

SUMMARY
N-terminal acetylation is a common and important protein modification catalysed by N-terminal acetyltransferases (NATs). Six human NATs (NatA-NatF) contain one catalytic subunit each, Naa10-Naa60, respectively. In contrast to the ribosome associated NatA-NatE, NatF/Naa60 specifically associates with Golgi membranes and acetylates transmembrane proteins. To gain insight into the molecular basis for function of Naa60, we developed a Naa60 bisubstrate CoA-peptide conjugate inhibitor, determined its X-ray structure when bound to CoA and inhibitor, and carried out biochemical experiments. We show that Naa60 adapts an overall fold similar to the catalytic subunits of ribosome-associated NATs, but with the addition of two novel elongated loops that play important roles in substrate-specific binding. One of these loops mediates a dimer to monomer transition upon substrate-specific binding. Naa60 employs a catalytic mechanism most similar to Naa50. Collectively, this data reveals the molecular basis for Naa60-specific acetyltransferase activity with implications for its Golgi-specific functions.
Keywords: acetyltransferase, crystal structure, Naa60, NAT, NatF, N-terminal acetylation
eTOC blurb
Protein N-terminal acetylation is catalysed by N-terminal acetyltransferases (NATs). Stove et al report the crystal structure of Naa60, the only NAT specifically associating with cellular membranes. Two Naa60-specific elongated loops play important roles in substrate-specific binding and in a dimer to monomer transition for substrate-specific binding and catalysis.
INTRODUCTION
Among all N-terminal protein modifications that occur in eukaryotes, N-terminal acetylation (Nt-acetylation) is by far the most common, occurring on approximately 50–85% of all soluble proteins (Arnesen et al., 2009; Bienvenut et al., 2012). Nt-acetylation is catalyzed by a group of enzymes called N-terminal acetyl transferases (NATs), which predominantly acetylate protein N-termini co-translationally as they emerge from the ribosomal exit tunnel (Aksnes et al., 2015a). Nt-acetylation has been shown to be important for protein-protein interactions (Scott et al., 2011), protein subcellular localization (Behnia et al., 2004; Forte et al., 2011; Setty et al., 2004), protein folding (Holmes et al., 2014), and to function as the degradation signal of the Ac/N-end rule pathway (Hwang et al., 2010; Shemorry et al., 2013). According to the Ac/N-end rule, Nt-acetylated protein N-termini are recognized by specific E3 ligases, ubiquitylated and degraded through the proteasome pathway. This degradation pathway was shown to regulate protein quality control and protein complex stoichiometries (Shemorry et al., 2013). NATs also have connections to human disease. Several NAT subunits have previously been shown to be over-expressed in different types of cancers (Kalvik and Arnesen, 2013), and recently several point mutations in the NAA10 gene (encoding the catalytic subunit of the NatA complex) have been shown to cause intellectual disabilities, Lenz-Micropthalmia syndrome and Ogden syndrome (Casey et al., 2015; Esmailpour et al., 2014; Myklebust et al., 2015; Popp et al., 2015; Rope et al., 2011; Saunier et al., 2016).
NATs are bisubstrate enzymes that catalyze the transfer of an acetyl group from acetyl CoA (Ac-CoA) to a protein N-terminal amine group. In humans, six NATs have been identified and characterized (Arnesen et al., 2005a; Evjenth et al., 2009; Hole et al., 2011; Starheim et al., 2008; Starheim et al., 2009; Van Damme et al., 2011b). NatA–NatC (catalytic subunits Naa10–Naa30) are protein complexes consisting of two or more subunits (Arnesen et al., 2005a; Mullen et al., 1989; Park and Szostak, 1992; Polevoda et al., 2003; Polevoda and Sherman, 2001; Starheim et al., 2008; Starheim et al., 2009), NatE (Naa50) is a monomeric enzyme that has been shown to associate with NatA (Gautschi et al., 2003; Williams et al., 2003), while NatD (Naa40) and NatF (Naa60) are monomeric enzymes believed to act independent of other subunits (Aksnes et al., 2015b; Hole et al., 2011; Magin et al., 2015; Song et al., 2003; Van Damme et al., 2011b). Each NAT has distinct substrate specificity, mainly based on the first two amino acids of the N-terminus of the substrate polypeptide. For one of the NATs, Naa10, the substrate specificity changes upon binding to the auxiliary subunit Naa15 (Liszczak et al., 2013). Upon binding to Naa15, Naa10 undergoes a conformational change altering the active site of the enzyme enabling acetylation of protein N-termini in which the initiator methionine has been removed by methionine aminopeptidases (MetAPs) exposing small amino acids as the new N-termini. Uncomplexed Naa10 on the other hand has been shown to mainly acetylate acidic N-termini (Glu- or Asp- starting) (Van Damme et al., 2011a). Naa20 acetylates protein N-termini with a methionine in position 1, followed by an acidic residue in position 2. Naa40 specifically acetylates the serine-starting N-termini of histones H2A and H4, while Naa30, Naa50 and Naa60 acetylate unprocessed methionine N-termini with either a positively charged or hydrophobic amino acid in position 2 (Arnesen et al., 2009; Evjenth et al., 2009; Hole et al., 2011; Magin et al., 2015; Polevoda et al., 1999; Song et al., 2003; Starheim et al., 2009; Tercero and Wickner, 1992; Van Damme et al., 2011a; Van Damme et al., 2015; Van Damme et al., 2011b; Van Damme et al., 2012). For the rest of this text, we will refer to NATs by the name of their catalytic subunit.
