

Abstract
The traditional view of protein-ligand binding treats a protein as comprising distinct binding epitopes on the surface of a degenerate structural scaffold, largely ignoring the impact of a protein’s energy landscape. To determine the robustness of this simplification, we compared two small helix-turn-helix transcription factors with different energy landscapes. λ-repressor is stable and well folded, while MarA appears to be marginally stable with multiple native conformations (molten). While λ-repressor is known to tolerate any hydrophobic mutation in the core, we find MarA drastically less tolerant to core mutation. Moreover, core mutations in MarA (distant from the DNA-binding interface) change the relative affinities of its binding partners, altering ligand specificity. These results can be explained by taking into account the effects of mutations on the entire energy landscape and not just the native state. Thus, for proteins with multiple conformations that are close in energy, such as many intrinsically disordered proteins, residues distant from the active site can alter both binding affinity and specificity.
Keywords: ligand binding, hydrophobic core, DNA binding, energy landscape
Graphical Abstract
Introduction
The informational content of a particular residue is a way of describing the degree to which a particular amino acid or sidechain contributes to a protein’s function. Sites of high informational content are more restrictive in terms of tolerable substitutions or mutations. Residues responsible for ligand binding are typically thought about in terms of their direct interactions with the ligand. Such clusters form local regions of high informational content that are superimposed upon the structural scaffold. The majority of binding energy is proposed to come from a minority of residues that function as binding hot spots[1]; these are often treated as functionally separable from an over-determined protein core with loose sequence requirements[2,3].
This assumption of separability between binding and fold has been hugely important for studies in protein design, engineering, and related areas. Techniques like protein grafting[4–6] explicitly treat proteins as collections of distinct modules that can be recombined to create new functions. Work investigating the evolution of protein folds or structure has also frequently been based on the assumption that a protein’s general fold can be treated as a scaffold for the active site, largely independent of the identity of active site residues[7,8]. Even the use of alanine-scanning mutagenesis to identify sites for biochemical study or drug design[9] works best for proteins that can be effectively modeled by separable functional epitopes. Many of these techniques rely on the ability to screen a large number of candidates, both in silico and via experimental means. The potential for engineering enzymes with new functions or new inhibitors for medically relevant targets would be greatly enhanced if we could better predict which residues are information rich and which are not.
One obvious limitation to this assumption of separability is that the native structure does not provide the entire picture. Proteins are not static; under native conditions they populate an ensemble of conformations, including unfolded conformations, according to their relative stabilities. Any experiment that measures binding will also be affected by the equilibrium between binding competent and binding incompetent conformations. Thus, it is more accurate to talk about an apparent dissociation constant, Kd,app = Kd ( 1 + Kf ), that incorporates both folding and binding energetics. This scenario is further complicated for proteins with promiscuity in binding targets, as these proteins are likely to have an ensemble of native conformations, all close in energy; interactions with each substrate may be differentially affected by mutation. Although this feature has been acknowledged, its exploitation or circumvention has, with few exceptions[10], remained largely elusive.
Here we characterize how two DNA-binding proteins with different energy landscapes – that is, differences in the relative stability and accessibility of all potential conformations[11] – vary in their abilities to modulate binding via residues distant from the binding site. Additionally, we investigate the degree to which differences in their energy landscapes affect ligand binding and specificity.
MarA and λ-repressor’s DNA-binding domain are both small helix-turn-helix transcription factors with very different energy landscapes (Fig. 1A). For both, the binding interface geometry and binding sites have been well documented. λ-repressor function is to bind to three different sites, all with sequences quite similar to one another[12]. It has a well-defined native conformation of archetypal stability (4.82 kcal/mol)[13]. MarA, on the other hand, regulates expression of over 60 genes[14] in vivo, with different genes regulated by different binding-site (marbox) permutations at their promoters. Marboxes are fairly degenerate; MarA is quite sensitive to their orientation and location relative to the transcriptional start site as well as the few sequence changes and thus binds different promoters with varying affinity[15]. Structural information about MarA in complex with DNA is available from both X-ray crystallography[16] and NMR[17,18], and its sequence logo has been experimentally determined[19]. In order to bind such a diverse array of sequences, MarA must have access to many similar but slightly reoriented conformation. In the absence of ligand, however, MarA is insoluble, suggesting that it is largely unfolded in isolation. Taken together, these data suggest that MarA is marginally stable with multiple native conformations and therefore in the absence of ligand its ground state may be best described as molten.
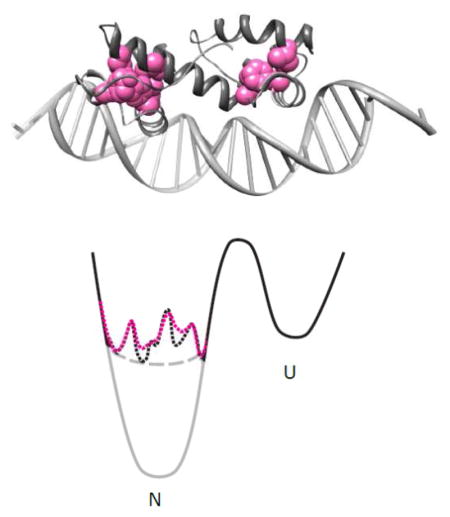
Figure 1.

