

Abstract
Background
Renovascular hypertension (RVH) impairs cardiac structure and left ventricular (LV) function, but whether mitochondrial injury is implicated in RVH‐induced myocardial damage and dysfunction has not been defined. We hypothesized that cardiac remodeling in swine RVH is partly attributable to cardiac mitochondrial injury.
Methods and Results
After 12 weeks of hypercholesterolemic (HC)‐RVH or control (n=14 each), pigs were treated for another 4 weeks with vehicle or with the mitochondrial‐targeted peptide (MTP), Bendavia (0.1 mg/kg subcutaneously, 5 days/week), which stabilizes mitochondrial inner‐membrane cardiolipin (n=7 each). Cardiac function was subsequently assessed by multidetector‐computed tomography and oxygenation by blood‐oxygen‐level–dependent magnetic resonance imaging. Cardiolipin content, mitochondrial biogenesis, as well as sarcoplasmic‐reticulum calcium cycling, myocardial tissue injury, and coronary endothelial function were assessed ex vivo. Additionally, mitochondrial cardiolipin content, oxidative stress, and bioenergetics were assessed in rat cardiomyocytes incubated with tert‐butyl hydroperoxide (tBHP) untreated or treated with MTP. Chronic mitoprotection in vivo restored cardiolipin content and mitochondrial biogenesis. Thapsigargin‐sensitive sarcoplasmic reticulum Ca2+‐ATPase activity that declined in HC‐RVH normalized in MTP‐treated pigs. Mitoprotection also improved LV relaxation (E/A ratio) and ameliorated cardiac hypertrophy, without affecting blood pressure or systolic function. Myocardial remodeling and coronary endothelial function improved only in MTP‐treated pigs. In tBHP‐treated cardiomyocytes, mitochondrial targeting attenuated a fall in cardiolipin content and bioenergetics.
Conclusions
Chronic mitoprotection blunted myocardial hypertrophy, improved LV relaxation, and attenuated myocardial cellular and microvascular remodeling, despite sustained HC‐RVH, suggesting that mitochondrial injury partly contributes to hypertensive cardiomyopathy.
Keywords: bendavia, heart failure, hypertension, mitochondria, renal artery stenosis, renovascular hypertension
Subject Categories: Hypertension, Heart Failure, Atherosclerosis, Animal Models of Human Disease, Cardiorenal Syndrome
Introduction
Renovascular hypertension (RVH) accompanied by kidney dysfunction is prevalent in elderly individuals and commonly associated with cardiac abnormalities and dysfunction.1 Importantly, patients with RVH also have increased mortality rate, warranting development of preventive and management strategies for these patients.2 We have previously shown that left ventricular (LV) structure and relaxation are selectively impaired in RVH compared with essential hypertensive patients.3 Alas, to date, few treatment options are available to improve cardiac structure and dysfunction in hypertension, possibly because of the lack of clinically applicable therapies targeting underlying biological mechanisms.
Accumulating evidence suggests that mitochondrial abnormalities and dysfunction are important contributors to hypertension‐induced damage in the heart and other major target organs.4, 5 For example, murine mitochondrial DNA mutations induce cardiac hypertrophy and dilatation, partly mediated by mitochondrial dysfunction and apoptosis.6 Cardiac mitochondria regulate calcium homeostasis, by capturing and recycling released calcium.7 However, in failing cardiomyocytes, mitochondrial calcium overload triggers opening of the mitochondrial permeability‐transition‐pore (mPTP), resulting in mitochondrial swelling and release of cytochrome‐c to the cytosol and reactive oxygen species (ROS), leading to apoptosis and oxidative stress. In agreement, we have previously shown that experimental RVH induces myocardial apoptosis, oxidative stress, fibrosis, LV remodeling, and dysfunction, associated with decreased mitochondrial biogenesis,8 underscoring a potential physiological role for mitochondria in cardiomyocyte homeostasis.
Mitochondria also regulate cardiac endothelial cell function through modulating intracellular concentrations of ROS, nitric oxide (NO), and calcium. For example, endothelial NO synthesis depends on the activity of the mitochondrial enzyme arginase‐II,9 whereas uncoupling protein (UCP)‐2, an inner mitochondrial membrane transporter, preserves endothelial function by preventing mitochondrial ROS‐induced NO deficiency.10 Excessive mitochondrial ROS contributes to impaired coronary vascular relaxation, triggering oxidative damage and mitochondrial dysfunction.11 When mitochondrial function is affected, endothelial cell migration, tube formation, and angiogenesis are compromised, resulting in microvascular remodeling and loss.12 Damage to the cardiovascular endothelium is a major risk factor for heart disease, thus strategies aimed to protect both cardiomyocyte and endothelial cell mitochondria may attenuate RVH‐induced myocardial remodeling and dysfunction.
Cardiolipin13 is a phospholipid found only in the inner mitochondrial membrane.14 Its unique conical structure, which imparts properties favoring membrane curvature, makes cardiolipin ideal for forming cristae and supercomplexes in the electronic transport chain and maintaining mitochondrial bioenergetics.15 Furthermore, cardiolipin anchors cytochrome‐c to the inner mitochondrial membrane, facilitating electron transfer between complexes III and IV.16 However, cardiolipin is vulnerable to increased cytosolic calcium and oxidative stress, which invoke its peroxidation and subsequent loss,17 leading to mitochondrial dysfunction underpinning various forms of cardiovascular disease.
Experimental studies have shown that cardiolipin protection adjunctive to reperfusion injury limits myocardial infarct size and improves cardiac function,18 implicating mitochondrial damage in acute cardiac insults. The mitochondria‐targeted peptide (MTP), Bendavia, targets the mitochondrial matrix independent of membrane potential, preventing peroxidation of cardiolipin.13 We have also previously shown that a single 3‐hour MTP infusion during swine renal revascularization that led to regression of hypertension expedited subsequent cardiac recovery.8 Yet, the ability to blunt hypertension‐induced cardiomyopathy by mitoprotection in the face of ongoing hypertension remained unknown. Therefore, the current study tested the hypothesis that mitochondrial injury mediates RVH‐induced cardiac damage, and that chronic mitoprotection can decrease cardiac remodeling and improve its function in hypertensive pigs.
Methods
Study Groups and Experimental Design
Animal procedures were approved by the Institutional Animal care and Use Committee. Female domestic pigs (n=28) were studied after 16 weeks of observation (Figure 1A).
Figure 1.
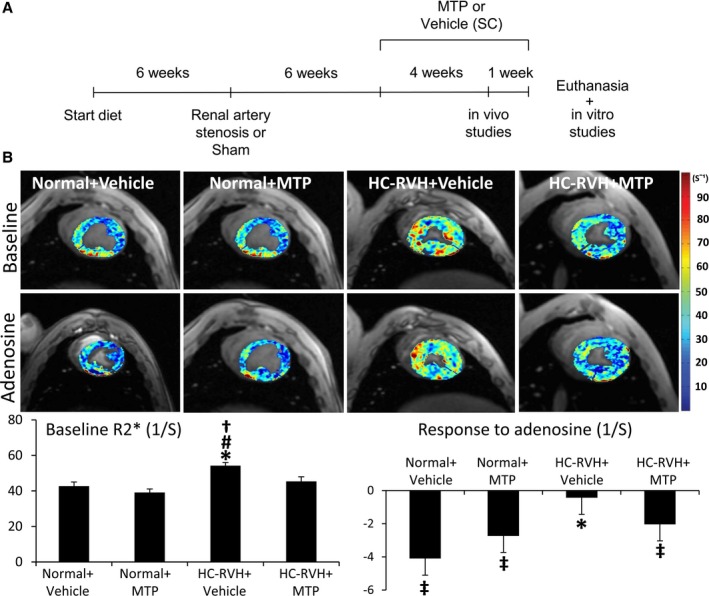
Mitochondrial‐targeted peptides (MTP) improved myocardial oxygenation. A, Schematic of the experimental protocol. B, Representative left ventricular cross‐sectional images of blood‐oxygen‐level–dependent magnetic resonance imaging (BOLD‐MRI), showing hypoxic myocardium (red) before and after adenosine (top). Quantification of R2* signal (bottom). *P<0.05 vs Normal+Vehicle; # P<0.05 vs Normal+MTP; † P<0.05 vs hypercholesterolemic renovascular hypertension (HC‐RVH)+MTP; ‡ P<0.05 vs baseline. HC indicates hypercholesterolemic; RVH, renovascular hypertension; SC, subcutaneous.
At baseline, pigs were randomized as hypercholecterolemic (HC)‐RVH or normal controls (n=14 each). To simulate concurrent atherosclerosis, HC‐RVH pigs started a high‐cholesterol diet (2% cholesterol and 15% lard (TD‐93296; Harlan‐Teklad, Madison, WI),19 whereas normal pigs consumed standard pig chow.
Six weeks later, renal artery stenosis and RVH were induced in pigs by placing a local‐irritant coil in the main renal artery, which increases arterial pressure within 7 to 10 days,20 whereas normal pigs underwent a sham procedure. Anesthesia was induced with 0.25 g of intramuscular tiletamine hydrochloride/zolazepam hydrochloride and 0.5 g of xylazine, and maintained with intravenous ketamine (0.2 mg/kg per minute) and xylazine (0.03 mg/kg per minute).
Six weeks after induction of RVH, animals were similarly anesthetized. Daily subcutaneous injections of the MTP Bendavia (Stealth BioTherapeutics, Inc. Newton, MA; 0.1 mg/kg in 1 mL of PBS, 1/day 5 days/week) or vehicle (PBS) were then initiated in both HC‐RVH and sham pigs (n=7 each group).
Four weeks later, the degree of stenosis was determined by angiography and systemic venous blood samples collected under similar anesthesia. Creatinine, plasma renin activity (PRA), cholesterol fractions and triglycerides levels were measured.8, 21 Multidetector computed tomography (MDCT) and blood oxygen‐level–dependent magnetic‐resonance imaging (BOLD‐MRI) scanning was conducted to assess cardiac function and myocardial oxygenation, respectively.
A few days after completion of in vivo studies, pigs were euthanized (intravenous sodium pentobarbital, 100 mg/kg, Fatal Plus; Vortech Pharmaceuticals, Dearborn, MI). LV tissue was harvested, frozen in liquid nitrogen (and maintained at −80°C), or preserved in formalin for ex vivo studies. Another segment was prepared for micro‐CT studies. Additionally, main branches of the coronary artery were dissected and placed in control solution for in vitro assessment of coronary endothelial function.
Cardiac Oxygenation and Function
BOLD‐MRI was performed to assess myocardial oxygenation, as we described.22 Briefly, pigs anesthetized with 1% to 2% isoflurane were positioned in the MRI scanner (3 Tesla, Signa Echo Speed; GE Medical Systems, Milwaukee, WI). Scans were performed during suspended respiration before and after a 5‐minute intravenous injection of adenosine (400 μg/kg per minute). The average relaxivity index R2*, a surrogate of myocardial hypoxia, was estimated in regions of interest traced in the septum in each slice, and images analyzed using MATLAB software (version 7.10; The MathWorks, Inc., Natick, MA).22
Two days after BOLD‐MRI studies, 64‐slice MDCT (Somatom Definition‐64; Siemens Medical Solution, Forchheim, Germany) studies were performed before and during a 5‐minute intravenous infusion of adenosine (400 μg/kg per minute). The entire LV was scanned throughout the cardiac cycle to obtain cardiac systolic function, end‐diastolic volume (EDV), and LV muscle mass (LVMM).23 Early (E) and late (A) LV filling rates were obtained from the volume/time curve, and myocardial perfusion from time‐attenuation curves obtained from the anterior cardiac wall, as described.24 Images were analyzed with the Analyze software package (Biomedical Imaging Resource; Mayo Clinic, Rochester, MN).