Naa60 is the most recently identified and least studied human NAT. Unlike Naa10–Naa50, which are conserved from yeast to man, Naa60 appears to be lost in the Fungi kingdom (Rathore et al., 2016; Van Damme et al., 2011b). Furthermore, Naa60 has a distinct subcellular localization pattern; while Naa10–Naa50 are located throughout the cytoplasm and in some cases also in the nucleus, Naa60 displays an organellular localization pattern associating to the cytosolic side of Golgi membranes (Aksnes et al., 2015b). Depletion of NAA60 results in Golgi fragmentation in HeLa cells (Aksnes et al., 2015b) and abnormal chromosome segregation in drosophila DmeI2 cells (Van Damme et al., 2011b), resembling the observed phenotypes of NAA50 depleted drosophila cells. However, while NAA60-depleted drosophila cells exhibited normal metaphase and abnormal anaphase, NAA50-depleted drosophila cells exhibited abnormal metaphase with mitotic arrest (Hou et al., 2007; Pimenta-Marques et al., 2008; Van Damme et al., 2011b; Williams et al., 2003).
Initial studies of human Naa60 (hNaa60) suggested that there are extensive redundancy in the substrate specificity profile of hNaa30, hNaa50 and hNaa60 when only considering the N-terminal sequence of the substrate (Van Damme et al., 2011b). However, recent data has also revealed that there is a large over-representation of transmembrane proteins in the hNaa60 acetylome, suggesting that hNaa60 has evolved to specifically acetylate the cytosolic N-termini of membrane proteins from its membrane-associated localization (Aksnes et al., 2015b).
In this study, we set out to investigate the structure and biochemical properties of human Naa60. To this end, we cloned, purified and crystallized hNaa60 both in complex with CoA and in complex with a newly developed bisubstrate analogue closely resembling CoA and an acetylated substrate polypeptide. We also carried out structure-based mutagenesis and biochemical and enzymatic studies in order to make structure-function correlations underlying hNaa60-specific activities.
RESULTS
Structure of hNaa60/CoA-Ac-MKAV7 reveals Naa60-specific regions
Sequence alignment of hNaa60 with other NAT homologues reveals sequence homology within the N-terminal 180 amino acids (Figure 1A), but sequence divergence of C-terminal residues 181–242 that has been shown to be responsible for the Golgi subcellular localization of hNaa60 (Figure S1) (Aksnes et al., 2015b). Within the conserved NAT region, hNaa60 shows the greatest regions of sequence divergence in the β3–β4 (residues 73–94) and β6–β7 (residues 165–173) loops, which are also longer in hNaa60 than in most other eukaryotic NATs. Interestingly, the β3–β4 loop of the NAT homologue from S. solfataricus (SsNAT) is also extended compared to the β3–β4 loops of other NATs, and was previously shown to be important for SsNAT function (Liszczak and Marmorstein, 2013).
Figure 1. Sequence alignment of selected NATs and overall structure of the binary hNaa60/CoA-Ac-MKAV7 complex.