A) Two-dimensional representations of idealized energy landscapes for a well-folded protein with a single native ground state (solid line), a marginally stable protein with a defined native state (dashed line), and a marginally stable protein with multiple minima (dotted line). B) Representation of the crystal structure of MarA binding to DNA. Cluster 1 residues I13 (76%), F48 (96%), and L56 (59%) are indicated in green; cluster 2 residues I68 (20%) and L94 (20%), in cyan; cluster 3 residues L72 (99%) and Y109 (75%), in red. F98 (98%), which is included in both clusters 2 and 3, is indicated in purple[16,48]. Numbers in parentheses after residue number indicate degree of conservation among homologues.
In this work, we show that MarA, in contrast to λ-repressor, is largely intolerant to mutations within the hydrophobic core. Furthermore, we show that these mutations effect differential changes in binding affinity for different MarA ligands. We use a simple two-state thermodynamic model to show that these results can be used to explain the general feature that if a protein is on the brink of stability, then mutations throughout the entire protein will have dramatic effects on function, e.g. ligand binding; sites distant from the active site play a role that goes beyond defining the fold. Moreover, a single site substitution within the hydrophobic core, distant from the binding site, is sufficient to alter binding specificity for promiscuous proteins (proteins with many high-affinity binding partners).
Results
Library Construction
To probe the functional impact of changes to the energy landscape, we created three libraries of MarA variants by changing residues within the hydrophobic core, distant from the ligand-binding surface. Each library was randomized at three clustered positions within the core, defined as comprising residues that are 95% or more solvent inaccessible (Fig. 1B): I13, F48, and L56 comprise cluster 1; I68, L94, and F98, cluster 2; and L72, F98, and Y109, cluster 3. The core positions chosen are diverse both in their location within the protein and in their conservation among homologues (20 – 99%); none contact DNA.
To confirm unbiased library compositions, the individual libraries were evaluated for their sequence diversity. Each library was transformed into cells and grown in the absence of MarA functional selection; 86 – 94 transformants from each library were then sequenced. The results were compared to a simulation of the above protocol; the simulation was performed with the assumption that there is no bias for any codon over another and repeated 1000 times (Fig. 2A). Our experimental data correspond with the results of the simulation, consistent with the presence of a diverse and unbiased library.
Figure 2.
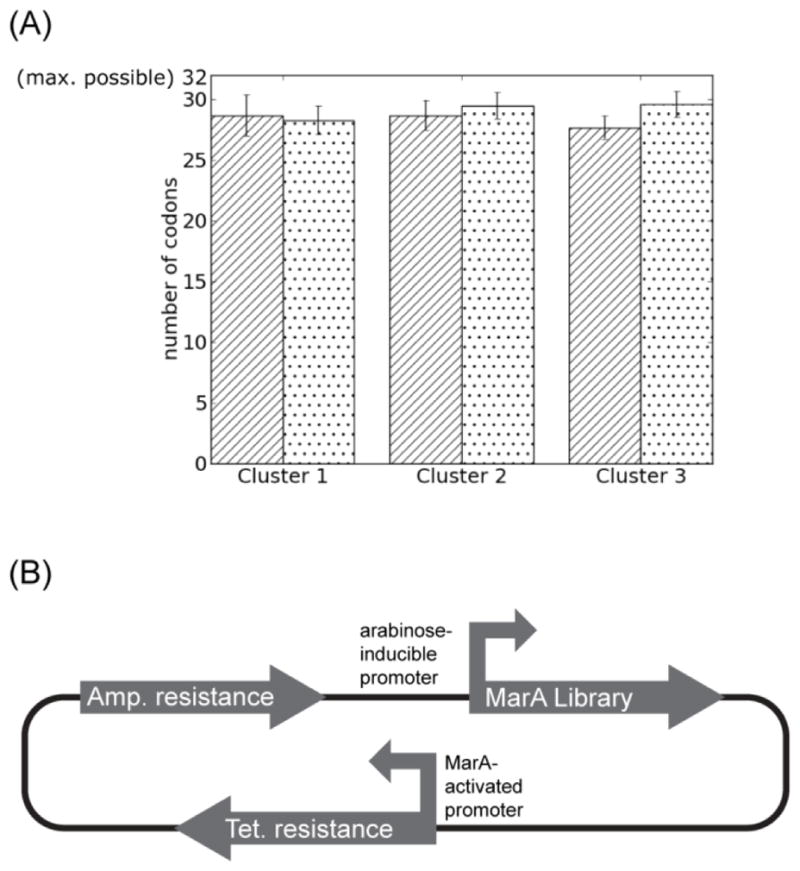
A) Libraries do not appear biased. Dotted bars indicate results of simulated sequencing of 86 (Cluster 1), 93 (Cluster 2), or 94 (Cluster 3) individual clones, repeated 1000 times, with error bars indicating one standard deviation. Striated bars show results of actual sequencing, with the results of three randomized positions averaged for each cluster and error bars indicating one standard deviation. B) The MarA selection system contains MarA mutants under control of an arabinose-inducible promoter and the MarA consensus binding sequence as the promoter for tetracycline resistance.
