

Abstract
Propofol is a widely used intravenous general anesthetic. We synthesized 2-fluoro-1,3-diisopropylbenzene, a compound that we call “fropofol”, to directly assess the significance of the propofol 1-hydroxyl for pharmacologically relevant molecular recognition in vitro and for anesthetic efficacy in vivo. Compared to propofol, fropofol had a similar molecular volume and only a small increase in hydrophobicity. Isothermal titration calorimetry and competition assays revealed that fropofol had higher affinity for a protein site governed largely by van der Waals interactions. Within another protein model containing hydrogen bond interactions, propofol demonstrated higher affinity. In vivo, fropofol demonstrated no anesthetic efficacy, but at high concentrations produced excitatory activity in tadpoles and mice; fropofol also antagonized propofol-induced hypnosis. In a propofol protein target that contributes to hypnosis, α1β2γ2L GABAA receptors, fropofol demonstrated no significant effect alone or on propofol positive allosteric modulation of the ion channel, suggesting an additional requirement for the 1-hydroxyl within synaptic GABAA receptor site(s). However, fropofol caused similar adverse cardiovascular effects as propofol by a dose-dependent depression of myocardial contractility. Our results directly implicate the propofol 1-hydroxyl as contributing to molecular recognition within protein targets leading to hypnosis, but not necessarily within protein targets leading to side effects of the drug.
Keywords: Anesthesia, molecular recognition, GABAA receptor, hydrogen bonding, propofol
Graphical abstract
The mechanisms underlying general anesthetic action leading to desired anesthesia endpoints or adverse effects have been a mystery for nearly two centuries. The first major proposal for a mechanism of anesthetic-induced hypnosis arose when a relationship between anesthetic lipophilicity and potency was independently observed by Meyer and Overton.1,2 This correlation led to various hypotheses for membrane mediated mechanisms of anesthesia; however, evidence that anesthetics bind and cause functional effects through specific sites on multiple protein targets has steadily emerged. Crystallized anesthetic–protein complexes,3–5 recognition of highly specific and selective responses by ion channels,6,7 and elucidation of receptor binding site character using photolabel analogues8–12 have propelled the concept of protein-mediated mechanisms to general acceptance.
The binding affinity of a drug for a protein site is generally mediated by multiple specific noncovalent interactions in a process known as molecular recognition. Drug occupancy of a target protein site results in alteration(s) in protein conformation and/or dynamics that reflect (or produce) changes in the protein’s activity. General anesthetic ligands are a unique class of drugs in that they share only broad physicochemical features, such as low molecular weight and hydrophobicity. While causing similar desired endpoints including hypnosis, immobility, and amnesia, and adverse effects including cardiovascular depression, nausea, and hyperthermia, the drug concentrations to achieve the effects can be considerably different between anesthetics. These observations suggest that a penchant of an individual anesthetic for a pharmacological effect may be reliant on distinctive chemical features giving rise to a relative higher affinity for particular protein target(s). It is therefore essential to characterize the molecular interactions between proteins and anesthetics to successfully design new anesthetic agents that selectively cause the desirable effects through specific targets.
Propofol (2,6-diisopropylphenol) is the most widely used intravenous general anesthetic for the induction and maintenance of anesthesia.13 While a fairly simple compound, propofol contains a particular feature within its chemical structure, a 1-hydroxyl, that is capable of distinctive intermolecular interactions.14–16 The hydrophilic group permits hydrogen bonding to both aqueous solvent and to amino acids within protein targets, which contribute to solvation and specific molecular recognition, respectively. Solvation is critical for hydrophobic ligands to gain access to protein target(s).17 Previous studies that examined various substitutions on the alkylphenol backbone could not rigorously attribute the changes in ligand efficacy to molecular recognition or to solvation.18,19 To achieve this, a modification of propofol that removes hydrogen bonding propensity while retaining solvation properties, and then a comparison of both ligand activity and binding, is essential.
Thus, we substituted the 1-hydroxyl of propofol with a fluorine atom to produce 2-fluoro-1,3-diisopropylbenzene, or “fropofol” (Figure 1). This otherwise physicochemically similar analogue to propofol allowed us to explicitly link the 1-hydroxyl to protein affinity and, most importantly, to multiple in vivo consequences. Our data indicates that hydrogen bonding plays a surprisingly dominant role in molecular recognition for propofol-protein interactions that contribute to hypnosis, whereas the loss of the hydrophilic feature did not prevent binding to targets that lead to less favorable endpoints.
Figure 1.
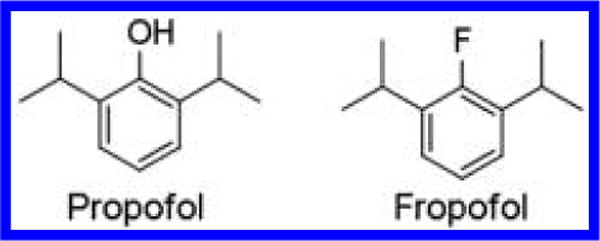
Chemical Structures. Chemical structures of 2,6-diisopropylphenol (propofol) and 2-fluoro-1,3-diisopropylbenzene (fropofol).
RESULTS AND DISCUSSION
To directly evaluate the contribution of the 1-hydroxyl in propofol molecular recognition, we designed an analogue that selectively weakens the hydrogen bond capability while retaining other physiochemical properties of propofol. The synthesis of fropofol involved the diazotization of 2,6-diisopropylaniline, precipitation of the tetrafluoroborate salt, and dediazotization-fluorination under mild vacuum overnight (Scheme 1). 1H, 13C, and 19F NMR spectra are presented in Supporting Information Figures 1–3, and the calculated mass/charge value was appropriate for the fropofol chemical structure.
Scheme 1.
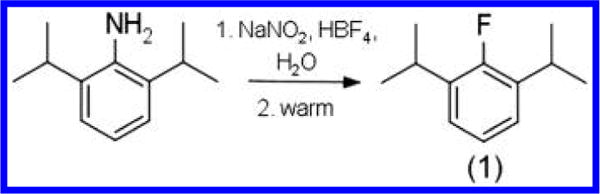
Synthesis
The physiochemical properties of propofol and fropofol are reported in Table 1. The calculated van der Waals molecular volumes were similar between the two compounds with fropofol (189 Å3) being marginally smaller than propofol (192 Å3). Based on the Meyer–Overton rule, the increase in hydrophobicity by calculated octanol/water partition coefficients for fropofol (3.96) relative to propofol (3.79) predicted a modest increase in the compound’s anesthetic potency.2 Consistent with the clogP value, the measured maximum aqueous concentration achieved by fropofol was 116 ± 4.4 μM, approximately 5-fold less than propofol; however, the solubility of fropofol exceeded or was within the same range of halogenated alkylphenols that retain activity; 2,4-diethylphenol bromide and 4-iodo-2,6-diisopropylphenol.19
Table 1.