Myocardial Injury and Remodeling
Myocardial apoptosis was evaluated in LV sections double stained with terminal deoxynucleotidyl transferase‐mediated dUTP nick end end‐labeling (TUNEL; Promega, Madison, WI) and connexin‐43 (Catalog No.: ab79010; Abcam, Cambridge, MA) fluorescent staining.8 Protein expression of the antiapoptotic, B‐cell lymphoma (Bcl)‐xl (1:1000; Lifespan BioSciences, Seattle, WA), was assessed by Western blotting.19 In situ production of superoxide anion was detected using dihydroethidium (DHE),21 and myocardial expression of the NAD(P)H‐oxidase subunit p47 and gp91 by Wester ‐blotting (both 1:200; Santa Cruz Biotechnology, Santa Cruz, CA).8 Additionally, total ROS burden was determined in LV samples. In brief, ≈50 mg of LV tissue were homogenized by polytron in 1 mL of PBS buffer and centrifuged at 16 099 g for 10 minutes. Clear supernatant was collected and protein determined by Bio‐Rad DC Protein Assay (Bio‐Rad Laboratories, Hercules, CA). A 100‐μL aliquot of the diluted samples in PBS was added into a 96‐well plate followed by 100 μL of SuperSignal West Pico Luminol solution (1 part of Luminol, Product No. 1856136+1 part of Stable Peroxide Solution, Product No. 1856135; Thermo Scientific, Waltham, MA). After the resulting mixture was incubated for 5 minutes at room temperature, luminescence reading was taken by a Tecan Saffire (Tecan Group Ltd., Männedorf, Switzerland). Values are expressed as relative luminescence units (RLU)/mg protein.
Thapsigargin‐sensitive sarcoplasmic reticulum (SR) Ca2+‐ATPase (SERCA‐2a) activity in membrane vesicles was determined at free Ca2+ concentrations ranging from 0.1 to 10 μmol/L in a total assay volume of 200 μL, as described previously.25, 26 Briefly, ≈200 mg of LV powder were homogenized in the presence of protease and phosphatase inhibitors (Sigma‐Aldrich, St. Louis, MO), membrane vesicles isolated from the LV homogenate, and protein determined using the Bio‐Rad DC Protein Assay (Bio‐Rad Laboratories). SERCA‐2a activity was determined in the absence and presence of 1 μmol/L of thapsigargin. In parallel, known concentrations of Pi between 0.1 and 0.5 μmol were run as standard for calculating the amount of Pi released during the enzyme reaction. The difference between the activities assayed in the presence and absence of thapsigargin was considered as the activity of SERCA‐2a associated with the SR. Maximal velocity (Vmax) of SERCA‐2a activity expressed as nmol Pi released/minutes per mg and affinity (K0.5) expressed as μmol/L were calculated.
Protein level of SERCA‐2a (Thermo Scientific), phosphorylated phospholamban (PLB) at serine 16 (pPLB‐S16; Badrilla Ltd., Leeds, UK), total PLB (t‐PLB; Badrilla), total ryanodine receptor (RyR2; Abcam), phosphorylated RyR2 at serine 2808 (p‐RyR2‐S2808; Abcam), and sodium‐calcium exchanger (NCX; Thermo scientific) in LV homogenate was measured by Western blotting. Briefly, LV homogenate was prepared from ≈100 mg LV powder as described previously,27, 28 and protein level was determined by Bio‐Rad DC Protein Assay (Bio‐Rad Laboratories). Approximately 10 to 100 μg of protein of each dog LV sample was separated on 4% to 20% SDS‐polyacrylamide gel (Bio‐Rad Laboratories), and the separated proteins were electrophoretically transferred to a PVDF membrane. Accuracy of the electrotransfer was confirmed by staining the membrane with 0.1% Panacea S dye. For identification of the desired protein, the blot was incubated with the appropriately diluted primary monoclonal or polyclonal antibody specific to each protein, based on the supplier's instructions. Antibody‐binding protein(s) was visualized by autoradiography after treating the blot with HRP‐conjugated secondary antibody (antirabbit) and enhanced chemiluminescence color developing reagents according to the supplier (Thermo Scientific). Band intensity was quantified using a Bio‐Rad GS‐670 imaging densitometer (Bio‐Rad Laboratories). Calsequestrin, a calcium‐binding protein of the SR whose levels remain unaltered during heart failure,25, 29 was used as a control for normalization of all proteins. In all cases, the antibody was in excess over the antigen and the density of each protein band was in the linear scale. Furthermore, immunofluorescence costaining of the mitochondrial membrane through a voltage‐dependent anion channel (VDAC) and the sarcoplasmic‐reticulum marker, RYR, was performed and colocalization assessed with the Mander's coefficient using the National Institutes of Health ImageJ software (version 1.44 for Windows) and the Colocalization Plugin,30 and reported as M1 (overlap between VDAC and RyR).31
In mid‐LV cross‐sections, myocyte cross‐sectional area (hematoxylin and eosin; H&E), interstitial collagen content (Sirius Red), and fibrosis (Masson's trichrome) were assessed using ZEN.32 Myocardial expression of plasminogen activator‐inhibitor (PAI)‐1 (Catalog No.: ab66705; Abcam), tissue‐inhibitor of metalloproteinases (TIMP)‐1 (sc‐5528; Santa Cruz Biotechnology), and transforming growth‐factor (TGF)‐β (sc‐52891; Santa Cruz Biotechnology) was determined by Western blot.8
Cardiolipin Content and Mitochondrial Biogenesis
Mitochondrial content was assessed by immunofluorescence staining with the mitochondrial outer membrane marker preprotein, translocases of the outer membrane (TOM)‐20 (1:50; Santa Cruz Biotechnology, Inc., Dallas, TX).33 Cardiolipin content was assessed by enhanced multidimensional mass spectrometry (MS)‐based shotgun lipidomics34 and immunofluorescence staining (10N‐nonyl acridine‐orange, A1372; Invitrogen, Carlsbad, CA),8 and results adjusted by mitochondrial density (TOM‐20 expression). Intensity and percentage of area stained were quantified semiautomatically in 10 to 15 random fields (ZEN 2012 blue edition; Carl Zeiss SMT, Oberkochen, Germany). For MS, lipids were extracted from the tissue sample with a chloroform/methanol solution (Bligh Dyer extraction). Individual lipid extracts were reconstituted with chloroform:methanol (1:1), flushed with N2, and then stored at −20°C before analysis by electrospray ionization MS using a triple‐quadruple mass spectrometer equipped with an automated nanospray apparatus. Enhanced multidimensional MS‐based shotgun lipidomics for cardiolipin was performed as previously described.34 Cardiolipin contains 2 phosphate heads groups, 3 glycerol moieties, and 4 fatty acid acyl chains. More than 100 molecular fatty acid species of cardiolipin have been identified including shorter chain saturated and monounsaturated fatty acids (C16:0, C18:0, and C18:1) and polyunsaturated fatty acids (C18:2, C20:4, and C22:6).35 In animal species, cardiolipin contains almost exclusively 18‐carbon fatty acids, and 80% of these are typically linoleic acid (tetra‐linoleoyl cardiolipin [C18:2]). MS also detects fragments of other cardiolipin molecular species including fatty acids palmitoleic (C16:1), palmitic (C16:0), oleic (C18:1), stearic (C18:0), and arachidonic (C20:4) acid.36
Additionally, myocardial mRNA expression of taffazin (Taz)‐1 was measured by quantitative real‐time polymerase chain reaction (RT‐PCR) using the delta‐delta CT method with validated TaqMan primers from Life Technologies (Carlsbad, CA) according to the Minimum Information for Publication of Quantitative Real‐Time PCR Experiments (MIQE),19 whereas protein expression of Taz‐1 (Catalog No.: ab105104; Abcam), cardiolipin synthase (CRLS)‐1 (Catalog No.: LS‐B12635; Lifespan Biosciences), and Acyl‐CoA:lysocardiolipin acyltransferase (ALCAT)‐1 (Catalog No.: NBP1‐59347; Novus Biologicals, LLC, Littleton, CO) was measured by Western blot.
In addition, mitochondria were isolated using the MITO‐ISO kit (Catalog No.: 8268; ScienCell, Carlsbad, CA),33 and protein expression of the mitochondrial biogenesis markers peroxisome proliferator‐activated receptor‐γ‐coactivator (PGC)‐1α (1:1000; Abcam), nuclear respiratory factor (NRF)‐1 (1:300; Abcam), GA‐binding protein (GABP; 1:1000; Abcam), and peroxisome proliferator‐activated receptor (PPAR)‐α (1:1000; Abcam) measured by Western blot.21
Microvascular Structure and Coronary Endothelial Function
The proximal left anterior descending artery was cannulated and perfused under physiological pressure with a radiopaque contrast agent (MV‐122; Flow Tech, Carver, MA). An LV transmural portion (2 cm3) was scanned, and images analyzed, as described.32 The spatial density and average diameter of microvessels in the subepicardium and subendocardium were calculated using Analyze, and vessel tortuosity (maturity index) calculated. For tissue analysis, media‐to‐lumen ratio was calculated in randomly selected intramyocardial vessels in α‐SMA‐stained sections (DakoCytomation A/S, Glostrup, Denmark), and myocardial expression of vascular endothelial growth factor (VEGF) measured by Western blot (1:200; Santa Cruz Biotechnology).21
To assess coronary vascular function ex vivo, coronary arteries were dissected immediately after euthanasia (1 ring/animal per chamber), precontracted with endothelin‐1 (10−7 mol/L), and the response to cumulative concentrations of the endothelium‐dependent bradykinin (10−10–10−5 mol/L) or endothelium‐independent sodium nitroprusside (SNP; 10−10–10−4 mol/L) studied. The method has been previously described in detail.37, 38, 39 Coronary artery sections (2–3 mm long) were dissected under a dissecting microscope and placed in 25‐mL organ chambers (1 ring from each animal per chamber) filled with Kreb's solution at 37°C (pH=7.4, 95% O2, and 5% CO2). Each ring was suspended by 2 stainless clips, 1 attached to a stationary post and the other to a strain gauge to measure isometric force (Statham Gould UC 2; Viggo Spectamed, Critical Care Division, Oxnard, CA). Rings were progressively stretched (potassium chloride 20 mmol/L) to achieve the optimal point for their length‐tension relationship. After an equilibration period of 30 minutes, the vessels were precontracted with endothelin‐1 (10−7 mol/L), and then the response to cumulative concentrations of bradykinin (10−10–10−5 mol/L) was recorded to evaluate endothelium dependent relaxation. The same experiments were repeated using increasing concentrations of SNP (10−10 to 10−4 mol/L) as an endothelium‐independent vasodilator. Cardiolipin content (10N‐nonyl‐acridine‐orange), mitochondrial UCP‐2 (Abcam, catalog# ab97931), apoptosis (TUNEL), superoxide anion production (DHE), and endothelial NO synthase (eNOS; Catalog No.: ab5589; Abcam) expression were assessed in coronary artery sections.19
Cardiomyoblast Studies
To affirm the ability of MTP to directly blunt mitochondrial injury, H9c2 rat cardiomyoblast (CM; ATCC CRL‐1446) were grown in staining chamber slides untreated or treated for 4 hours with the MTP, Bendavia, 1 μmol/L and/or tert‐butyl hydroperoxide (tBHP) 25 mmol/L13 (6 wells/group). Cells were cultured and maintained at 37°C in MEM 10% culture media supplemented with 10% FBS, as previously described.40 Then, CM were divided into 4 groups (6 wells per group), which were grown in staining chamber slides untreated or treated with either the MTP, Bendavia, 1 μmol/L, tert‐butyl hydroperoxide (tBHP) 25 mmol/L that causes lipid peroxidation and mitochondrial dysfunction13, 41 or tBHP+MTP for 4 hours. CM were subsequently stained for cardiolipin (10N‐nonyl acridine orange)19, 21 and Mito‐SOX for detection of mitochondrial ROS production. In addition, an XF24 Analyzer (Seahorse Biosciences, North Billerica, MA) was used to measure bioenergetic function in intact CM, CM+MTP, CM+tBHP, and CM+tBHP+MTP in real time. Cells were seeded into Seahorse Bioscience XF24 cell culture plates at a population density of approximately 100 000/well in 250 μL of media and allowed to adhere and grow for 24 hours in a 37°C humidified incubator with 5% CO2. Media were changed 1 hour before the start of the extracellular flux assay to unbuffered (pH 7.4) DMEM containing 2 mmol/L of GlutaMax, 1 mmol/L of sodium pyruvate, and supplemented with 25 mmol/L of glucose and incubated at 37°C without CO2. The XF24 protocol consists of 4 measurements (2 minutes mix/2 minutes measure) of basal oxygen consumption rate (OCR; an indicator of mitochondrial respiration) followed by 3 measures after addition of 9 μmol/L of Oligomycin (ATP uncoupler), 0.3 μmol/L of carbonyl cyanide p‐trifluoromethoxy‐phenylhydrazone (FCCP; electron transport chain accelerator), and a combination of 11 μmol/L of Antimycin‐A (Complex‐III inhibitor) and 11 μmol/L of Rotenone (Complex‐I inhibitor). Basal respiration, ATP production (Basal respiration‐Oligomycin response), Proton leak (Oligomycin response‐Antimycin‐A and Rotenone response), and Maximal respiration (Basal respiration‐non‐mitochondrial respiration) were calculated, following standard procedures.42 On completion of the XF assay, cells were lysed with CellLytic MT lysis reagent (200 μL/well) and protein concentration determined using the Bradford reagent. OCR data are expressed as pmol/minutes per mg of protein.
