte dimensional increase in female HHR equivalent to male HHR. Under basal conditions, a lower twitch amplitude in female myocytes was prominent in normal (NHR) but not in hypertrophic (HHR) myocytes. The cardiomyocyte Ca2+ responses to β-adrenergic challenge differed in hypertrophic male and female cardiomyocytes, with the accentuated response in males abrogated in females—even while contractile responses were similar. In simulated ischemia, the Ca2+ transient responses of hypertrophic male and female myocytes were markedly different—though again, all groups demonstrated similar shifts in myocyte contractile performance. A prominent and selective elevation of end-ischemia Ca2+ in female NHR was completely suppressed in female hypertrophic myocytes. Similarly, after the 30-min simulated reperfusion, male and female cardiomyocytes evinced different Ca2+ response patterns indicating an absence of the Ca2+ desensitization characterizing the male response.
Our data demonstrate that cardiac hypertrophy produces dramatically different basal and stress-induced pathophenotypes in female- and male-origin cardiomyocytes. The lower Ca2+ operational status characteristic of female (vs male) cardiomyocytes comprising normal hearts is not exhibited by myocytes of hypertrophic hearts. After ischemia/reperfusion, availability of activator Ca2+ is suppressed in female hypertrophic myocytes, whereas sensitivity to Ca2+ is blunted in male hypertrophic myocytes. These findings demonstrate that selective intervention strategies should be pursued to optimize post-ischemic electromechanical support for male and female hypertrophic hearts.
Normal female-male cardiomyocyte contractile performance differential is eliminated with concomitant hypertrophy
The present study assessed male/female cardiomyocyte function and stress responses in a novel pathological hypertrophic HHR strain (vs control NHR strain). Significantly lower body weight in female rats is consistent with previous reports [23, 37–39], as are our findings that cardiomyocyte dimensions are smaller in normal females (vs male NHR) [24, 40–42]. This male/female cell size difference is not always evident and may be dependent on animal strain, aging/disease context and the experimental conditions under which myocyte dimensions are measured (e.g. resting Ca2+ concentration) [37, 43]. The possibility that there is a fundamental relationship between cardiomyocyte size and electromechanical characteristics which may partly reflect a sex-dependent size-scaling effect merits future investigation. The present finding that under standardized basal conditions, normal female cardiomyocytes contract at a lower operating level than male myocytes is consistent with our earlier report (subsequently confirmed by others) [16, 39]. This feature of female cardiomyocyte performance suggests a relatively ‘economical’ basal metabolic state. There are discrepancies within the literature regarding the mechanisms driving this female phenotype, though changes in Ca2+ transporter function are undoubtedly implicated. While differences in protein expression levels of various Ca2+ transporters have been reported between males and females in some contexts [15, 30], oestrogen regulation of the activity of some key Ca2+ handling effectors is likely to be of most functional importance [15, 44–48]. These previous studies assessing Ca2+ transporter expression/activity have utilized a series of different species (e.g. mouse, rat, guinea pig) and animal ages, which may explain variability in findings. We have recently shown that expression of the sarcoplasmic reticulum (SR) Ca2+ release channel (RyR2) does not differ in male/female Sprague Dawley (SpD) rats (12–16 weeks) [30], similar to that reported in 5–10-month-old C57Bl/6 mice [25]. This contrasts to previous studies in SpD and Wistar rats (age not stated), reporting an increased expression of the SR Ca2+ release channel (RyR2) [49, 50]. We also showed no difference in sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA2a) and phospholamban (PLB) expression in male/female SpD rats, consistent with previous reports [49]. Directly relevant to the present study, we have shown that expression of RyR2, SERCA2a and PLB was not different in female HHR and NHR ventricular homogenates [30]. Our data indicate that it is the regulation of Ca2+ transporter activity, and not the expression, that differs in males/females. In particular, we have shown that basal Ca2+/calmodulin-dependent kinase II (CaMKII) phosphorylation (and hence, CaMKII-mediated phosphorylation of RyR2 and PLB) is lower in females, a mediator which exerts major influence on numerous Ca2+ transporter properties [30].
Estrogenic influence on cardiomyocyte Ca2+ handling includes voltage-gated Ca2+ current suppression, internal Ca2+ store release modulation and myofilament Ca2+ sensitivity alteration [25, 46, 51–59]. The sex-specific distinguishing characteristics in contractile phenotypes in non-hypertrophic myocytes can be partially attributed to a Ca2+-dependent mechanism, but likely non-Ca2+ dependent estrogenic actions (cell energetics/gene expression) are involved [5, 60] as the extent of contractility performance difference between males and females was more prominent than the Ca2+ transient amplitude differences.
