

Abstract
AIM: To determine if calnexin (CANX), RAB1 and alpha-tubulin were involved in the production of hepatitis C virus (HCV) particles by baby hamster kidney-West Nile virus (BHK-WNV) cells.
METHODS: Using a siRNA-based approach complemented with immuno-fluorescence confocal microscope and Western blot studies, we examined the roles of CANX, RAB1 and alpha-tubulin in the production of HCV particles by permissive BHK-WNV cells expressing HCV structural proteins or the full-length genome of HCV genotype 1a. Immuno-fluorescence studies in producer cells were performed with monoclonal antibodies against HCV structural proteins, as well as immunoglobulin from the serum of a patient recently cured from an HCV infection of same genotype. The cellular compartment stained by the serum immunoglobulin was also observed in thin section transmission electron microscopy. These findings were compared with the JFH-1 strain/Huh-7.5 cell model.
RESULTS: We found that CANX was necessary for the production of HCV particles by BHK-WNV cells. This process involved the recruitment of a subset of HCV proteins, detected by immunoglobulin of an HCV-cured patient, in a compartment of rearranged membranes bypassing the endoplasmic reticulum-Golgi intermediary compartment and surrounded by mitochondria. It also involved the maturation of N-linked glycans on HCV envelope proteins, which was required for assembly and/or secretion of HCV particles. The formation of this specialized compartment required RAB1; upon expression of HCV structural genes, this compartment developed large vesicles with viral particles. RAB1 and alpha-tubulin were required for the release of HCV particles. These cellular factors were also involved in the production of HCVcc in the JFH-1 strain/Huh-7.5 cell system, which involves HCV RNA replication. The secretion of HCV particles by BHK-WNV cells presents similarities with a pathway involving caspase-1; a caspase-1 inhibitor was found to suppress the production of HCV particles from a full-length genome.
CONCLUSION: Prior activity of the WNV subgenomic replicon in BHK-21 cells promoted re-wiring of host factors for the assembly and release of infectious HCV in a caspase-1-dependent mechanism.
Keywords: Membrane rearrangements, Hepatitis C virus, Flavivirus replicon, Virus assembly and secretion, Host cellular factors
Core tip: Our system for production of authentic infectious hepatitis C virus (HCV) in non-humanized, non-hepatic cells involves the rearrangement of inner cellular membranes triggered by the replication of flaviviruses. The present results suggest that this feature relies on the re-wiring of host factors that usually contribute to the secretion of glycoproteins to generate an unusual secretory pathway. This model offers a new way to study the properties of free HCV particles, i.e., independently from lipoproteins.
INTRODUCTION
Hepatitis C virus (HCV) genotype 1 has accounted for up to 70% of HCV infections worldwide and is more often resistant than the other genotypes to the combination of pegylated interferon plus ribavirin, the standard of care until recently[1]. New antiviral drugs specifically target either the viral protease NS3, the protein NS5A or the RNA polymerase NS5B and are able to clear HCV infections in a higher number of individuals with a shorter duration of treatment than the standard of care[2]. However, a significant proportion of patients requiring treatment still cannot be cured[3], does not have access to treatment[4] and/or will need the development of a vaccine[5]. Experimental models are being developed[6], but may not cover all aspects of the pathogenesis of hepatitis C, such as the mechanism by which antibodies prevent the spread of infection[7].
In an effort to develop alternative systems, we have established a model in which infectious HCV production is independent from its replicon-mediated RNA replication[8], hence circumventing limitations inherent to existing cell culture models[9]. For instance, the replication of HCV genotype 1 isolates in hepatocellular carcinoma cell lines is inefficient or generates adaptive mutations interfering with viral fitness in vivo. Briefly, our system relies on the amplification provided by a dual bacteriophage RNA polymerase plasmid system (referred to as P2B) that generates large amounts of HCV RNA transcripts from a T7 promoter-driven plasmid in the cytoplasm of baby hamster kidney (BHK)-21 cells conditioned by a lineage II West Nile virus (WNV) subgenomic replicon[8]. We observed that the WNV replicon in this cell line created an environment permissive for the assembly and release of infectious HCV of various genotypes, including virions of strains H77 (genotype 1a)[10] or Con1 (genotype 1b)[11]; these virions infected human liver slices ex vivo[8].
BHK-WNV cells produce infectious HCV particles independently from lipoprotein biosynthesis. The fact that these particles retain the possibility to interact with lipoproteins in vitro[8] is in line with previous results[12-14] and supports the view that HCV particles may interact with lipoproteins in a second step (e.g.,[15,16]) and not necessarily co-assemble with them (e.g.,[17]). Potential mechanisms for WNV-conditioned BHK cells producing highly infectious HCV virions could relate to common genomic features between the Flavivirus and Hepacivirus genera within the Flaviviridæ family[18]. In addition, several flaviviruses infect hepatocytes[19,20] and may use similar host factors as HCV for their production[21-25].
Our previous results showed that after curing BHK cells from the WNV subgenomic replicon, the production and release of infectious HCV particles were still observed for a while[8]. In addition, although recombinant expression of HCV structural genes in cultured cells, including in human hepatocytes[26], usually leads to their retention in the endoplasmic reticulum (ER)[27], BHK-WNV cells released infectious HCV particles even in the absence of HCV non-structural genes[8]. These findings suggested that, while the viral replication machineries played no direct role in the secretion process, the reorganization of intracellular membranes induced by the WNV subgenomic replicon contributed to the permissiveness of BHK cells.
In mammalian cells, conventional protein traffic from the ER to the Golgi complex passes through the membrane clusters of the ER-Golgi intermediate compartment (ERGIC), the marker of which is the lectin ERGIC-protein of 53 kDa (ERGIC-53). ER-derived cargo is first shuttled to the ERGIC in a coat protein (COP) II-dependent step and subsequently to the Golgi apparatus in a second vesicular transport step involving COPI-coated vesicles, RAB and ARF GTPases, as well as cytoskeletal networks; incoming vesicles can also be recycled to the ER in a COPI-mediated process[28]. The ERGIC contributes to the concentration, folding, and quality control of newly synthesized proteins and is required for the production of several viral pathogens[29]. N-linked glycosyl antenna are matured by Golgi-resident enzymes along with glycoproteins’ progression from the proximal to the distal Golgi cisternæ, then across the plasma membrane for their secretion, via the trans-Golgi/endosomal network.
In the present work, we studied the potential involvement of components of this secretory machinery in the production of HCV particles by BHK-WNV cells and compared this model with the JFH-1 strain/Huh-7.5 cells model. We show that, upon expression of an HCV genome of genotype 1a in these cells, a subpopulation of HCV proteins were recruited through calnexin (CANX) to a cytoplasmic compartment of rearranged membranes. The small GTPase RAB1 was involved in the formation of this compartment. The secretion of HCV particles produced from a full-length genome required also the N-linked glycosylation of HCV envelope glycoproteins and alpha-tubulin (α-TUB), a component of microtubules and, surprisingly, the activity of cysteine protease caspase-1. As our understanding of the HCV virus life cycle has recently widened to alternative routes of transmission, elucidating mechanisms at work in BHK-WNV cells could shed some light on the production of HCV in vivo.
MATERIALS AND METHODS
Cell cultures
BHK-21 cells were grown in E-MEM supplemented with 10% fetal bovine serum (FBS; HyClone, United States), Glutamax-I (Gibco, Life Technologies, United States); BHK cells harboring WNV lineage II SG-replicon encoding Renilla luciferase[30], herein called BHK-WNV cells, were propagated in D-MEM supplemented with 10% FBS, Glutamax-I and 5 μg/mL blasticidin. Huh-7.5 cells were maintained in D-MEM supplemented with 10% FBS, Glutamax-I, non-essential amino acid mix (Gibco, Life Technologies, United States).
Plasmid constructs
A previously described system of two plasmids (P2B = dual phage RNA polymerases plasmid system for generation of T7 RNA polymerase in the cytoplasm) was used to amplify the cytoplasmic transcription of a plasmid encoding HCV bicistronic particles (HCVbp) under the control of bacteriophage T7 DNA-dependent RNA polymerase’s cognate promoter[8]; a sequence encoding an HDV antigenomic ribozyme[31] was added at its C termini; as a consequence, HCV transcripts were uncapped and have correct 5’- and 3’-ends. p90 HCVconFLlongpU[10] and pH-SGNeo (L + I) encoding a SG-replicon of the same strain with cell-culture adaptive mutations[32] were used as templates to construct HCVbp-coding plasmids. pHCV STp7 is a pcDNA3.1(+)-based plasmid (Life Technologies, United States) encoding the structural genes (core, E1, E2) plus p7 of HCV genotype 1a linked to the human cytomegalovirus (CMV) immediate early promoter. HCVbp-4cys are HCVbp particles with a sequence encoding a tetracysteine tag[33] inserted in the part of their genome encoding NS5A[8].
Antibodies and cellular markers
Anti-E2 (ALP98 and AP33)[34], and anti-E1 (A4) monoclonal antibodies were used for Western blot (WB) analysis. Conformational AP33 monoclonal antibody, human serum from an HCV patient (genotype 1a) that recognizes conformational HCV core and E2 protein subspecies by WB and anti-HCV core peptides 9-21 (C1) or 7-50 (Thermo Scientific, United States) monoclonal antibodies were used for confocal microscopy analysis. Neutralizing monoclonal antibody E16 recognizes WNV E by WB[35]. Antibodies against various cellular proteins are as followed: ERGIC-53 (Alexis Biochemicals, United States), GDI (Life Technologies, United States); RAB1 and atlastin (Santa Cruz Biotechnology, United States); CANX and GM130 (Abcam, United States); and calreticulin (Cell Signaling Technology, United States). For immunofluorescence analysis, the secondary antibodies used were Alexa Fluor 488-, 594- or 635-conjugated goat anti-mouse and anti-human antibodies, and Alexa Fluor 594-, 635- or -680 conjugated goat anti-rabbit antibodies from Molecular Probes (Life Technologies, United States). Mito Tracker Orange CMTMRos, TC-ReASH II In-Cell Tetracysteine Tag detection kit and Paclitaxel (Taxol) Oregon Green® 488 Conjugate were obtained from Molecular Probes (Life Technologies, United States). Nuclei were counterstained with DAPI (blue). The cells were observed with a laser-scanning confocal microscope and the pictures were deconvoluted. Bar scales = 5 μm.
Production of HCV particles in mammalian cells
One day before transfection, BHK-WNV cells were seeded at a density of 6 × 106 cells per 162 cm2 flask. Plasmids encoding the HCV sequence under the control of the CMV early promoter or the bacteriophage T7 promoter were transfected using Lipofectamine® LTX with PlusTM Reagent according to the manufacturer’s protocol (Life Technologies, United States). Culture medium after transfection was D-MEM supplemented with 10% FBS, non-essential amino acid mix, Glutamax-I, 25 mmol/L Hepes and 3.7 g/L sodium bicarbonate. Cells were incubated at 37 °C for 3 d in an incubator with a 95% air/5% CO2 atmosphere saturated in humidity. Culture media were harvested, centrifuged at 30000 × g for 30 min at 4 °C to remove cell debris; then clarified supernatants (SN) were filtered with 0.45 μm PVDF membrane (Millipore, United States) and centrifuged at 100000 × g for 3 h at 4 °C. Pellets were suspended in ice-cold Tris-buffered saline solution (TBS; Quality Biologicals, United States) containing protease inhibitor cocktail (Roche, United States). HCVcc (Huh-7.5-produced JFH-1) was obtained by electroporating IVT RNA into Huh-7.5 cells as described[36]; the SN was concentrated, and aliquots of the virus stock were stored at -80 °C.