Physicochemical Parameters and Binding Properties of Propofol and Fropofol
| physicochemical properties | propofol (2,6-diisopropylphenol) | fropofol (2-fluoro-1,3-diisopropylbenzene) | ||
|---|---|---|---|---|
| molecular weight | 178.27 Da | 180.26 Da | ||
| van der Waals volume | 192 Å | 189 Å | ||
| cLogP | 3.79 | 3.96 | ||
| density | 0.96 g/cm3 | 0.9 g/cm3 | ||
|
| ||||
| propofol (2,6-diisopropylphenol)
|
fropofol (2-fluoro-1,3-diisopropylbenzene)
|
|||
| protein affinities (μM)a | HSAFb | hSA | HSAFb | hSA |
|
| ||||
| ITC | 9 (7.1–11) | 43 (36–50) | 1.7 (0.5–2.9) | 91 (72–110) |
| 1-AMA competitionc | 10 (7–15) | 0.7 (0.3–1.5) | ||
Values are represented as mean (95% CI).
Stoichiometry of HSAF sites were fixed at N = 1.
KD Fluorescence data derived from Cheng–Prusoff equation.
We initiated studies of fropofol binding to model proteins. Previously, anesthetic binding to horse spleen apoferritin (HSAF) had been found to correlate strongly with GABAA receptor potentiation and tadpole loss of righting reflex (LORR).20,21 Anesthetic binding to a single site on HSAF is exothermic and mediated by electrostatic and hydrophobic interactions;21 a hydrogen bond was not apparent in the crystal structure of the complex with propofol. Propofol and fropofol bound HSAF with low micromolar KD values (Figure 2A, B; Table 1); however, fropofol had a 4- to 5-fold increase in affinity (KD = 1.7 μM (0.5–2.9) (95% CI)) relative to propofol (KD = 9.0 μM (7.1–11) (95% CI)).
Figure 2.
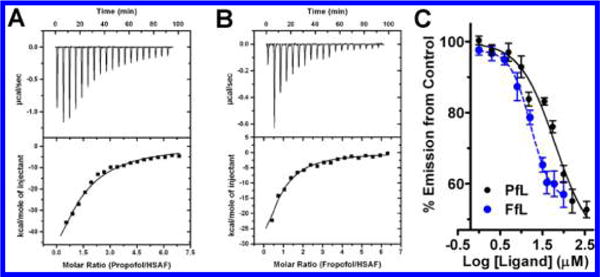
Propofol and fropofol horse spleen apoferrition (HSAF) binding. Isothermal titration calorimetry profiles of HSAF(A-B) interaction with propofol (A) or fropofol (B) using sequential titrations. Top: time response heat change from addition of ligand. Bottom: best fit attained from a single site binding model (best χ2 statistic) fitted to a 1:1 stoichiometry for HSAF. Drug affinities (KD) for HSAF site were 9 and 1.7 μM for propofol and fropofol, respectively. (C) HSAF fluorescence competition using 1-aminoanthracene with titrations of either propofol (PfL; black circles) or fropofol (FfL; blue circles). Intensity was corrected for ambient ligand and protein fluorescence (see Table 1).
The propofol photoactive analogue meta-azi-propofol (AziPm) has been shown to photolabel the crystallographically determined propofol site on HSAF.22 Therefore, we used [3H]AziPm photoradiolabeling with and without competing ligands to definitively determine the fropofol-HSAF binding site. One μM propofol or fropofol caused a 31% and 61% reduction in [3H]AziPm binding to HSAF, respectively. 1-Aminoanthracene (1-AMA) also binds the same site on HSAF.23 1-AMA decreases in fluorescence when displaced from the HSAF site, and this feature allows calculation of ligand KD values through competition experiments. Calculated KD values from 1-AMA fluorescence competition (Figure 3A) correlated well with ITC and photoradiolabel competition studies for both propofol (KD = 10 μM (7–15) (95% CI)) and fropofol (KD = 0.7 μM (0.3–1.5) (95% CI)). Cumulatively, the data show that propofol and fropofol bind within the same hydrophobic cavity on HSAF;21 however, fropofol consistently demonstrated an approximately 3- to 5-fold increase in affinity, presumably due to the modest increase in hydrophobicity.
Figure 3.
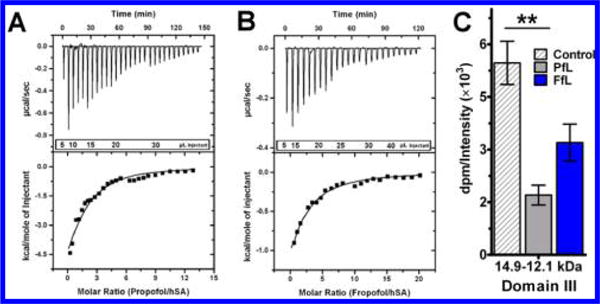
Propofol and fropofol human serum albumin (hSA) binding. Isothermal titration calorimetry profiles of hSA interaction with propofol (A) or fropofol (B) using incremental titrations. Top: time response heat change from addition of ligand. Bottom: best fit attained from a single site binding model (best χ2 statistic) resulting in roughly 1:2 stoichiometry for hSA for fropofol (n = 1.3 ± 0.9) and propofol (n = 1.4 ± 0.5). Affinity (KD) for the hSA sites were 43 and 91 μM for propofol and fropofol respectively (see Table 1). (C) Competition binding assay with [3H]AziPm photoradiolabeling of hSA hSA CNBr 14.9–12.1 kDa domain III digestion fragment with DMSO control or 75 μM propofol (PfL) or fropofol (FfL) competition. Quantitation of dpm (disintegrations per minute) was normalized to averaged relative lane intensities of Commassie blue (CB) stain. Data sets are represented as normalized mean ± normalized SEM and were analyzed by one-way ANOVA with Bonferroni’s post hoc test (p < 0.05) comparing significance in competition to fragment control (*).
Propofol has also been shown with crystallography to bind two sites within domain III of human serum albumin (hSA).4 Within each site there resides hydrogen bonding partners that facilitate propofol binding.4,24 Similar to HSAF, ITC measurements showed propofol and fropofol bound to hSA exothermically (Figure 3A, B; Table 1). The cumulative KD of propofol binding to hSA was determined as 43 μM (36–50) (95% CI), with stoichiometry being dominated by the higher affinity complex.25 Fropofol demonstrated similar stoichiometry but with a cumulative KD of 91 μM (72–110) (95% CI).
[3H]AziPm photoradiolabeling competition experiments were also performed with hSA, and these experiments were supplemented with CNBr digestion to specifically study binding to domain III. Within the isolated domain III fragments, propofol significantly decreased radiolabel incorporation to about 67 ± 5% of the control [3H]AziPm photolabeling, while fropofol inhibited it by only half as much (37 ± 12% (mean ± SEM); Figure 3C). This suggests that the substitution of the 1-hydroxyl results in lower affinity to the specific propofol hSA binding sites that contain hydrogen bonding partners.
Together, our data with HSAF and hSA demonstrate a contribution of the propofol hydroxyl for specific molecular recognition of model proteins. Our results demonstrate that propofol binds with higher affinity than fropofol to sites that contain hydrogen bond interactions, but that in the absence of hydrogen bond partners, fropofol binds with higher affinity. The relatively high affinities of these interactions also suggest that this result is independent of the solubility of the ligands.