Genetic selection to identify functional core variants of MarA
To identify functional variants within each library, we employed a genetic selection system wherein growth on tetracycline reports on the presence of functional MarA protein able to activate transcription from the consensus promoter (Fig. 2.2B, see Materials and Methods). Surprisingly, of the roughly 24,000 MarA library variants evaluated, only 17 retain wild-type function sufficient to pass the selection; these 17 feature almost exclusively conservative substitutions (Table I). The number of molecules subjected to selection is more than 3.4 times the size of our library, which corresponds to a greater than 95% chance of having evaluated any random mutant at least once. Additionally, most positive hits have been picked up repeatedly, suggesting that we have effectively sampled the available sequence space.
Table I.
MarA core variants surviving functional selection.
| 13 | 48 | 56 | class | 68 | 94 | 98 | class |
| I | F | L | wt | I | L | F | wt |
| V | F | L | 1 | I | F | F | 1 |
| A | F | V | 2 | L | L | F | 1 |
| V | F | E | 3 | V | F | F | 1 |
| A | F | F | 1 | ||||
| 72 | 98 | 109 | class | C | F | F | 1 |
| L | F | Y | wt | F | L | F | 1 |
| L | F | F | 1 | M | F | F | 1 |
| L | L | F | 2 | V | L | F | 1 |
| I | F | F | 3 | L | F | F | 2 |
| P | I | I | 3 | V | M | F | 2 |
Classes are described in Figure 3A. ‘wt’ denotes wild-type sequence.
The positive hits identified in our selection can be divided into functional classes based on growth on solid media (Fig. 3A). Class 1 variants (10 members) are defined as those that demonstrate wild type-like growth on plates following a 24-hour incubation when transformed into E. coli. The four class 2 variants similarly result in numerous colonies but require a 48-hour incubation. Finally, the three class 3 variants exhibit the least robust growth, resulting in very few colonies after a 24-hour incubation and showing no inclination to be rescued by extended incubation.
Figure 3.

Characterization of functional MarA variants. A) Class 1 variants exhibit wild type-like growth on plates following a 24-hour incubation when transformed into E. coli. Class 2 variants also result in numerous colonies but require a 48-hour incubation. Class 3 variants exhibit the least robust growth, resulting in very few colonies regardless of incubation time. B) Mean log-phase growth rates of E. coli transformed with the variant indicated. Standard errors for wild type, Y109F, and I68V/L94M replicates were 0.033, 0.051, and 0.011, respectively. Standard error among all growth rates was 0.025. C) Increasing concentrations of wild type or L72I/Y109F (class 3) MarA were mixed with radioactively labeled target DNA, equilibrated for 30 minutes, and passed through Tuffryn size exclusion membrane. Percentages indicate amount of total signal present on the Tuffryn membrane.
These functional differences, however, are not reflected in growth rates obtained from growth-curve analyses. Measured in this way, even the most dramatic differences in growth rates among variants are less than two fold, and the standard error of all measured growth rates is smaller than that of any one variant’s replicates (Fig. 3B). In vitro, class 3 variants have a dramatically increased propensity for aggregation compared to class 1 or even class 2 (Fig. 3C). The qualitative correlation between the penetrance of tetracycline resistance and protein aggregation suggests the latter as a possible explanation of the phenotypic differences between functional variants.
Destabilized λ-repressor recapitulates MarA’s intolerance to core substitution
Our results with MarA seem at odds with previous published studies using the protein λ-repressor. If hydrophobicity were the sole or nearly sole requirement for core residues, as has been reported for proteins like λ-repressor[20], we would expect 1,022 variants to pass the selection. MarA, however, is strikingly sensitive to core mutation: our selection recovered only 17 variants. We hypothesized that these differences might be due to differences in one aspect of the energy landscape: global stability.
To evaluate this hypothesis, we first aimed to confirm that MarA is marginally stable. Since in the absence of ligand, MarA is insoluble in standard aqueous buffer, we carried out urea-induced denaturation studies in 20% glycerol monitoring the circular dichroism (CD) signal at 222nm (Fig. 4a). These experiments indicate that in the presence of the stabilizing agent, glycerol, MarA has a free energy of unfolding (ΔGu) = 1.3 ± 0.4 kcal/mol. Even under these conditions, MarA has a propensity for aggregation; the limitations of the CD, however, prohibited higher concentrations of glycerol. Thus, our measurements were not truly at equilibrium and can serve only as an estimated value. Never-the-less, this low value is consistent with our model of MarA as marginally stable.
Figure 4.

Global stability of MarA and λ-repressor. A) Urea denaturation of MarA in 20% glycerol. B) Urea denaturation of wild type (triangles) and R17A/S77A (diamonds) λ-repressor N-terminal domain. Denaturation was measured by CD [25°C, 0.2 μM tris(2-carboxyethyl)phosphine (TCEP), 50 mM sodium chloride, 50 mM sodium phosphate, pH 7. For MarA, glycerol was added to 20%.]. Data were fit with a two-state model [47] and converted to fraction folded.
To determine whether sensitivity to mutation is a result of lower overall stability, we tested a subset of previously identified functional λ-repressor variants in a destabilized background using a similar functional screen[20]. To create a destabilized background, we used the λ-repressor variant R17A/S77A[13]. This variant destabilizes the protein by 2.8 ± 0.3 kcal/mol with a resulting ΔGu = 2.5 ± 0.1 kcal/mol (Fig. 4b). The mutations disrupt a salt bridge interaction distant from the active site; the resulting protein is folded and active. We evaluated the effects of three previously studied core variants V36I, V36I/M40F, and V36I/M40I/V47I in both the wild-type and destabilized λ-repressor backgrounds. While V36I was functional in both backgrounds, V36I/M40F and V36I/M40I/V47I yielded functional proteins only in the wild-type background (Table II). Taken together, these data indicate that decreasing the global stability of λ-repressor is sufficient to alter its tolerance to core mutations.