Hypertrophy is linked with accentuated response to β-adrenoceptor stimulation in male, but not female myocytes
Sex-specific aspects of altered β-adrenoceptor response in cardiac disease pathogenesis are not well characterized. Experimentally, evidence indicates that the β-adrenergic response is less pronounced in healthy cardiac tissues/myocytes of females compared with males under some circumstances. We and others have previously shown that Ca2+ transients are smaller in female cardiomyocytes (vs males) treated with isoproterenol [39, 61–63]. These male/female differences do not appear due to changes in β-adrenergic expression/density [64, 65], and suppressed responsiveness in females is more likely attributable to downstream signaling intermediates (i.e. adenylyl cyclase/PKA/cAMP) [25, 62].
We did not observe male/female differences in the extent of increase in twitch or Ca2+ transient amplitude in isoproterenol-treated NHR myocytes—suggesting that pacing under near-physiological conditions minimizes response difference [65]. Notably, male/female differences in response to isoproterenol were evident in hypertrophic myocytes. Hypertrophic female myocytes did not exhibit the accentuated elevation in Ca2+ transient amplitude observed in male HHR myocytes, even though both female and male HHR had similarly augmented contractile responses. This suggests a Ca2+-independent component determining adrenergic response selectively in females. The mechanism is unclear, but may be related to differential cellular metabolism profile—with hypertrophic hearts drawing on sex-selective energy reserves to effect an adrenergic-driven contractile response [5, 66].
Cardiomyocyte Ca2+ handling and responsiveness during ischemia and reperfusion is different in hypertrophy in females and males
In this investigation, distinct mechanisms underlying vulnerability to ischemia/reperfusion in female- and male-origin cardiomyocytes are identified. These findings build on our prior observations involving isolated ex vivo hearts where we established that the more robust functional recovery of female hearts to ischemic insult was abrogated in hypertrophy [23]. In that study, HHR male and female hearts performed equally poorly, displayed similar necrotic damage (lactate dehydrogenase release) and the enhanced activation of the pro-survival reperfusion injury survival kinase (RISK; [23]) pathway seen in NHR female hearts was absent in HHR female hearts. In the two heart strains, coronary flow during ischemia/reperfusion in that study was also not different—eliminating vascular perfusion status as an underlying causative agent in sex- and strain-related performance. Our new in vitro finding presented in this investigation, that overall isolated cardiomyocyte viability loss through simulated ischemia/reperfusion was not different between sexes or strains, indicates that the measures of ex vivo heart damage reflect primarily cell rupture injury attributable to in situ tissue torsion (likely during contracture) and not cell-origin oncotic lesion.
During ischemia, female NHR showed a surprising tolerance for elevated cardiomyocyte Ca2+ levels—suggesting that the interplay between Ca2+, Na+ and pH which is an important determinant of electromechanical status at end reperfusion operates differently in normal male and female myocytes. Related, but qualitatively different, observations have been previously reported. The sex differences in ionic homeostatic responses to simulated ischemia may depend on precise experimental conditions [24]. Sex-comparative data relating to hypertrophic cardiomyocytes have not previously been presented. An exploration of how these ionic flux responses may be differentially manipulated via pharmacologic means may provide a valuable therapeutic lead for new post-ischemia sex- and disease-specific intervention approaches.
Cell shortening is typically reduced by approximately 50 % relative to basal in the first 5 min of simulated ischemia, though this reduction was attenuated in female HHR cardiomyocytes (Fig. 4a). This 50 % reduction is similar to that described previously [24, 67], though not as extensive as other studies describing near cessation of myocyte shortening [35, 36, 68–70]. As was observed in isolated HHR hearts [23], at the end of ischemia, male and female isolated cells showed similar levels of contractile recovery (whereas in NHR sex-specific performance, differences were apparent). Most dramatically, different underlying Ca2+ handling and response characteristic produced this similar net effect in HHR cell types. In the male HHR myocytes, a significant elevation of Ca2+ transient amplitude occurred with a major offsetting reduction in myofilament sensitivity to Ca2+. In the female HHR myocytes, this combination of Ca2+ adaptations was not apparent. In relation to adverse outcomes in early reperfusion, these findings may be extrapolated to suggest that in acute reperfusion, Ca2+ overload arrhythmogenic predisposition presents as a male myocyte vulnerability. In contrast, the blunted female myocyte Ca2+ transient response to β-adrenoceptor inotropic influence has the potential for compromised functional performance. Thus, ischemic recovery is similarly impaired in both males and females—but via different cellular mechanisms. Clinically, the importance of gender differences in arrhythmia substrate is increasingly recognized—and advancing the understanding of underlying cellular processes involved is essential [71].