Statistical Analysis
Statistical analysis was performed using JMP software (version 10.0). Results are expressed as mean±SD or median and interquartile range (IQR) and compared with ANOVA/Kruskal Wallis followed by Student t test/Wilcoxon. Statistical significance was accepted if P≤0.05.
Results
At 16 weeks, all HC‐RVH animals developed significant and similar levels of hypertension (Table). Serum creatinine was higher in HC‐RVH+Vehicle compared to normal, but normalized in HC‐RVH+MTP. PRA levels were similar among the groups. Cholesterol levels were elevated in HC‐RVH, whereas triglycerides were unaltered (Table).
Table 1.
Systemic Characteristics, Renal Hemodynamics, and Function of Study Groups (n=7 Each) at 16 Weeks
| Parameter | Normal+Vehicle | Normal+MTP | HC‐RVH+Vehicle | HC‐RVH+MTP |
|---|---|---|---|---|
| Degree of stenosis (%) | 0 (0–0) | 0 (0–0) | 95.0 (77.5–99.5)a , b | 85.0 (62.5–87.5)a , b |
| Body weight, kg | 49.3±1.5 | 49.8±1.7 | 47.8±1.3 | 48.3±2.8 |
| Mean blood pressure, mm Hg | 78.7 (75.9–82.8) | 81.3 (77.0–84.0) | 123.0 (117.7–125.0)a , b | 123.7 (119.2–127.8)a , b |
| Serum creatinine, mg/dL | 1.4±0.2 | 1.5±0.1 | 1.9±0.2a | 1.5±0.3 |
| Plasma renin activity, ng/mL/h | 0.13±0.07 | 0.13±0.15 | 0.17±0.10 | 0.16±0.08 |
| Cholesterol, mg/dL: Total | 88.0 (86.0–122.0) | 88.5 (79.5–91.5) | 519.0 (426.0–545.0)a , b | 491.0 (443.0–653.0)a , b |
| HDL | 37.0 (28.0–66.0) | 46.0 (41.5–55.0) | 169.0 (147.0–180.0)a , b | 166.0 (154.0–180.0)a , b |
| LDL | 54.0 (47.6–58.0) | 37.3 (33.4–40.8) | 324.6 (298.8–375.0)a , b | 287.4 (213.8–385.0)a , b |
| Triglycerides, mg/dL | 9.0±1.7 | 9.3±4.3 | 6.5±2.9 | 5.8±2.8 |
| Heart rate, bpm | 71.7±9.5 | 77.0±8.1 | 79.8±10.1 | 75.0±2.2 |
| Stroke volume, mL | 42.6.3±5.5 | 44.3±5.9 | 47.2±10.8 | 45.1±5.8 |
| Ejection fraction, % | 58.5±10.0 | 58.1±4.4 | 60.0±7.0 | 60.8±6.1 |
| LVMM, g/kg | 1.7 (1.6–1.8) | 1.6 (1.5–1.7) | 2.5 (1.7–3.2)a , b , c | 1.8 (1.7–1.9)b |
| E/A ratio | 1.2±0.3 | 1.0±0.1 | 0.7±0.3a , c | 1.0±0.2 |
| EDV, mL | 85.0±12.5 | 79.7±10.2 | 68.2±8.4a , c | 83.7±11.4 |
| Myocardial perfusion, mL/min per g | ||||
| Baseline | 0.8 (0.7–1.3) | 0.8 (0.7–0.9) | 0.7 (0.6–0.8)a , c | 0.7 (0.7–0.8) |
| Response to adenosine | 0.9 (0.8–1.5)d | 0.8 (0.8–1.0)d | 0.7 (0.6–0.8)a , b , c | 0.8 (0.7–1.0)d |
E/A indicates early and late left ventricular filling ratio; EDV, end‐diastolic volume; HC‐RVH, hypercholesterolemic renovascular hypertension; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; LVMM, left ventricular muscle mass; MTP, mitochondrial‐targeted peptides.
P<0.05 vs Normal+Vehicle.
P<0.05 vs Normal+MTP.
P<0.05 vs HC‐RVH+MTP.
P<0.05 vs baseline.
Cardiac Oxygenation and Function
Heart rate, stroke volume, and ejection fraction were similar among the groups (Table; P>0.05, ANOVA). LVMM was higher in HC‐RVH+Vehicle compared to normal, but restored in HC‐RVH+MTP. E/A ratio, which decreased in HC‐RVH+Vehicle, was also normalized in HC‐RVH+MTP (P=0.03 vs HC‐RVH+Vehicle; P=0.21 vs normal), as was EDV (P=0.02 vs HC‐RVH+Vehicle, P=0.86 vs normal).
Baseline myocardial perfusion and its response to adenosine were decreased in HC‐RVH+Vehicle, but normalized in HC‐RVH+MTP (Table). Furthermore, baseline R2* was elevated in HC‐RVH+Vehicle compared to normal, suggesting decreased oxygenation, but normalized in HC‐RVH+MTP (Figure 1B). Similarly, R2* response to adenosine was blunted in RVH+Vehicle, but restored to normal levels in HC‐RVH+MTP.
Myocardial Injury and Remodeling
The number of TUNEL‐positive cells was elevated in both HC‐RVH groups, yet lower in HC‐RVH+MTP compared to HC‐RVH+Vehicle (Figure 2A). TUNEL‐positive staining colocalized with connexin‐43, suggesting apoptotic cardiomyocytes. Myocardial expression of Bcl‐xl was upregulated in HC‐RVH+MTP compared to HC‐RVH+Vehicle and to normal pigs (Figure 2B).
Figure 2.
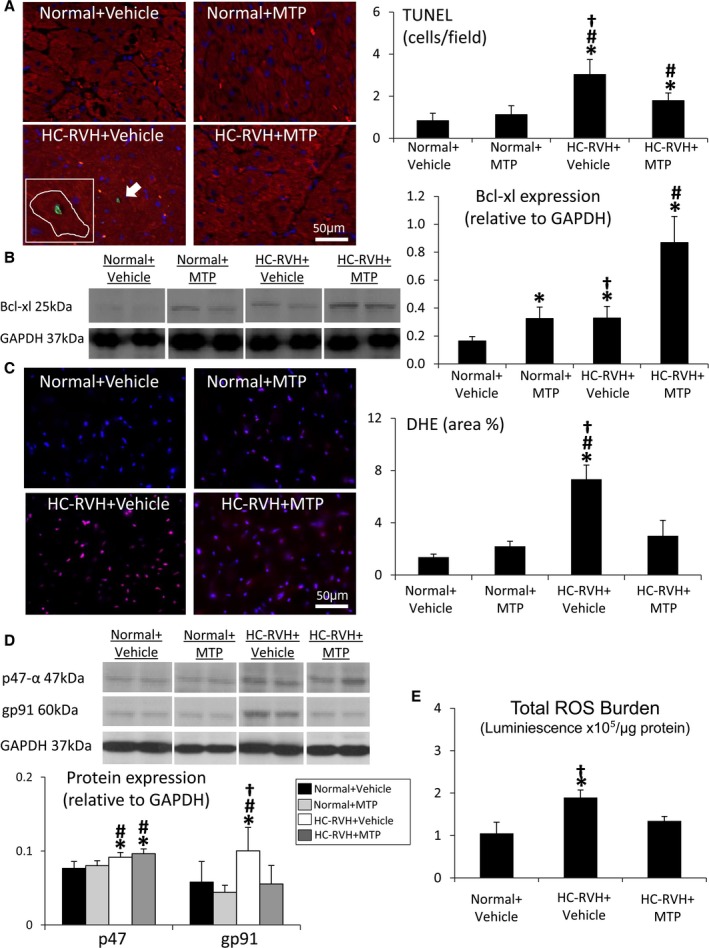
Mitochondrial‐targeted peptides (MTP) attenuate myocardial apoptosis and oxidative stress. A, The number of cells positive for terminal deoxynucleotidyl transferase dUTP nick‐end labeling (TUNEL; green) was elevated in renovascular hypertension (RVH), but decreased in RVH+MTP. Red: connexin‐43. B, Myocardial protein expression of B‐cell lymphoma (Bcl)‐xl was upregulated in RVH+MTP pigs. C, In situ myocardial production of superoxide anion (dihydroethidium; DHE) decreased in RVH pigs treated with MTP. D, Myocardial expression of the nicotinamide adenine dinucleotide phosphate (NADPH)‐oxidase subunits, p47 and gp91, were elevated in RVH, but gp91 expression normalized in RVH+MTP. E, Total reactive oxygen species (ROS) burden was higher in RVH+Vehicle, yet normalized in MTP‐treated RVH. *P<0.05 vs Normal+Vehicle; # P<0.05 vs Normal+MTP; † P<0.05 vs hypercholesterolemic (HC)‐RVH+MTP. GAPDH indicates glyceraldehyde‐3‐phosphate dehydrogenase; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Superoxide anion production was higher in the HC‐RVH myocardium compared to normal, yet restored in HC‐RVH+MTP (Figure 2C). Myocardial expression of p47 and gp91 was elevated in HC‐RVH+Vehicle compared to normal, but only gp91 expression was downregulated in MTP‐treated HC‐RVH (Figure 2D), as was total ROS burden (Figure 2E).