It bears mentioning that substitution for the 1-hydroxyl would also result in electronic changes that could modulate binding. It is very difficult to entirely separate this possibility from the hydrogen bonding hypothesis, but it seems an unlikely explanation for the large differences measured and the generally weaker van der Waals interactions that would be influenced. Halogens can also serve as weak hydrogen bond acceptors. However, fluorine, due to its high electronegativity and lack of polarizability,26,27 is generally excluded from this form of interaction.28
Next, to characterize the relevance of the hydroxyl on pharmacological activity, we performed several in vivo experiments. The pharmacological activity of fropofol within albino Xenopus laevis tadpoles was evaluated over the course of 60 and 90 min exposure periods. When administered 3–100 μM fropofol, none of the X. laevis tadpoles within any dose group exhibited the standard loss-of-mobility endpoints. In contrast, excitatory phenotypes29 were observed in some tadpoles (~11%) at 40–60 min with greater than 30 μM concentrations, and longer (80–90 min) exposures to these high concentrations produced this behavior in ~70% of the tadpoles. This excitatory behavior included continuous tight circular and/or “darting” swimming patterns, previously reported as indicators of lower class seizures in X. laevis.29 When fropofol-containing water was exchanged for fresh pond water, normal swimming behaviors resumed within 10–15 min.
The excitatory activity was similarly observed within a mammalian model. Wild type C57/Bl6 mice were given bolus tail vein injections of fropofol dissolved in 10% lipid emulsion. At low dose (96 mg/kg), no observable effects were noted. At higher dose (180 mg/kg), electroencephalography (EEG) recordings and physical observation showed generalized tonic-clonic seizure-like activity 2 min post injection (Figure 4A), after which a lethargic postictal state was observed. Mice resumed normal activity within 2–3 h, and no toxicity was observed within the following days postinjection. Loss of righting reflex, the standard endpoint for general anesthetics, was produced with 20 mg/kg of propofol, but not with even the highest dose of fropofol.
Figure 4.
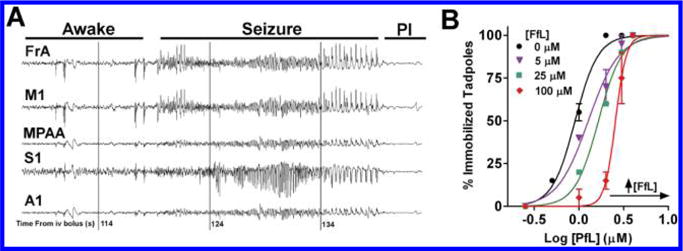
Ligand in vivo activity. (A) EEG recording 1.75–2.4 min after injection of 200 mg/kg fropofol. Phenotypical seizure activity observed within frontal association (FrA), primary motor (M1), medial parietal association area (MPAA), primary somatosensory (S1), and primary auditory (A1) traces. Postictal state (PI) was observed after seizing activity. (B) Propofol dose–response curves with the coadministration of 0 μM (black circles), 5 μM (violet triangles), 25 μM (green squares), and 100 μM (red diamonds) of fropofol within X. laevis tadpoles. Immobility was measured as loss of spontaneous movement.
To ensure that fropofol accessed the brain, some mice were euthanized at 45 s and at 10 min after IV bolus injection of 96 and 200 mg/kg, respectively, and fropofol content in the brain was assayed by reverse phase-high performance liquid chromatography. At both time points, fropofol was detectable within processed brain samples at 38 μg/gram of brain tissue and 51 μg/gram of brain tissue for the lower and higher doses, respectively. These concentrations are higher than propofol concentrations that result in hypnosis.30 When combined with the obvious central nervous system-derived behavioral change, this confirms that exclusion by either the blood-brain barrier or through active pumps does not explain the absence of a fropofol hypnotic action.
Finally, in order to determine whether fropofol has subphenotypic hypnotic activity,31 tadpoles were exposed to increasing concentrations of propofol in the presence of four fixed concentrations of fropofol (0, 5, 25, and 100 μM). Rather than demonstrating an additive effect, fropofol induced a right-shift in the propofol dose–response curve (Figure 4B), indicating antagonism toward propofol induced hypnosis at all concentrations.
To test our hypothesis and determine a potential mechanism for fropofol excitatory activity and antagonism of propofol induced hypnosis, we investigated the influence of these agents on recombinantly expressed GABAA receptors. Propofol has been shown to be a strong positive modulator of GABAA receptors,33 which likely contributes to its hypnotic action. Perfusion with propofol in the absence of GABA caused minor direct activation, consistent with previous literature (Figure 5A).33 Also consistent with previous studies, coexposure to propofol and GABA resulted in a concentration-dependent increase in current up to 80 μM (Figure 5B); positive modulation began to decline at propofol concentrations of 100 μM.33,34 In contrast, 5 and 50 μM fropofol elicited no significant direct activation of the receptor (Figure 5A). Furthermore, fropofol, at any concentration, demonstrated no significant modulation or inhibition of the α1β2γ2L GABAA receptor at solubility permitted concentrations (Figure 5).
Figure 5.
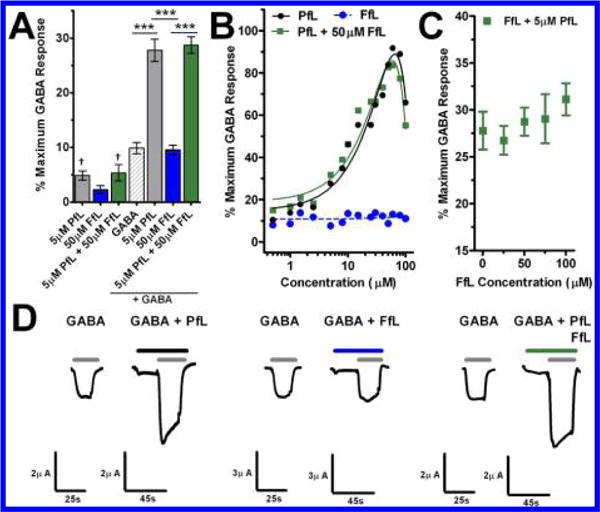
GABAA receptor activity. (A) Effect on current by concentrations of propofol (PfL; n = 3), fropofol (FfL; n = 5), or propofol and fropofol (n = 5) without GABA EC10 and propofol (n = 5), fropofol (n = 8), or propofol and fropofol with (n = 6) GABA EC10 within X. laevis oocytes expressing α1β2γ2L GABAA receptor. GABA represents initial control EC10 exposure for modulation studies (n = 19). Data is normalized to maximum GABA response and represented as mean ± SEM. Data were analyzed by one-way ANOVA with Bonferroni’s post hoc test (p < 0.05) showing significant differences in fropofol or propofol or propofol and fropofol modulation (*) or significance from EC00 (†). (B) Propofol (PfL; black circles), fropofol (FfL; blue circles), or propofol with 50 μM fropofol (green squares) concentration–response curves for α1β2γ2L GABAA receptor positive modulation in the presence of GABA EC10. Each point represents individually tested oocytes. Lines represent polynomial (PfL/black solid; PfL+ 50 μM FfL/green solid) and linear (FfL/blue dash) best fit curves. (C) Response to fropofol (FfL) concentrations in the presence of 5 μM propofol represented as mean ± SEM (n = 3–6). (D) Representative traces of evoked current by GABA EC10 control and following combined of GABA EC10 and 5 μM propofol (PfL; left); GABA EC10 control and following combined of GABA EC10 and 50 μM fropofol (FfL; middle); GABA EC10 control and following combined of GABA EC10, 5 μM propofol and 50 μM fropofol (PfL+FfL; right) exposures within individual X. laevis oocytes expressing α1β2γ2L GABAA receptor.