The present study, of young adult rodents at prime estrogenic life stage, demonstrates the absence of a female ‘protected’ state in the context of hypertrophy. The conventional view of female oestrogen cardioprotection may require refinement [15, 72]. Oestrogen receptors (ERα, oestrogen receptor β (ERβ), G-protein-coupled oestrogen receptor (GPER)) are expressed in male and female myocardium. When expression/activity is manipulated genetically or pharmacologically, dual sex effects may be observed [73]—yet oestrogen exposure is not always favourable [15]. Experimentally, where female hearts have shown more ischemic resilience than male hearts, this has been most evident when the context involved baseline elevated Ca2+ loading levels [20–22]. New information suggests that local myocardial androgen-to-oestrogen conversion may be an element in determining the sex specificity of responses to cardiac challenge [74–76], and in hypertrophy, local steroid levels may become more important in modulating function under stress conditions. More detailed studies of the specific molecular processes involved in regulating ionic homeostasis in male- and female-origin cardiomyocytes in defined disease hypertrophic states and acute stress settings are warranted. A limitation of the present study is the in vitro and ‘unloaded’ conditions under which myocyte recordings are undertaken. However, the advantage of the approach is the capacity to achieve a high level of control over cellular ambience to resolve key functional differences.
Conclusions
In summary, this experimental investigation demonstrates that the responses of hypertrophic male and female cardiomyocytes to β-adrenoceptor stimulation and ischemia/reperfusion challenges are fundamentally different at the cellular level—and that hypertrophy in females confers a distinctive liability. Our findings indicate that therapeutic interventions targeted to the early reperfusion period may have differential sex-specific efficacy depending on the presence of an underlying comorbid hypertrophic state. Where clinical trials have previously failed to identify benefit in early reperfusion manipulation of cellular processes impacting on myofilament Ca2+ sensitivity, there may be a case for review in selected at-risk female subgroups. This study provides a basis to support relevant focused pre-clinical research to pursue this possibility.
Abbreviations
ANOVA, analysis of variance; CaMKII, Ca2+/calmodulin-dependent kinase II; CWI, cardiac weight index; ERα, oestrogen receptor α; ERβ, oestrogen receptor β; GPER, G-protein-coupled oestrogen receptor; HEPES, (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; HHR, hypertrophic heart rat; NHR, normal heart rat; RISK, reperfusion injury salvage kinase; SHR, spontaneously hypertensive rat
Acknowledgements
Career support was provided through the National Heart Foundation of Australia and the University of Melbourne R. Douglas Wright Faculty Trust (JRB). Research support was provided through Grant-in-Aid from the National Heart Foundation of Australia (LMDD, JRB and CLC) and the National Health and Medical Research Council of Australia (LMDD, SBH and JRB).
Authors’ contributions
JRB, CLC, MVP and LMD conceived and designed the experiments. JRB, CLC, TWH and MVP performed the experiments and analysed the data. SBH and LMD generated the HHR model. JRB, CLC and LMD wrote the manuscript. All authors contributed to the discussion, critical revision and approved the final manuscript.
Authors’ information
None
Competing interests
The authors declare that they have no competing interests.
Additional files
Basal sex differences in cardiomyocyte performance suppressed with hypertrophy. (*sex p < 0.05, #strain p < 0.05; mean ± SEM, n = hearts or animals in brackets for each group). (PPTX 68 kb)
Sex differences in cardiomyocyte performance following either β-adrenoceptor stimulation or stimulated ischemia. (Values expressed as % basal. *sex p < 0.05, # strain p < 0.05; mean ± SEM, n = cells in brackets for each group). (PPTX 62 kb)
Contributor Information
James R. Bell, Email: belljr@unimelb.edu.au
Claire L. Curl, Email: ccurl@unimelb.edu.au
Tristan W. Harding, Email: tristanharding@me.com
Martin Vila Petroff, Email: mvila@atlas.med.unlp.edu.ar.