SERCA‐2a protein levels and activity fell in HC‐RVH and its affinity for calcium was lower than normal (higher SERCA‐2a activity K0.5), but both were restored in HC‐RVH+MTP (Figure 3A through C). PLB activation (p‐PLB‐S16/total) decreased in HC‐RVH+Vehicle, but restored in HC‐RVH+MTP (Figure 3D), whereas activation of RYR2 (p‐RYR2/Total RYR2) and NCX protein levels were similar among the groups (Figure 3E through F). Although total VDAC and RYR fluorescence did not differ among the groups, their overlap coefficient (M1) decreased in HC‐RVH groups, but improved in HC‐RVH+MTP (Figure 4).
Figure 3.
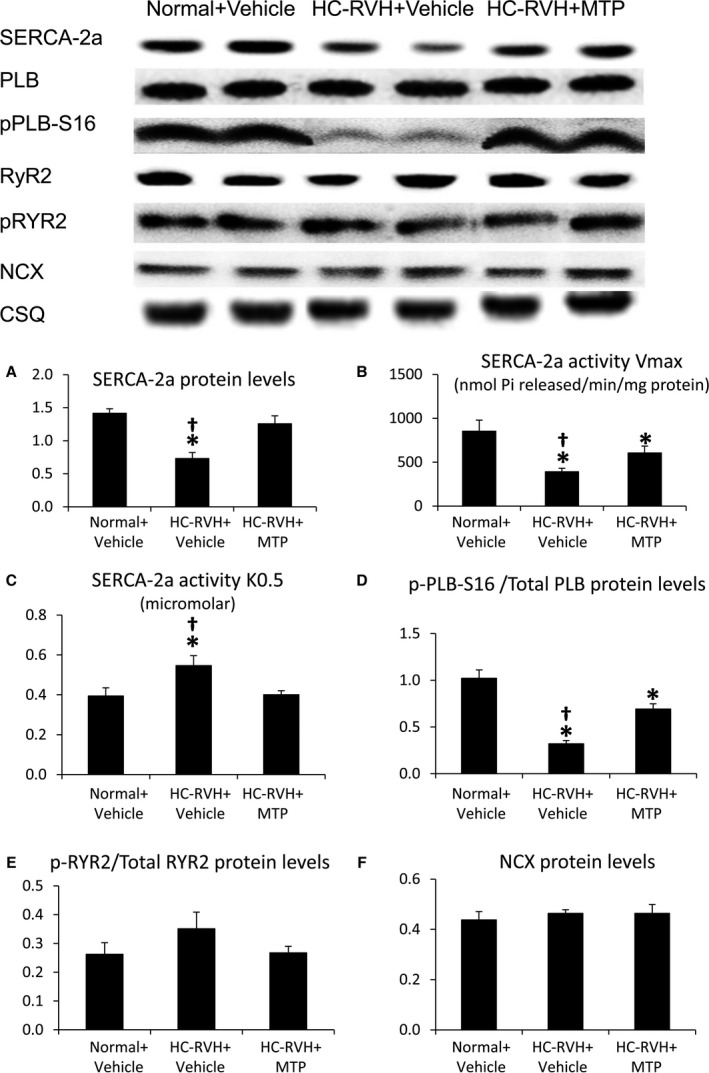
Mitoprotection preserved calcium cycling activity. Thapsigargin‐sensitive sarcoplasmic reticulum (SR) Ca2+‐ATPase (SERCA)‐2a protein levels (A) and activity (B) were reduced in renovascular hypertensive (RVH) pigs, whereas SERCA‐2a activity K0.5 was elevated (C), but all were improved in RVH+Mitochondrial‐targeted peptides (MTP) pigs. Phospholamban (PLB) activation (p‐PLB‐S16/total) decreased in RVH+Vehicle, but restored in RVH+MTP (D), whereas activation of ryanodine receptor (RYR)2 (p‐RYR2/Total‐RYR2) and sodium‐calcium exchanger (NCX) protein levels remained unaltered (E and F). *P<0.05 vs Normal+Vehicle; † P<0.05 vs hypercholesterolemic (HC)‐RVH+MTP.
Figure 4.
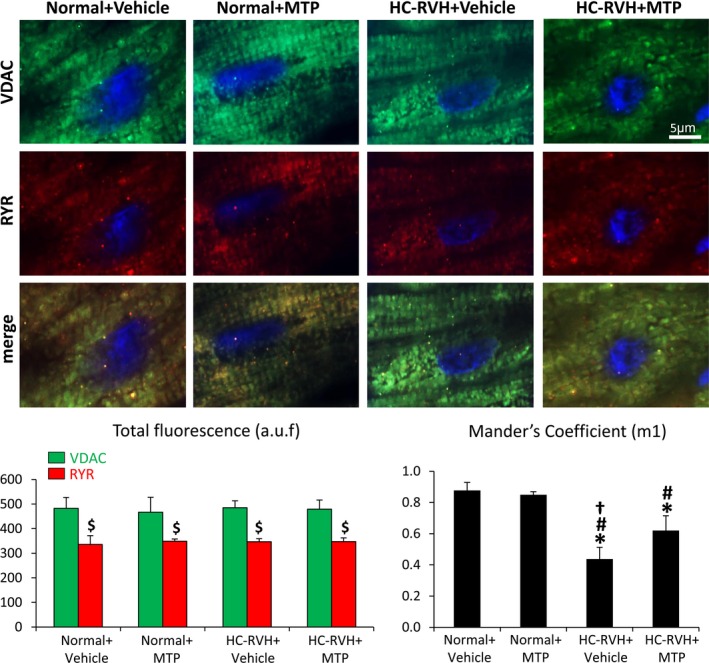
Mitochondrial‐targeted peptides (MTP) improved mitochondria‐sarcoplasmic reticulum functional coupling. Double immunofluorescence of the mitochondrial membrane through a voltage‐dependent anion channel (VDAC) and the sarcoplasmic reticulum marker, ryanodine receptor (RYR, top), and quantification of total VDAC and RYR fluorescence, and Mander's coefficient colocalization. $ P<0.05 vs VDAC; *P<0.05 vs Normal+Vehicle; # P<0.05 vs Normal+MTP; † P<0.05 vs hypercholesterolemic renovascular hypertension (HC‐RVH)+MTP. a.u.f. indicates relative arbitrary units.
Myocyte cross‐sectional area in HC‐RVH was higher than normal, and myocardial collagen deposition (Sirius Red) and fibrosis were increased, but all improved in HC‐RVH+MTP (Figure 5A). Myocardial PAI‐1 expression was similarly upregulated in both HC‐RVH groups, whereas TIMP‐1 and TGF‐β expression improved in HC‐RVH+MTP (Figure 5B).
Figure 5.
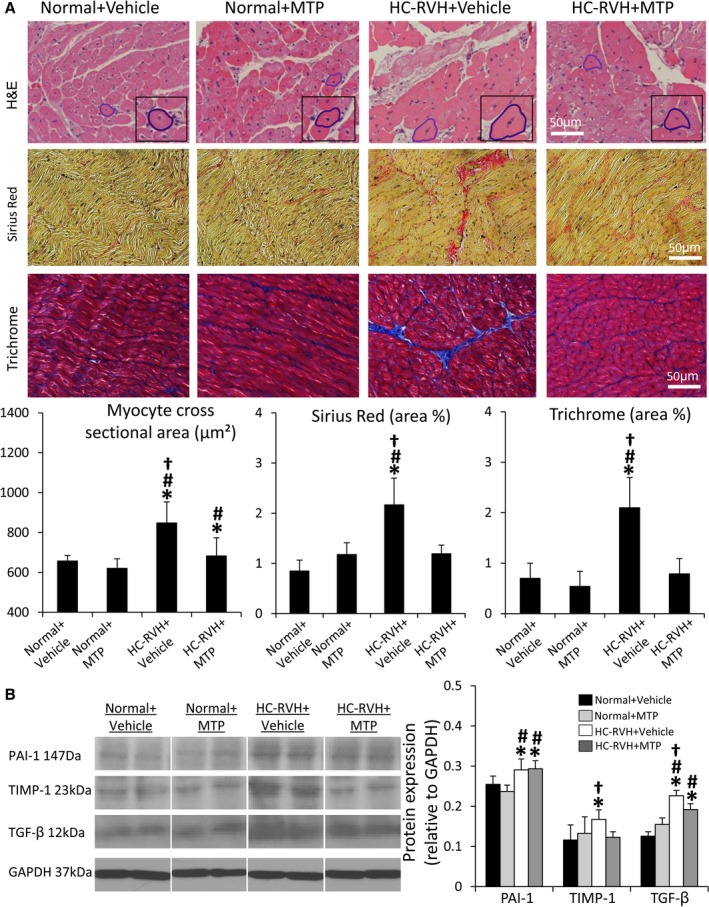
Mitoprotection ameliorates myocardial remodeling and fibrosis in renovascular hypertension (RVH). A, Myocyte cross‐sectional area (hematoxylin and eosin; H&E), collagen deposition (Sirius Red), and fibrosis (Trichrome) were attenuated in mitochondrial‐targeted peptides (MTP)‐treated pigs. B, Myocardial expression of plasminogen activator inhibitor (PAI)‐1 was higher in all RVH pigs, but expression of tissue inhibitor of metalloproteinase (TIMP)‐1 and transforming growth factor (TGF)‐β was decreased in RVH+MTP. *P<0.05 vs Normal+Vehicle; # P<0.05 vs Normal+MTP; † P<0.05 vs hypercholesterolemic (HC)‐RVH+MTP.
Cardiolipin Content and Mitochondrial Biogenesis
Myocardial expression of the outer mitochondrial membrane protein, TOM‐20, was similar among groups, suggesting preserved mitochondrial content. Contrarily, cardiolipin content was decreased in HC‐RVH+Vehicle, but normalized in HC‐RVH+MTP (Figure 6A and 6B). The most prominent molecular species, tetra‐linoleoyl cardiolipin (C18:2) followed by C20:4, both decreased in HC‐RVH+Vehicle, but normalized in HC‐RVH+MTP (Figure 6C), as was mRNA expression of taz‐1 (Figure 6D). Myocardial protein expression of Taz‐1 was similarly blunted in HC‐RVH+Vehicle and HC‐RVH+MTP (Figure 6E), whereas expression of ALCAT‐1 and CRLS‐1 did not differ among the groups. Furthermore, expression of PGC‐1α, decreased in HC‐RVH+Vehicle, was normalized in HC‐RVH+MTP (Figure 7). Although NFR‐1 expression was unchanged, GABP and PPAR‐α expression was downregulated in HC‐RVH+Vehicle, but not in HC‐RVH+MTP.
Figure 6.