Gene knockdown in BHK-WNV cells using siRNAs
CANX, melanocortin 5 receptor (MC5R), α-TUB were analyzed for their effects on HCV release. BHK-WNV cells were treated with the corresponding siRNA (2 siRNAs per target gene; Dharmacon, United States) for 2 d, re-seeded and transfected the next day with HCV-coding plasmid. Cells and supernatants were harvested 48 h later, and analyzed by WB. To verify the effects of these genes on HCV release, CANX and α-TUB cDNAs were synthesized from BHK-WNV mRNA, and cloned into pTracer-CMV/Bsd (Invitrogen, United States). The corresponding pTracer plasmid was co-transfected with HCV-coding plasmid in siRNA-treated BHK-WNV cells to show the specificity of their knockdown.
Effect of RAB1 on HCV production by BHK-WNV cells
To study co-localization of HCV and RAB1 in the producer cells, BHK and BHK-WNV cells were transfected with HCVbp-coding plasmid, and the following day, re-seeded on 8-well chambered coverglass (5 × 103 cells/well). Two days later, cells were fixed and permeabilized as above, then incubated with serum from an HCV-infected patient (HCV genotype 1a) and anti-RAB1 antibodies. BHK-WNV cells were then treated with siRNA against RAB1 (2 siRNA per target gene; Dharmacon, United States) for 2 d, re-seeded, and the following day were transfected with HCVbp-coding plasmid. Cells and SN were harvested 3 d after transfection; cell lysates and particles released into SN were analyzed by WB.
Electron microscopy
BHK-WNV cells seeded in a 6-well plate (2.5 × 105 cells) were transfected with HCVbp-coding plasmid. Three days later, cells were fixed in 2% glutaraldehyde in 0.1 mol/L sodium cacodylate for 1 h at RT, then at 4 °C, overnight. Cells were subsequently processed for transmission electron microscopy (TEM) as described[37].
Effects of siRNAs on HCVcc production and release in Huh-7.5 cells
Cells were treated with the siRNAs (CANX, α-TUB, RAB1A, RAB1B, or non-target) for 48 h; the same number of cells was reseeded on 24-well plates, and the next day was inoculated with HCVcc at MOI 0.5. Cells were harvested daily, total RNA was extracted using lysis buffer of TaqMan Gene Expression Cells-to-ct kit (Life Technologies, United States) and HCV RNA was analyzed by RT-TaqManTM PCR with a StepOne Plus thermocycler (Applied Biosystems, United States).
IF experiments
For live staining after inoculation with HCVbp-4cys, Huh-7.5 cells were incubated with two cell-permeant reagents: The arsenical ReASH-ethane dithiol (Life Technologies) that fluoresces upon binding a tetracysteine tag[33], and Oregon Green 488 paclitaxel bis-acetate (TubulinTracker Green Reagent; Life Technologies, United States) that binds to polymerized alpha tubulin. Immuno-fluorescence studies on fixed cells were performed on stacks of images (a dozen cells per IF condition); co-localization was analyzed using Coloc 2 Plugin (http://imagej.net/Coloc_2) with ImageJ software (https://imagej.nih.gov/ij/).
Experimental reproducibility
Unless specified otherwise, all shown results are representative of at least three independent experiments.
RESULTS
Identification of a compartment possibly linked to the assembly of HCV particles in the cytoplasm of BHK-WNV cells
To gain insight regarding the mechanisms by which BHK-WNV cells produce HCV particles, we transfected the cells with either of three different plasmids based on HCV genotype 1a (HCV1a): (1) HCV structural and p7 genes[38] linked to a CMV promoter (Str + p7); (2) HCV subgenomic replicon (SG repl); or (3) full-length HCV genome (FL) linked to the bacteriophage T7 promoter; for (2) and (3), the cells were also co-transfected with the P2B (dual phage RNA polymerases) plasmid system for generation of T7 RNA polymerase in the cytoplasm. The cells were then permeabilized and incubated with monoclonal antibodies (mAbs) against HCV1a core (C1), E1 (A4) and E2 (AP33) or a control mAb. Like in hepatic cells expressing a HCV genome[22], we observed that the three HCV1a mAbs stained a large and heterogeneous area in the cytoplasm of BHK-WNV cells expressing the structural genes, while the control mAb did not stain these cells (Figure 1A). AP33 is a neutralizing mAb that targets a linear epitope within a flexible region of E2[34] and, as expected, did not stain cells not expressing HCV1a structural genes (Figure 1A). In contrast, a neutralizing IgG from the serum of a patient (HCV1a sIgG) who had been cured from hepatitis C of this genotype[8] stained a more limited and heterogeneously shaped compartment of the cytoplasm of BHK-WNV cells expressing a full-length genome of HCV1a, which was also stained by AP33 but not by control human sIgG (Figure 1A); of note, HCV1a sIgG did not stain BHK-WNV cells expressing a HCV1a subgenomic replicon. A mitochondrion-specific staining surrounded this compartment (Figure 2A). By WB, beside non-specific bands (NS), HCV1a sIgG specifically recognized bands with apparent molecular weights of 21 kDa and 75 kDa (Figure 1B) whose sizes coincided with those reported for HCV core and E2, both known to display most HCV epitopes recognized by human B cell (http://www.iedb.org/).
Figure 1.
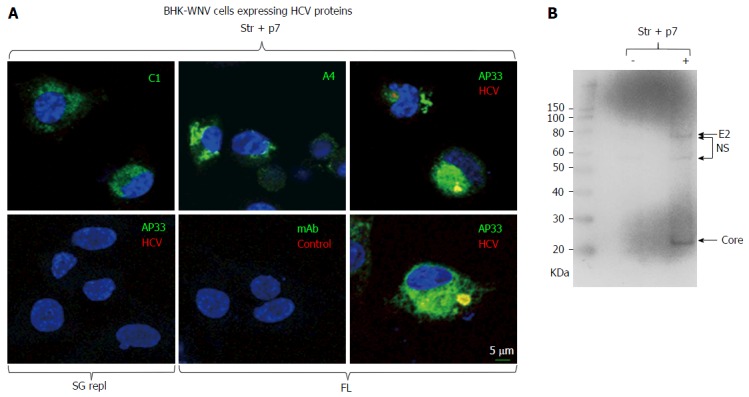
Immunodetection of hepatitis C virus proteins of genotype 1a expressed in baby hamster kidney-West Nile virus cells. A: A plasmid encoding Str + p7 of HCV strain H77 (genotype 1a) from an early human cytomegalovirus promoter or a system of plasmids (P2B) expressing a subgenomic replicon or genome (FL) of same genotype in the cytoplasm (8) were transfected in BHK-WNV cells; after 2 d, IF study was performed with monoclonal antibodies (green) targeting core 9-21 (C1), envelope E1 (A4) and E2 (AP33) (28) glycoproteins of strain H77 (all IgG1a), or anti-rabbit IgG mouse Ab (mAb) of same isotype; and with human serum IgG (red) obtained from a patient recently cured from an infection of same genotype (HCV) or uninfected (control). Nuclei were counterstained with DAPI (blue) and cells were observed with a laser-scanning confocal microscope; B: BHK-WNV cells were either mock transfected (-) or transfected with a system of plasmids expressing the genome of H77 strain (+); after 3 d, cell lysates were prepared and human anti-HCV IgG tested in (a) were used as a Western blot probe. The scale on the left shows molecular weight markers. HCV: Hepatitis C virus; BHK-WNV: Baby hamster kidney-West Nile virus; Str + p7: Structural and p7 genes.
Figure 2.
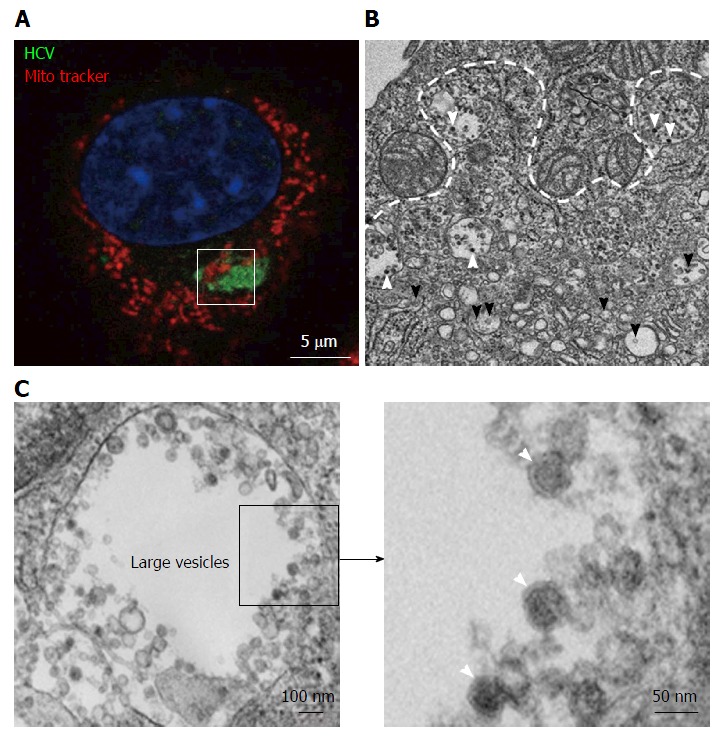
Transmission electron microscopy view of a hepatitis C virus compartment forming in baby hamster kidney-West Nile virus cells cells. BHK-WNV cells were transfected with a mix of HCVbp-expressing and P2B plasmids (8). A: IF analysis showing a compartment recognized by human serum HCV IgG (green) surrounded by mitochondria (red); nucleus is counterstained with DAPI (blue). The white square delimits an area similar to that displayed in the next panel; B: Thin section observed by transmission electron microscopy: White arrowheads: Electron-dense viral particles in large vesicles; black arrowheads: Nascent viral particles in traffic vesicles; white dotted line: Limit of mitochondria surrounding the compartment containing viral particles (cf. area within white square in previous panel); C: Left panel: Example of large vesicle that develops in the cytoplasm of permissive BHK-WNV cells upon expression of full-length HCV genome; Right panel: Magnification of viral particles. HCV: Hepatitis C virus; BHK-WNV: Baby hamster kidney-West Nile virus.