Stephen B. Harrap, Email: s.harrap@unimelb.edu.au
Lea M. D. Delbridge, Phone: +61 3 8344 5821, Email: lmd@unimelb.edu.au
References
- 1.Lam CSP, Little MS. Sex and cardiovascular risk. Circ. 2012;126:913–5. doi: 10.1161/CIRCULATIONAHA.112.121582. [DOI] [PubMed] [Google Scholar]
- 2.Go AS, Mozaffarian D, Roger VL, et al. Executive summary: heart disease & stroke statistics. Circ. 2014;129:399–410. doi: 10.1161/01.cir.0000442015.53336.12. [DOI] [PubMed] [Google Scholar]
- 3.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–6. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 4.Deo R, Vittinghoff E, Lin F, Tseng ZH, Hulley SB, Shlipak MG. Risk factor and prediction modeling for sudden cardiac death in women with coronary artery disease. Arch Int Med. 2011;171:1703–9. doi: 10.1001/archinternmed.2011.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Regitz-Zagrosek V, Dworatzek E, Kintscher U, Dragun D. Sex and sex hormone-dependent cardiovascular stress responses. Hypertension. 2013;61:270–7. doi: 10.1161/HYPERTENSIONAHA.111.189233. [DOI] [PubMed] [Google Scholar]
- 6.Vaccarino V, Badimon L, Corti R, et al. Ischaemic heart disease in women: are there sex differences in pathophysiology and risk factors? Cardiovasc Res. 2011;90:9–17. doi: 10.1093/cvr/cvq394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mehta LS, Beckie TM, DeVon HA, Grines CL, Krumholz HM, Johnson MN, Lindley KJ, Vaccarino V, Wang TY, Watson KE, Wenger NK, American Heart Association Cardiovascular Disease in Women and Special Populations Committee of the Council on Clinical Cardiology, Council on Epidemiology and Prevention, Council on Cardiovascular and Stroke Nursing, and Council on Quality of Care and Outcomes Research Acute myocardial infarction in women: a scientific statement from the American Heart Association. Circulation. 2016;133:916–47. doi: 10.1161/CIR.0000000000000351. [DOI] [PubMed] [Google Scholar]
- 8.Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics—2012 update. A report from the American Heart Association. Circulation. 2012;125:e2–220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaw LJ, Bugiardini R, Merz CN. Women and ischemic heart disease: evolving knowledge. J Am Coll Cardiol. 2009;54:1561–75. doi: 10.1016/j.jacc.2009.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaccarino V. Ischemic heart disease in women. Circ Cardiovsc Qual Outcomes. 2010;3:111–5. doi: 10.1161/CIRCOUTCOMES.109.925313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller VM. Why are sex and gender important to basic physiology and translational and individualized medicine. Am J Physiol Heart Circ Physiol. 2014;306:H781–8. doi: 10.1152/ajpheart.00994.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O'Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty SE, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E, RELAX Trial Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. J Am Med Assoc. 2013;309:1268–77. doi: 10.1001/jama.2013.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sasaki H, Nagayama T, Blanton RM, Seo K, Zhang M, Zhu G, Lee DI, Bedja D, Hsu S, Tsukamoto O, Takashima S, Kitakaze M, Mendelsohn ME, Karas RH, Kass DA, Takimoto E. PDE5 inhibitor efficacy is estrogen dependent in female heart disease. J Clin Invest. 2014;124:2464–71. doi: 10.1172/JCI70731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murphy E, Steenbergen C. Sex, drugs, and trial design: sex influences the heart and drug responses. J Clin Invest. 2014;124(6):237S–2377. doi: 10.1172/JCI76262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bell JR, Bernasochi GB, Varma U, Raaijmakers AJ, Delbridge LM. Sex and sex hormones in cardiac stress—mechanistic insights. J Steroid Biochem Mol Biol. 2013;137:125–35. doi: 10.1016/j.jsbmb.2013.05.015. [DOI] [PubMed] [Google Scholar]