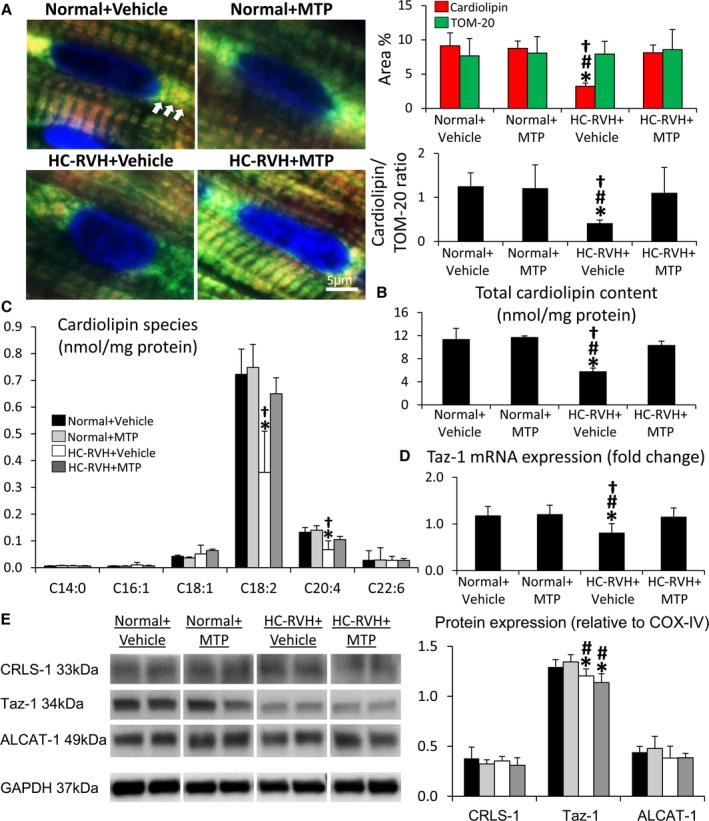
Mitochondrial‐targeted peptides (MTP) restored cardiolipin content. A, Representative immunofluorescent staining of the mitochondrial outer membrane marker pre‐protein, translocases of the outer membrane (TOM)‐20 (green), and cardiolipin (red) showing decreased myocardial cardiolipin expression in renovascular hypertension (RVH) despite preserved mitochondrial content, which was normalized in RVH+MTP. B and C, Total cardiolipin content and its prominent molecular species (mass spectrometry) were restored in MTP‐treated RVH animals. D, Tafazzin (Taz)‐1 mRNA expression was downregulated in RVH, yet normalized in RVH+MTP. E, Myocardial expression of cardiolipin synthase (CRLS)‐1, Taz‐1, and Acyl‐CoA:lysocardiolipin acyltransferase (ALCAT)‐1 in study groups. *P<0.05 vs Normal+Vehicle; # P<0.05 vs Normal+MTP; † P<0.05 vs hypercholesterolemic (HC)‐RVH+MTP.
Figure 7.
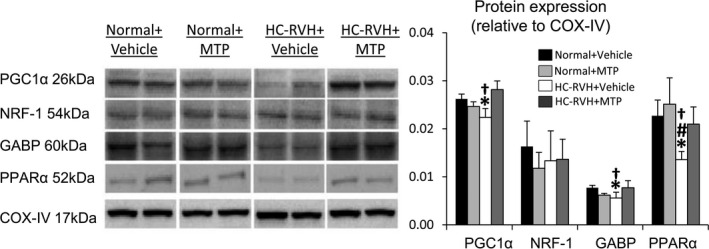
Mitochondrial‐targeted peptides (MTP) induce mitochondrial biogenesis. Myocardial protein expression of the mitochondrial biogenesis markers, peroxisome proliferator‐activated receptor‐γ‐coactivator (PGC)‐1α, nuclear respiratory factor (NFR), GA‐binding protein (GABP), and peroxisome proliferator–activated receptor (PPAR)‐α. *P<0.05 vs Normal+Vehicle; # P<0.05 vs Normal+MTP; † P<0.05 vs hypercholesterolemic (HC)‐renovascular hypertension (RVH)+MTP.
Vascular Structure and Function
Spatial density of subepicardial microvessels was similar among the groups, whereas density of small (0.02–0.02 mm) subendocardial microvessels decreased in HC‐RVH, but improved in HC‐RVH+MTP (Figure 8A). Similarly, transmural vessel tortuosity was higher in both RVH groups compared to normal, but lower in HC‐RVH+MTP pigs (Figure 8B). Myocardial expression of VEGF was upregulated in HC‐RVH+MTP (Figure 8C), whereas vessel wall‐to‐lumen ratio, which was higher in both HC‐RVH groups compared to normal, improved in MTP‐treated pigs (Figure 8D).
Figure 8.
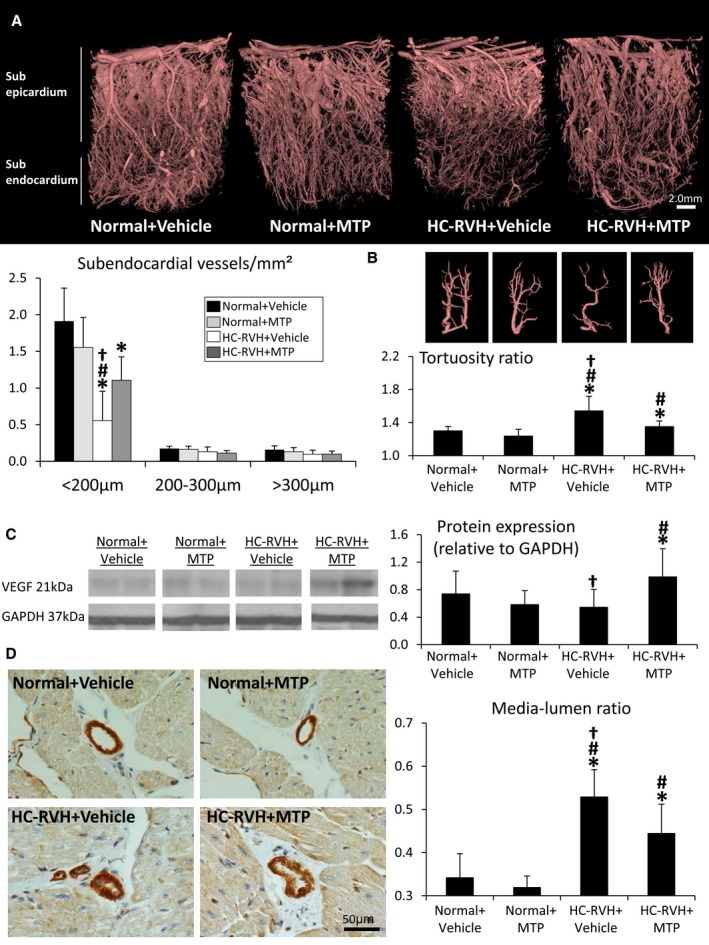
Mitoprotection preserved microvascular structure. A, Representative 3‐dimensional micro‐computed‐tomography images of the left ventricle and quantification of spatial density of subendocardial microvessels. B, Vessel tortuosity was higher in renovascular hypertension (RVH) compared to normal, but slightly decreased in RVH+ Mitochondrial‐targeted peptides (MTP). C, Myocardial protein expression of vascular endothelial growth factor (VEGF) was upregulated in RVH+MTP. D, Myocardial vessel wall‐to‐lumen ratio in α‐smooth muscle actin stained slides. *P<0.05 vs Normal+Vehicle; # P<0.05 vs Normal+MTP; † P<0.05 vs hypercholesterolemic (HC)‐RVH+MTP.
The vasorelaxation response to bradykinin in excised HC‐RVH coronary vessels was attenuated compared with normal, but improved (although not normalized) in HC‐RVH+MTP, whereas response to SNP was unchanged (Figure 9A). Cardiolipin staining in coronary artery sections was blunted in HC‐RVH+Vehicle, but restored in HC‐RVH+MTP (Figure 9B), as were the number of TUNEL‐positive cells and the immunoreactivity of eNOS and UCP‐2. Finally, superoxide anion production was elevated in HC‐RVH+Vehicle, but decreased in HC‐RVH+MTP animals (Figure 9B).
Figure 9.
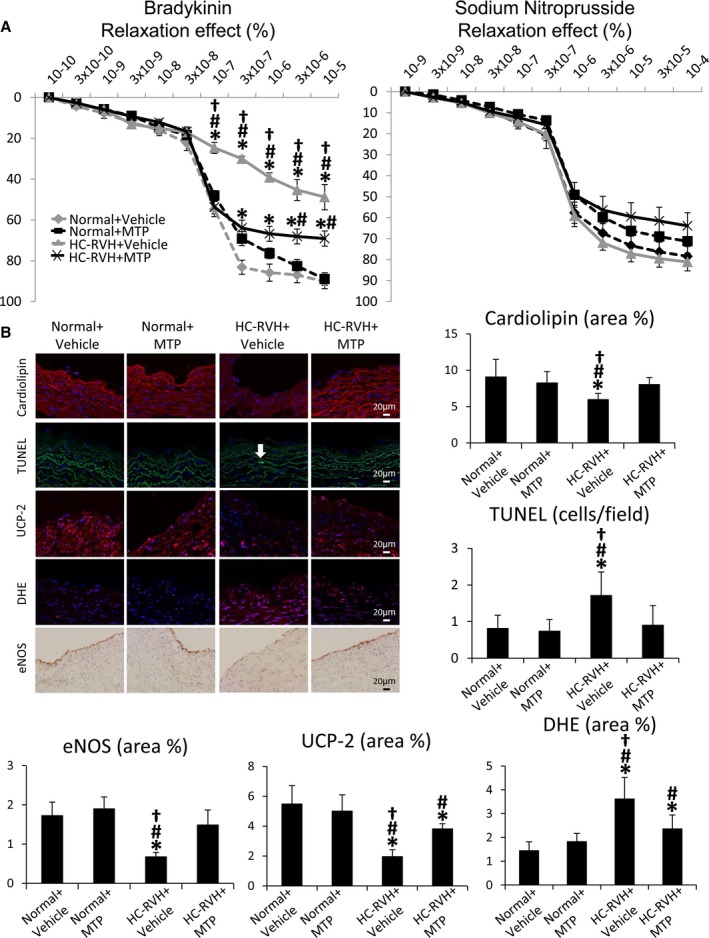
Mitochondrial‐targeted peptides (MTP) improve coronary endothelial function. A, Endothelial‐dependent (left) and ‐independent (right) relaxation of coronary segments from normal and renovascular hypertensive (RVH) pigs 4 weeks after chronic subcutaneous injections of MTP or Vehicle. B, Representative images and quantification of cardiolipin expression (10N‐nonyl‐acridine‐orange), apoptosis (terminal deoxynucleotidyl transferase dUTP nick‐end labeling; TUNEL), uncoupling protein (UCP)‐2 expression, in situ production of superoxide anion (dihydroethidium; DHE), and endothelial nitric oxide synthase (eNOS) immunoreactivity in coronary artery sections. *P<0.05 vs Normal+Vehicle; # P<0.05 vs Normal+MTP; † P<0.05 vs hypercholesterolemic (HC)‐RVH+MTP.
CM Studies
Incubation with tBHP decreased CM cardiolipin expression, which normalized upon coincubation with MTP (Figure 10A). Mitochondrial ROS levels were upregulated in both tBHP‐treated groups, but improved in CM+tBHP+MTP (Figure 10B). Furthermore, basal respiration, ATP production, and maximal respiration were blunted in CM+tBHP, but improved in CM+tBHP+MTP (Figure 10C), whereas proton leak remained unaltered.
Figure 10.