To determine whether the antagonistic action of fropofol on propofol hypnosis was mediated through GABAA receptors, we exposed all three ligands (GABA, propofol, and fropofol) to the recombinant ion channels. The introduction of 50 μM fropofol resulted in no significant change of propofol positive modulation of the GABAA receptor (Figure 5A, B, D), and exposure of saturating concentrations of fropofol displayed no significant alteration of 5 μM propofol potentiation of GABAA receptor currents (Figure 5C, D).
In total, fropofol had no influence whatsoever on propofol positive modulation of the α1β2γ2L GABAA receptor. Because propofol has shown similar potency across most synaptic GABAA receptor subtypes,34,35 these data suggest that fropofol in vivo excitatory activity and antagonism of propofol hypnosis were via a non-GABAergic mechanism. However, we examined only a single subtype, and thus cannot completely rule out GABAergic antagonism as underlying fropofol excitation. Fropofol might be a useful tool to characterize GABAA receptor specificity. In addition, these data suggest that our previous demonstration of the correlation of affinity for HSAF “anesthetic site”, GABAA receptor potentiation, and LORR21 represents only a portion of the molecular recognition features required for transducing a likely major component for the pharmacological effect. While HSAF provides a convenient model amenable to high throughput screening, which is a considerable improvement over previous approaches, like the Meyer–Overton rule,32 these fropofol data strongly indicate an additional requirement for a hydrogen bond within synaptic GABAA receptor site(s).
As fropofol does not partake in the molecular recognition features that lead to hypnosis, we decided to examine whether the propofol 1-hydroxyl was similarly vital for mechanisms resulting in an alternative pharmacological endpoint. A known adverse effect of propofol is cardiovascular depression, which previous reports suggest is at least partially caused by a direct effect on myocardial contraction.36,37 The influence of propofol and fropofol on myocardial contractility was measured by the change in force development of isolated, intact rat trabecular muscle. A concentration of 100 μM propofol depressed maximum force development by 49 ± 4% (Figure 6), and 100 μM fropofol exposure resulted in a similar effect with a 35 ± 1% reduction (Figure 6). These results suggest that the molecular interactions that lead to the decrease in myocardial contractility are less dependent on the 1-hydroxyl, and likely hydrogen bond interactions, in contrast to GABAA receptor potentiation. These data indicate that fropofol would be an effective tool to dissect interactions within these different molecular targets and tissues.
Figure 6.
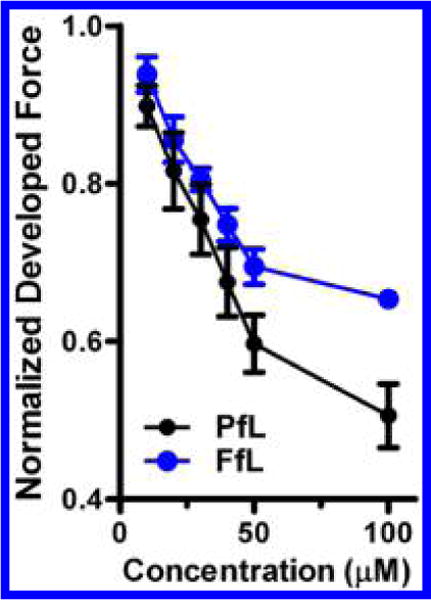
Influence on trabecular muscle generation. Force development by intact trabecular muscle over a range of propofol (PfL; n = 8) and fropofol (FfL; n = 3) concentrations. Force development was normalized to initial force development without agent exposure. Values are represented at normalized mean ± SEM.
The cardiovascular and excitatory activity of fropofol emphasizes the importance of distinguishing different forms of molecular recognition involved in the pharmacology of propofol. Our evidence suggests that more degenerate, apolar binding sites may transduce either no effect or are associated with certain adverse effects. Furthermore, we demonstrate that these alternative non-hydrogen bonding dependent pathways that fropofol unveils are probably non-GABAergic. By expanding the repertoire of recognized propofol targets, and relating molecular recognition features with the functional effect, further progress in anesthetic development is possible.
In summary, we synthesized a propofol analogue with fluorine replacing the 1-hydroxyl to result in the loss of hydrogen bond capabilities. The compound, fropofol, displayed analogous physiochemical properties and specific binding to commonly employed anesthetic-site protein models. Within a protein site defined primarily by hydrophobic forces, fropofol showed greater affinity than propofol. However, within a model containing cavities with clear hydrogen bonding residues, fropofol demonstrated lower affinity. Within animal models, fropofol administration resulted in no hypnotic activity, but rather weak excitatory activity. The excitatory activity and antagonism of propofol efficacy was determined not to be mediated by direct postsynaptic GABAergic signaling. On the other hand, fropofol induced myocardial depression like that of propofol. These data indicate that hydrogen bonding is a critical molecular recognition feature for propofol protein binding sites that transduce hypnosis, and that fropofol may be used to identify and distinguish these sites.
METHODS
General Synthetic Procedures
Solvents and reagents used were purchased from commercial sources unless otherwise noted. 1H, 13C, and 19F nuclear magnetic resonance (NMR) spectra are provided in the Supporting Information. 1H and 13C NMR spectra were recorded using a Bruker DMX 500 MHz NMR spectrometer, and 19F NMR spectra was recorded using Bruker DMX 360 MHz NMR spectrometer. Purity of Fropofol was determined using reverse phase-high performance liquid chromatography (rpHPLC) with C-18 analytical column. An isocratic gradient (67:28:5:0.1 acetonitrile/ddH2O/isopropanol/trifluoroacetic acid) with a 1 mL/min flow at ambient temperature (21–22 °C) was applied, and fropofol was monitored for UV–vis absorbance at 210 and 205 nm. The retention time for fropofol was observed at 8.5 min with a purity of >98%.
Preparation of 2-Fluoro-1,3-diisopropylbenzene (Fropofol)
A 250 mL round-bottom flask (rbf) with stir bar was filled with 2.0 g (11.3 mmol) of >99% pure 2,6-diisopropylaniline, water (39 mL), and 48% HBF4 (5.7 g; 31.1 mmol). This clear, homogeneous solution was cooled to 0 °C in an ice–water bath. A solution of NaNO2 (0.78 g; 11.3 mmol) in water (1.7 mL) was added dropwise over the course of 5 min, while keeping the temperature of the stirred solution below 3 °C. After stirring for a few additional minutes, the resulting yellow crystals were suction filtered on a fritted glass funnel and then transferred to a 250 mL rbf, which was evacuated under aspirator pressure overnight. The next day, the resulting brown liquid residue was extracted with hexanes (3 × 25 mL) and the combined organic layers were washed with 1 N KOH solution (4 × 35 mL), followed by water (3 × 15 mL). The organic layer was dried over magnesium sulfate and concentrated in vacuo to yield 1.53 g of a yellow oil. The product was purified by passing it through a plug of silica gel (33 cc) using hexane. Evaporation of the solvent followed by bulb-to-bulb transfer of the residue under dynamic vacuum gave 1.15 g (57%) of fropofol as a clear, colorless liquid. 1H NMR (500 MHz, CDCl3): δ 7.14–7.23 (3H, m), 3.41 (2H, sep, J = 7 Hz), 1.40 (12H, d, J = 7 Hz). 13C NMR (125 MHz, CDCl3): δ 158.5 (d, JC–F = 243 Hz), 135.1 (d, JC–F = 15 Hz), 124.4 (d, JC–F = 6.3 Hz), 123.8 (d, JC–F = 3.8 Hz), 27.2 (d, J = 3.8 Hz), 22.8 ppm. 19F NMR (360 MHz, CDCl3): δ −126.14 ppm (t, J = 3.6 Hz). HRMS m/z calculated for C12H17F (M)+ 180.1314; found 180.1311.