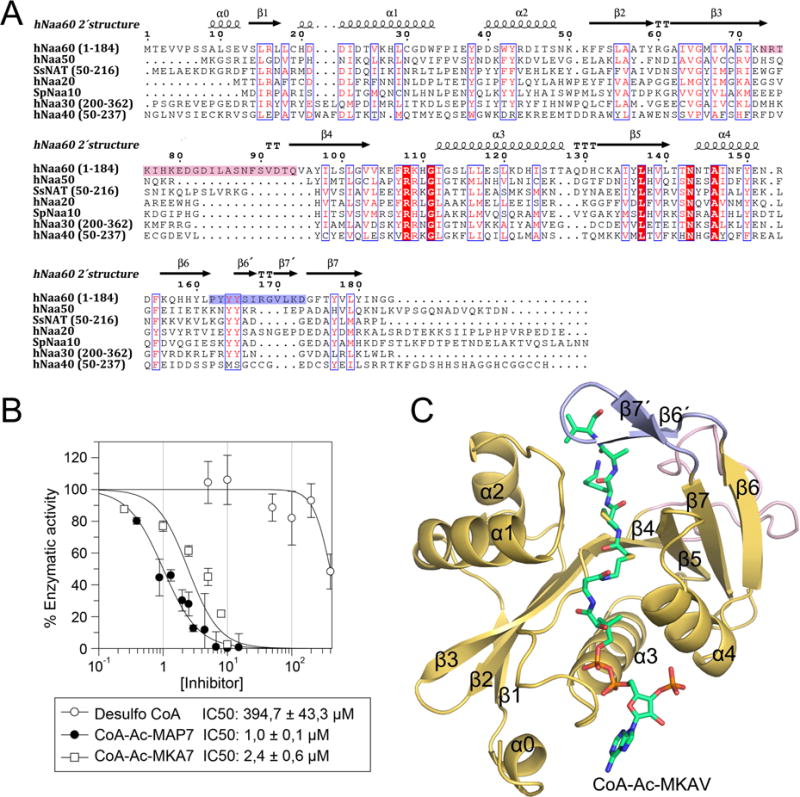
A) Sequence alignment of hNaa60 (1–184) and NAT homologues hNaa50, hNaa40 (50–237), SsNAT (50–216) and SpNaa10 for which crystal structures already have been determined, and the human homologues hNaa20 and hNaa30 (200–362). Sequence identity is shown with white letters in red boxes and sequence similarity with red letters in blue frames. The secondary structure elements of hNaa60 are shown on top of the alignment. The β3–β4 loop is marked in pink and the β6–β7 loop in light blue. B) IC50 values of the two bisubstrate analogues CoA-Ac-MAPL7 and CoA-Ac-MKAV7 in addition to the control compound desulfoCoA. C) Overall structure of the binary hNaa60/CoA-Ac-MKAV7 complex. The β3–β4 loop is shown in pink (on the back side of the structure), the β6–β7 loop in light blue, and the CoA-Ac-MKAV7 inhibitor in green sticks. See also Figure S1–S3.
In order to structurally and biochemically examine hNaa60, we cloned, expressed and purified different N-terminal hNaa60 variants and were able to prepare constructs containing residues 1–184 and 5–184, which were amenable to biochemical and structural analysis. In vitro enzymatic assays confirmed that hNaa60 1–184 has a similar substrate specificity profile as full length hNaa60 in vivo (Figure S2) (Aksnes et al., 2015b; Van Damme et al., 2011b). We also prepared two bisubstrate analogues, CoA-Ac-MAPLDLD-NH2 (CoA-Ac-MAPL7) and CoA-Ac-MKAVQAD-NH2 (CoA-Ac-MKAV7) derived from previously identified N-terminal hNaa60 in vivo substrates (Aksnes et al., 2015b; Van Damme et al., 2011b) which were tested as inhibitors of Naa60 1–184 using in vitro acetylation assays. In these experiments, desulfoCoA (Figure S3A), which previously has been identified as a competitive inhibitor of the N-terminal acetyltransferase Naa50 (Evjenth et al., 2012), and also other acetyltransferases (De Angelis et al., 1998; Thompson et al., 2001), was used as a control.
Both bisubstrate analogues were very potent inhibitors, with a half maximum inhibitor concentration (IC50) of 1.0 ± 0.1 μM and 2.44 ± 0.6 μM for CoA-Ac-MAPL7 and CoA-Ac-MKAV7 respectively whereas the IC50 of desulfoCoA was 394.7 ± 43.3 μM (Figure 1B). Further analysis of CoA-Ac-MAPL7 confirmed that it was a tight binding, competitive inhibitor (Copeland and Dr. William McDowell Mastin Fund., 2000) compared to Ac-CoA with a Ki of 0.23 ± 0.42 μM (Figure S3 B and C).
We were able to obtain crystals of hNaa60 5–184 in binary complex with CoA-Ac-MKAVQAD and to determine its structure to a resolution of 1.53 Å, using molecular replacement with SsNAT (PDB ID: 4LX9) as the initial search model. The crystals formed in spacegroup P41 and contained two hNaa60/CoA-Ac-MKAV7 complexes per asymmetric unit. The resulting electron density allowed for the tracing of residues 5–184 of hNaa60, and the CoA-Ac portion and residues 1 to 4 of the bisubstrate inhibitor (herein called CoA-Ac-MKAV7). Both complexes in the asymmetric unit overlay almost perfectly except for a slight adjustment of the side chain positions of M1p and K2p of the bisubstrate inhibitor. The final model was refined to a Rwork/Rfree of 0.1905/0.2084 with good geometry (Table 1). We also obtained crystals and determined the structure of Naa60 in complex with CoA as will be more thoroughly described below. The structure of hNaa60 adopts the characteristic GNAT (Gcn5-related N-acetyltransferase) fold (Vetting et al., 2005) found in all NATs, consisting of a mixed α/β fold with a conserved Ac-CoA binding site (Figure 1C). However, the β3–β4 and β6–β7 loops adopt hNaa60-specific structures. The β3–β4 loop folds back towards the peptide and core domain, and the β6–β7 loop, contains a β-hairpin structure with two shorts strands that we call β6′ and β7′ that hang over the peptide binding site in a manner that is highly characteristic for all NATs (Figure 1C).