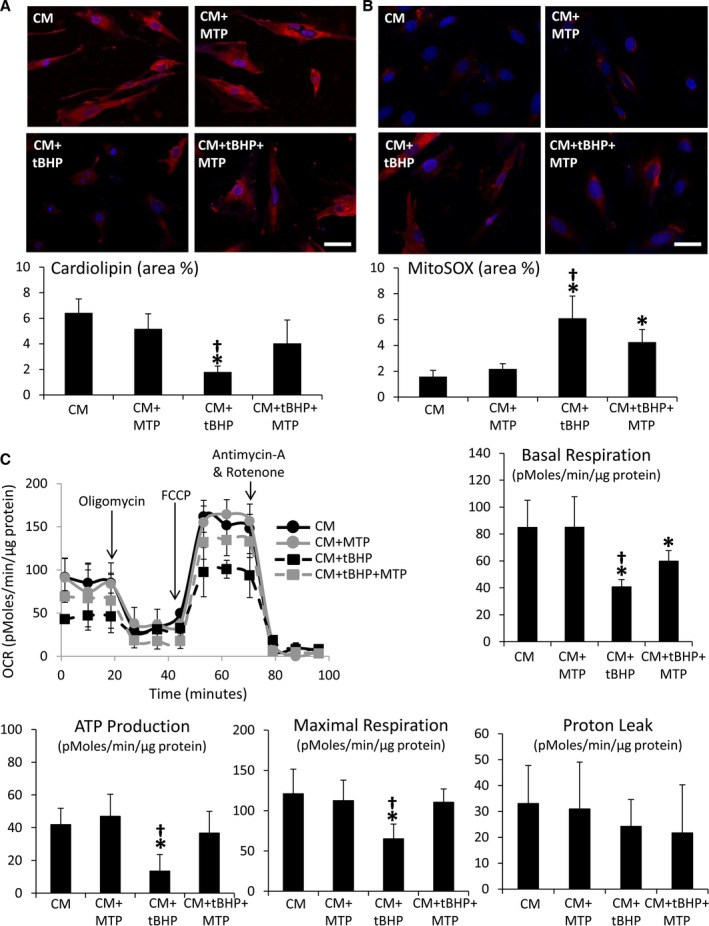
Mitochondrial‐targeted peptides (MTP) improved mitochondrial bioenergetics in cultured cardiomyocytes. Cardiolipin expression decreased (A) and mitochondrial reactive oxygen species production (B) increased in normal cardiomyoblasts (CM) treated with tert‐butyl hydroperoxide (tBHP), but improved in cells coincubated with MTP. C, Representative tracing of oxygen consumption in CM, CM+MTP, CM+tBHP, and CM+tBHP+MTP under basal conditions, and after the addition of Oligomycin (ATP uncoupler), 0.3 μmol/L of carbonyl cyanide p‐trifluoromethoxy‐phenylhydrazone (FCCP, electron transport chain accelerator), and a combination of 11 μmol/L of Antimycin‐A (Complex‐III inhibitor) and 11 μmol/L of Rotenone (Complex‐I inhibitor), and quantification of basal respiration, ATP production, maximal respiration, and proton leak in study groups. *P<0.05 vs CM; # P<0.05 vs CM+MTP; † P<0.05 vs CM+tBHP+MTP.
Discussion
The current study demonstrates that experimental HC‐RVH induces cardiolipin loss, myocardial injury, and impaired LV function, and that hypertension notwithstanding, stabilization of mitochondrial cardiolipin can attenuate these changes. These findings support the concept that myocardial cardiolipin loss partly contributes to hypertension‐induced LV remodeling and impaired relaxation, and that mitochondrial‐targeted peptides can effectively ameliorate them in swine HC‐RVH.
We have previously shown that adjunct short‐term MTP therapy during renal revascularization in swine HC‐RVH improved diastolic function and myocardial tissue integrity, supporting a role for MTP for cardiac protection during regression of HC‐RVH.8 The current study extends our previous observations demonstrating that chronic MTP delivery restored cardiolipin content and cardiac integrity despite sustained RVH.
RVH can initially induce cardiac remodeling associated with isolated LV diastolic dysfunction, increasing cardiovascular morbidity and mortality.43, 44 Patients with RVH exhibit a high prevalence of LV hypertrophy and progressive LV dilatation.45 Indeed, LV remodeling and relaxation are worse in RVH compared with essential hypertensive patients,3 warranting development of preventive strategies in these patients.
Mitochondria comprise one third of cardiomyocyte volume and regulate ATP and calcium homeostasis required for sustaining cardiomyocyte viability and contractility.46 Furthermore, endothelial cell mitochondria modulate intracellular ROS, NO, and calcium, which regulate endothelial cell function.47 Importantly, mitochondria also control cell proliferation and death (apoptosis and necrosis), cellular oxidative stress, and intracellular calcium homeostasis. However, the involvement of mitochondrial injury in RVH‐induced LV dysfunction remains incompletely understood.
In the present study, we found that despite preserved mitochondrial content (TOM‐20 expression), HC‐RVH induced mitochondrial injury, characterized by decreased cardiolipin content, and mRNA and protein expression of its main remodeling enzyme, taz‐1. Contrarily, myocardial expression of ALCAT‐1 and CRLS‐1 remained unaltered, suggesting that HC‐RVH affects primary (rather than salvages) cardiolipin remodeling (ALCAT‐1)48 or de novo synthesis (CRLS‐1).49 The most prominent molecular species that decreased in HC‐RVH+Vehicle was the tetra‐linoleoyl, cardiolipin (C18:2). This contrasts with the increase in C20:4 and C22:6 reported in murine models of heart failure,50, 51 possibly attributed to differences in animal model, disease duration, or interspecies variability. Although MTP improved Taz‐1 mRNA expression, this did not suffice to restore its protein expression, arguing against a major role of MTP in preserving primary cardiolipin remodeling.
Furthermore, loss of cardiolipin impaired mitochondrial biogenesis, reflected in decreased expression of PGC‐1α, a major regulator of mitochondrial biogenesis that activates several transcription factors, including NRF‐1, GABP, and PPAR‐α.52, 53 In our study, although HC‐RVH downregulated PGC‐1α, GABP, and PPAR‐α myocardial expression, NRF‐1 levels remained unchanged, suggesting early changes in expression of mitochondrial biogenesis proteins. Given that cardiolipin content remained unchanged, defects in mitochondrial biogenesis might precede changes in their content. Furthermore, HC‐RVH‐induced primary cardiolipin remodeling may trigger mPTP formation and cytochrome‐c release (Figure 11), promoting cardiomyocyte apoptosis (by TUNEL). Interestingly, HC‐RVH increased myocardial expression of the antiapoptotic, Bcl‐xl, possibly as a compensatory mechanism,54 which did not suffice to alleviate apoptosis. Moreover, mPTP opening favors release of mitochondrial‐ROS to the cytosol, triggering cardiomyocyte oxidative stress (increased total ROS burden, superoxide anion production, and gp91 expression).
Figure 11.
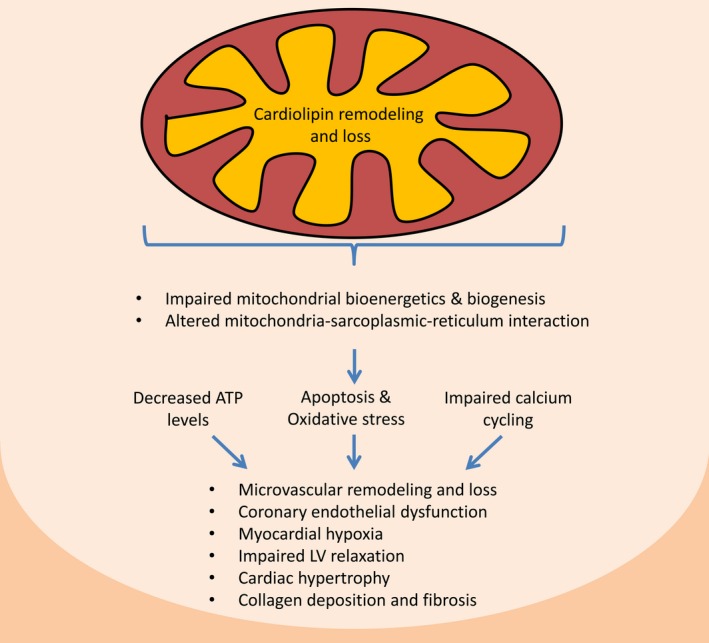
Proposed mechanisms implicated in renovascular hypertension–induced myocardial injury. Cardiolipin remodeling and loss impairs mitochondrial biogenesis and bioenergetics. Furthermore, loss of cardiolipin triggers mitochondria permeability transition pore opening and release of cytochrome‐c and reactive oxygen species to the cytosol, increasing myocardial cell apoptosis and oxidative stress. This instigates microvascular remodeling and endothelial dysfunction, as well as myocardial hypoxia, collagen deposition, and fibrosis. Mitochondrial dysfunction also alters mitochondria‐sarcoplasmic‐reticulum interaction, impairing calcium cycling. Finally, myocardial remodeling and altered sarcoplasmic‐reticulum calcium cycling leads to impaired left ventricular (LV) relaxation.
Furthermore, MTP ameliorated HC‐RVH‐induced cardiomyocyte apoptosis, possibly by preventing mPTP formation and consequent cytochrome‐c release. Inhibition of mPTP opening and increased electron transport chain efficiency might have also prevented mitochondrial ROS release, thereby decreasing myocardial oxidative stress in HC‐RVH+MTP. Nevertheless, myocardial hypoxia and microvascular rarefaction might have also contributed to myocardial oxidative damage in HC‐RVH+Vehicle pigs, and, in turn, damaged the mitochondria, creating a vicious cycle of myocardial injury and oxidative stress.
Taken together, our observations suggest that HC‐RVH‐induced cardiomyopathy involves decreased cardiolipin content and impaired mitochondrial biogenesis, leading to increased myocardial oxidative stress and apoptosis, which can be attenuated by cardiolipin restoration. Furthermore, studies in CM exposed to lipid peroxidation and mitochondrial injury confirmed that loss of cardiolipin increased mitochondrial ROS production and impaired basal respiration, ATP production, and maximal respiration, and that MTP directly attenuates mitochondria‐derived oxidative stress and restores bioenergetics. However, unlike other mitochondria‐targeted compounds, such as mitoQ, mitoTempol, and mitoE, that inhibit proton leak,55 MTP had no effect on proton leak. These observations suggest that mechanisms other than inhibiting mPTP opening, such as improved efficiency of energy production and utilization, might have mediated its cardioprotective effect.
Interestingly, mitoprotection restored intracellular calcium cycling in HC‐RVH. Mitochondria are closely associated to the calcium release sites of the sarcoplasmic reticulum, facilitating calcium entry through the mitochondrial channel VDAC,56 and uncoupled sarcoplasmic reticulum‐mitochondria communication amplifies myocardial oxidative stress.31, 57 The current study applied the Manders overlap coefficient to quantify the degree of overlap and codistributions of mitochondria (VDAC) and sarcoplasmic reticulum (RYR) probes in each pixel of fluorescence staining images. This method examines mathematically the degree to which the variability in red and green co‐occurrence (i.e., for each pixel, both are above background) can be explained with a simple, linear relationship between the two.58 We found that HC‐RVH interferes with mitochondrial and sarcoplasmic reticulum juxtaposition (VDAC and RYR), which MTP improved, preserving myocardial calcium cycling enzymes. Indeed, the fall in myocardial SERCA‐2a and PLB activities and SERCA‐2a expression59 in our HC‐RVH model support the functional MDCT findings of impaired LV relaxation. Importantly, mitochondria can sequester calcium, impacting its cytosolic concentration and modulating the activity of membrane channels and transporters,60 particularly SERCA‐2a.61 Despite sustained hypertension, mitoprotection restored SERCA‐2a levels, activity, and affinity for calcium, and improved p‐PLB‐S16 expression. In contrast, RYR2 remained unaltered, suggesting preserved systolic function, underscoring the early stage of RVH.