To further characterize the cytoplasmic compartment stained by HCV1a sIgG, thin sections of BHK-WNV cells expressing HCV genome of genotype 1a were observed by TEM. The sub-compartment was easily identified due to the presence of mitochondria in its periphery (Figure 2B)[39]. As previously, it was comprised of large vesicles containing viral-like particles - not observed in cells expressing an HCV1a subgenomic replicon, i.e., without structural genes - as well as membranous web with vesicle packets, both typical of membrane rearrangements triggered by WNV replication[8]. This time, we also observed a mix of rearranged cellular organelles (ER, Golgi cisternæ) with a few additional viral-like particles (Figure 2B). Altogether, these results support the view that HCV particles assembled in BHK-WNV cells were specifically recognized by sIgG from a cured patient.
CANX recruits a subset of HCV proteins into a compartment of BHK-WNV cells for the release of HCV particles
Classically, the secretion of glycoproteins involves lectins of the ER, CANX and calreticulin (CRT), microtubule-dependent ER-to-Golgi vesicular traffic[40,41], maturation of their carbohydrates in the Golgi apparatus and the secretion machinery of the trans-Golgi network[42]. We had previously observed that brefeldin A prevented the secretion of HCV by BHK-WNV cells, while it did not affect that of exosomes[8]. Brefeldin A is an inhibitor of GBF1 that activates small-sized GTPase ARF1 and, in turn, COPI-dependent traffic[43]. The effect of brefeldin A was consistent with HCV particles being released via a classical secretion path. Two components of the classical secretion path, CANX and microtubules, have been previously implicated in HCV life cycle in other systems[27,44,45]. Since what appeared to be the assembly compartment displayed major membrane rearrangements, we explored whether a classical secretion path was involved in the production of HCV particles by BHK-WNV cells.
In BHK-WNV cells expressing an HCV1a genome, most CANX was unexpectedly detected in the same compartment as HCV proteins stained by HCV1a sIgG (Figure 3A, left panels). After a treatment of BHK-WNV cells by CANX siRNA, which decreased the level of CANX (Figure 3A, right panels, and Figure 3C, top panels), HCV1a sIgG staining was no longer localized in this compartment but, instead, displayed a scattered pattern in the cytoplasm (Figure 3A, right panels). The effect of knocking down CANX and α-TUB expression on HCV production was analyzed. Figure 3B shows WB analysis of E2 glycoprotein in particles purified from the supernatants and cell lysates as a marker for HCV production. Treatment of BHK-WNV cells by control or MC5R siRNA did not alter the production or release of HCV (Figure 3B). In contrast, siRNA-mediated down-regulation of CANX or α-TUB significantly reduced the secretion of HCV (Figure 3B). Albeit incomplete, the siRNA effect on CANX expression was sufficient to abolish BHK-WNV permissiveness for HCV production (Figure 3C, top panels).
Figure 3.
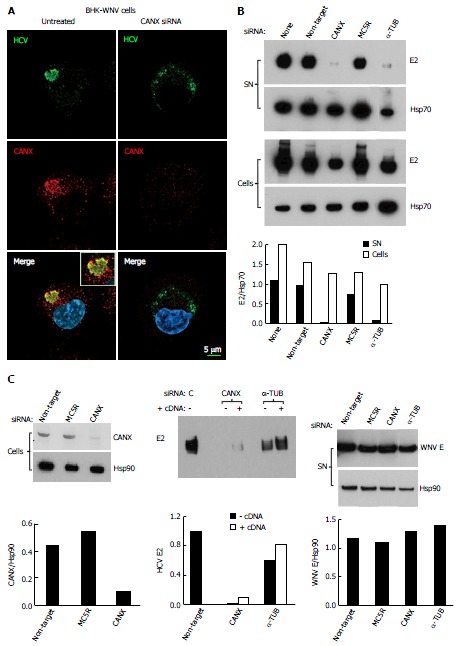
Involvement of calnexin and alpha-tubulin in the release of hepatitis C virus particles by baby hamster kidney-West Nile virus cells. A: BHK-WNV cells were treated, or not, with CANX siRNA and, three days later, transfected with the HCV-coding and P2B plasmids. Two days later, IF was performed with anti-HCV serum of Figure 1 (green) and anti-CANX antibody (red) followed by confocal microcopy analysis; nucleus were counterstained with DAPI (blue); B: Top panels: BHK-WNV cells were treated with the indicated siRNA for 2 d, then transfected with a plasmid encoding full length HCV (HCVbp) in the cytoplasm. Contents in E2 envelope protein of both supernatant (SN) and cell lysate (Cells) were analyzed 2 d later by Western blot (WB); Bottom panel: Densitometry analysis; C: Left panels: BHK-WNV cells were treated with the indicated siRNA for 2 d and content in CANX was analyzed by WB; Hsp90 was used as a control; Middle panel: BHK-WNV cells were treated with siRNA targeting CANX or a-TUB transcript for 2 d; cells were then reseeded and transfected the next day with the HCV-coding plasmid together with a control plasmid (-) or one expressing the cDNA of the knocked-down transcript (+). Two days later, HCV materials released in SN were analyzed by WB; Right panels: BHK-WNV treated as in (B) were transfected with a plasmid encoding West Nile virus (WNV) structural genes (core, prM and E). Two days later, materials released in the SN were analyzed by WB with an antibody recognizing WNV E (29); Hsp90 was used as a control; Bottom panels: Densitometry analyses. CANX: Calnexin; α-TUB: Alpha-tubulin; HCV: Hepatitis C virus; BHK-WNV: Baby hamster kidney-West Nile virus; C: Control siRNA.
Hsp70 or Hsp90 were chosen as controls since they associate with exosomes[46,47], which are particulate materials secreted by BHK cells yet distinct from HCV particles[8]. After a treatment of BHK-WNV cells with α-TUB siRNA, the releases of Hsp70- and HSp90-containing particles in the supernatant decreased (Figure 3B and C, bottom panels), in accordance with the requirement for a functional cellular traffic to release exosomes and consistent with an accumulation of Hsp70 in the producer cells. In contrast, HCV E2 did not accumulate in BHK-WNV cells (Figure 3B).
To evaluate whether inhibition of HCV release could result from an off-target effect, CANX and α-TUB cDNAs cloned from BHK-WNV cells were inserted into mammalian expression plasmids. In BHK-WNV cells treated by CANX siRNA, expression of recombinant CANX gene partially restored both CANX levels (Figure 4) and HCV secretion (Figure 3C, middle panels). A similar result was observed with α-TUB cDNA expressed in α-TUB siRNA-treated BHK-WNV cells (Figure 3C, middle panels). The release of WNV particles upon expression of WNV structural genes was minimally affected by either siRNA treatment (Figure 3C, bottom panels). These results show that HCV production in BHK-WNV cells specifically involved CANX and α-TUB.
Figure 4.
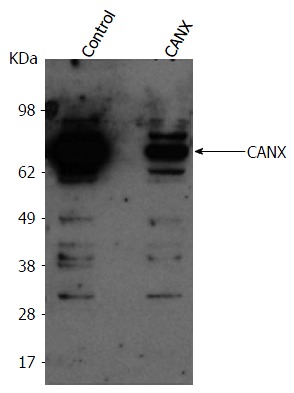
Baby hamster kidney-West Nile virus cells treated with the indicated siRNA (on top). After 2 d, cells from both conditions were reseeded and transfected the next day with either a control plasmid or plasmid expressing CANX cDNA, respectively in control or CANX siRNA-treated BHK-WNV cells. The following day, content in CANX was analyzed by Western blot in cell lysates. CANX: Calnexin; BHK-WNV: Baby hamster kidney-West Nile virus.
N-glycans of HCV envelope proteins are required for the non-classical secretion of HCV particles by BHK-WNV cells
Together with CANX, CRT usually ensures the proper conformation of nascent glycoproteins in the ER lumen. However, CRT expression was lower in BHK-WNV cells than in parental cells (Figure 5A) and did not co-localize with HCV proteins detected by the immune sIgG (Figure 5B). An additional C-type lectin, ERGIC-53, transports properly folded glycoproteins from the ER to the Golgi apparatus. Its expression was also slightly lower in BHK-WNV cells (Figure 5A) and did not co-localize with this pool of HCV proteins (Figure 5B).
Figure 5.
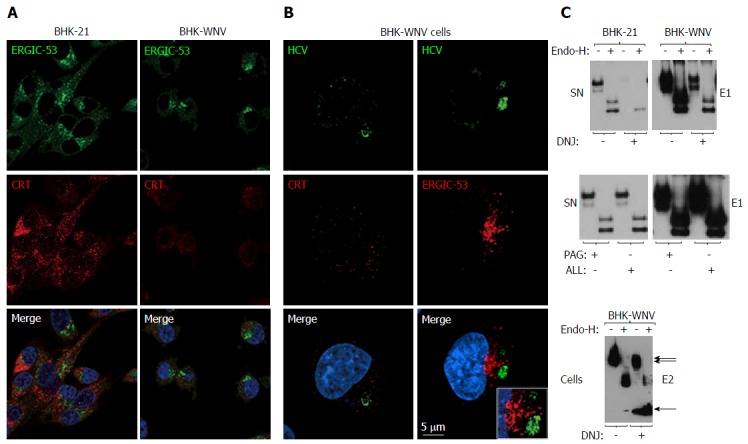
Calnexin and N-linked glycosylation are involved in the release of hepatitis C virus particles via a non-classical secretion path in baby hamster kidney-West Nile virus cells. A: BHK-21 and BHK-WNV cells were transfected with the HCV expression plasmid system. IF was performed three days later with anti-ERGIC-53 (green) and anti-CRT (red) antibodies, followed by confocal microscopy analysis; B: Same protocol as in (A) with BHK-WNV cells transfected with the HCV-coding plasmids; C: Twelve hours after transfection with the HCV expression plasmid mix, parental BHK cells or BHK-WNV cells were treated, or not, with N- (DNJ) or O- (PAG, ALL) glycosylation inhibitors; materials released in SN (top and middle panels) or cell lysates (bottom panel) were collected, incubated with or without Endo-H and analyzed by Western blot. HCV: Hepatitis C virus; BHK-WNV: Baby hamster kidney-West Nile virus; ERGIC-53: Endoplasmic reticulum-Golgi intermediate compartment-protein of 53 kDa; CRT: Calreticulin; SN: Supernatants; DNJ: Deoxynojirimycin; Endo-H: Endo-β-N-acetylglucosaminidase H.
Treatment of producer cells with deoxynojirimycin (DNJ), which prevents the binding of nascent glycoproteins to CANX[42], modified the glycosylation and stability of HCV envelope proteins in BHK-WNV cells (Figure 5C, bottom panel), which was accompanied by a lower amount of HCV particles released (Figure 5C, top panels). As expected, DNJ treatment resulted in decreased resistance of carbohydrates on E1 to a digestion by endo-β-N-acetylglucosaminidase H (Endo-H). In contrast, O-glycosylation[48] inhibitors PAG and ALL did not display any effect on HCV production or E1 glycosylation (Figure 5C, middle panels). A similar pattern was observed with BHK cells bearing a Dengue 2 subgenomic replicon (Figure 6). Therefore, the decreased production of HCV in the presence of DNJ resulted from a lack of maturation of N-linked glycosyl antenna on HCV envelope proteins, consistent with the effect of CANX siRNA observed.