Table 1.
Data collection and refinement statistics
| hNaa60/CoA-Ac-MKAV7 | hNaa60/CoA | |
|---|---|---|
|
| ||
| PDB ID | 5ICV | 5ICW |
| Crystal Parameters | ||
|
| ||
| Space group | P41 | P21 |
| Unit cell dimension | 48.448 (90) | 63.840 (90) |
| 48.448 (90) | 84.298 (91.097) | |
| 148.37 (90) | 68.485 (90) | |
|
| ||
| Data collection | ||
|
| ||
| Resolution (Å) | 40.56−1.53 | 46.249−1.951 |
| Unique reflections | 51410 | 52859 |
| Rmerge | 0.122 (0.635) | 0.098 (0.527) |
| I/σ | 18.62 (4.49) | 15.17 (1.04) |
| Completeness | 99.8 (99.9) | 95 (63.1) |
| Redundancy | 13.0 (11.8) | 2.9 (1.9) |
|
| ||
| Refinement | ||
|
| ||
| Rwork/Rfree | 0.1905/0.2084 | 0.2072/0.2547 |
| r.m.s.d. | ||
| Bonds (Å) | 0.007 | 0.008 |
| Angeles (°) | 1.284 | 1.278 |
| Average B-factors (Å2) | ||
| Protein | 25.62 | 32.89 |
| CoA-Ac-MKAV/CoA | 28.10 | 31.02 |
| Solvent | 38.51 | 38.68 |
| Ramachandran statistics (%) | ||
| Favored | 99.45 | 99.57 |
| Allowed | 0.55 | 0.43 |
| Outlier | 0 | 0 |
Structural and functional similarities and differences between hNaa60 and other NATs
A superposition of hNaa60/CoA-Ac-MKAV7 with other NAT structures shows the highest degree of structural similarity to hNaa50 with an rmsd of 1.59 Å for backbone atoms, although there is still significant structural superposition with S. pombe Naa10 (SpNaa10) (rmsd = 1.96 Å) and SsNAT (rmsd = 1.70 Å) (Figure 2A). Consistent with the sequence divergence between these proteins, hNaa60 shows the greatest structural divergence with these proteins in the β3–β4 and β6–β7 loops (Figures 2B and 2C). Interestingly, the β3–β4 loop of SsNAT is most similar to the corresponding loop of hNaa60 and was previously shown to be important for SsNAT function by hanging over and contacting the β5, β6, β7 strands positioning residues in these strands to make peptide substrate interactions (Liszczak and Marmorstein, 2013). As will be discussed below, this loop is carrying out a similar role in hNaa60. Interestingly, two tyrosine residues in the β6–β7 loop of hNaa60 (Y164 and Y165) are conserved in other NATs (except for hNaa40) and play a role in peptide anchoring (Figure 2C).
Figure 2. Structural alignment highlighting hNaa60 specific features.
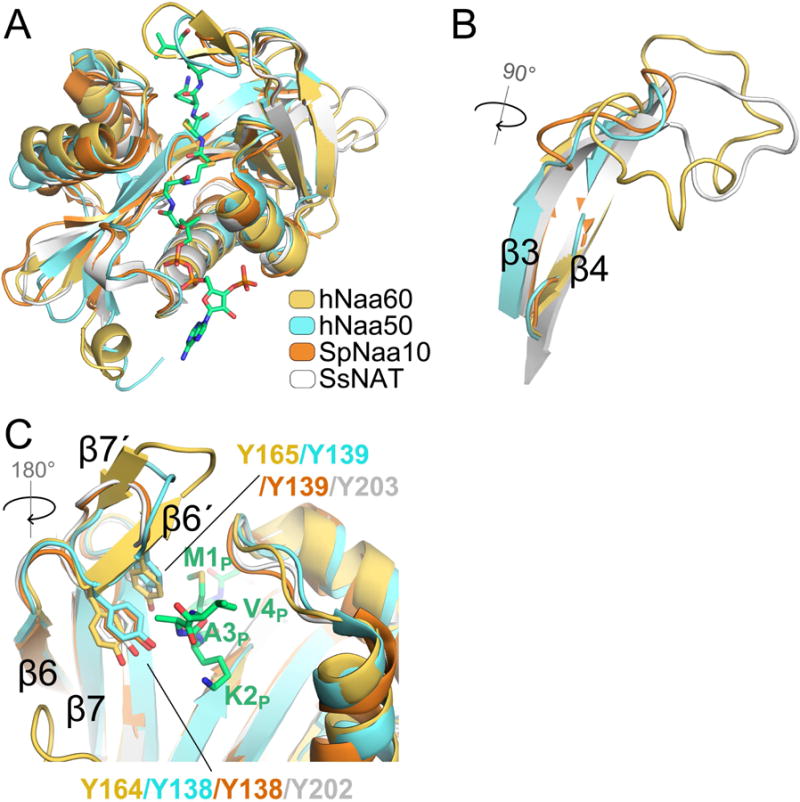
A) Superimposition of hNaa60 (yellow) with hNaa50 (cyan), SpNaa10 (orange) and ssNAT (white). CoA-Ac-MKAV7 bound to hNaa60 is shown in green sticks. B) Superimposition of hNaa60 (yellow), hNaa50 (cyan), SpNaa10 (orange) and SsNAT (white) β3–β4 loop. C) Superimposition of hNaa60 (yellow), hNaa50 (cyan), SpNaa10 (orange) and SsNAT (white) β6–β7 loop containing a hairpin structure with two shorts strands (β6′ and β7′).