Notably, our study implicates mitochondrial injury in the pathogenesis of RVH‐induced myocardial microvascular rarefaction. Mitochondria orchestrate essential aspects of control of vascular tone and angiogenesis by regulating ROS, cell proliferation, apoptosis, and calcium signaling. For example, mitochondrial injury regulates eNOS activation in endothelial cells,62, 63 apoptosis, and endothelial dysfunction,64 underscoring our findings. Therefore, endothelial cell apoptosis and oxidative stress attributed to cardiolipin loss might have accounted for HC‐RVH‐induced microvascular loss and endothelial dysfunction. Furthermore, myocardial apoptosis and oxidative stress modulate microvascular architecture65 and might have thereby affected microvascular remodeling and function. Strikingly, spatial density and morphology of small subendocardial microvessels improved in MTP‐treated HC‐RVH pigs, despite unaffected blood pressure possibly owing to PGC‐1α‐stimulated secretion of VEGF and other angiogenic factors.66
Possibly consequent to impaired angiogenesis and microvascular architecture, myocardial oxygenation, perfusion, and its response to intravenous adenosine were slightly attenuated in HC‐RVH+Vehicle. Given that both perfusion and oxygenation normalized in HC‐RVH+MTP, mitochondrial injury likely contributes to functionally consequent vascular dysfunction. Indeed, chronic mitoprotection improved coronary endothelial function ex vivo, possibly by restoring cardiolipin, decreasing superoxide anion production, improving eNOS immunoreactivity, or other mechanisms regulated by mitochondria of endothelial cells.67 Furthermore, this approach improved expression of the mitochondrial transporter UCP‐2, which regulates mitochondrial ROS formation in ischemia.68
Mitochondrial ROS has been shown to contribute directly to the development of cardiac hypertrophy and fibrosis.69 We found that mitochondrial injury and increased ROS, which exert direct effects on cardiac fibroblasts,70 were associated with upregulated expression of the profibrotic mediators, TGF‐β, PAI‐1, and TIMP‐1. Additionally, myocardial apoptosis, oxidative stress, and hypoxia might have also contributed to the development of fibrosis in our model. Although blood pressure remained elevated, myocardial hypertrophy, fibrosis, and, in turn, LV relaxation and diastolic filling, all improved in HC‐RVH+MTP, underscoring a link between myocardial fibrosis and mitochondrial damage.71 Importantly, expression of TIMP‐1 and TGF‐β, major contributors of myocardial fibrosis,72, 73 decreased in HC‐RVH+MTP.
Our study is limited by the use of relatively young animals and by the short duration of HC‐RVH compared to the human disease. Nevertheless, myocardial structure and function in the swine model mimic those observed in human RVH, providing a unique opportunity to evaluate the role of the mitochondria in a preclinical setting. Additional traditional parameters of LV compliance and diastolic function remain to be assessed in future studies. Some of the benefits observed in the myocardium might have also been secondary to renal improvement.19 However, the unchanged plasma renin activity argues against systemic activation of the renin/angiotensin system. Additionally, some measurements were not available in Normal+MTP animals. Yet, similar to the normal kidney,5, 19 treatment of normal animals with MTP did not affect myocardial structure and function, suggesting that MTP does not measurably affect normal organs. Finally, mitochondrial membrane potential and respiration rate may interfere with cardiolipin staining.74 Therefore, we also assessed cardiolipin content by MS.
In summary, our study demonstrates that HC‐RVH leads to cardiolipin loss and impaired mitochondrial biogenesis, possibly secondary to cellular oxidative stress, which, in turn, mediates cellular and vascular injury. Despite unchanged blood pressure, mitochondrial protection restores cardiolipin levels, improves cardiac relaxation and structure, normalizes calcium cycling activity, and improves coronary vascular remodeling and endothelial function. These findings underscore the vulnerability of the mitochondria and the contribution of mitochondrial injury to RVH‐induced cardiac remodeling and dysfunction. Yet, future studies are needed to establish a cause‐effect relationship and test the cardioprotective properties of MTP as a novel therapeutic strategy to decrease cardiac damage and improve LV relaxation in human RVH.
Sources of Funding
This study was supported by grants from Stealth Biopharmaceuticals, Inc., and from the NIH (DK106427, HL123160, DK104273, HL092621, and DK76308).
Disclosures
Dr. Amir Lerman is a consultant to Stealth Biopharmaceuticals, Inc.
(J Am Heart Assoc. 2016;5:e003118 doi: 10.1161/JAHA.115.003118)
References
- 1. Chrysochou C, Kalra PA. Atherosclerotic renovascular disease and the heart. J Ren Care. 2010;36(suppl 1):146–153. [DOI] [PubMed] [Google Scholar]
- 2. Green D, Kalra PA. The heart in atherosclerotic renovascular disease. Front Biosci (Elite Ed). 2012;4:856–864. [DOI] [PubMed] [Google Scholar]
- 3. Khangura KK, Eirin A, Kane GC, Misra S, Textor SC, Lerman A, Lerman LO. Cardiac function in renovascular hypertensive patients with and without renal dysfunction. Am J Hypertens. 2014;27:445–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eirin A, Lerman A, Lerman LO. Mitochondrial injury and dysfunction in hypertension‐induced cardiac damage. Eur Heart J. 2014;35:3258–3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eirin A, Lerman A, Lerman LO. Mitochondria: a pathogenic paradigm in hypertensive renal disease. Hypertension. 2015;65:264–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dai DF, Chen T, Wanagat J, Laflamme M, Marcinek DJ, Emond MJ, Ngo CP, Prolla TA, Rabinovitch PS. Age‐dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell. 2010;9:536–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arnaudeau S, Kelley WL, Walsh JV Jr, Demaurex N. Mitochondria recycle Ca(2+) to the endoplasmic reticulum and prevent the depletion of neighboring endoplasmic reticulum regions. J Biol Chem. 2001;276:29430–29439. [DOI] [PubMed] [Google Scholar]
- 8. Eirin A, Williams BJ, Ebrahimi B, Zhang X, Crane JA, Lerman A, Textor SC, Lerman LO. Mitochondrial targeted peptides attenuate residual myocardial damage after reversal of experimental renovascular hypertension. J Hypertens. 2014;32:154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Topal G, Brunet A, Walch L, Boucher JL, David‐Dufilho M. Mitochondrial arginase ii modulates nitric‐oxide synthesis through nonfreely exchangeable l‐arginine pools in human endothelial cells. J Pharmacol Exp Ther. 2006;318:1368–1374. [DOI] [PubMed] [Google Scholar]
- 10. Tian XY, Wong WT, Xu A, Lu Y, Zhang Y, Wang L, Cheang WS, Wang Y, Yao X, Huang Y. Uncoupling protein‐2 protects endothelial function in diet‐induced obese mice. Circ Res. 2012;110:1211–1216. [DOI] [PubMed] [Google Scholar]
- 11. Cho YE, Basu A, Dai A, Heldak M, Makino A. Coronary endothelial dysfunction and mitochondrial reactive oxygen species in type 2 diabetic mice. Am J Physiol Cell Physiol. 2013;305:C1033–C1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schleicher M, Shepherd BR, Suarez Y, Fernandez‐Hernando C, Yu J, Pan Y, Acevedo LM, Shadel GS, Sessa WC. Prohibitin‐1 maintains the angiogenic capacity of endothelial cells by regulating mitochondrial function and senescence. J Cell Biol. 2008;180:101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, Seshan SV, Pardee JD, Szeto HH. The mitochondrial‐targeted compound SS‐31 re‐energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol. 2013;24:1250–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol. 2011;192:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Szeto HH. First‐in‐class cardiolipin‐protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 2014;171:2029–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rytomaa M, Kinnunen PK. Evidence for two distinct acidic phospholipid‐binding sites in cytochrome c. J Biol Chem. 1994;269:1770–1774. [PubMed] [Google Scholar]
- 17. Paradies G, Petrosillo G, Paradies V, Ruggiero FM. Mitochondrial dysfunction in brain aging: role of oxidative stress and cardiolipin. Neurochem Int. 2011;58:447–457. [DOI] [PubMed] [Google Scholar]
- 18. Kloner RA, Hale SL, Dai W, Gorman RC, Shuto T, Koomalsingh KJ, Gorman JH III, Sloan RC, Frasier CR, Watson CA, Bostian PA, Kypson AP, Brown DA. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria‐targeting cytoprotective peptide. J Am Heart Assoc. 2012;1:e001644 doi:10.1161/JAHA.112.001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eirin A, Ebrahimi B, Zhang X, Zhu XY, Woollard JR, He Q, Textor SC, Lerman A, Lerman LO. Mitochondrial protection restores renal function in swine atherosclerotic renovascular disease. Cardiovasc Res. 2014;103:461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chade AR, Zhu X, Mushin OP, Napoli C, Lerman A, Lerman LO. Simvastatin promotes angiogenesis and prevents microvascular remodeling in chronic renal ischemia. FASEB J. 2006;20:1706–1708. [DOI] [PubMed] [Google Scholar]
- 21. Eirin A, Li Z, Zhang X, Krier JD, Woollard JR, Zhu XY, Tang H, Herrmann SM, Lerman A, Textor SC, Lerman LO. A mitochondrial permeability transition pore inhibitor improves renal outcomes after revascularization in experimental atherosclerotic renal artery stenosis. Hypertension. 2012;60:1242–1249. [DOI] [PubMed] [Google Scholar]
- 22. Ebrahimi B, Gloviczki M, Woollard JR, Crane JA, Textor SC, Lerman LO. Compartmental analysis of renal bold MRI data: introduction and validation. Invest Radiol. 2012;47:175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rodriguez‐Porcel M, Lerman A, Herrmann J, Schwartz RS, Sawamura T, Condorelli M, Napoli C, Lerman LO. Hypertension exacerbates the effect of hypercholesterolemia on the myocardial microvasculature. Cardiovasc Res. 2003;58:213–221. [DOI] [PubMed] [Google Scholar]
- 24. Urbieta Caceres VH, Lin J, Zhu XY, Favreau FD, Gibson ME, Crane JA, Lerman A, Lerman LO. Early experimental hypertension preserves the myocardial microvasculature but aggravates cardiac injury distal to chronic coronary artery obstruction. Am J Physiol Heart Circ Physiol. 2011;300:H693–H701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gupta RC, Shimoyama H, Tanimura M, Nair R, Lesch M, Sabbah HN. Sr Ca(2+)‐atpase activity and expression in ventricular myocardium of dogs with heart failure. Am J Physiol. 1997;273:H12–H18. [DOI] [PubMed] [Google Scholar]
- 26. Lowry OH, Lopez JA. The determination of inorganic phosphate in the presence of labile phosphate esters. J Biol Chem. 1946;162:421–428. [PubMed] [Google Scholar]
- 27. Gupta RC, Mishra S, Yang XP, Sabbah HN. Reduced inhibitor 1 and 2 activity is associated with increased protein phosphatase type 1 activity in left ventricular myocardium of one‐kidney, one‐clip hypertensive rats. Mol Cell Biochem. 2005;269:49–57. [DOI] [PubMed] [Google Scholar]