- 16.Parks RJ, Howlett SE. Sex differences in mechanisms of cardiac excitation-contraction coupling. Pflugers Arch. 2013;465:747–63. doi: 10.1007/s00424-013-1233-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Kishimoto I, Saito Y, Harada M, Kuwahara K, Izumi T, Hamanaka I, Takahashi N, Kawakami R, Tanimoto K, Nakagawa Y, Nakanishi M, Adachi Y, Garbers DL, Fukamizu A, Nakao K. Androgen contributes to gender-related cardiac hypertrophy and fibrosis in mice lacking the gene encoding guanylylcyclase-A. Endocrinol. 2004;145:951–8. doi: 10.1210/en.2003-0816. [DOI] [PubMed] [Google Scholar]
- 18.Skavdahl M, Steenbergen C, Clark J, Myers P, Demianenko T, Mao L, Rockman HA, Korach KS, Murphy E. Estrogen receptor-beta mediates male-female differences in the development of pressure overload hypertrophy. Am J Physiol Heart Circ Physiol. 2005;288:H469–76. doi: 10.1152/ajpheart.00723.2004. [DOI] [PubMed] [Google Scholar]
- 19.Fliegner D, Schubert C, Penkalla A, Witt H, Kararigas G, Dworatzek E, Staub E, Martus P, Ruiz Noppinger P, Kintscher U, Gustafsson JA, Regitz-Zagrosek V. Female sex and estrogen receptor-beta attenuate cardiac remodeling and apoptosis in pressure overload. Am J Physiol Reg Integr Comp. 2010;298:R1597–606. doi: 10.1152/ajpregu.00825.2009. [DOI] [PubMed] [Google Scholar]
- 20.Cross HR, Lu L, Steenbergen C, Philipson KD, Murphy E. Overexpression of the cardiac Na+/Ca2+ exchanger increases susceptibility to ischemia/reperfusion injury in male, but not female, transgenic mice. Circ Res. 1998;83:1215–23. doi: 10.1161/01.RES.83.12.1215. [DOI] [PubMed] [Google Scholar]
- 21.Cross HR, Murphy E, Steenbergen C. Ca(2+) loading and adrenergic stimulation reveal male/female differences in susceptibility to ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2002;283:H481–9. doi: 10.1152/ajpheart.00790.2001. [DOI] [PubMed] [Google Scholar]
- 22.Gabel SA, Walker VR, London RE, Steenbergen C, Korach KS, Murphy E. Estrogen receptor beta mediates gender differences in ischemia/reperfusion injury. J Mol Cell Cardiol. 2005;38:289–97. doi: 10.1016/j.yjmcc.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 23.Bell JR, Porrello ER, Huggins CE, Harrap SB, Delbridge LMD. The intrinsic resistance of female hearts to an ischemic insult is abrogated in primary cardiac hypertrophy. Am J Physiol. 2008;294:H1514–22. doi: 10.1152/ajpheart.01283.2007. [DOI] [PubMed] [Google Scholar]
- 24.Ross JL, Howlett SE. Age and ovariectomy abolish beneficial effects of female sex on rat ventricular myocytes exposed to simulated ischemia and reperfusion. PLoS One. 2012;7:e38425. doi: 10.1371/journal.pone.0038425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parks RJ, Ray G, Bienvenu LA, Rose RA, Howlett SE. Sex differences in (SR) Ca(2+) release in murine ventricular myocytes are regulated by the cAMP/PKA pathway. J Mol Cell Cardiol. 2014;75:162–73. doi: 10.1016/j.yjmcc.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 26.Thorgeirsson G, Sigvaldason H, Witteman J. Risk factors for out-of-hospital cardiac arrest: the Reykjavik Study. Eur Heart J. 2005;26:1499–505. doi: 10.1093/eurheartj/ehi179. [DOI] [PubMed] [Google Scholar]
- 27.Vaccarino V, Badimon L, Corti R, de Wit C, Dorobantu M, Manfrini O, et al. Presentation, management, and outcomes of ischaemic heart disease in women. Nat Rev Cardiol. 2013;10:508–18. doi: 10.1038/nrcardio.2013.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porrello ER, Bell JR, Schertzer JD, Curl CL, McMullen JR, Mellor KM, et al. Heritable pathologic cardiac hypertrophy in adulthood is preceded by neonatal cardiac growth restriction. Am J Physiol. 2009;296:R672–80. doi: 10.1152/ajpcell.00004.2009. [DOI] [PubMed] [Google Scholar]
- 29.Harrap SB, Danes VR, Ellis JA, Griffiths CD, Jones EF, Delbridge LM. The hypertrophic heart rat: a new normotensive model of genetic cardiac and cardiomyocyte hypertrophy. Physiol Genomics. 2002;9:43–8. doi: 10.1152/physiolgenomics.00006.2002. [DOI] [PubMed] [Google Scholar]
- 30.Bell JR, Raaijmakers AJ, Curl CL, Reichelt ME, Harding TW, Bei A, Ng DC, Erickson JR, Vila Petroff M, Harrap SB, Delbridge LM. Cardiac CaMKIIδ splice variants exhibit target signalling specificity and confer sex-selective arrhythmogenic actions in the ischemic-reperfused heart. Int J Cardiol. 2015;181:288–96. doi: 10.1016/j.ijcard.2014.11.159. [DOI] [PubMed] [Google Scholar]