Table II.
λ repressor variant activity monitored in vivo by immunity to lambda phage infection.
| mutation(s): | background: | |
|---|---|---|
| wild type | R17A/S77A | |
| V36I | + | + |
| V36I/M40F | + | − |
| V36I/M40I/V47I | + | − |
+ indicates resistance to KH54 and KH54h80 lambdaphage strains; - indicates sensitivity to both. All were sensitive to imm21c; none were resistant to KH54 and not KH54h80.
Core mutation differentially affects MarA binding to different promoters, altering binding specificity
The specificity of an interaction reflects the preference for a protein to bind one particular ligand over another; specificity is a measure of a protein’s relative affinity for different ligands[21]. Because mutations within MarA’s hydrophobic core can abolish binding by destabilizing the native conformation relative to the unfolded conformation, we hypothesized that core mutations could actually alter binding specificity by destabilizing certain native conformations relative to others. If so, then some of the core variants should show altered specificity against the known marbox promoters.
Although we are able to detect binding of purified MarA protein to fluorescently labeled oligonucleotides via electrophoretic mobility shift assay (EMSA) (Fig. 5), we were unable to obtain the needed quantitative information via these in vitro assays due to MarA’s propensity for aggregation, weak binding (Kd ~ 150 nM[15]), and small size relative to its ligand. Instead, we employed, a β-galactosidase expression assay developed and used in previous studies to investigate the relative affinity of MarA to different promoters[22]. This assay employs E. coli strains engineered with one of three different naturally occurring MarA promoters – mar, micF, or zwf14 – regulating β-galactosidase expression. A subset of our MarA variants were transformed into each of these strains, and β-galactosidase activity was assayed as a measure of the variant’s affinity for the particular promoter[23]. We evaluated a random subset of variants isolated during our initial selection (I68V, I13V, L94F, Y109F, I68F, L94F, I68V/L94F) as well as designed single-site alanine variants I68A and F98A (Fig. 6). The F98A substitution abolishes expression at all three promoters tested; I68A maintains some ability to activate expression at all three promoters but is consistently worse than wild type. In contrast, all of the variants isolated in our library selection are able to activate expression at the mar promoter as well as or better than the wild type variant. Strikingly, their ability to bind the micF or zwf promoters is quite varied, supporting our hypothesis that core mutations differentially affect the different native conformations required for binding different promoters. These data demonstrate that single-site substitutions within the core can alter DNA-binding specifity, decreasing affinity for one subset of promoters while maintaining wild-type levels of activity or better for a different subset.
Figure 5.

Wild type MarA binding to A) consensus, B) micF, or C) zwf promoters. For each gel, the concentration of protein in each lane, from left to right: 20 μM, 10 μM, 5 μM, 2 μM, 1 μM, 500 nM, 200 nM, 100 nM, 50 nM, 20 nM, 10 nM, none.
Figure 6.

Units of β-galactosidase activity, where β-galactosidase expression is driven by MarA variant activation of A) mar, B) micF, or C) zwf promoters. The same data are shown in D) expressed relative to wild-type MarA-driven expression at the same promoter. Empty refers to relevant strains untransformed with marA-containing plasmid. Each value is the average of 3–5 biological replicates; error bars indicate standard error; dotted line serves as guide for activity levels of empty cells.
2.4 Discussion
We have demonstrated that for MarA, a protein with multiple “ground-state” conformations close in energy, substitutions at sites distant from the active site can have dramatic ramifications for protein function. Whereas canonical well-folded globular proteins like λ-repressor[3,20] and barnase[2] have been shown to have hydrophobic cores tolerant of many amino acid substitutions (little informational content), MarA possesses a hydrophobic core that is intensely intolerant to substitution. Moreover, conservative substitutions within MarA’s hydrophobic core can alter its relative ligand specificity.
In specific circumstances, changes in global stability can drive differences in tolerance to amino acid substitutions. Mutations in the hydrophobic core will modulate protein stability[24]. For a stable globular protein, these small changes in stability do not change the concentration of the binding-competent state to a meaningful degree. For a marginally stable protein, however, the same small changes can appreciably shift the equilibrium towards binding-incompetent states and, in so doing, have profound functional consequences[25,26]. Consider, for example, a simple two-state system:
where L is the ligand, U is the unfolded polypeptide, and N is the native protein,
The apparent dissociation constant of the system, Kd,app, is:
For ordered proteins, Kf is small and the above effect becomes largely invisible experimentally: for a hypothetical well-ordered globular protein with Kd = 500 nM and ΔGf = 8.2 kcal/mol (and thus KF = 10−7),
If this protein is destabilized by 3 kcal/mol, the resultant variant has a KF = 1.6 × 10−5 and still,
Conversely, proteins that are marginally stable will have KF’s that are not insignificant. For a protein with Kd = 500nM but ΔGf = 0 kcal/mol (and thus KF = 1),
Destabilizing this protein by 3 kcal/mol leads to a KF = 160 and now,
This simple two-state thermodynamic perspective outlines how the ability to retain function changes drastically depending on global stability, one aspect of the energy landscape. However, more needs to be considered for proteins that are marginally stable and have the potential to populate multiple native conformations, such as those that are known to bind multiple targets or are better described as molten. For these proteins, the above analysis can be extended to changes in stability between any functionally distinct conformations; for conformations that are close in energy, mutations at a distance can alter the population of one native conformation relative to others[27,28], expanding the discussion above from merely maintaining binding to actually altering specificity. It is important to note that the model outlined above makes no assumptions about the mode of binding, as the distinction between conformational selection and induced fit lies in the ordering of the folding and binding steps rather than in the overall energetics.