Figure 6.
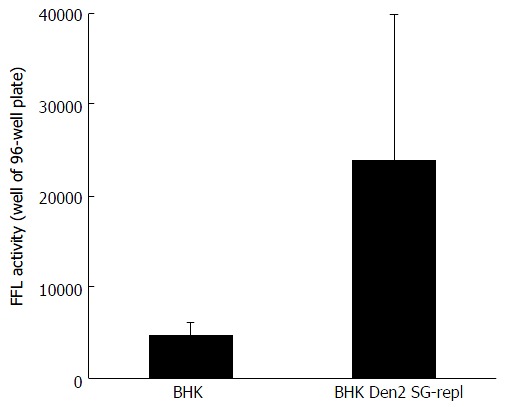
Huh-7.5 cells were incubated with hepatitis C virus reporter particles particles produced either in parental baby hamster kidney cells or in baby hamster kidney cells chronically replicating a dengue 2 subgenomic replicon (similar to the West Nile virus’s). Infectivity was measured in target cells with a Firefly luciferase (FFL)-based reporter system, as previously decribed[8]. Error bars represent the SD in a representative experiment. BHK: Baby hamster kidney.
RAB1 and conformational HCV protein subspecies co-localize within a compartment of reorganized ER and Golgi components
We looked for additional cellular factors that could contribute to the secretion of HCV particles. RAB1 exerts a key control on the ER-to-Golgi traffic. In parental BHK-21 cells, RAB1 was spread in the cytoplasm, but co-localized with HCV proteins expressed after transfection (Figure 7A). The WNV subgenomic replicon enhanced RAB1 staining, coalescing into a few membranous-like spots within which most HCV proteins detected by HCV1a sIgG were localized (Figure 7B). Treatment of BHK-WNV cells with RAB1 siRNA greatly reduced HCV protein expression (Figure 7D); concomitantly, fewer HCV particles were released (Figure 7D).
Figure 7.
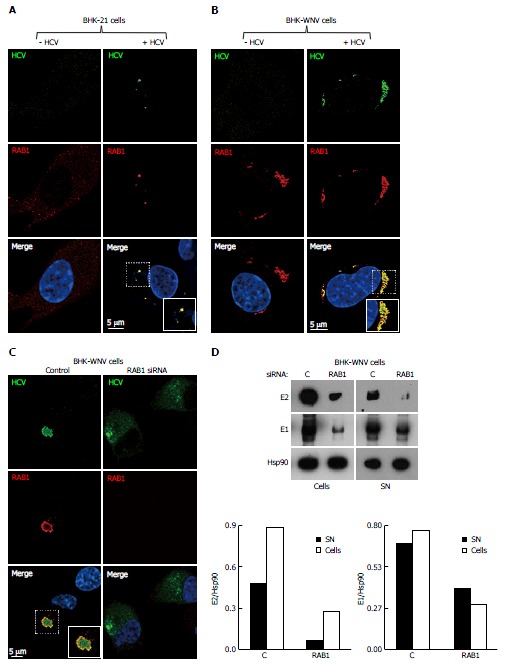
Release of hepatitis C virus particles by baby hamster kidney-West Nile virus cells requires RAB1 in a cytoplasmic subcompartment. Three days after transfection with the HCV-coding and P2B plasmids, or not, IF of BHK-21 (A) or BHK-WNV (B) cells was performed with anti-HCV serum (green) and anti-RAB1 antibody (red), followed by confocal microscopy analysis; C: BHK-WNV cells treated (right panels) or not (left panels) with RAB1 siRNA were transfected with the HCV expression plasmid system; IF was performed as in (B); A-C: Nuclei were counterstained with DAPI (blue); D: BHK-WNV cells treated with RAB1 siRNA were transfected with the HCV expression plasmid system. Cells and SN were harvested 3 d later, and analyzed by Western blot; Hsp90 = control; bottom: Densitometry analysis. HCV: Hepatitis C virus; BHK-WNV: Baby hamster kidney-West Nile virus; SN: Supernatants.
Although maturation of HCV envelope glycoproteins did not involve a classical secretory pathway, some carbohydrate residues at the surface of released HCV particles became resistant to a treatment by Endo-H[42]. This indicates that the ER-to-Golgi machinery as well as the Golgi apparatus contributed to HCV production in BHK-WNV cells. Interestingly, in these cells, the expression of RAB1 GDP dissociation inhibitor (GDI)[49] was enhanced and co-localized with HCV proteins in a pattern reminiscent of RAB1’s (Figure 8). In addition, the expression of Atlastin 1, a high molecular weight GTPase that is involved in maintenance of the ER compartment[50] and fusion of ER tubules[51], as well as in ER-to-Golgi trafficking[52], was up-regulated in BHK-WNV cells compared to parental BHK-21 cells (Figure 9). We detected its presence within the CANX-enriched compartment formed in BHK-WNV cells (Figure 10), as well as that of p115/USO1[53], required for membrane fusion of ERGIC vesicles with the Golgi apparatus. We also detected the presence of GM130/GOLGA2 (Figure 11), which contributes to the progression of glycoproteins between Golgi cisternæ[41], which in turn correlates with the maturation of their carbohydrate residues[54].
Figure 8.
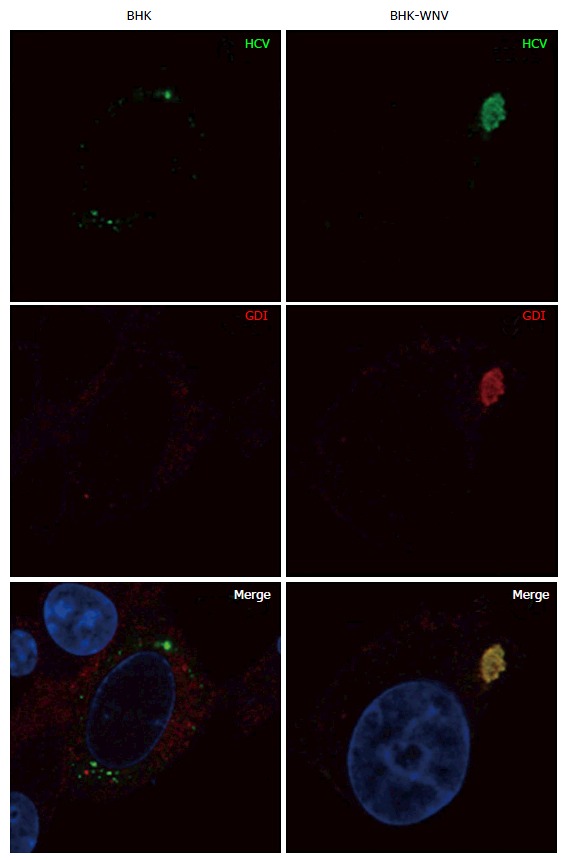
Parental or baby hamster kidney-West Nile virus cells were transfected to express hepatitis C virus genome in the cytoplasm and IF experiment was performed and analyzed as described throughout the manuscript. HCV: Hepatitis C virus; BHK-WNV: Baby hamster kidney-West Nile virus; GDI: GDP dissociation inhibitor.
Figure 9.
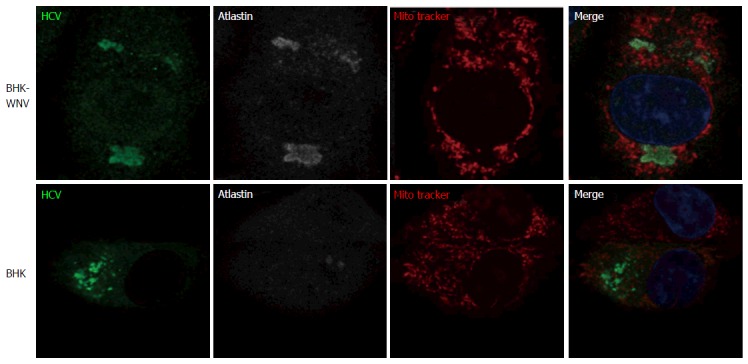
Parental or baby hamster kidney-West Nile virus cells were transfected to express hepatitis C virus genome in the cytoplasm and IF experiment was performed and analyzed as described throughout the manuscript. HCV: Hepatitis C virus; BHK-WNV: Baby hamster kidney-West Nile virus.
Figure 10.
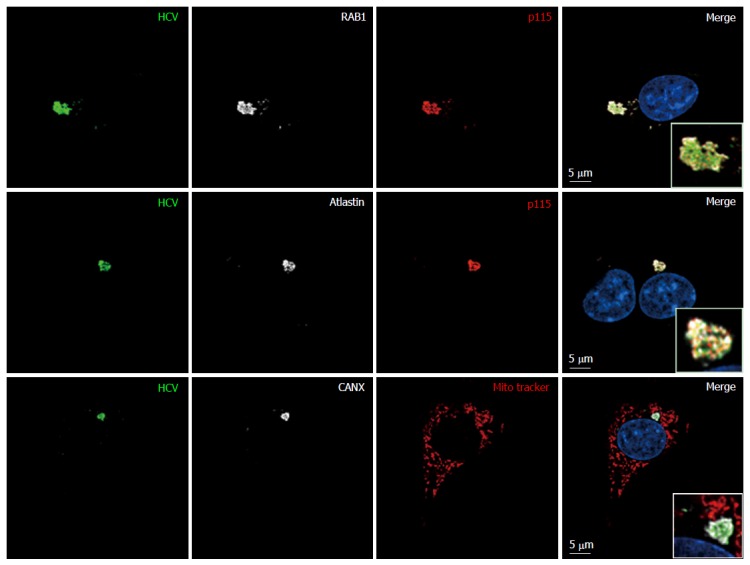
Endoplasmic reticulum-Golgi membrane remodelers (RAB1, p115 and atlastin) are recruited into the hepatitis C virus assembly compartment in baby hamster kidney-West Nile virus cells. Three days after transfection of BHK-WNV cells with the HCV expression plasmid system, IF was performed with anti-HCV serum (green) and anti-p115 (red), RAB1, Atlastin-1 or CANX (white) antibodies. Mitochondria were labeled with Mito-Tracker-Orange-CMTMRos (red). HCV: Hepatitis C virus; BHK-WNV: Baby hamster kidney-West Nile virus; CANX: Calnexin.
Figure 11.
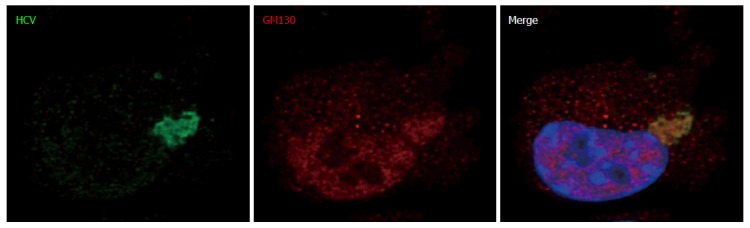
This experiment was performed with baby hamster kidney-West Nile virus cells transfected to express hepatitis C virus genome in the cytoplasm and IF experiment was performed and analyzed as described throughout the manuscript (anti-GM130 monoclonal antibodies have been reported to cross-react with an unidentified protein of lower molecular weight). HCV: Hepatitis C virus.