- 31.Delbridge LM, Satoh H, Yuan W, Bassani JW, Qi M, Ginsburg KS, Samarel AM, Bers DM. Cardiac myocyte volume, Ca2+ fluxes, and sarcoplasmic reticulum loading in pressure-overload hypertrophy. Am J Physiol. 1997;272:H2425–35. doi: 10.1152/ajpheart.1997.272.5.H2425. [DOI] [PubMed] [Google Scholar]
- 32.Satoh H, Delbridge LM, Blatter LA, Bers DM. Surface:volume relationship in cardiac myocytes studied with confocal microscopy and membrane capacitance measurements: species-dependence and developmental effects. Biophys J. 1996;70:1494–504. doi: 10.1016/S0006-3495(96)79711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mellor KM, Wendt IR, Ritchie RH, Delbridge LM. Fructose diet treatment in mice induces fundamental disturbance of cardiomyocyte Ca2+ handling and myofilament responsiveness. Am J Physiol Heart Circ Physiol. 2012;302:H964–72. doi: 10.1152/ajpheart.00797.2011. [DOI] [PubMed] [Google Scholar]
- 34.Delbridge LM, Connell PJ, Morgan TO, Harris PJ. Contractile function of cardiomyocytes from the spontaneously hypertensive rat. J Mol Cell Cardiol. 1996;28:723–33. doi: 10.1006/jmcc.1996.0067. [DOI] [PubMed] [Google Scholar]
- 35.O'Brien JD, Ferguson JH, Howlett SE. Effects of ischemia and reperfusion on isolated ventricular myocytes from young adult and aged Fischer 344 rat hearts. Am J Physiol. 2008;294:H2174–83. doi: 10.1152/ajpheart.00058.2008. [DOI] [PubMed] [Google Scholar]
- 36.Vila-Petroff M, Salas MA, Said M, Valverde CA, Sapia L, Portiansky E, Hajjar RJ, Kranias EG, Mundiña-Weilenmann C, Mattiazzi A. CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovasc Res. 2007;73:689–98. doi: 10.1016/j.cardiores.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 37.Farrell SR, Ross JL, Howlett SE. Sex differences in mechanisms of cardiac excitation-contraction coupling in rat ventricular myocytes. Am J Physiol. 2010;299:H36–45. doi: 10.1152/ajpheart.00299.2010. [DOI] [PubMed] [Google Scholar]
- 38.Ren J. Influence of gender on oxidative stress, lipid peroxidation, protein damage and apoptosis in hearts and brains from spontaneously hypertensive rats. Clin Exp Pharmacol Physiol. 2007;34:432–8. doi: 10.1111/j.1440-1681.2007.04591.x. [DOI] [PubMed] [Google Scholar]
- 39.Curl CL, Wendt IR, Kotsanas G. Effects of gener on intracellular Ca2+ in rat cardiac myocytes. Pflugers Arch. 2001;441:709–16. doi: 10.1007/s004240000473. [DOI] [PubMed] [Google Scholar]
- 40.Boutin-Ganache I, Picard S, Deschepper CF. Distinct gene-sex interactions regulate adult rat cardiomyocyte width and length independently. Physiol Genomics. 2002;12:61–7. doi: 10.1152/physiolgenomics.00121.2002. [DOI] [PubMed] [Google Scholar]
- 41.Haroon J, Foureaux G, Martins AS, Ferreira AJ, Reis AM, Javed Q. Gender differences in normal left ventricle of adult FVB/N mice due to variation in interleukins and natriuretic peptides expression levels. Cytokine. 2015;71:54–9. doi: 10.1016/j.cyto.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 42.Mahmoodzadeh S, Leber J, Zhang X, Jaisser F, Messaoudi S, Morano I, Furth PA, Dworatzek E, Regitz-Zagrosek V. Cardiomyocyte-specific estrogen receptor alpha increases angiogenesis, lymphangiogenesis and reduces fibrosis in the female mouse heart post-myocardial infarction. J Cell Sci Ther. 2014;5:153. doi: 10.4172/2157-7013.1000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parks RJ, Fares E, Macdonald JK, Ernst MC, Sinal CJ, Rockwood K, Howlett SE. A procedure for creating a frailty index based on deficit accumulation in aging mice. J Gerentol A Biol Sci Med Sci. 2012;67:217–27. doi: 10.1093/gerona/glr193. [DOI] [PubMed] [Google Scholar]
- 44.Murphy E. Estrogen signalling and cardiovascular disease. Circ Res. 2011;109:687–96. doi: 10.1161/CIRCRESAHA.110.236687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Curl CL, Wendt IR, Canny BJ, Kotsanas G. Effects of ovariectomy and 17 beta-oestradiol replacement on [Ca2+] in female rat cardiac myocytes. Clin Exp Pharmacol Physiol. 2003;30:489–94. doi: 10.1046/j.1440-1681.2003.03864.x. [DOI] [PubMed] [Google Scholar]