- 28. Mishra S, Gupta RC, Tiwari N, Sharov VG, Sabbah HN. Molecular mechanisms of reduced sarcoplasmic reticulum Ca(2+) uptake in human failing left ventricular myocardium. J Heart Lung Transplant. 2002;21:366–373. [DOI] [PubMed] [Google Scholar]
- 29. Gupta RC, Mishra S, Mishima T, Goldstein S, Sabbah HN. Reduced sarcoplasmic reticulum Ca(2+)‐uptake and expression of phospholamban in left ventricular myocardium of dogs with heart failure. J Mol Cell Cardiol. 1999;31:1381–1389. [DOI] [PubMed] [Google Scholar]
- 30. Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. [DOI] [PubMed] [Google Scholar]
- 31. Fernandez‐Sanz C, Ruiz‐Meana M, Miro‐Casas E, Nunez E, Castellano J, Loureiro M, Barba I, Poncelas M, Rodriguez‐Sinovas A, Vazquez J, Garcia‐Dorado D. Defective sarcoplasmic reticulum‐mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis. 2014;5:e1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eirin A, Zhu XY, Ferguson CM, Riester SM, van Wijnen AJ, Lerman A, Lerman LO. Intra‐renal delivery of mesenchymal stem cells attenuates myocardial injury after reversal of hypertension in porcine renovascular disease. Stem Cell Res Ther. 2015;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang X, Li ZL, Crane JA, Jordan KL, Pawar AS, Textor SC, Lerman A, Lerman LO. Valsartan regulates myocardial autophagy and mitochondrial turnover in experimental hypertension. Hypertension. 2014;64:87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Palacios R, Merchante N, Macias J, Gonzalez M, Castillo J, Ruiz J, Marquez M, Gomez‐Mateos J, Pineda JA, Santos J. Incidence of and risk factors for insulin resistance in treatment‐naive HIV‐infected patients 48 weeks after starting highly active antiretroviral therapy. Antivir Ther. 2006;11:529–535. [PubMed] [Google Scholar]
- 35. Ta NL, Jia X, Kiebish M, Seyfried TN. Autosomal dominant inheritance of brain cardiolipin fatty acid abnormality in VM/DK mice: association with hypoxic‐induced cognitive insensitivity. Lipids. 2014;49:113–117. [DOI] [PubMed] [Google Scholar]
- 36. Valianpour F, Wanders RJA, Barth PG, Overmars H, van Gennip AH. Quantitative and compositional study of cardiolipin in platelets by electrospray ionization mass spectrometry: application for the identification of barth syndrome patients. Clin Chem. 2002;48:1390–1397. [PubMed] [Google Scholar]
- 37. Wilson SH, Simari RD, Best PJ, Peterson TE, Lerman LO, Aviram M, Nath KA, Holmes DR Jr, Lerman A. Simvastatin preserves coronary endothelial function in hypercholesterolemia in the absence of lipid lowering. Arterioscler Thromb Vasc Biol. 2001;21:122–128. [DOI] [PubMed] [Google Scholar]
- 38. Wilson SH, Best PJ, Lerman LO, Holmes DR Jr, Richardson DM, Lerman A. Enhanced coronary vasoconstriction to oxidative stress product, 8‐epi‐prostaglandinf2 alpha, in experimental hypercholesterolemia. Cardiovasc Res. 1999;44:601–607. [DOI] [PubMed] [Google Scholar]
- 39. Herrmann J, Lerman LO, Rodriguez‐Porcel M, Holmes DR Jr, Richardson DM, Ritman EL, Lerman A. Coronary vasa vasorum neovascularization precedes epicardial endothelial dysfunction in experimental hypercholesterolemia. Cardiovasc Res. 2001;51:762–766. [DOI] [PubMed] [Google Scholar]
- 40. Aly A, Peterson K, Lerman A, Lerman L, Rodriguez‐Porcel M. Role of oxidative stress in hypoxia preconditioning of cells transplanted to the myocardium: a molecular imaging study. J Cardiovasc Surg (Torino). 2011;52:579–585. [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao K, Luo G, Giannelli S, Szeto HH. Mitochondria‐targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert‐butyl hydroperoxide in neuronal cell lines. Biochem Pharmacol. 2005;70:1796–1806. [DOI] [PubMed] [Google Scholar]
- 42. Aravamudan B, Kiel A, Freeman M, Delmotte P, Thompson M, Vassallo R, Sieck GC, Pabelick CM, Prakash YS. Cigarette smoke‐induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2014;306:L840–L854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296–1305. [DOI] [PubMed] [Google Scholar]
- 44. Edwards MS, Craven TE, Burke GL, Dean RH, Hansen KJ. Renovascular disease and the risk of adverse coronary events in the elderly: a prospective, population‐based study. Arch Intern Med. 2005;165:207–213. [DOI] [PubMed] [Google Scholar]
- 45. Wright JR, Shurrab AE, Cooper A, Kalra PR, Foley RN, Kalra PA. Progression of cardiac dysfunction in patients with atherosclerotic renovascular disease. QJM. 2009;102:695–704. [DOI] [PubMed] [Google Scholar]
- 46. Goffart S, von Kleist‐Retzow JC, Wiesner RJ. Regulation of mitochondrial proliferation in the heart: power‐plant failure contributes to cardiac failure in hypertrophy. Cardiovasc Res. 2004;64:198–207. [DOI] [PubMed] [Google Scholar]
- 47. Szewczyk A, Jarmuszkiewicz W, Koziel A, Sobieraj I, Nobik W, Lukasiak A, Skup A, Bednarczyk P, Drabarek B, Dymkowska D, Wrzosek A, Zablocki K. Mitochondrial mechanisms of endothelial dysfunction. Pharmacol Rep. 2015;67:704–710. [DOI] [PubMed] [Google Scholar]
- 48. Li J, Romestaing C, Han X, Li Y, Hao X, Wu Y, Sun C, Liu X, Jefferson LS, Xiong J, Lanoue KF, Chang Z, Lynch CJ, Wang H, Shi Y. Cardiolipin remodeling by alcat1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 2010;12:154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sakamoto T, Inoue T, Otomo Y, Yokomori N, Ohno M, Arai H, Nakagawa Y. Deficiency of cardiolipin synthase causes abnormal mitochondrial function and morphology in germ cells of caenorhabditis elegans. J Biol Chem. 2012;287:4590–4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moulin M, Solgadi A, Veksler V, Garnier A, Ventura‐Clapier R, Chaminade P. Sex‐specific cardiac cardiolipin remodelling after doxorubicin treatment. Biol Sex Differ. 2015;6:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sparagna GC, Johnson CA, McCune SA, Moore RL, Murphy RC. Quantitation of cardiolipin molecular species in spontaneously hypertensive heart failure rats using electrospray ionization mass spectrometry. J Lipid Res. 2005;46:1196–1204. [DOI] [PubMed] [Google Scholar]
- 52. Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold‐inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. [DOI] [PubMed] [Google Scholar]
- 53. Virbasius JV, Scarpulla RC. Activation of the human mitochondrial transcription factor a gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc Natl Acad Sci USA. 1994;91:1309–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Janumyan YM, Sansam CG, Chattopadhyay A, Cheng N, Soucie EL, Penn LZ, Andrews D, Knudson CM, Yang E. Bcl‐xL/Bcl‐2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. EMBO J. 2003;22:5459–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD. Mitochondrial proton and electron leaks. Essays Biochem. 2010;47:53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Franzini‐Armstrong C. Er‐mitochondria communication. How privileged? Physiology (Bethesda). 2007;22:261–268. [DOI] [PubMed] [Google Scholar]
- 57. Kohlhaas M, Maack C. Adverse bioenergetic consequences of Na+‐Ca2+ exchanger‐mediated Ca2+ influx in cardiac myocytes. Circulation. 2010;122:2273–2280. [DOI] [PubMed] [Google Scholar]
- 58. Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol. 2011;300:C723–C742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Marks AR. Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Invest. 2013;123:46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Demaurex N, Poburko D, Frieden M. Regulation of plasma membrane calcium fluxes by mitochondria. Biochem Biophys Acta. 2009;1787:1383–1394. [DOI] [PubMed] [Google Scholar]
- 61. De Marchi U, Castelbou C, Demaurex N. Uncoupling protein 3 (UCP3) modulates the activity of sarco/endoplasmic reticulum Ca2+‐atpase (SERCA) by decreasing mitochondrial ATP production. J Biol Chem. 2011;286:32533–32541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lugus JJ, Ngoh GA, Bachschmid MM, Walsh K. Mitofusins are required for angiogenic function and modulate different signaling pathways in cultured endothelial cells. J Mol Cell Cardiol. 2011;51:885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Csiszar A, Labinskyy N, Pinto JT, Ballabh P, Zhang H, Losonczy G, Pearson K, de Cabo R, Pacher P, Zhang C, Ungvari Z. Resveratrol induces mitochondrial biogenesis in endothelial cells. Am J Physiol Heart Circ Physiol. 2009;297:H13–H20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pangare M, Makino A. Mitochondrial function in vascular endothelial cell in diabetes. J Smooth Muscle Res. 2012;48:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhu XY, Daghini E, Chade AR, Rodriguez‐Porcel M, Napoli C, Lerman A, Lerman LO. Role of oxidative stress in remodeling of the myocardial microcirculation in hypertension. Arterioscler Thromb Vac Biol. 2006;26:1746–1752. [DOI] [PubMed] [Google Scholar]
- 66. Arany Z, Foo SY, Ma Y, Ruas JL, Bommi‐Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A, Spiegelman BM. HIF‐independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC‐1alpha. Nature. 2008;451:1008–1012. [DOI] [PubMed] [Google Scholar]
- 67. Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res. 2013;112:1171–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xu MJ, Song P, Shirwany N, Liang B, Xing J, Viollet B, Wang X, Zhu Y, Zou MH. Impaired expression of uncoupling protein 2 causes defective postischemic angiogenesis in mice deficient in AMP‐activated protein kinase alpha subunits. Arterioscler Thromb Vac Biol. 2011;31:1757–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves‐Cintron M, Chen T, Marcinek DJ, Dorn GW II, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS. Mitochondrial oxidative stress mediates angiotensin II‐induced cardiac hypertrophy and Galphaq overexpression‐induced heart failure. Circ Res. 2011;108:837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol. 2001;280:C53–C60. [DOI] [PubMed] [Google Scholar]
- 71. Joshi M, Kotha SR, Malireddy S, Selvaraju V, Satoskar AR, Palesty A, McFadden DW, Parinandi NL, Maulik N. Conundrum of pathogenesis of diabetic cardiomyopathy: role of vascular endothelial dysfunction, reactive oxygen species, and mitochondria. Mol Cell Biochem. 2014;386:233–249. [DOI] [PubMed] [Google Scholar]
- 72. Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, Imaizumi T. Transforming growth factor‐beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure‐overloaded rats. Circulation. 2002;106:130–135. [DOI] [PubMed] [Google Scholar]
- 73. Lopez B, Gonzalez A, Diez J. Role of matrix metalloproteinases in hypertension‐associated cardiac fibrosis. Curr Opin Nephrol Hypertens. 2004;13:197–204. [DOI] [PubMed] [Google Scholar]
- 74. Jacobson J, Duchen MR, Heales SJ. Intracellular distribution of the fluorescent dye nonyl acridine orange responds to the mitochondrial membrane potential: implications for assays of cardiolipin and mitochondrial mass. J Neurochem. 2002;82:224–233. [DOI] [PubMed] [Google Scholar]