A superposition of the hNaa60/CoA-Ac-MKAV7 structure with other NAT structures also reveals that the active site of hNaa60 most closely resembles the active site of hNaa50, in which two residues contribute to general base catalysis. In hNaa50, these residues are Y73 and H112 and the corresponding residues in hNaa60 are Y97 and H138 (Figure 3A). Both structures also show an ordered water molecule in a similar position to facilitate proton extraction from the target amine. The water molecule is coordinated by the side chain of Y97, backbone carbonyl of L98, backbone nitrogen of H138, and the M1p N-terminus (Figure 3A).
Figure 3. Catalytic mechanism and peptide binding site of hNaa60.
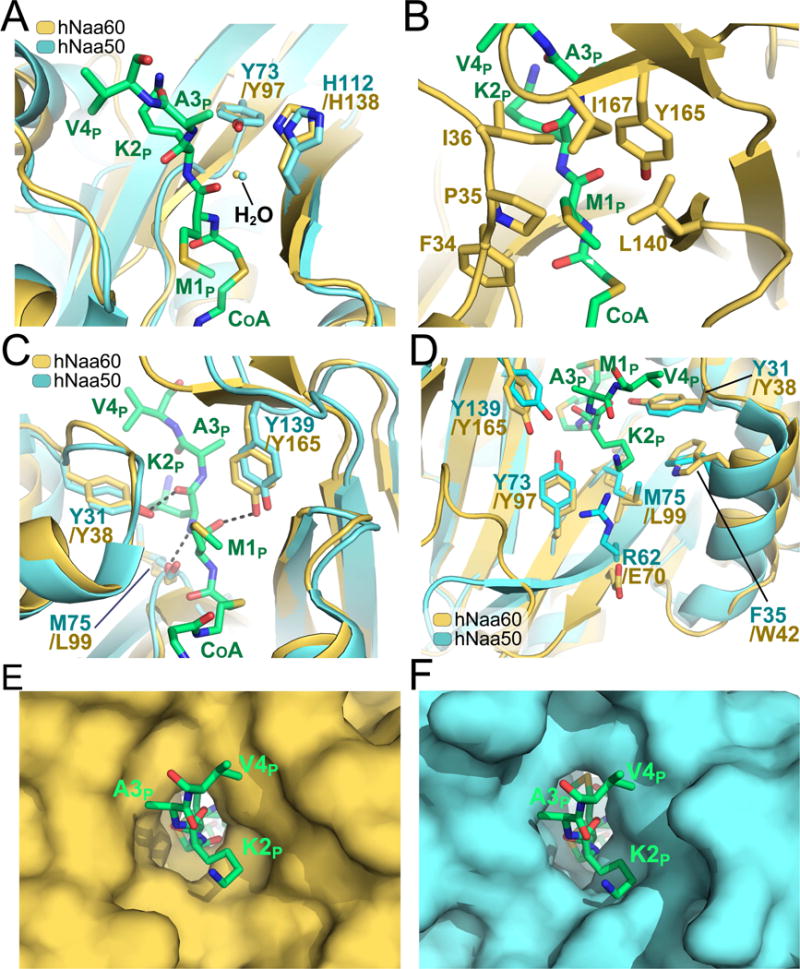
A) Superimposition of the hNaa60 and hNaa50 active site (shown in yellow and cyan respectively). Residues associated with catalysis (Y97, H138 of hNaa60 and Y73, H112 of Naa50) are shown in sticks. An ordered water molecule involved in catalysis is shown in yellow and cyan for the hNaa60 and hNaa50 structure respectively. The bisubstrate analogue CoA-Ac-MKAV7 is shown in green sticks in all figures (A–F). B) Residues F34, P35 I36, L140, Y165 and I167 (yellow sticks) are forming a hydrophobic pocket surrounding the substrate peptide M1p side chain. C) Structural alignment of hNaa60 and hNaa50 active sites. Residues L99, Y38, and Y165 of hNaa60 that plays important roles in peptide anchoring and which are structurally conserved between hNaa60 and hNaa50 are shown as yellow sticks. D) Superimposition of the cavity surrounding the hNaa60 and hNaa50 peptide binding site. E) Active site surface representation of hNaa60. F) Active site surface representation of hNaa50.