Similarities between HCV productions in BHK-WNV and Huh-7.5 cells
We next tested whether host factors assisting HCV production in BHK-WNV cells also play a role in the JFH-1 strain of genotype 2a/Huh-7.5 hepatocarcinoma cell paradigm. Mock transfection and non-target siRNA slightly decreased HCV production in Huh-7.5 cells infected with HCVcc. All other siRNAs tested further decreased the amount of HCV RNA, the inhibition ranging between 1 and 3 logs; HCV particle release in the supernatant was diminished as well (Figure 12A).
Figure 12.
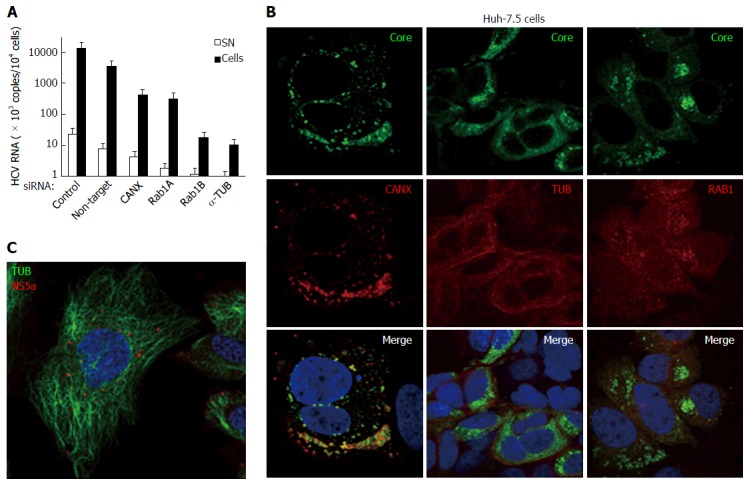
Hepatitis C virus produced in cultured human hepatic cells involves same cellular factors as those enhancing hepatitis C virus production in baby hamster kidney-West Nile virus cells. A: Huh-7.5 cells were seeded on 24-well plates and the next day were transfected, or not, with siRNA, as indicated. After 2 d, cells were reseeded into 24-well plates and, the next day, incubated at a MOI = 0.5 with HCVcc produced with the JFH-1 strain in Huh-7.5 cells. At 3 dpi, HCV RNA contents were determined in the cells (closed bars) and SN (open bars) by RT-qPCR (TaqMan). Results are plotted on a log-scale; errors bars represent maximum variations observed in this assay; B: Huh-7.5 cells were electroporated with in vitro-transcribed genome of the JFH-1 strain and passaged for two weeks, then were seeded onto coverslips. Two days later, IF was performed with anti-HCV core 7-50 (green) and anti-human CANX, tubulin (TUB) or RAB1 antibodies (red); C: Huh-7.5 cells were inoculated with HCVbp-4cys produced in permissive BHK-WNV cells (8); 2 d later, the cells were incubated with both ReASH (red) and Taxol fluorophore conjugate (green); result representative of two independent experiments; B and C: Nuclei were counterstained with DAPI and cells were observed by confocal microscopy. CANX: Calnexin; HCV: Hepatitis C virus; BHK-WNV: Baby hamster kidney-West Nile virus; SN: Supernatants.
The human HCV serum used in BHK-WNV cells was of a different genotype than JFH-1; hence, for our IF studies we used a different antibody recognizing HCV core 7-50 peptide. There was not obvious co-localization of the staining it elicited in Huh-7.5 cells inoculated with HCVcc and that of the studied cellular factors. It has been reported that the localization of HCV proteins changed with time[55]. Therefore, we examined Huh-7.5 cells infected with the JFH-1 strain for over two weeks. HCV core staining was more widely spread within the cytoplasm and several hot spots were observed, similar to HCV proteins detected with the immune human serum in BHK-WNV cells. Core preferentially co-localized with CANX and RAB1 (Figure 12B); it did less so with tubulin.
Huh-7.5 cells were then inoculated with HCVbp-4cys particles (encoding a tetra-cysteine tag on NS5A) of genotype 1a produced in BHK-WNV cells (Figure 12C). In a live-cell immunofluorescence study, the pattern of NS5A-4cys was reminiscent of that observed with the same construct expressed in BHK-WNV cells[8]. However, this pattern was different from the staining of NS5A (without a tag) detected with an in-house antibody against a 48 aa-long peptide[8], suggesting that distinct NS5A protein species were observed with two detection methods; each species could differentially affect HCV particle assembly and secretion. Interestingly, in Huh-7.5 cells, NS5A-4cys closely associated with microtubules (Figure 12C)[56].
With minor differences, these results suggest that cellular factors involved in permissiveness of BHK-WNV cells are also required for long term HCV production in Huh-7.5 cells.
Caspase-1 is required for the release of HCV particles via interplay with viral non-structural genes
Caspase-1 is a cysteine protease and the common denominator of some twenty NLR/ALR inflammasome complexes identified so far and that contribute to host defenses against infections[57]. Cleavage sites for caspase-1 are present on HCV E2, NS2 and NS3 (http://web.expasy.org/peptide_cutter/). They are well conserved between genotypes, but those on E2 and NS2 are generally missing in genotypes 6 and 2, respectively. Additional sites on E1, NS3, NS4B or NS5A are found only in particular HCV strains/isolates. Overall, an average of 3 (2 to 5) cleavage sites are found in HCV polyproteins.
The HCV strain H77 polyprotein displays cleavage sites for caspase-1 at aspartate residues 728 (E2), 989 (NS2) and 1302 (NS3), but none for other caspases. This prompted us to test whether this protease could interfere with the production of HCV particles by BHK-WNV cells. We tested the effect of an inhibitor preferentially targeting caspase-1 (ZYVAD-FMK) on the production of this strain of HCV by BHK-WNV cells. While neither production of envelope E1 protein nor its release by parental BHK cells changed with the inhibitor, the release of HCVbp particles by BHK-WNV cells was dramatically reduced (Figure 13A). This was associated with a different processing of the core protein, with a predominant size of 21 kDa in treated vs 23 kDa in untreated cells (Figure 13A). This observation is reminiscent of the shorter core protein in HCVcc released by Huh7.5.1 cells, compared to that of HCV progenies produced in human liver slices or primary hepatocytes[58]. In contrast, no clear effect of the inhibitor was observed on HCV production with BHK-WNV cells expressing only HCV structural genes, with or without p7 (Figure 13B).
Figure 13.
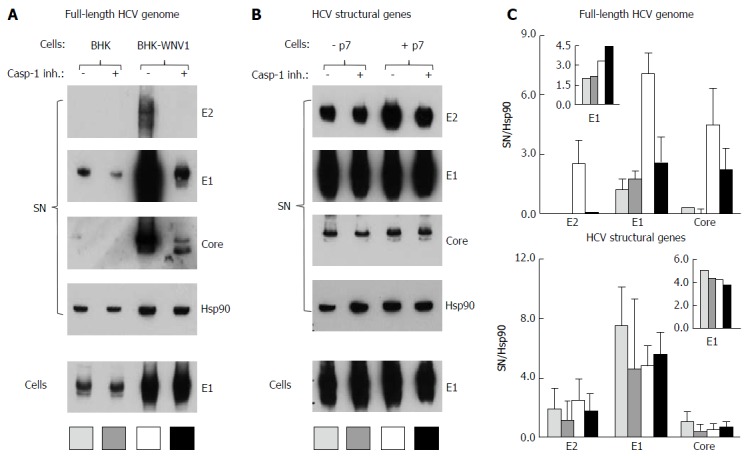
Caspase-1 inhibitor conditionally inhibits the secretion of hepatitis C virus particles by baby hamster kidney-West Nile virus cells. A: BHK-21 and BHK-WNV cells were transfected with a mix of HCVbp-coding and P2B plasmids; the next day, a caspase-1 inhibitor was added in the culture medium and the cells were incubated for 2 more days; cell lysates and HCV particles were harvested and analyzed by Western blot (WB); B: BHK-WNV cells were transfected with a plasmid coding for the structural (core, E1, E2) genes of HCV H77 strain, plus (+) or minus (-) p7, then were analyzed as in (A); C: Quantification of WBs: Top panels for (A) and bottom panels for (B); bar inside patterns are displayed underneath corresponding data; error bars represent standard deviations; inserts = results in cells. HCV: Hepatitis C virus; BHK-WNV: Baby hamster kidney-West Nile virus.
DISCUSSION
A classical secretion pathway has been implicated in the release of HCV virions[59] that are associated with apolipoprotein E (apoE) in Huh-7.5 cells[60] or other cell types[61]. In spite of a fully functional secretion pathway, parental BHK-21 cells, which do not express apoE, released only trace amounts of HCV particles[8]. Lack of apoE was overcome by the prior reorganization of intracellular trafficking by the replication of WNV (or dengue virus) for the production of infectious HCV particles (Figure 14). Epitopes detected by IgG from a patient cured of an infection of same genotype as the HCV strain used in this study (H77), are likely to be amongst those displayed on native HCV virions. This pool of HCV protein isoforms/conformers represented only a fraction of the HCV structural proteins stained by monoclonal antibodies. It overlapped with a compartment of rearranged membranes. The presence of virus particles in this compartment supports the hypothesis that it could be the HCV assembly site in permissive BHK-WNV cells.
Figure 14.
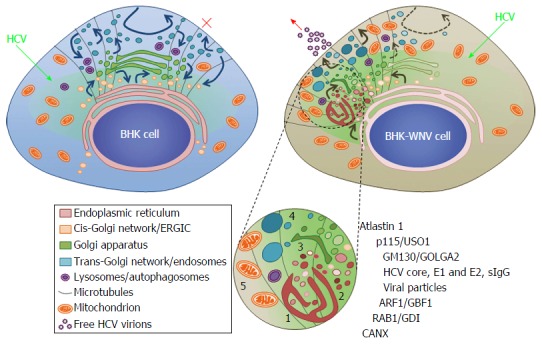
Model of a hepatitis C virus assembly compartment in baby hamster kidney-West Nile virus cells. Schematic organization of cell traffic in parental BHK-21 (top left) and BHK-WNV (top right) cells; curved arrows represent cell traffic; HCV genome is produced by the P2B plasmid system (green arrows) and expressed (greenish areas) in the cytoplasm of BHK cells. Bottom, sketches’ legend and close up of a HCV assembly site (cf. also Figure 2B): (1) convoluted membranes; (2) vesicular packets; (3) Golgi cisternæ; (4) large vesicles filled with viral particles; and (5) mitochondrion; host and viral factors identified within this compartment. SIgG: Antibodies from the serum of a cured HCV patient; CANX: Calnexin; HCV: Hepatitis C virus; BHK-WNV: Baby hamster kidney-West Nile virus; ERGIC: Endoplasmic reticulum-Golgi intermediate compartment; GDI: GDP dissociation inhibitor.