Our results for MarA are consistent with this model. As previously mentioned, MarA binds to a number of different targets and is thus likely to have a number of distinct native conformations that are likely to be differentially affected by core mutations. The MarA core variants identified in our selection exhibit varying degrees of activity relative to wild type at the three naturally occurring promoters tested, demonstrating that specificity can be altered by mutations located solely within the hydrophobic core. Our results are consistent with a previous alanine-scanning study on MarA which found several sites distant from the binding surface where mutation to alanine differentially impact binding to different promoters[23]. The disparate impact of core or other mutations on different native states is, of-course, not limited to marginally stable proteins[29]. The existence of multiple conformations close in energy to the “ground state” (multiple minima) means that even subtle changes that would be functionally invisible in stable globular proteins can alter specificity in a protein whose energy landscape is akin to that of MarA.
A major protein class likely to be enriched in MarA-like energy landscapes is intrinsically disordered proteins (IDPs)[30], a significant percentage of the eukaryotic proteome[31]. How IDPs respond to substitutions remains unclear. Some studies argue that the lack of a well-defined native state renders them more tolerant to mutation than well-folded proteins[32,33], while other studies suggest that relatively weak, transient contacts may play a disproportionately critical functional role[34]. Consistent with the functional importance of residues outside the binding interface in these proteins, IDPs appear to have rates of evolution and patterns of conservation distinct from those of globular, single-ligand proteins[35]. As such, it has been argued that the evolution of disordered proteins and regions is subject to a distinct set of biophysical constrains[35]. Since the energy landscapes of IDPs more closely resemble that of MarA than of λ-repressor, our data indicate that those proteins whose function requires a disorder-to-order transition may actually be less tolerant to mutation. Thus, this work provides a potential functional role for a subset of the high percent of the human genome that encodes disorder. Additionally, it offers some tantalizing clues as to what factors may have favored the evolution of such proteins, as several studies have implicated functional plasticity as a critical factor in the molecular evolution of new function[35–38], with selective stabilization of some subset of states from a molten native ensemble specifically posited as a driving factor[39]. In this work, we’ve shown that marginally stable proteins have a broad distribution of informational content over their amino acid sequence, increasing the probability that a random mutation will have an impact on binding. The increased ease of effecting appreciable change may explain the favorability of marginal stability to the evolution of novel binding targets.
It has been proposed that the maintenance of organismal evolvability may depend on the ability to have large-scale functional impact with a small number of mutations[40]. Multi-gene regulators like MarA present the possibility to accomplish exactly this, as even small changes in specificity for promiscuous proteins could affect multiple pathways and result in global changes within a cell. Biophysical and biological plasticity thus represent two related but distinct nodes upon which evolution can act to rapidly achieve phenotypic diversity. This may be part of the reason why transcription factors, which frequently bind multiple targets[41], are enriched for intrinsically disordered regions[42,43].
Our experimental results demonstrate that the shape of the energy landscape and, in particular, global stability are indicative of the distribution of informational content over a protein’s sequence. In essence, stable proteins are overdetermined and can accommodate mutations more easily. This relationship between energy landscape and informational content can guide protein design in two ways. Firstly, it can increase protein design efficiency by identifying proteins with distinct regions high in informational content that can be treated as functionally modular. Secondly, it suggests a path for engineering proteins with dispersed informational content that have previously been problematic for protein engineering, highlighting the importance of incorporating non-interface residues into design strategies.
2.5 Materials & Methods
Gene synthesis
Libraries of MarA genes with selected codons randomized were purchased from GeneArt®, PCR-amplified, and inserted into the selection plasmid[19]. These were transformed into XL10-Gold cells for library propagation, plated on 30 μg/ml ampicillin LB plates. ~200,000 colonies per library were resuspended in liquid media, and the plasmids were extracted using the Qiagen Midi-prep Kit. All genes were subcloned into pET28 plasmid for purification.
We used the λ-repressor N-terminal domain from plasmid pWL104[20]. Specific point mutations were introduced using QuikChange mutagenesis.
Functional selections
MarA libraries were transformed into E. coli strain N8453 (Δmar, Δsox-8::cat, Δrob::kan variant of GC4468) made by J.L. Rosner and R.G. Martin and obtained from the M.B. Eisen laboratory. MarA expression was induced with 0.1% L-arabinose after one hour of recovery; cells were allowed to grow for an additional four hours before plating on 30 μg/ml tetracycline plates + 0.1% L-arabinose and incubated at 37°C for 48 hours.
λ-repressor functional assays were performed in X90 cells. Single colonies were streaked sequentially across lambda phage strains KH54, KH54h80, and imm21c on 200 μg/ml ampicillin plates + 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG).