In addition to similarities in the active site, hNaa60 and hNaa50 both contain a conserved hydrophobic core that recognizes Met1 of the cognate N-terminal substrate. Specifically, F34, P35, I36, L140, Y165 and I167 of hNaa60, which are conserved in hNaa50, surround the side chain of M1p (Figure 3B). The peptide is anchored in the active site of the enzyme through several hydrogen bonds. Y38 and Y165, both of which are conserved between hNaa50 and hNaa60, form hydrogen bonds to the carbonyl groups of M1p and K2p respectively, while the backbone carbonyl of L99 forms a hydrogen bond to the backbone amide of M1p (Figure 3C).
There are also major differences between these enzymes. While the hNaa50 structure reveals a second hydrophobic pocket for residue 2p, the corresponding region of hNaa60 is more solvent exposed and thus can accommodate more variability in position 2p (Figure 3D, E, F). Indeed, K2p of the MKAV7 peptide region of the CoA-Ac-MKAV7 bisubstrate inhibitor, as well as A3p and V4p side chains are not making significant interactions to the protein. The larger and more solvent exposed cavity of hNaa60 suggest that this enzyme is able to accommodate larger and more polar residues at position 2p and 3p compared to hNaa50 which previously have been shown to prefer methionine starting substrates with hydrophobic amino acids like leucine or valine in position 2p.
Mutational analysis of hNaa60 shows good correlation with the structure
In order to examine the importance of specific residues on the catalytic function and substrate binding of hNaa60, several hNaa60 point mutants were prepared and tested for activity using the substrate peptide MKAV24, representing a substrate that previously were shown to be acetylated by hNaa60 in vivo (Aksnes et al., 2015b). Mutation of residues predicted to be directly involved in the catalysis of hNaa60 resulted in severe impairments. An Y97F point mutant showed complete loss of catalytic activity (Figure 4) confirming that this residue is important for hNaa60 catalysis. The H138F and H138A mutants were unstable and could not be purified (data not shown) suggesting that this residue is not only involved in catalysis, but is also important for maintaining the overall structure of hNaa60.
Figure 4. Catalytic parameters of hNaa60 WT and mutants.
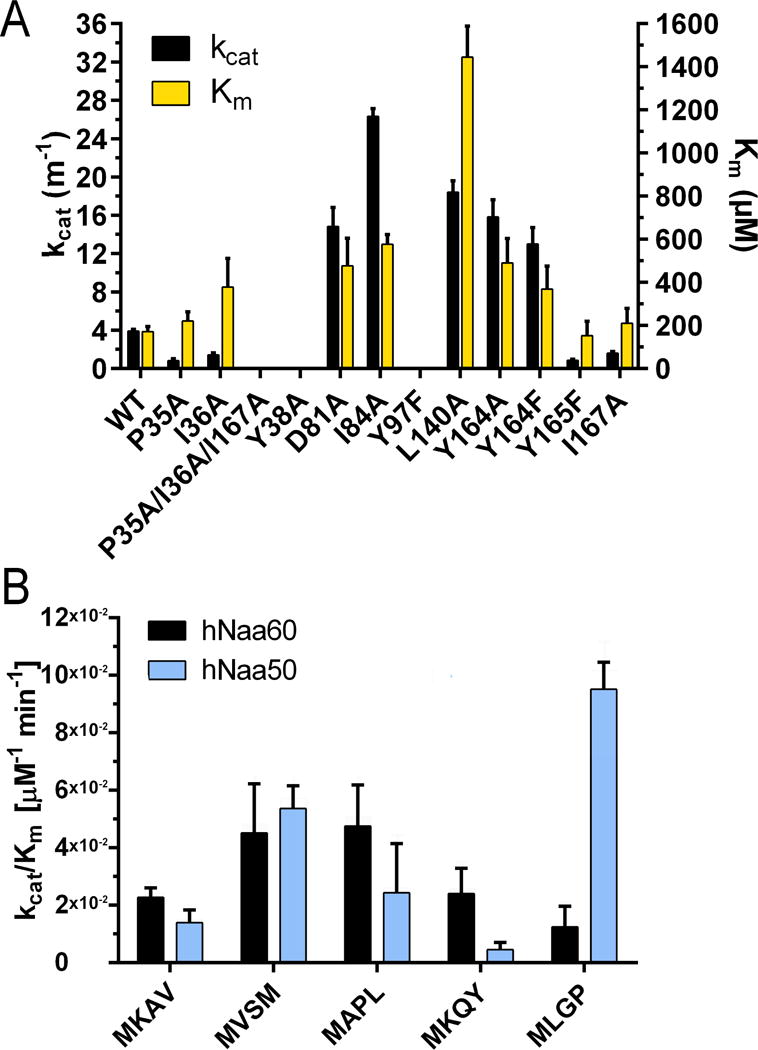
A) kcat values (black bars) and Km values (yellow bars) of wild-type hNaa60 and selected hNaa60 mutants toward the substrate polypeptide MKAV24. B) Catalytic efficiency of hNaa60 (black bars) and hNaa50 (blue bars) towards different substrate polypeptides representing previously identified hNaa60 or hNaa50 substrates in vivo. See also Tables S1 and S2.