Some arguments are consistent with the involvement of a conventional secretion pathway in the production of HCV particles by BHK-WNV cells; other arguments suggest otherwise. On one hand, an involvement of the ERGIC implies a requirement for RAB1, which was the case for the formation of the assembly compartment, as well as for the production of HCV particles. In accordance, RAB1 and p115/USO1 were co-enriched in this compartment[43,62]. GBF1, an additional component of the ERGIC that activates small-sized GTPase ARF1 and, in turn, COPI-dependent traffic[43], was also involved in the production of HCV particles[8]. On the other hand, another feature of the ERGIC, the lectin ERGIC-53, was excluded from the assembly compartment. As observed in TEM, the latter directly brought together membranes from the ER and the Golgi apparatus, which was consistent with an enrichment of this compartment in, respectively, Atlastin 1[50-52] and GM130/GOLGA2[41], both reported to reorganize and/or maintain membranes of their organelle. In addition, an enrichment in both RAB1 and its GDI, like what was observed in the assembly compartment, has been reported to initiate a cascade of events involving GBF1, ARF1 and phosphatidylinositol 4-kinase III alpha (PI4KIIIα)[43], which may increase the local proportion of phosphatidylinositol 4-phosphate[63] and subvert endocytic trafficking[64]. In Huh-7.5 cells, this role is normally devoted to HCV NS5A that brings together PI4KIIIα and TBC1D20, an activator of RAB1[65]. Therefore, replication of the genomic RNA of flaviviruses in BHK-21 cells may pre-position components - or their equivalent - HCV establishes by itself in the cells in which it usually replicates. Although excluding ERGIC as we know it in the classical secretion pathway, such re-organization of the secretion machinery in BHK-21 cells (Figure 14) could explain why a BFA treatment inhibited HCV particle production.
Finally, if CANX was required for the production of HCV particles, most of it shuttled a pool of HCV protein subspecies to the assembly compartment of BHK-WNV cells, instead of co-localizing with CRT for the proper folding of glycoproteins. How CANX really assists these cells to produce HCV virions is not fully understood. CANX still probably requires binding N-linked glycans on the HCV envelope proteins. The reported slow dissociation rate between CANX and HCV envelope proteins[27] could contribute to unusual involvements of CANX, such as what is observed with proteins targeted to mitochondria-ER associated membranes (MAMs) for the formation of inflammasome and autophagy compartments[66]. HCV has previously been shown to target MAMs in hepatocytes[67]. However, mitochondria were excluded from the assembly compartment in BHK-WNV cells. And, upon expression of HCV structural genes, this coincided with the appearance within the assembly compartment of large vesicles containing particles[8].
Oxidative stress, inflammation and/or infection can lead to the appearance in the cytoplasm of large vesicles, often indiscriminately referred to as multivesicular bodies, releasing extracellular particles of very different compositions depending on their origin. They are formed by the fusion of late endosomal vesicles with membranes from lysosomal and/or autophagosomal compartments, or originating from the periphery of the ER-Golgi complex assembling - independently from a COPII- and COPI-mediated membrane transport - into a compartment of unconventional protein secretion[68]. Unconventional pathways are used by intracellular pathogens reorganizing the vesicular traffic to secrete proteins (e.g.,[69]). The reorganization of the secretion machinery required for the production of HCV particles by BHK-WNV cells displayed similar features.
Knocking down α-TUB expression not only resulted in a lesser release of HCV particles, but also of particulate materials containing Hsp70 and Hsp90, a feature of exosomes. Such extracellular vesicles have been shown to participate in the transfer of viral materials[46]. Recently, exosomes have also been implicated in the propagation/dissemination of HCV genome, proteins and replication complexes, although in the former case with a weaker effectiveness than lipoprotein-associated virions[70,71]. However, the production of HCV particles by BHK-WNV cells cannot be reduced to the secretion of exosomes. We had previously shown that, albeit overlapping, the peaks of particles containing HCV structural and heat-shock proteins released by BHK-WNV cells did not coincide after ultracentrifugation on a density gradient[8]. In addition, while the secretion of HCV particles was blocked, the release of heat-shock proteins was insensitive to a treatment by brefeldin A (BFA), an inhibitor of GBF1[8]. In this work, the secretions of Hsp70 and Hsp90-containing particles and HCV particles displayed different patterns. Therefore, the dual effect of α-TUB siRNA on the secretions of exosomes and HCV particles probably reflects separate needs for microtubules taking place at different steps. For exosomes, it could relate to effects on the BFA-insensitive trans-Golgi network, which is consistent with an accumulation of hsp70 inside producer cells following knockdown of α-TUB expression. Conversely, the production of HCV particles could be inhibited at the level of a GBF1/ARF1-dependent traffic, within the assembly compartment, or later during their secretion.
Treatment of BHK-WNV cells with a caspase-1 inhibitor abolished the release of HCV particles. Caspase-1 has been shown to activate IL-1β and IL-18 proinflammatory cytokine precursors[57]. It has long remained unknown how their matured species translocate across the plasma membrane. Recent results support the idea that their secretion involves a cytosolic compartment containing vesicles and their exocytosis[72]. It could be the mechanism by which a caspase-1-dependent secretory pathway contributed to the production of HCV particles by BHK-WNV cells. However, inhibition of HCV production with the caspase inhibitor occurred only in the context of a full-length genome, suggesting the existence of interaction(s) between caspase-1 and HCV non-structural genes, here NS2 and/or NS3. Although the catalytic activity of NS2 is dispensable, this viral cysteine protease is required for the production of HCV particles[73], as is the helicase/serine protease NS3[74]. Therefore, the cleavage(s) of NS2/3 by caspase-1, in addition, could prevent the maturation of non-structural proteins required for RNA replication and/or remove interactions, either of them or both being detrimental to the formation and secretion of HCV particles.
At variance with the JFH-1/Huh-7.5 cell model[59,75], infectious HCV particles released by BHK-WNV cells did not require lipoproteins or exosomes. It is still unclear whether discrepancies between models[9] could not also relate to active HCV replication in a non-physiological environment[76], differences between viral genotypes[12] and/or differential processing of some HCV proteins[58]. Nevertheless, host factors involved in BHK-WNV cells were also required in Huh-7.5 cells that, over time, developed a cytoplasmic compartment[26] enriched in HCV core, CANX and RAB1. Infectious HCV particles are assembled in the cytoplasm of Huh-7.5 cells[13] before their association to lipoproteins[8,77] or exosomes[75,78]. The associations to lipoprotein particles and extracellular particles have been proposed as mechanisms to regulate vertebrate Hedgehog dispersion during development[79]. In addition, ApoE[25,80] and extracellular particles[47] are suspected to contribute also to the pathogenesis of viruses unrelated to HCV. Therefore, rather than having evolved a unique mechanism of viral propagation, HCV may have instead subverted existing cellular processes. It could involve intrinsic properties of its envelope proteins, since HCV E1 interacts with apoE[81] and possibly lipids[82], which is believed to help the virus escape from immune recognition[83].
The release of HCV particles by BHK-WNV cells, instead, involved an unusual, if not unconventional secretion pathway. The present data suggest that their mechanism of assembly and egress could be a mean by which HCV circumvents intracellular defenses. These infectious particles are not those usually observed as such in infected patients. Therefore, as for other HCV particles produced in vitro[9], we are not sure of their relationship to the normal viral cycle. Are they merely a precursor for the main HCV species, which will eventually associate to lipoproteins, or do they represent different viral species of higher buoyant density? In patients, the proportion of circulating HCV particles associated to lipoproteins varies between individuals and during the course of infection. Denser viral species can amount up to half of the circulating HCV genome[84]; they are usually opsonized by IgG in the serum of infected patients, making the study of free virions difficult. At least during the assembly process in BHK-WNV cells, HCV particles exposed epitopes that were recognized by neutralizing sIgG from a patient previously chronically infected by HCV of same genotype. We propose that the BHK-WNV cell model could be useful to study the structure of free HCV virions and their immunological properties.
ACKNOWLEDGMENTS
We thank Charles M Rice (Center for the Study of Hepatitis C, the Rockefeller University) for his generosity in providing plasmids encoding HCV RNA of genotype 1a (wild type and adaptive mutants) and Huh-7.5 cells; Theodore C Pierson (LVD, NIAID, NIH) for kindly provided BHK-21 cells bearing a WNV subgenomic replicon as well as a plasmid encoding WNV structural genes; Takaji Wakita (Department of Virology, National Institute of Infectious Diseases, Tokyo) provided plasmids encoding HCV RNA of genotype 2a; anti-HCV antibodies were generous gifts from Arvind H Patel (anti-E2; MRC Virology Unit, Institute of Virology, University of Glasgow), Ramsey C Cheung (anti-E1; Division of Gastroenterology and Hepatology, Stanford University School of Medicine), Stanislas Pol (HCV serum; Hôpital and Institut Cochin, Paris). We are indebted to the staff of the Biological Imaging Section (RTB, NIAID, NIH) for their assistance with the confocal microscopy and Kunio Nagashima (SAIC/NCI, NIH Frederick) for the EM analyses.
COMMENTS
Background
The production of infectious hepatitis C virus (HCV) particles by model human hepatocellular carcinoma Huh-7.5 cells is believed to involve a classical secretion pathway; it has also been reported to involve lipoproteins and exosomes. The authors previously demonstrated that the intracellular environment generated by a West-Nile virus (WNV) subgenomic replicon rendered non-human, non-hepatic mammalian cells [here referred to as baby hamster kidney-WNV (BHK-WNV) cells] permissive for assembly and release of infectious HCV particles of various genotypes wherein the HCV genome is generated in the cytoplasm independently of its replication machinery. HCV production did not require ongoing activity of the WNV replicon, but instead was associated with persisting replicon-induced changes in the cellular environment. Since secretion of HCV particles by BHK-WNV cells neither involved lipoproteins nor exosomes, could it still follow a conventional pathway? Activity of the small GTPase ARF1 and the maturation of carbohydrates on envelope proteins of released HCV particles were both required for the production of infectious HCV by permissive BHK-WNV cells, suggesting that, indeed, it followed a classical secretion pathway. The authors, therefore, examined the possibility that the endoplamic reticulum (ER)-bound lectin calnexin (CANX), the small GTPase RAB1 and microtubule-associated alpha-tubulin, which all contribute to the secretion of several glycoproteins by BHK-21 cells, were involved in the production of HCV particles by BHK-WNV cells. Surprisingly, the results show that secretion of HCV particles went through a re-organized and re-wired pathway bypassing the conventional ER-to-Golgi intermediary compartment and involving components of the inflammasome.
Research frontiers
The structure of HCV virions/progenies remains elusive. The fact that infectious HCV particles are produced independently from lipoprotein biosynthesis, yet retain the possibility to interact with lipoproteins in vitro supports the view that, in vivo, HCV particles may interact with lipoproteins in a second step and not necessarily co-assemble with them. This has probably important consequences regarding the way HCV interacts with producer cells and the immune system.