- 46.Fares E, Parks RJ, Macdonald JK, Egar JM, Howlett SE. Ovariectomy enhances SR Ca2+ release and increases Ca2+ spark amplitudes in isolated ventricular myocytes. J Mol Cell Cardiol. 2012;52:32–42. doi: 10.1016/j.yjmcc.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 47.Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–63. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 48.Shao Q, Fallica J, Casin KM, Murphy E, Steenbergen C, Kohr MJ. Characterization of the sex-dependent myocardial S-nitrosothiol proteome. Am J Physiol Heart Circ Physiol. 2015:ajpheart.00681.2015. doi: 10.1152/ajpheart.00681.2015. [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 49.Chu SH, Sutherland K, Beck J, Kowalski J, Goldspink P, Schwertz D. Sex differences in expression of calcium-handling proteins and beta-adrenergic receptors in rat heart ventricle. Life Sci. 2005;76:2735–49. doi: 10.1016/j.lfs.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 50.Yaras N, Tuncay E, Purali N, Sahinoglu B, Vassort G, Turan B. Sex-related effects on diabetes-induced alterations in calcium release in the rat heart. Am J Physiol. 2007;293:H3584–92. doi: 10.1152/ajpheart.00619.2007. [DOI] [PubMed] [Google Scholar]
- 51.Meyer R, Linz KW, Surges R, Meinardus S, Vees J, Hoffmann A, Windholz O, Grohé C. Rapid modulation of L-type calcium current by acutely applied oestrogens in isolated cardiac myocytes from human, guinea-pig and rat. Exp Physiol. 1998;83:305–21. doi: 10.1113/expphysiol.1998.sp004115. [DOI] [PubMed] [Google Scholar]
- 52.Patterson E, Ma L, Szabo B, Robinson CP, Thadani U. Ovariectomy and estrogen-induced alterations in myocardial contractility in female rabbits: role of the L-type calcium channel. J Pharmacol Exp Ther. 1998;284:586–91. [PubMed] [Google Scholar]
- 53.Grohé C, Kahlert S, Löbbert K, Meyer R, Linz KW, Karas RH, Vetter H. Modulation of hypertensive heart disease by estrogen. Steroids. 1996;61:201–4. doi: 10.1016/0039-128X(96)00014-1. [DOI] [PubMed] [Google Scholar]
- 54.Kravtsov GM, Kam KW, Liu J, Wu S, Wong TM. Altered Ca(2+) handling by ryanodine receptor and Na(+)-Ca(2+) exchange in the heart from ovariectomized rats: role of protein kinase A. Am J Physiol Cell Physiol. 2007;292:C1625–35. doi: 10.1152/ajpcell.00368.2006. [DOI] [PubMed] [Google Scholar]
- 55.Wattanapermpool J. Increase in calcium responsiveness of cardiac myofilament activation in ovariectomized rats. Life Sci. 1998;63:955–64. doi: 10.1016/S0024-3205(98)00353-1. [DOI] [PubMed] [Google Scholar]
- 56.Wattanapermpool J, Riabroy T, Preawnim S. Estrogen supplement prevents the calcium hypersensitivity of cardiac myofilaments in ovariectomized rats. Life Sci. 2000;66:533–43. doi: 10.1016/S0024-3205(99)00623-2. [DOI] [PubMed] [Google Scholar]
- 57.MacDonald JK, Pyle WG, Reitz CJ, Howlett SE. Cardiac contraction, calcium transients, and myofilament calcium sensitivity fluctuate with the estrous cycle in young adult female mice. Am J Physiol Heart Circ Physiol. 2014;306:H938–53. doi: 10.1152/ajpheart.00730.2013. [DOI] [PubMed] [Google Scholar]
- 58.Pandit S, Woranush W, Wattanapermpool J, Bupha-Intr T. Significant role of female sex hormones in cardiac myofilament activation in angiotensin II-mediated hypertensive rats. J Physiol Sci. 2014;64:269–77. doi: 10.1007/s12576-014-0316-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Curl CL, Bell JR, Delbridge LM. 'Gain'-ful insight into the cardiomyocyte Ca2+ seX factor. J Mol Cell Cardiol. 2012;52:7–9. doi: 10.1016/j.yjmcc.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 60.Murphy E, Lagranha C, Deschamps A, Kohr M, Nguyen T, Wong R, Sun J, Steenbergen C. Mechanism of cardioprotection: what can we learn from females? Pediatr Cardiol. 2011;32:354–9. doi: 10.1007/s00246-010-9877-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen J, Petranka J, Yamamura K, London RE, Steenbergen C, Murphy E. Gender differences in sarcoplasmic reticulum calcium loading after isoproterenol. Am J Physiol Heart Circ Physiol. 2003;285:H2657–62. doi: 10.1152/ajpheart.00557.2003. [DOI] [PubMed] [Google Scholar]