Physicochemical Properties
The density of fropofol was determined from triplicate measurements of the volume/mass relationship. The extinction coefficient (Σ270 = 10611/M) was calculated through the UV (Varian Cary 300 Bio UV–vis spectrophotometer) benzene absorption at 270 nm within a methanolic solution of known concentrations. The maximal water solubility (116 ± 4.4 μM; mean ± SD) was calculated from the extinction coefficient after 24 h titration and incubation in double distilled water (ddH2O). Octanol/water partition coefficients were calculated using XLOGP3.38 Molecular volume was calculated using NAMD program developed by the Theoretical Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana–Champaign.39
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) permits the calculation of the binding affinity and entropy based on measurement of binding enthalpy. Propofol and fropofol injections into the soluble protein models horse spleen apoferritin (HSAF) and human serum albumin (hSA) were conducted similar to previously reported procedures21 and were resolved using a VP-ITC microcalorimeter (MicroCal, Inc., Northampton, MA). For all ITC studies, 20 mM sodium phosphate buffer containing 130 mM NaCl (pH 7.0) was used and referenced against ddH2O. The sample cell (1.43 mL) contained either 5 or 2.5 μM HSAF or 20 μM hSA solution at 20 or 26 °C based on pilot studies, respectively. The injectate solution (286 μL) was either propofol (160 μM) or fropofol (75 μM). Injections were titrated (15 μL) into the sample cell for HSAF. Because of the low-affinity interaction(s) with hSA, and the limited solubility of the ligand(s), sequential titrations were performed to achieve near complete occupancy of the binding site(s). This was accomplished by loading and titrating (10–40 μM) with the same ligand, propofol (0.55 mM), or fropofol (85 μM) without removal from the sample cell until the titration signal was near constant. The titrations were linked together prior to data analysis using ConCat32 software provided by MicroCal, Inc. The signals of buffer into buffer, ligand to buffer, and buffer to protein were subtracted after separate titrations. Origin 5.0 software was used to best fit thermodynamic parameters to the heat profiles.
Fluorescence Competition with HSAF
A fluorescence competition assay utilizing 1-aminoanthracine (1-AMA) allowed comparison of ligand–protein binding in HSAF. The extent of 1-AMA fluorescence inhibition has been previously reported as a reliable measurement of anesthetic occupation of hydrophobic protein cavities.20,23 All solutions were prepared in 20 mM sodium phosphate buffer containing 130 mM NaCl (pH 7.2) in 1 mL quartz cuvettes. For competition with the HSAF anesthetic site,21 samples containing 5 μM HSAF and 5 μM 1-AMA were mixed with increasing concentrations of propofol (1–350 μM) or fropofol (1–100 μM). The 1-AMA fluorescence was determined with 380 nm excitation and emission monitoring between 400 and 700 nm. The fluorescence curves were corrected by subtraction of the 1-AMA/protein, ligand/protein and ligand/1-AMA baseline emission post acquisition. The fluorescence intensity versus concentration data were fitted to variable slope Hill models to obtain the IC50 and Hill slope. The KD was calculated using the Cheng–Prusoff equation23 to correct for the presence of the 1-AMA competitors.
[3H]m-Azipropofol Photolabel Competition with HSAF
In addition to 1-AMA, we employed radiophotolabel competition using tritiated m-azipropofol ([3H]AziPm) to confirm occupancy of the HSAF propofol site. In 1 mm quartz cuvettes, 3 μM HSAF and 1 μM [3H]AziPm, respectively, in ddH2O were combined with 10 μM fropofol or propofol, or vehicle control (DMSO). After 5 min equilibration, the sample was irradiated for 10 min with ~340–375 nm light generated by filtering a 100 W arc mercury lamp through broadband (~340–625 nm) and UV bandpass (~250–375 nm) filters (lamp and filters from Newport, Stratford, CT). After precipitation with 4× volume cold acetone and two additional cold acetone washes (1 mL each), the dried pellet was suspended in 1% SDS, 1% Triton-X, and 5 mM Tris (pH 7.6) to achieve 12 μM HSAF (BCA Protein Assay Kit using HSAF as standard). A sample volume of 5 uL was scintillation counted using Ecolite (+) liquid scintillation cocktail (MP Biomedicals) with a PerkinElmer Tri-Carb 2800TR instrument. The final dpm (disintegrations per minute) were normalized to protein content.
[3H]m-Azipropofol Photolabel Competition with has
Similar competition experiments were used for hSA, except that CNBr protein digestion was used after photolabeling. Thus, after irradiation of 5 μM hSA (>98%; Fluka) and 2 μM [3H]AziPm with 75 μM fropofol or propofol or vehicle control (DMSO), samples were diluted to 1.5 μM hSA with 0.1 M Tris-HCl (pH 8.0) and 3% SDS. DTT was then added to achieve 2 mM and samples were heated for 2 min at 96 °C. CNBr and formic acid were added to produce a 9 mM CNBr, 70% formic acid solution. These samples were left at room temperature for 24 h, followed by the addition of 200 μL of N-ethylmorpholine (97%, Sigma). Following acetone precipitation and drying under nitrogen, the pellets were resuspended in 1.5% SDS, 50 mM Tris pH 7.0. Protein content was determined with the BCA Protein Assay Kit using hSA as standard. A total of 30 μg of digested protein was separated on 4–15% SDS gels. After electrophoresis, the gel was stained with Commassie blue G-250 and imaged using Kodak Image Station 4000 mm Pro. Bands were excised and the polyacrylamide dissolved with 30% H2O2 for ~3 h at 65–70 °C. The sample volume was scintillation counted using Ecolite (+) liquid scintillation cocktail with a PerkinElmer Tri-Carb 2800TR instrument. The final dpm were normalized to the Commassie blue stain intensity relative to the total sample lane. In a parallel study, bands were excised and submitted to the Proteomics Core Facility at the University of Pennsylvania for mass spectrometry to verify band peptide content.