Innovations and breakthroughs
The authors’ results suggest that the HCV production made possible by the prior WNV replication in BHK cells could be related to that observed in human hepatocytes. It is unclear whether this reflects an organization of HCV production more complex than expected in hepatocytes or the existence of additional route(s) of HCV secretion.
Applications
Although no immediate application of these findings is foreseen, the production of free HCV virions could pave the way for identifying the most important epitopes for HCV neutralization and the development of an effective vaccine.
Terminology
Subgenomic replicon: Fragment of a viral RNA genome encoding the non structural genes required for its own replication in the cytoplasm, but not the structural genes required for the formation of virions.
Peer-review
In this manuscript, the authors aimed to examine whether the cellular factors including CANX, RAB1 and alpha-tubulin were involved in the production of HCV particles by BHK-WNV cells. Moreover, it brings an interesting contribution to the current literature.
Footnotes
Supported by Intramural Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases (Project No. 1 ZIA AI000733-15: Enveloped Virus Glycoprotein/Receptor Interactions) to Edward A Berger, PhD (MSS, LVD, DIR, NIAID); and ORISE Senior Fellow award (Award No. 1238-1238-03: Department of Energy/Oak Ridge Institute for Science and Education) to Bertrand Saunier, MD, PhD.
Institutional review board statement: Approved the use of serum IgG of a patient cured from an HCV infection for in vitro studies (Hôpital and Institut Cochin, Paris).
Institutional animal care and use committee statement: Not applicable.
Conflict-of-interest statement: Saunier B, Triyatni M and Berger EA are co-inventors on NIH-owned patent #US 9052321 B2 on the HCV particle production system.
Data sharing statement: No additional data are available.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Manuscript source: Invited manuscript
Peer-review started: February 1, 2016
First decision: March 25, 2016
Article in press: June 3, 2016
P- Reviewer: Melhem NM, Temel HE, Uyanik M S- Editor: Ji FF L- Editor: A E- Editor: Li D
References
- 1.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5:558–567. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 2.Webster DP, Klenerman P, Dusheiko GM. Hepatitis C. Lancet. 2015;385:1124–1135. doi: 10.1016/S0140-6736(14)62401-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sølund C, Krarup H, Ramirez S, Thielsen P, Røge BT, Lunding S, Barfod TS, Madsen LG, Tarp B, Christensen PB, et al. Nationwide experience of treatment with protease inhibitors in chronic hepatitis C patients in Denmark: identification of viral resistance mutations. PLoS One. 2014;9:e113034. doi: 10.1371/journal.pone.0113034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feeney ER, Chung RT. Antiviral treatment of hepatitis C. BMJ. 2014;348:g3308. doi: 10.1136/bmj.g3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baumert TF, Fauvelle C, Chen DY, Lauer GM. A prophylactic hepatitis C virus vaccine: a distant peak still worth climbing. J Hepatol. 2014;61:S34–S44. doi: 10.1016/j.jhep.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 6.de Jong YP, Rice CM, Ploss A. New horizons for studying human hepatotropic infections. J Clin Invest. 2010;120:650–653. doi: 10.1172/JCI42338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mina MM, Luciani F, Cameron B, Bull RA, Beard MR, Booth D, Lloyd AR. Resistance to hepatitis C virus: potential genetic and immunological determinants. Lancet Infect Dis. 2015;15:451–460. doi: 10.1016/S1473-3099(14)70965-X. [DOI] [PubMed] [Google Scholar]
- 8.Triyatni M, Berger EA, Saunier B. A new model to produce infectious hepatitis C virus without the replication requirement. PLoS Pathog. 2011;7:e1001333. doi: 10.1371/journal.ppat.1001333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saunier B, Triyatni M, Berger EA. Culturing HCV: challenges and progress. Future Virol. 2011;6:1169–1178. [Google Scholar]
- 10.Kolykhalov AA, Agapov EV, Blight KJ, Mihalik K, Feinstone SM, Rice CM. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science. 1997;277:570–574. doi: 10.1126/science.277.5325.570. [DOI] [PubMed] [Google Scholar]
- 11.Lohmann V, Körner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 12.Pietschmann T, Zayas M, Meuleman P, Long G, Appel N, Koutsoudakis G, Kallis S, Leroux-Roels G, Lohmann V, Bartenschlager R. Production of infectious genotype 1b virus particles in cell culture and impairment by replication enhancing mutations. PLoS Pathog. 2009;5:e1000475. doi: 10.1371/journal.ppat.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gastaminza P, Kapadia SB, Chisari FV. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J Virol. 2006;80:11074–11081. doi: 10.1128/JVI.01150-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Felmlee DJ, Sheridan DA, Bridge SH, Nielsen SU, Milne RW, Packard CJ, Caslake MJ, McLauchlan J, Toms GL, Neely RD, et al. Intravascular transfer contributes to postprandial increase in numbers of very-low-density hepatitis C virus particles. Gastroenterology. 2010;139:1774–1783, 1783.e1-e6. doi: 10.1053/j.gastro.2010.07.047. [DOI] [PubMed] [Google Scholar]
- 15.Lindenbach BD, Rice CM. The ins and outs of hepatitis C virus entry and assembly. Nat Rev Microbiol. 2013;11:688–700. doi: 10.1038/nrmicro3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grassi G, Di Caprio G, Fimia GM, Ippolito G, Tripodi M, Alonzi T. Hepatitis C virus relies on lipoproteins for its life cycle. World J Gastroenterol. 2016;22:1953–1965. doi: 10.3748/wjg.v22.i6.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartenschlager R, Penin F, Lohmann V, André P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011;19:95–103. doi: 10.1016/j.tim.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Murray CL, Jones CT, Rice CM. Architects of assembly: roles of Flaviviridae non-structural proteins in virion morphogenesis. Nat Rev Microbiol. 2008;6:699–708. doi: 10.1038/nrmicro1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fredericksen BL, Smith M, Katze MG, Shi PY, Gale M. The host response to West Nile Virus infection limits viral spread through the activation of the interferon regulatory factor 3 pathway. J Virol. 2004;78:7737–7747. doi: 10.1128/JVI.78.14.7737-7747.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 2009;5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye J. Reliance of host cholesterol metabolic pathways for the life cycle of hepatitis C virus. PLoS Pathog. 2007;3:e108. doi: 10.1371/journal.ppat.0030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol. 2007;9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 23.Mackenzie JM, Khromykh AA, Parton RG. Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe. 2007;2:229–239. doi: 10.1016/j.chom.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Samsa MM, Mondotte JA, Iglesias NG, Assunção-Miranda I, Barbosa-Lima G, Da Poian AT, Bozza PT, Gamarnik AV. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 2009;5:e1000632. doi: 10.1371/journal.ppat.1000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faustino AF, Martins IC, Carvalho FA, Castanho MA, Maurer-Stroh S, Santos NC. Understanding Dengue Virus Capsid Protein Interaction with Key Biological Targets. Sci Rep. 2015;5:10592. doi: 10.1038/srep10592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rouillé Y, Helle F, Delgrange D, Roingeard P, Voisset C, Blanchard E, Belouzard S, McKeating J, Patel AH, Maertens G, et al. Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J Virol. 2006;80:2832–2841. doi: 10.1128/JVI.80.6.2832-2841.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dubuisson J, Rice CM. Hepatitis C virus glycoprotein folding: disulfide bond formation and association with calnexin. J Virol. 1996;70:778–786. doi: 10.1128/jvi.70.2.778-786.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dodonova SO, Diestelkoetter-Bachert P, von Appen A, Hagen WJ, Beck R, Beck M, Wieland F, Briggs JA. VESICULAR TRANSPORT. A structure of the COPI coat and the role of coat proteins in membrane vesicle assembly. Science. 2015;349:195–198. doi: 10.1126/science.aab1121. [DOI] [PubMed] [Google Scholar]
- 29.Klaus JP, Eisenhauer P, Russo J, Mason AB, Do D, King B, Taatjes D, Cornillez-Ty C, Boyson JE, Thali M, et al. The intracellular cargo receptor ERGIC-53 is required for the production of infectious arenavirus, coronavirus, and filovirus particles. Cell Host Microbe. 2013;14:522–534. doi: 10.1016/j.chom.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pierson TC, Sánchez MD, Puffer BA, Ahmed AA, Geiss BJ, Valentine LE, Altamura LA, Diamond MS, Doms RW. A rapid and quantitative assay for measuring antibody-mediated neutralization of West Nile virus infection. Virology. 2006;346:53–65. doi: 10.1016/j.virol.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 31.Wadkins TS, Been MD. Ribozyme activity in the genomic and antigenomic RNA strands of hepatitis delta virus. Cell Mol Life Sci. 2002;59:112–125. doi: 10.1007/s00018-002-8409-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blight KJ, McKeating JA, Marcotrigiano J, Rice CM. Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J Virol. 2003;77:3181–3190. doi: 10.1128/JVI.77.5.3181-3190.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Griffin BA, Adams SR, Tsien RY. Specific covalent labeling of recombinant protein molecules inside live cells. Science. 1998;281:269–272. doi: 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- 34.Owsianka A, Clayton RF, Loomis-Price LD, McKeating JA, Patel AH. Functional analysis of hepatitis C virus E2 glycoproteins and virus-like particles reveals structural dissimilarities between different forms of E2. J Gen Virol. 2001;82:1877–1883. doi: 10.1099/0022-1317-82-8-1877. [DOI] [PubMed] [Google Scholar]
- 35.Oliphant T, Engle M, Nybakken GE, Doane C, Johnson S, Huang L, Gorlatov S, Mehlhop E, Marri A, Chung KM, et al. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat Med. 2005;11:522–530. doi: 10.1038/nm1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dussupt V, Javid MP, Abou-Jaoudé G, Jadwin JA, de La Cruz J, Nagashima K, Bouamr F. The nucleocapsid region of HIV-1 Gag cooperates with the PTAP and LYPXnL late domains to recruit the cellular machinery necessary for viral budding. PLoS Pathog. 2009;5:e1000339. doi: 10.1371/journal.ppat.1000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saunier B, Triyatni M, Ulianich L, Maruvada P, Yen P, Kohn LD. Role of the asialoglycoprotein receptor in binding and entry of hepatitis C virus structural proteins in cultured human hepatocytes. J Virol. 2003;77:546–559. doi: 10.1128/JVI.77.1.546-559.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Castro IF, Volonté L, Risco C. Virus factories: biogenesis and structural design. Cell Microbiol. 2013;15:24–34. doi: 10.1111/cmi.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tomás M, Martínez-Alonso E, Ballesta J, Martínez-Menárguez JA. Regulation of ER-Golgi intermediate compartment tubulation and mobility by COPI coats, motor proteins and microtubules. Traffic. 2010;11:616–625. doi: 10.1111/j.1600-0854.2010.01047.x. [DOI] [PubMed] [Google Scholar]