- 62.Vizgirda VM, Wahler GM, Sondgeroth KL, Ziolo MT, Schwertz DW. Mechanisms of sex differences in rat cardiac myocyte response to beta-adrenergic stimulation. Am J Physiol Heart Circ Physiol. 2002;282:H256–63. doi: 10.1152/ajpheart.2002.282.1.H256. [DOI] [PubMed] [Google Scholar]
- 63.Maurice DH, Palmer D, Tilley DG, Dunkerley HA, Netherton SJ, Raymond DR, Elbatarny HS, Jimmo SL. Cyclic nucleotide phosphodiesterase activity, expression, and targeting in cells of the cardiovascular system. Mol Pharmacol. 2003;64:533–46. doi: 10.1124/mol.64.3.533. [DOI] [PubMed] [Google Scholar]
- 64.McIntosh VJ, Chandrasekera PC, Lasley RD. Sex differences and the effects of ovariectomy on the beta-adrenergic contractile response. Am J Physiol Heart Circ Physiol. 2011;301:H1127–34. doi: 10.1152/ajpheart.00711.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoeker GS, Hood AR, Katra RP, Poelzing S, Pogwizd SM. Sex differences in β-adrenergic responsiveness of action potentials and intracellular calcium handling in isolated rabbit hearts. PLoS One. 2014;9:e111411. doi: 10.1371/journal.pone.0111411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Foryst-Ludwig A, Kreissl MC, Sprang C, et al. Sex differences in physiological cardiac hypertrophy are associated with exercise-mediated changes in energy substrate availability. Am J Physiol Heart Circ Physiol. 2011;301:H115–22. doi: 10.1152/ajpheart.01222.2010. [DOI] [PubMed] [Google Scholar]
- 67.Ross JL, Howlett SE. Beta-adrenoceptor stimulation exacerbates detrimental effects of ischemia and reperfusion in isolated guinea pig ventricular myocytes. Eur J Pharmacol. 2009;602:364–72. doi: 10.1016/j.ejphar.2008.11.034. [DOI] [PubMed] [Google Scholar]
- 68.Cordeiro JM, Ferrier GR, Howlett SE. Effects of adenosine in simulated ischemia and reperfusion in guinea pig ventricular myocytes. Am J Physiol. 1995;269:H121–9. doi: 10.1152/ajpheart.1995.269.1.H121. [DOI] [PubMed] [Google Scholar]
- 69.Louch WE, Ferrier GR, Howlett SE. Losartan improves recovery of contraction and inhibits transient inward current in a cellular model of cardiac ischemia and reperfusion. J Pharmacol Exp Ther. 2000;295:697–704. [PubMed] [Google Scholar]
- 70.Louch WE, Ferrier GR, Howlett SE. Changes in excitation-contraction coupling in an isolated ventricular myocyte model of cardiac stunning. Am J Physiol. 2002;283:H800–10. doi: 10.1152/ajpheart.00020.2002. [DOI] [PubMed] [Google Scholar]
- 71.Tadros R, Ton AT, Fiset C, Nattel S. Sex differences in cardiac electrophysiology and clinical arrhythmias: epidemiology, therapeutics, and mechanisms. Can J Cardiol. 2014;30:783–92. doi: 10.1016/j.cjca.2014.03.032. [DOI] [PubMed] [Google Scholar]
- 72.Bell JR, Mellor KM, Wollermann AC, Delbridge LM. Cardiac ischaemic stress: cardiomyocyte Ca2+, sex and sex steroids. Clin Exp Pharmacol Physiol. 2011;38:717–23. doi: 10.1111/j.1440-1681.2011.05567.x. [DOI] [PubMed] [Google Scholar]
- 73.Schuster I, Mahmoodzadeh S, Dworatzek E, Jaisser F, Messaoudi S, Morano I, Regitz-Zagrosek V. Cardiomyocyte-specific overexpression of estrogen receptor beta improves survival and cardiac function after myocardial infarction in female and male mice. Clin Sci (Lond) 2015;130(5):365–76. doi: 10.1042/CS20150609. [DOI] [PubMed] [Google Scholar]
- 74.Bell JR, Mellor KM, Wollermann AC, Ip WT, Reichelt ME, Meachem SJ, Simpson ER, Delbridge LM. Aromatase deficiency confers paradoxical postischemic cardioprotection. Endocrinology. 2011;152:4937–47. doi: 10.1210/en.2011-1212. [DOI] [PubMed] [Google Scholar]
- 75.Bell JR, Bernasochi GB, Varma U, Boon WC, Ellem SJ, Risbridger GP, Delbridge LM. Aromatase transgenic upregulation modulates basal cardiac performance and the response to ischemic stress in male mice. Am J Physiol Heart Circ Physiol. 2014;306:H1265–74. doi: 10.1152/ajpheart.00012.2014. [DOI] [PubMed] [Google Scholar]
- 76.Bell JR, Bernasochi GB, Wollermann AC, Raaijmakers AJ, Boon WC, Simpson ER, Curl CL, Mellor KM, Delbridge LM. Myocardial and cardiomyocyte stress resilience is enhanced in aromatase-deficient female mouse hearts through CaMKIIδ activation. Endocrinology. 2015;156:1429–40. doi: 10.1210/en.2014-1700. [DOI] [PubMed] [Google Scholar]