We also prepared several point mutants of residues that form the hydrophobic pocket surrounding the M1p side chain. P35A, I36A and I167A all showed an approximate 2.5–4 fold fold reduction in kcat and a small increase in Km when tested towards the substrate polypeptide MKAV24 (Figure 4A). Surprisingly, the hNaa60 L140A mutant had a 4–5 fold increase in kcat but at the same time a 5–10 fold increase in Km (Figure 4A). Thus, despite the increased kcat, the catalytic efficiency (kcat/Km) of this mutant is approximately 0.5 fold of hNaa60 WT (Table 2). In comparison, the catalytic efficiency of the P35A, I36A and I167A point mutants had an approximate 6-fold, 6-fold and 3-fold reduction compared to the hNaa60 WT, respectively (Table 2). We also created a P35A/I36A/I167A triple mutant, but we were not able to detect any catalytic activity for this mutant in our assay (Table 2). Together these data show that the M1p hydrophobic pocket is important for substrate-specific acetylation.
Table 2.
Catalytic efficiency for hNaa60 WT and mutants
| Enzyme | Kcat/Km (μM−1 min−1) | Wild-type efficiency (%) |
|---|---|---|
| WT | 2. 27 ×10−2 | 100 |
| P35A | 0.36 ×10−2 | 16.5 |
| I36A | 0.36 ×10−2 | 16.6 |
| P35A/I36A/I167A | ND | <10 |
| Y38A | ND | <10 |
| D81A | 3.11 ×10−2 | 136.9 |
| I84A | 4.55 ×10−2 | 200.4 |
| Y97F | ND | <10 |
| L140A | 1.26 ×10−2 | 56.0 |
| Y164A | 3.21 ×10−2 | 141.7 |
| Y164F | 3.51 ×10−2 | 154.9 |
| Y165F | 0.54 ×10−2 | 24.4 |
| I167A | 0.74 ×10−2 | 33.3 |
In order to probe the importance of substrate polypeptide anchoring in the active site, we mutated tyrosines Y38 and Y165 that showed hydrogen bonds with the peptide substrate backbone (Figure 3C). Y165 also forms parts of the hydrophobic pocket surrounding the side chain peptide substrate M1p, however by creating an Y165F point mutant only the hydroxyl group involved in peptide anchoring will be removed and the hydrophobic pocket will remain unaffected. The hNaa60 Y38A variant resulted in a complete loss of catalytic activity, while the hNaa60 Y165F mutant had approximately 4 fold reduction in catalytic efficiency (Table 2). Together, these studies show that Y38 and Y165 play important roles in peptide binding.
To probe the substrate selectivity of hNaa60 for comparison with hNaa50, we determined kinetic constants for both hNaa60 and hNaa50 towards several different peptides representing N-termini that were previously identified as substrates of either hNaa60 or hNaa50 in vivo (Aksnes et al., 2015b; Van Damme et al., 2011a). The catalytic efficiency of the two enzymes is similar for several of the tested substrates but there are also differences (Figure 4B). As expected hNaa50 has the highest catalytic efficiency for the substrate polypeptides MLGP24 and MVSM24. hNaa60 on the other hand has the highest catalytic efficiency for peptide substrates MAPL24, MVSM24, MKAV24 and MKQY24. Interestingly, while hNaa60 had approximately the same catalytic efficiency for MKAV24 and MKQY24 with larger more bulky side chains in position 3p and 4p, hNaa50 had a 3-fold lower catalytic efficiency for MKQY24 compared to MKAV24. This is consistent with the larger and more solvent exposed cavity seen in hNaa60 relative to hNaa50 around the K2p, A3p and V4p residues of the bisubstrate analogue.