- 41.Rivero S, Cardenas J, Bornens M, Rios RM. Microtubule nucleation at the cis-side of the Golgi apparatus requires AKAP450 and GM130. EMBO J. 2009;28:1016–1028. doi: 10.1038/emboj.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 43.Dumaresq-Doiron K, Savard MF, Akam S, Costantino S, Lefrancois S. The phosphatidylinositol 4-kinase PI4KIIIalpha is required for the recruitment of GBF1 to Golgi membranes. J Cell Sci. 2010;123:2273–2280. doi: 10.1242/jcs.055798. [DOI] [PubMed] [Google Scholar]
- 44.de Chassey B, Navratil V, Tafforeau L, Hiet MS, Aublin-Gex A, Agaugué S, Meiffren G, Pradezynski F, Faria BF, Chantier T, et al. Hepatitis C virus infection protein network. Mol Syst Biol. 2008;4:230. doi: 10.1038/msb.2008.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roohvand F, Maillard P, Lavergne JP, Boulant S, Walic M, Andréo U, Goueslain L, Helle F, Mallet A, McLauchlan J, et al. Initiation of hepatitis C virus infection requires the dynamic microtubule network: role of the viral nucleocapsid protein. J Biol Chem. 2009;284:13778–13791. doi: 10.1074/jbc.M807873200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lancaster GI, Febbraio MA. Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J Biol Chem. 2005;280:23349–23355. doi: 10.1074/jbc.M502017200. [DOI] [PubMed] [Google Scholar]
- 47.Chahar HS, Bao X, Casola A. Exosomes and Their Role in the Life Cycle and Pathogenesis of RNA Viruses. Viruses. 2015;7:3204–3225. doi: 10.3390/v7062770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bräutigam J, Scheidig AJ, Egge-Jacobsen W. Mass spectrometric analysis of hepatitis C viral envelope protein E2 reveals extended microheterogeneity of mucin-type O-linked glycosylation. Glycobiology. 2013;23:453–474. doi: 10.1093/glycob/cws171. [DOI] [PubMed] [Google Scholar]
- 49.Chen CY, Balch WE. The Hsp90 chaperone complex regulates GDI-dependent Rab recycling. Mol Biol Cell. 2006;17:3494–3507. doi: 10.1091/mbc.E05-12-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Orso G, Pendin D, Liu S, Tosetto J, Moss TJ, Faust JE, Micaroni M, Egorova A, Martinuzzi A, McNew JA, et al. Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature. 2009;460:978–983. doi: 10.1038/nature08280. [DOI] [PubMed] [Google Scholar]
- 51.Morin-Leisk J, Saini SG, Meng X, Makhov AM, Zhang P, Lee TH. An intramolecular salt bridge drives the soluble domain of GTP-bound atlastin into the postfusion conformation. J Cell Biol. 2011;195:605–615. doi: 10.1083/jcb.201105006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu J, Shibata Y, Zhu PP, Voss C, Rismanchi N, Prinz WA, Rapoport TA, Blackstone C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell. 2009;138:549–561. doi: 10.1016/j.cell.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Allan BB, Moyer BD, Balch WE. Rab1 recruitment of p115 into a cis-SNARE complex: programming budding COPII vesicles for fusion. Science. 2000;289:444–448. doi: 10.1126/science.289.5478.444. [DOI] [PubMed] [Google Scholar]
- 54.Rothman JE, Fine RE. Coated vesicles transport newly synthesized membrane glycoproteins from endoplasmic reticulum to plasma membrane in two successive stages. Proc Natl Acad Sci USA. 1980;77:780–784. doi: 10.1073/pnas.77.2.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boson B, Granio O, Bartenschlager R, Cosset FL. A concerted action of hepatitis C virus p7 and nonstructural protein 2 regulates core localization at the endoplasmic reticulum and virus assembly. PLoS Pathog. 2011;7:e1002144. doi: 10.1371/journal.ppat.1002144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wölk B, Büchele B, Moradpour D, Rice CM. A dynamic view of hepatitis C virus replication complexes. J Virol. 2008;82:10519–10531. doi: 10.1128/JVI.00640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Zoete MR, Palm NW, Zhu S, Flavell RA. Inflammasomes. Cold Spring Harb Perspect Biol. 2014;6:a016287. doi: 10.1101/cshperspect.a016287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lagaye S, Shen H, Saunier B, Nascimbeni M, Gaston J, Bourdoncle P, Hannoun L, Massault PP, Vallet-Pichard A, Mallet V, et al. Efficient replication of primary or culture hepatitis C virus isolates in human liver slices: a relevant ex vivo model of liver infection. Hepatology. 2012;56:861–872. doi: 10.1002/hep.25738. [DOI] [PubMed] [Google Scholar]
- 59.Goueslain L, Alsaleh K, Horellou P, Roingeard P, Descamps V, Duverlie G, Ciczora Y, Wychowski C, Dubuisson J, Rouillé Y. Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication. J Virol. 2010;84:773–787. doi: 10.1128/JVI.01190-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Steinmann E, Whitfield T, Kallis S, Dwek RA, Zitzmann N, Pietschmann T, Bartenschlager R. Antiviral effects of amantadine and iminosugar derivatives against hepatitis C virus. Hepatology. 2007;46:330–338. doi: 10.1002/hep.21686. [DOI] [PubMed] [Google Scholar]
- 61.Hueging K, Doepke M, Vieyres G, Bankwitz D, Frentzen A, Doerrbecker J, Gumz F, Haid S, Wölk B, Kaderali L, et al. Apolipoprotein E codetermines tissue tropism of hepatitis C virus and is crucial for viral cell-to-cell transmission by contributing to a postenvelopment step of assembly. J Virol. 2014;88:1433–1446. doi: 10.1128/JVI.01815-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guo Y, Linstedt AD. Binding of the vesicle docking protein p115 to the GTPase Rab1b regulates membrane recruitment of the COPI vesicle coat. Cell Logist. 2013;3:e27687. doi: 10.4161/cl.27687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dorobantu CM, Albulescu L, Harak C, Feng Q, van Kampen M, Strating JR, Gorbalenya AE, Lohmann V, van der Schaar HM, van Kuppeveld FJ. Modulation of the Host Lipid Landscape to Promote RNA Virus Replication: The Picornavirus Encephalomyocarditis Virus Converges on the Pathway Used by Hepatitis C Virus. PLoS Pathog. 2015;11:e1005185. doi: 10.1371/journal.ppat.1005185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berger KL, Cooper JD, Heaton NS, Yoon R, Oakland TE, Jordan TX, Mateu G, Grakoui A, Randall G. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc Natl Acad Sci USA. 2009;106:7577–7582. doi: 10.1073/pnas.0902693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nevo-Yassaf I, Yaffe Y, Asher M, Ravid O, Eizenberg S, Henis YI, Nahmias Y, Hirschberg K, Sklan EH. Role for TBC1D20 and Rab1 in hepatitis C virus replication via interaction with lipid droplet-bound nonstructural protein 5A. J Virol. 2012;86:6491–6502. doi: 10.1128/JVI.00496-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta. 2014;1837:461–469. doi: 10.1016/j.bbabio.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 67.Horner SM, Liu HM, Park HS, Briley J, Gale M. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci USA. 2011;108:14590–14595. doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nickel W, Rabouille C. Mechanisms of regulated unconventional protein secretion. Nat Rev Mol Cell Biol. 2009;10:148–155. doi: 10.1038/nrm2617. [DOI] [PubMed] [Google Scholar]
- 69.Shoji JY, Kikuma T, Kitamoto K. Vesicle trafficking, organelle functions, and unconventional secretion in fungal physiology and pathogenicity. Curr Opin Microbiol. 2014;20:1–9. doi: 10.1016/j.mib.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 70.Bukong TN, Momen-Heravi F, Kodys K, Bala S, Szabo G. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog. 2014;10:e1004424. doi: 10.1371/journal.ppat.1004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Longatti A, Boyd B, Chisari FV. Virion-independent transfer of replication-competent hepatitis C virus RNA between permissive cells. J Virol. 2015;89:2956–2961. doi: 10.1128/JVI.02721-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Monteleone M, Stow JL, Schroder K. Mechanisms of unconventional secretion of IL-1 family cytokines. Cytokine. 2015;74:213–218. doi: 10.1016/j.cyto.2015.03.022. [DOI] [PubMed] [Google Scholar]
- 73.Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J Virol. 2007;81:8374–8383. doi: 10.1128/JVI.00690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ma Y, Yates J, Liang Y, Lemon SM, Yi M. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J Virol. 2008;82:7624–7639. doi: 10.1128/JVI.00724-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gastaminza P, Dryden KA, Boyd B, Wood MR, Law M, Yeager M, Chisari FV. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J Virol. 2010;84:10999–11009. doi: 10.1128/JVI.00526-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lindenbach BD, Meuleman P, Ploss A, Vanwolleghem T, Syder AJ, McKeating JA, Lanford RE, Feinstone SM, Major ME, Leroux-Roels G, et al. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc Natl Acad Sci USA. 2006;103:3805–3809. doi: 10.1073/pnas.0511218103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Triyatni M, Saunier B, Maruvada P, Davis AR, Ulianich L, Heller T, Patel A, Kohn LD, Liang TJ. Interaction of hepatitis C virus-like particles and cells: a model system for studying viral binding and entry. J Virol. 2002;76:9335–9344. doi: 10.1128/JVI.76.18.9335-9344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Masciopinto F, Giovani C, Campagnoli S, Galli-Stampino L, Colombatto P, Brunetto M, Yen TS, Houghton M, Pileri P, Abrignani S. Association of hepatitis C virus envelope proteins with exosomes. Eur J Immunol. 2004;34:2834–2842. doi: 10.1002/eji.200424887. [DOI] [PubMed] [Google Scholar]
- 79.Vyas N, Walvekar A, Tate D, Lakshmanan V, Bansal D, Lo Cicero A, Raposo G, Palakodeti D, Dhawan J. Vertebrate Hedgehog is secreted on two types of extracellular vesicles with different signaling properties. Sci Rep. 2014;4:7357. doi: 10.1038/srep07357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang L, Yesupriya A, Chang MH, Teshale E, Teo CG. Apolipoprotein E and protection against hepatitis E viral infection in American non-Hispanic blacks. Hepatology. 2015;62:1346–1352. doi: 10.1002/hep.27938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mazumdar B, Banerjee A, Meyer K, Ray R. Hepatitis C virus E1 envelope glycoprotein interacts with apolipoproteins in facilitating entry into hepatocytes. Hepatology. 2011;54:1149–1156. doi: 10.1002/hep.24523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.El Omari K, Iourin O, Kadlec J, Sutton G, Harlos K, Grimes JM, Stuart DI. Unexpected structure for the N-terminal domain of hepatitis C virus envelope glycoprotein E1. Nat Commun. 2014;5:4874. doi: 10.1038/ncomms5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wahid A, Dubuisson J. Virus-neutralizing antibodies to hepatitis C virus. J Viral Hepat. 2013;20:369–376. doi: 10.1111/jvh.12094. [DOI] [PubMed] [Google Scholar]
- 84.André P, Komurian-Pradel F, Deforges S, Perret M, Berland JL, Sodoyer M, Pol S, Bréchot C, Paranhos-Baccalà G, Lotteau V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J Virol. 2002;76:6919–6928. doi: 10.1128/JVI.76.14.6919-6928.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]