Protein purification
Proteins were overexpressed in Rosetta2 DE3 pLysS cells induced with 1 mM IPTG and purified using nickel column chromatography. For MarA purification, cells were lysed by sonication in 50 mM HEPES, pH 8, 500 mM NaCl, and 1 mM TCEP. MarA was expressed in inclusion bodies, which were isolated by centrifugation and resuspended in lysis buffer supplemented with 8 M urea and 10 mM imidazole. Imidazole concentration was increased to 350 mM to elute, and protein was dialyzed into storage buffer (50% glycerol, 50 mM HEPES, pH 8, 500 mM NaCl, 1 mM TCEP). For λ-repressor purification, cells were lysed by sonication in 50 mM sodium phosphate, pH 8, 500 mM NaCl, 20 mM imidazole. λ-repressor was found entirely in supernatant; to elute, imidazole concentration was increased to 250 mM.
Binding assays
For in vitro binding assays, serial dilutions of MarA variants were incubated for 30 minutes at room temperature with 0.5 nM oligo in 50 mM Tris, pH 8.5, 50 mM NaCl, and 10% glycerol. Experiments using dot-blot apparatus were performed as described in Chakravarthy et al.[44] with 5’ 32P-radiolabeled double-stranded consensus oligo (5’ – ATT CGA TTT AGC AAA ACG TGC CAT CGG T – 3’)[19]. EMSA used 3’ FAM-labeled consensus, micF (5’ – ATT CGA CAG CAC TGA ATG TCA AAA CCG GT – 3’), or zwf (5’ – ATT CGA TCG CAC GGG TGG ATA AGC GCG GT – 3’) MarA-binding sites[23]; after equilibration, samples were loaded onto 5% PAGE gel (29:1 acrylamide:bis-acrylamide and 10% glycerol in TAE) and run at approximately 80V at 4°C.
In vivo binding assays were performed as described in Gillete et al[23]. Briefly, marA mutants derived from pRGM9818 were transformed into E. coli strains N8795, N9212, and N9214, which contain a single-copy (mar, micF, or zwf, respectively)::lacZ transcriptional fusion at the attB site[45]. Overnight cultures were diluted 1:50, grown for 2 hours at 32°C, and assayed for β-galactosidase activity[46].
Circular dichroism
The N-terminal domain of λ-repressor was dialyzed into 50 mM sodium phosphate, pH 7, 50 mM NaCl, and 0.2 mM TCEP. MarA was dialyzed into the same but with 20% glycerol. Circular dichroism studies were carried out at 25 °C on an Aviv 410 spectrapolarimeter with Peltier temperature control using a Microlab titrator. For λ-repressor, ~30 μg/ml protein in ~10 M urea was titrated into 0 M urea sample of identical protein concentration, maintaining 2.5 ml total volume. After each titration step, sample was equilibrated for 5 minutes and CD signal was monitored at 222, averaged over a 60-second time period. For MarA, individual samples containing approximately 50 μg/mL protein and 20% glycerol at varying urea concentrations were equilibrated overnight. As with λ-repressor, the signal was monitored at 222 nm and averaged over 60 seconds. The change in Gibbs free energy upon unfolding (ΔG) was obtained by plotting the CD signal as a function of urea concentration and fitting the data using a two-state linear extrapolation model to obtain ΔG in the absence of urea[47].
Growth curves
MarA mutants were transformed into N8453 cells as described for the library selections. 5 ml 30 μg/ml tetracycline LB + 0.1% L-arabinose were inoculated with resulting colonies and allowed to grow at 37°C for approximately 20 hours. For each growth curve, 200 ml 30 μg/ml tetracycline LB + 0.1% L-arabinose was inoculated with 2 ml of overnight cultures and incubated at 37°C with agitation. Cell growth was monitored by measuring optical density (OD) at 600 nm, with samples taken every half hour during log phase. For OD600 between 0.12 and 2, the log10 of the OD was plotted against the time and fit to a line; the slope of the line corresponds to the growth rate in tennings (number of tenfold increases) per hour.
Highlights.
Ligand binding and specificity is attributed to a small number of residues comprising the ligand-binding interface of the native structure. This simple model, however, does not take into account features of a protein’s energy landscape – the energetics of the native conformation and the energetics of alternative conformations.
We evaluate the importance of buried residues in the hydrophobic core using two model DNA-binding proteins: the well-folded stable N-terminal domain of lambda repressor and the more molten, marginally stable MarA.
We show that the tolerance to mutation of different regions of the protein depends on its energy landscape. Binding hot spots are localized at the binding interface for proteins like lambda repressor, while residues throughout the protein can be functionally crucial, even altering specificity, in the case of MarA.
These results suggest that the underlying energy landscape is crucial for manipulating ligand binding/specificity and that energy landscape profiles can help identify promising candidates for protein engineering. Additionally, they have implications for evolutionary studies, as proteins like MarA may be poised to maintain both molecular and organismal evolvability.
Acknowledgments
We thank Ryan Shultzaberger and Judah Lee Rosner for MarA plasmids and strains; Jim Hu and Adrienne Zweifel for lambda phage strains and protocols; Sam Sternberg, David Steakley, and Robert Martin for technical assistance; Mike Botchan for comments on the manuscript; and the entire Marqusee lab for helpful discussions. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR001081). This work was supported by a grant from the NIH and NSF.