Activity in Tadpoles
Behavioral activity was initially determined in albino X. laevis tadpoles (stages 45–47) as previously described.22,29,40 Tadpoles (n = 240) were incubated in Petri dishes (10 tadpoles/dish) with concentrations (3, 30, 70, and 100 μM) of fropofol dissolved in pond water, containing <0.01% DMSO vehicle, for 60 or 90 min. Because loss of righting was not observed with any fropofol concentration, fropofol was coadministered with propofol to look for pharmacological additivity. Tadpoles (n = 630) were incubated with varying concentrations of fropofol (0, 5, 25, and 100 μM) and propofol (0.25–3 μM) dissolved in pond water containing <0.01% DMSO vehicle and were evaluated after 30 min. Hypnosis was defined as the percentage of tadpoles that did not demonstrate spontaneous movement over the course of a 30 s period preceding each time point. After both study conditions, the tadpoles were transferred to fresh pond water and observed overnight for signs of toxicity. The water temperature remained between 21 and 22 °C throughout the experiments. All animal care and experimental procedures involving X. laevis tadpoles were carried out according to protocol approved by the IACUC of University of Pennsylvania.
Pharmacological Activity in Mice
Fropofol was dissolved in 10% lipid emulsion to 30 g/L and two dosages, 96 mg/kg (n = 2) and 180 mg/kg (n = 2), were introduced into 12–20 week C57/B6 mice via tail vein bolus injection. Mice were then monitored for changes in behavior immediately and over the following days post injection. Some mice were euthanized by cervical dislocation within 45 s or 10 min post injection, and the brain was rapidly removed and frozen for subsequent fropofol extraction. Fropofol extraction proceeded according to methods published for propofol.41 Briefly, 2 volumes of 0.22 μm filtered PBS buffer was added to weighed brain samples. The brain was homogenized for 5 s using a Polytron PT 1300D hand-held homogenizer (Kinematica), vortexed for 30 s, and centrifuged for 20 min at 14 000g. The supernatant was removed and 2 volumes of HPLC-grade acetonitrile was added. Samples were vortexed for 30 s and centrifuged for 20 min at 14 000g. Fropofol amount was quantified using the same rpHPLC method as mentioned above. All brain tissue extractions were conducted within 12 h of rpHPLC quantification and the fropofol peak was clearly distinguishable from tissue peaks with the determined retention time of 8.5 min. Generated standard curves with neat fropofol in methanol provided absolute concentration values. Based on an average (n = 4) of fropofol spiked brain tissue samples, recovery from tissue was determined to be 4.8 ± 0.2% for both 210 and 205 nm wavelengths (similar for propofol).
Electroencephalography (EEG) Recording
12–20 week C57/B6 male mice (n = 2) were placed under general anesthesia maintained with isoflurane and implanted with five right-sided chronically indwelling silver ball EEG electrodes over frontal association cortex (2.6 mm anterior to bregma, 1.0 mm lateral), primary motor cortex (2.0 mm anterior to bregma, 2.0 mm lateral), the medial parietal association area (1.7 mm posterior to bregma, 1.2 mm lateral), primary somatosensory cortex (2.0 mm posterior to bregma, 2.6 mm lateral), and primary auditory cortex (2.3 mm posterior to bregma, 4.0 mm lateral); the leads were secured with dental cement. After a minimum of a 2 week recovery, a tail vein catheter was placed in the lateral tail vein of an implanted mouse and secured. After recording a 5 min baseline EEG, 200 mg/kg fropofol in 10% lipid emulsion was injected over 3 s through the catheter, and the catheter was flushed with 100 μL of normal saline. Acqknowledge (Biopac Systems Inc., Golea, CA) was used for processing with a 0.8–59 Hz software bandpass filter. All animal care and experimental procedures involving mice were carried out according to protocol approved by the IACUC of the University of Pennsylvania.
Electrophysiology
α1β2γ2L γ-Aminobutyric Acid Type A (GABAA) Receptor Expression within Oocytes
cDNAs for GABAA receptor α1, β2, and γ2L subunits were kindly provided by Dr. Robert Pearce (University of Wisconsin). Defolliculated stage V–VI X. laevis oocytes were microinjected with 2.8 ng of in vitro transcribed cRNA (mMessage mMachine kit, Ambion, Austin, TX) of α1/β2/γ2L subunits at a 1:1:10 weight ratio, respectively. Oocytes were incubated at 18 °C in a gentamycin supplemented ND96 solution (in mM: 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 5 HEPES, 2.5 Na-Pyr, pH 7.4 with NaOH) for 16–24 before use. All animal care and experimental procedures involving X. laevis frogs were carried out according to a protocol approved by the IACUC of Thomas Jefferson University.
Occyte Electrophysiology Recordings
GABAA receptor whole-oocyte currents were recorded at room temperature (21–23 °C) under two electrode voltage clamp (TEVC) conditions (OC-725C, Warner Instrument, Hamden, CT). All recordings were made at a holding voltage of −80 mV. Oocytes were continuously perfused with ND96-based solutions using gravity-driven perfusion system with an approximate perfusion rate 2–4 mL/min. The perfusion system was outfitted with Teflon tubing for drug exposure studies. γ-Aminobutyric acid (GABA; Sigma) solutions were prepared daily in ND96. Propofol and fropofol were directly dissolved in ND96 facilitated by sonication. Initially each oocyte was exposed to 2.5–5 μM GABA for the effective concentration (EC) 7–13 of maximum GABAA receptor activation. Maximum GABA response was determined by a 10 mM GABA perfusion post drug exposure and washout. To determine modulatory activity, oocytes were perfused for 20 s with the test compound(s) immediately followed by 20 s perfusion with the test compound and GABA at determined EC10. Oocytes continuously perfused in ND96 solution or fropofol ND96 based solution prepared as noted above. Data acquisition and initial analysis were performed using pClamp 9.2/10.3 (Molecular Devices, Sunnyvale, CA). Macroscopic currents were low-pass filtered at 1 kHz and digitized at 2 kHz. Expression of α1β2γ2L cRNA within X. laevis oocytes generated GABAA receptors that demonstrated a GABA EC50 of 33 μM (95% CI, 29–38) with a Hill coeffficient of 0.87 ± 0.04 (mean ± SEM) within 18–24 h post microinjection.
Trabeculae Preparation
Drug effects on myocardial contractility were conducted as previously reported.42 Briefly LBN/F1 rats (250–300g, Harlan Laboratories, Indianapolis, IN) were anesthetized by intraperitoneal injection of pentobarbital (100 mg/kg); the heart was exposed by sternotomy and rapidly removed. After transfer to a dissection dish, the aorta was cannulated and the heart perfused in a retrograde fashion with dissecting Krebs-Henseleit (K–H) solution (in mM: 120 NaCl, 20 NaHCO3, 5 KCl, 1.2 MgCl2, 10 glucose, 0.5 CaCl2, and 20 2,3-butanedione monoxime (BDM) (pH 7.35–7.45); equilibrated with 95% O2 and 5% CO2). The trabecular muscle was dissected from the right ventricle and mounted between a force transducer and a motor arm. The muscle was superfused with K–H solution without BDM at ~10 mL/min, and stimulated at 0.5 Hz. A transducer (KG7, Scientific Instruments GmbH, Heidelberg, Germany) was used to measure the developed force, and expressed as millinewtons per square millimeter of cross-sectional area. The muscles underwent isometric contractions with a set resting muscle length that was set at 15% of the total force development corresponding to resting sarcomere length of 2.20–2.30 μm as determined by laser diffraction.43 Propofol or fropofol was added to non-BDM containing K–H solution at desired concentrations during the experiments. Similar force depression was observed at both 37 °C and room temperature (20–22 °C); the experiments reported herein were performed at room temperature. Animal care and experimental protocols were approved by the Animal Care and Use Committee of The Johns Hopkins University School of Medicine. Data is represented as mean ± SEM of experiments normalized to initial force development without drug exposure.