Footnotes
No competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
2.7 References
- 1.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 2.Axe DD, Foster NW, Fersht AR. Active barnase variants with completely random hydrophobic cores. Proc Natl Acad Sci USA. 1996;93:5590–5594. doi: 10.1073/pnas.93.11.5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lim WA, Sauer RT. Alternative packing arrangements in the hydrophobic core of lambda repressor. Nature. 1989;339:31–36. doi: 10.1038/339031a0. [DOI] [PubMed] [Google Scholar]
- 4.Sia SK, Kim PS. Protein grafting of an HIV-1-inhibiting epitope. Proc Natl Acad Sci USA. 2003;100:9756–9761. doi: 10.1073/pnas.1733910100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang L, Althoff EA, Clemente FR, Doyle L, Röthlisberger D, et al. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kritzer JA, Zutshi R, Cheah M, Ran FA, Webman R, et al. Miniature protein inhibitors of the p53-hDM2 interaction. Chembiochem. 2006;7:29–31. doi: 10.1002/cbic.200500324. [DOI] [PubMed] [Google Scholar]
- 7.Anantharaman V, Aravind L, Koonin EV. Emergence of diverse biochemical activities in evolutionarily conserved structural scaffolds of proteins. Curr Opin Chem Biol. 2003;7:12–20. doi: 10.1016/s1367-5931(02)00018-2. [DOI] [PubMed] [Google Scholar]
- 8.Iyer LM, Zhang D, Rogozin IB, Aravind L. Evolution of the deaminase fold and multiple origins of eukaryotic editing and mutagenic nucleic acid deaminases from bacterial toxin systems. Nucleic Acids Res. 2011;39:9473–9497. doi: 10.1093/nar/gkr691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thorn KS, Bogan AA. ASEdb: a database of alanine mutations and their effects on the free energy of binding in protein interactions. Bioinformatics. 2001;17:284–285. doi: 10.1093/bioinformatics/17.3.284. [DOI] [PubMed] [Google Scholar]
- 10.Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, et al. Exploiting a natural conformational switch to engineer an interleukin-2 'superkine'. Nature. 2012;484:529–533. doi: 10.1038/nature10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 12.Ptashne M, Jeffrey A, Johnson AD, Maurer R, Meyer BJ, et al. How the lambda repressor and cro work. Cell. 1980;19:1–11. doi: 10.1016/0092-8674(80)90383-9. [DOI] [PubMed] [Google Scholar]
- 13.Marqusee S, Sauer RT. Contributions of a hydrogen bond/salt bridge network to the stability of secondary and tertiary structure in lambda repressor. Protein Sci. 1994;3:2217–2225. doi: 10.1002/pro.5560031207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barbosa TM, Levy SB. Differential expression of over 60 chromosomal genes in Escherichia coli by constitutive expression of MarA. J Bacteriol. 2000;182:3467–3474. doi: 10.1128/jb.182.12.3467-3474.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin RG, Gillette WK, Rhee S, Rosner JL. Structural requirements for marbox function in transcriptional activation of mar/sox/rob regulon promoters in Escherichia coli: sequence, orientation and spatial relationship to the core promoter. Molecular Microbiology. 1999;34:431–441. doi: 10.1046/j.1365-2958.1999.01599.x. [DOI] [PubMed] [Google Scholar]
- 16.Rhee S, Martin RG, Rosner JL, Davies DR. A novel DNA-binding motif in MarA: the first structure for an AraC family transcriptional activator. Proc Natl Acad Sci USA. 1998;95:10413–10418. doi: 10.1073/pnas.95.18.10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dangi B, Pelupessey P, Martin RG, Rosner JL, Louis JM, et al. Structure and dynamics of MarA-DNA complexes: an NMR investigation. Journal of Molecular Biology. 2001;314:113–127. doi: 10.1006/jmbi.2001.5106. [DOI] [PubMed] [Google Scholar]
- 18.Dangi B, Gronenborn AM, Rosner JL, Martin RG. Versatility of the carboxy-terminal domain of the α subunit of RNA polymerase in transcriptional activation: use of the DNA contact site as a protein contact site for MarA. Molecular Microbiology. 2004;54:45–59. doi: 10.1111/j.1365-2958.2004.04250.x. [DOI] [PubMed] [Google Scholar]
- 19.Shultzaberger RK, Maerkl SJ, Kirsch JF, Eisen MB. Probing the informational and regulatory plasticity of a transcription factor DNA-binding domain. PLoS Genet. 2012;8:e1002614. doi: 10.1371/journal.pgen.1002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim WA, Sauer RT. The role of internal packing interactions in determining the structure and stability of a protein. Journal of Molecular Biology. 1991;219:359–376. doi: 10.1016/0022-2836(91)90570-v. [DOI] [PubMed] [Google Scholar]
- 21.Kuriyan J, Konforti B, Wemmer D. The molecules of life: KURIYAN John, KONFORTI Boyana, WEMMER David: Librairie Lavoisier. 2013. [Google Scholar]
- 22.Martin RG, Rosner JL. Fis, an accessorial factor for transcriptional activation of the mar (multiple antibiotic resistance) promoter of Escherichia coli in the presence of the activator MarA, SoxS, or Rob. J Bacteriol. 1997;179:7410–7419. doi: 10.1128/jb.179.23.7410-7419.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gillette WK, Martin RG, Rosner JL. Probing the Escherichia coli transcriptional activator MarA using alanine-scanning mutagenesis: residues important for DNA binding and activation. Journal of Molecular Biology. 2000;299:1245–1255. doi: 10.1006/jmbi.2000.3827. [DOI] [PubMed] [Google Scholar]