Statistics
GraphPad Prism5, unless otherwise noted, was used for preparation and statistical data analysis. Details are given in the figure legends.
Supplementary Material
Acknowledgments
We thank Qing Cheng Meng of the Department of Anesthesiology and Critical Care Core Laboratory for rpHPLC assistance.
Funding
This work was supported by the National Institutes of Health [Grants GM055876, NS080519, GM008076] and the National Science Foundation Graduate Research Fellowship program [Grant DGE-1321851].
Footnotes
Supporting Information
1H, 13C, and 19F NMR spectra for 2-fluoro-1,3-diisopropylbenzene, or “fropofol” in Figures 1–3. This material is available free of charge via the Internet at http://pubs.acs.org/.
Author Contributions
Participated in research design: K.A.W., Q.L., B.P.W., A.M.-W., R.G.E., M.C., W.D.G., and W.P.D. Conducted experiments: K.A.W., Q.L., B.P.W., T.M., A.M.-W., B.P., W.P.D., and W.D.G. Contributed new reagents or analytic tools: B.P. and W.P.D. Performed data analysis: K.A.W., Q.L., B.P.W., T.M., A.M.-W., W.D.G., and W.P.D. Wrote or contributed to the writing of the manuscript: K.A.W., Q.L., B.P.W., A.M.-W., R.G.E., M.C., W.D.G., and W.P.D.
Notes
The authors declare no competing financial interest.
References
- 1.Meyer H. Zur Theorie der Alkoholnarkose. Arch Exp Pathol Pharmakol. 1899;42:109–118. [Google Scholar]
- 2.Overton C. Studien über die Narkose zugleich ein Beitrag zur allgemeinen Pharmakologie. Gustav Fischer; Jena, Switzerland: 1901. [Google Scholar]
- 3.Franks NP, Jenkins A, Conti E, Lieb WR, Brick P. Structural basis for the inhibition of firefly luciferase by a general anesthetic. Biophys J. 1998;75:2205–2211. doi: 10.1016/S0006-3495(98)77664-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhattacharya AA, Curry S, Franks NP. Binding of the general anesthetics propofol and halothane to human serum albumin. High resolution crystal structures. J Biol Chem. 2000;275:38731–38738. doi: 10.1074/jbc.M005460200. [DOI] [PubMed] [Google Scholar]
- 5.Liu R, Loll PJ, Eckenhoff RG. Structural basis for high-affinity volatile anesthetic binding in a natural 4-helix bundle protein. FASEB J. 2005;19:567–576. doi: 10.1096/fj.04-3171com. [DOI] [PubMed] [Google Scholar]
- 6.Franks NP, Lieb WR. Molecular and cellular mechanisms of general anaesthesia. Nature. 1994;367:607–614. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- 7.Rudolph U, Antkowiak B. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci. 2004;5:709–720. doi: 10.1038/nrn1496. [DOI] [PubMed] [Google Scholar]
- 8.Jayakar SS, Dailey WP, Eckenhoff RG, Cohen JB. Identification of Propofol Binding Sites in a Nicotinic Acetylcholine Receptor with a Photoreactive Propofol Analog. J Biol Chem. 2013;288:6178–6189. doi: 10.1074/jbc.M112.435909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yip GM, Chen ZW, Edge CJ, Smith EH, Dickinson R, Hohenester E, Townsend RR, Fuchs K, Sieghart W, Evers AS, Franks NP. A propofol binding site on mammalian GABA receptors identified by photolabeling. Nat Chem Biol. 2013;9:715–720. doi: 10.1038/nchembio.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiara DC, Dangott LJ, Eckenhoff RG, Cohen JB. Identification of nicotinic acetylcholine receptor amino acids photolabeled by the volatile anesthetic halothane. Biochemistry. 2003;42:13457–13467. doi: 10.1021/bi0351561. [DOI] [PubMed] [Google Scholar]
- 11.Ishizawa Y, Pidikiti R, Liebman PA, Eckenhoff RG. G protein-coupled receptors as direct targets of inhaled anesthetics. Mol Pharmacol. 2002;61:945–952. doi: 10.1124/mol.61.5.945. [DOI] [PubMed] [Google Scholar]
- 12.Jayakar SS, Zhou X, Chiara DC, Dostalova Z, Savechenkov PY, Bruzik KS, Dailey WP, Miller KW, Eckenhoff RG, Cohen JB. Multiple Propofol Binding Sites in a gamma-Aminobutyric Acid Type A Receptor (GABAAR) Identified Using a Photoreactive Propofol Analog. J Biol Chem. 2014;1:581728. doi: 10.1074/jbc.M114.581728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weiser TG, Regenbogen SE, Thompson KD, Haynes AB, Lipsitz SR, Berry WR, Gawande AA. An estimation of the global volume of surgery: A modelling strategy based on available data. Lancet. 2008;372:139–144. doi: 10.1016/S0140-6736(08)60878-8. [DOI] [PubMed] [Google Scholar]
- 14.Leon I, Cocinero EJ, Millan J, Jaeqx S, Rijs AM, Lesarri A, Castano F, Fernandez JA. Exploring microsolvation of the anesthetic propofol. Phys Chem Chem Phys. 2012;14:4398–4409. doi: 10.1039/c2cp23583h. [DOI] [PubMed] [Google Scholar]
- 15.Leon I, Cocinero EJ, Rijs AM, Millan J, Alonso E, Lesarri A, Fernandez JA. Formation of water polyhedrons in propofol-water clusters. Phys Chem Chem Phys. 2013;15:568–575. doi: 10.1039/c2cp42304a. [DOI] [PubMed] [Google Scholar]
- 16.Leon I, Millan J, Cocinero EJ, Lesarri A, Castano F, Fernandez JA. Mimicking anaesthetic-receptor interaction: a combined spectroscopic and computational study of propofol⋯phenol. Phys Chem Chem Phys. 2012;14:8956–8963. doi: 10.1039/c2cp40656j. [DOI] [PubMed] [Google Scholar]
- 17.Eckenhoff RG, Tanner JW, Johansson JS. Steric hindrance is not required for n-alkanol cutoff in soluble proteins. Mol Pharmacol. 1999;56:414–418. doi: 10.1124/mol.56.2.414. [DOI] [PubMed] [Google Scholar]
- 18.Krasowski MD, Hong X, Hopfinger AJ, Harrison NL. 4D-QSAR analysis of a set of propofol analogues: Mapping binding sites for an anesthetic phenol on the GABA(A) receptor. J Med Chem. 2002;45:3210–3221. doi: 10.1021/jm010461a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL. General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of gamma-aminobutyric acid (GABA) current at the GABA(A) receptor but not with lipid solubility. J Pharmacol Exp Ther. 2001;297:338–351. [PubMed] [Google Scholar]
- 20.Rai G, Bu W, Lea WA, Liang D, Weiser B, Setola V, Austin CP, Simeonov A, Jadhav A, Eckenhoff R, Maloney DJ. Probe Reports from the NIH Molecular Libraries Program. National Center for Biotechnology Information; Bethesda, MD: 2012. Discovery of Novel General Anesthetics Using Apoferritin as a Surrogate System. [PubMed] [Google Scholar]
- 21.Vedula LS, Brannigan G, Economou NJ, Xi J, Hall MA, Liu R, Rossi MJ, Dailey WP, Grasty KC, Klein ML, Eckenhoff RG, Loll PJ. A Unitary Anesthetic Binding Site at High Resolution. J Biol Chem. 2009;284:24176–24184. doi: 10.1074/jbc.M109.017814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hall MA, Xi J, Lor C, Dai SP, Pearce R, Dailey WP, Eckenhoff RG. m-Azipropofol (AziPm) a Photoactive Analogue of the Intravenous General Anesthetic Propofol. J Med Chem. 2010;53:5667–5675. doi: 10.1021/jm1004072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butts CA, Xi J, Brannigan G, Saad AA, Venkatachalan SP, Pearce RA, Klein ML, Eckenhoff RG, Dmochowski IJ. Identification of a fluorescent general anesthetic, 1-aminoanthracene. Proc Natl Acad Sci U S A. 2009;106:6501–6506. doi: 10.1073/pnas.0810590106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu R, Meng Q, Xi J, Yang J, Ha CE, Bhagavan NV, Eckenhoff RG. Comparative binding character of two general anaesthetics for sites on human serum albumin. Biochem J. 2004;380:147–152. doi: 10.1042/BJ20031652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu R, Yang J, Ha CE, Bhagavan NV, Eckenhoff RG. Truncated human serum albumin retains general anaesthetic binding activity. Biochem J. 2005;388:39–45. doi: 10.1042/BJ20041224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardegger LA, Kuhn B, Spinnler B, Anselm L, Ecabert R, Stihle M, Gsell B, Thoma R, Diez J, Benz J, Plancher JM, Hartmann G, Banner DW, Haap W, Diederich F. Systematic investigation of halogen bonding in protein-ligand interactions. Angew Chem, Int Ed Engl. 2011;50:314–318. doi: 10.1002/anie.201006781. [DOI] [PubMed] [Google Scholar]
- 27.Pavan MS, Durga Prasad K, Row TN. Halogen bonding in fluorine: experimental charge density study on intermolecular F⋯F and F⋯S donor-acceptor contacts. Chem Commun. 2013;49:7558–7560. doi: 10.1039/c3cc43513j. [DOI] [PubMed] [Google Scholar]
- 28.Politzer P, Lane P, Concha MC, Ma Y, Murray JS. An overview of halogen bonding. J Mol Model. 2007;13:305–311. doi: 10.1007/s00894-006-0154-7. [DOI] [PubMed] [Google Scholar]
- 29.Hewapathirane DS, Dunfield D, Yen W, Chen S, Haas K. In vivo imaging of seizure activity in a novel developmental seizure model. Exp Neurol. 2008;211:480–488. doi: 10.1016/j.expneurol.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 30.Simons PJ, Cockshott ID, Douglas EJ, Gordon EA, Knott S, Ruane RJ. Distribution in female rats of an anaesthetic intravenous dose of 14C-propofol. Xenobiotica. 1991;21:1325–1335. doi: 10.3109/00498259109043207. [DOI] [PubMed] [Google Scholar]
- 31.Fang Z, Sonner J, Laster MJ, Ionescu P, Kandel L, Koblin DD, Eger EI, 2nd, Halsey MJ. Anesthetic and convulsant properties of aromatic compounds and cycloalkanes: implications for mechanisms of narcosis. Anesth Analg. 1996;83:1097–1104. doi: 10.1097/00000539-199611000-00035. [DOI] [PubMed] [Google Scholar]
- 32.McKinstry-Wu AR, Bu W, Rai G, Lea WA, Weiser BP, Liang DF, Simeonov A, Jadhav A, Maloney DJ, Eckenhoff RG. Discovery of a Novel General Anesthetic Chemotype Using High-throughput Screening. Anesthesiology. 2015;122:325–333. doi: 10.1097/ALN.0000000000000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orser BA, Wang LY, Pennefather PS, MacDonald JF. Propofol modulates activation and desensitization of GABAA receptors in cultured murine hippocampal neurons. J Neurosci. 1994;14:7747–7760. doi: 10.1523/JNEUROSCI.14-12-07747.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hill-Venning C, Belelli D, Peters JA, Lambert JJ. Subunit-dependent interaction of the general anaesthetic etomidate with the gamma-aminobutyric acid type A receptor. Br J Pharmacol. 1997;120:749–756. doi: 10.1038/sj.bjp.0700927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lam DW, Reynolds JN. Modulatory and direct effects of propofol on recombinant GABAA receptors expressed in xenopus oocytes: influence of alpha- and gamma2-subunits. Brain Res. 1998;784:179–187. doi: 10.1016/s0006-8993(97)01334-6. [DOI] [PubMed] [Google Scholar]
- 36.Fujinaka W, Shimizu J, Iribe G, Imaoka T, Oshima Y, Kiyooka T, Morita K, Mohri S. Effects of propofol on left ventricular mechanoenergetics in the excised cross-circulated canine heart. Acta Med Okayama. 2012;66:435–442. doi: 10.18926/AMO/49039. [DOI] [PubMed] [Google Scholar]
- 37.Royse CF, Liew DF, Wright CE, Royse AG, Angus JA. Persistent depression of contractility and vasodilation with propofol but not with sevoflurane or desflurane in rabbits. Anesthesiology. 2008;108:87–93. doi: 10.1097/01.anes.0000296077.32685.26. [DOI] [PubMed] [Google Scholar]
- 38.Cheng TJ, Zhao Y, Li X, Lin F, Xu Y, Zhang XL, Li Y, Wang RX, Lai LH. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J Chem Inf Model. 2007;47:2140–2148. doi: 10.1021/ci700257y. [DOI] [PubMed] [Google Scholar]
- 39.Kale L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, Schulten K. NAMD2: Greater scalability for parallel molecular dynamics. J Comput Phys. 1999;151:283–312. [Google Scholar]
- 40.Weiser BP, Kelz MB, Eckenhoff RG. In vivo activation of azipropofol prolongs anesthesia and reveals synaptic targets. J Biol Chem. 2013;288:1279–1285. doi: 10.1074/jbc.M112.413989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gredell JA, Turnquist PA, MacIver MB, Pearce RA. Determination of diffusion and partition coefficients of propofol in rat brain tissue: implications for studies of drug action in vitro. Br J Anaesth. 2004;93:810–817. doi: 10.1093/bja/aeh272. [DOI] [PubMed] [Google Scholar]
- 42.Ding W, Li Z, Shen X, Martin J, King SB, Sivakumaran V, Paolocci N, Gao WD. Reversal of isoflurane-induced depression of myocardial contraction by nitroxyl via myofilament sensitization to Ca2+ J Pharmacol Exp Ther. 2011;339:825–831. doi: 10.1124/jpet.111.185272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao WD, Liu YG, Marban E. Selective effects of oxygen free radicals on excitation-contraction coupling in ventricular muscle – Implications for the mechanism of stunned myocardium. Circulation. 1996;94:2597–2604. doi: 10.1161/01.cir.94.10.2597. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.