- 24.Munson M, Balasubramanian S, Fleming KG, Nagi AD, O'Brien R, et al. What makes a protein a protein? Hydrophobic core designs that specify stability and structural properties. Protein Sci. 1996;5:1584–1593. doi: 10.1002/pro.5560050813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borcherds W, Theillet F-X, Katzer A, Finzel A, Mishall KM, et al. Disorder and residual helicity alter p53-Mdm2 binding affinity and signaling in cells. Nat Chem Biol. 2014;10:1000–1002. doi: 10.1038/nchembio.1668. [DOI] [PubMed] [Google Scholar]
- 27.Hilser VJ, Dowdy D, Oas TG, Freire E. The structural distribution of cooperative interactions in proteins: analysis of the native state ensemble. Proc Natl Acad Sci USA. 1998;95:9903–9908. doi: 10.1073/pnas.95.17.9903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu T, Whitten ST, Hilser VJ. Functional residues serve a dominant role in mediating the cooperativity of the protein ensemble. Proc Natl Acad Sci USA. 2007;104:4347–4352. doi: 10.1073/pnas.0607132104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haririnia A, Verma R, Purohit N, Twarog MZ, Deshaies RJ, et al. Mutations in the hydrophobic core of ubiquitin differentially affect its recognition by receptor proteins. Journal of Molecular Biology. 2008;375:979–996. doi: 10.1016/j.jmb.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flock T, Weatheritt RJ, Latysheva NS, Babu MM. ScienceDirectControlling entropy to tune the functions of intrinsically disordered regions. Curr Opin Struct Biol. 2014;26:62–72. doi: 10.1016/j.sbi.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 31.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Babu MM, Kriwacki RW, Pappu RV. Structural biology. Versatility from protein disorder. Science. 2012;337:1460–1461. doi: 10.1126/science.1228775. [DOI] [PubMed] [Google Scholar]
- 33.Schlessinger A, Schaefer C, Vicedo E, Schmidberger M, Punta M, et al. Protein disorder--a breakthrough invention of evolution? Curr Opin Struct Biol. 2011;21:412–418. doi: 10.1016/j.sbi.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 34.Mittag T, Orlicky S, Choy W-Y, Tang X, Lin H, et al. Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor. Proceedings of the National Academy of Sciences. 2008;105:17772–17777. doi: 10.1073/pnas.0809222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sikosek T, Chan HS. Biophysics of protein evolution and evolutionary protein biophysics. J R Soc Interface. 2014;11:20140419. doi: 10.1098/rsif.2014.0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tokuriki N, Tawfik DS. Protein dynamism and evolvability. Science. 2009;324:203–207. doi: 10.1126/science.1169375. [DOI] [PubMed] [Google Scholar]
- 37.Meier S, Ozbek S. A biological cosmos of parallel universes: does protein structural plasticity facilitate evolution? Bioessays. 2007;29:1095–1104. doi: 10.1002/bies.20661. [DOI] [PubMed] [Google Scholar]
- 38.Ancel LW, Fontana W. Plasticity, evolvability, and modularity in RNA. J Exp Zool. 2000;288:242–283. doi: 10.1002/1097-010x(20001015)288:3<242::aid-jez5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 39.Mannige RV, Brooks CL, Shakhnovich EI. A universal trend among proteomes indicates an oily last common ancestor. PLoS Comput Biol. 2012;8:e1002839. doi: 10.1371/journal.pcbi.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jarosz DF, Lindquist S. Hsp90 and environmental stress transform the adaptive value of natural genetic variation. Science. 2010;330:1820–1824. doi: 10.1126/science.1195487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MacQuarrie KL, Fong AP, Morse RH, Tapscott SJ. Genome-wide transcription factor binding: beyond direct target regulation. Trends Genet. 2011;27:141–148. doi: 10.1016/j.tig.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu J, Perumal NB, Oldfield CJ, Su EW, Uversky VN, et al. Intrinsic disorder in transcription factors. Biochemistry. 2006;45:6873–6888. doi: 10.1021/bi0602718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minezaki Y, Homma K, Kinjo AR, Nishikawa K. Human transcription factors contain a high fraction of intrinsically disordered regions essential for transcriptional regulation. Journal of Molecular Biology. 2006;359:1137–1149. doi: 10.1016/j.jmb.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 44.Chakravarthy S, Sternberg SH, Kellenberger CA, Doudna JA. Substrate-specific kinetics of Dicer-catalyzed RNA processing. Journal of Molecular Biology. 2010;404:392–402. doi: 10.1016/j.jmb.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin RG, Gillette WK, Rosner JL. Promoter discrimination by the related transcriptional activators MarA and SoxS: differential regulation by differential binding. Molecular Microbiology. 2000;35:623–634. doi: 10.1046/j.1365-2958.2000.01732.x. [DOI] [PubMed] [Google Scholar]
- 46.Miller JH. Experiments in molecular genetics. Cold Spring Harbor Laboratory Pr; 1972. p. 1. [Google Scholar]
- 47.Greene RF, Pace CN. Urea and guanidine hydrochloride denaturation of ribonuclease, lysozyme, alpha-chymotrypsin, and beta-lactoglobulin. J Biol Chem. 1974;249:5388–5393. [PubMed] [Google Scholar]
- 48.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]