The hNaa60 specific β3–β4 loop is essential for protein stability
The elongated 22-residue β3–β4 loop (residues 73–94) is a structure not found in most NATs. In SpNaa10, hNaa50, and hNaa40, β3 and β4 are linked together with only a short beta hairpin. In contrast, for both SsNAT and hNaa60, β3 and β4 are linked together by a large elongated loop, with the loop being significantly larger in hNaa60 (Figure 2B). The β3–β4 loop of hNaa60 folds back towards the enzyme core and peptide portion of the bisubstrate inhibitor and make multiple h-bonds and van der Waals contacts to residues in β-strands 5, 6 and 7 (Figure 5). D81 and D83 of the β3–β4 loop make h-bonds to H138 and H157 of the β5 and β7 strands respectively (Figure 5A) and the backbone amide nitrogen of I77 makes a water mediated hydrogen bond to the phenol oxygen of Y136 (not shown). In addition, E80, D81 and the backbone carbonyl of I84 form hydrogen bonds with Y164, T176 and Y180 of the β6 strand, respectively. Several residues of the β3–β4 loop (I77, I84, L85,) also participate in van der Waals interactions with residues of the β5–β6–β7 core (V95, A134, Y136, V178 (Figure 5B). Interestingly, despite the large number of contacts between the β3–β4 loop and β5, β6 and β7 strands, mutational analysis suggests that the positioning of this loop is easily disrupted. Mutating some of the residues that form these contacts (D81A, I84A or Y164A/F) all result in altered catalytic parameters. Surprisingly, these mutants resulted in a 3–6 fold increase in kcat, but also a 2–4 fold increase in the Km of the reaction (Figure 4A), resulting in a modest overall effect on catalytic efficiency (Table 2). This could indicate that mutating residues involved in the contact between β3–β4 loop and β5, β6 and β7 strands is altering the structure of the enzyme in a way that only affects substrate peptide binding and catalysis in subtle ways.
Figure 5. The hNaa60-specific β3–β4 loop.
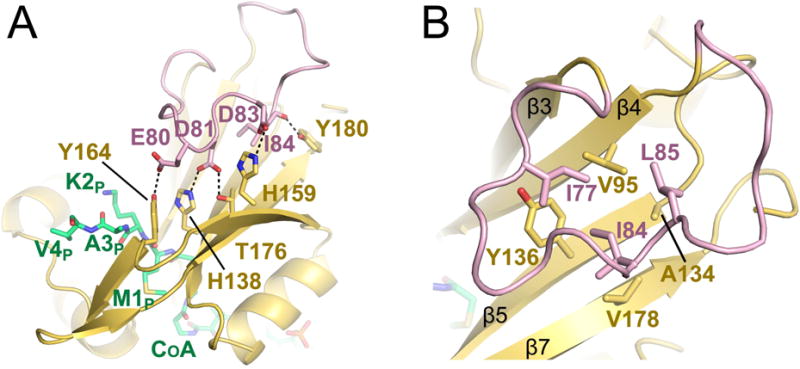
A) Residues D81 and D83 (pink sticks) of the extended β3–β4 loop of hNaa60 (shown in pink) that form salt bridges with residues H138 and H159 (yellow sticks) of β5 and β7 are shown. E80 and D81 (pink sticks) that form hydrogen bonds with Y164 and T176 (yellow sticks) of β6 and the carbonyl backbone of I84 (pink sticks) that form a hydrogen bond with Y180 (yellow sticks) are also shown. The CoA-Ac-MKAV is shown in green sticks. B) Residues I77, I84 and L85 (pink sticks) that form a hydrophobic interior of the loop and participate in van der Waals interactions with residues V95, A134, Y136 and V178 (yellow sticks) of β5, β6 and β7 strands is shown. See also Figure S4.
In order to further investigate the effects of mutating these residues, we analyzed the stability of these point mutants by differential scanning fluorimetry (DSF) as have been described previously (Figure S4) (Bustad et al., 2012). Apo hNaa60 WT had a melting point of 47.9°C and was clearly stabilized both by the addition of Ac-CoA (Tm increased gradually from 48 °C to 54.7 °C by addition of 0.16×–10× Ac-CoA) and the bisubstrate analogue CoA-Ac-MAPL7 (Tm ranging from 47.6 – 53.2 °C with increasing bisubstrate analogue concentrations) (Figure S4A and B). As expected, the I36A point mutant that had severe catalytic effects due to disruption of the M1p hydrophobic pocket, did not show any signs of destabilization while hNaa60 mutants involved in forming contacts between the β3–β4 loop and β5, β6 and β7 strands, D81A, I84A and Y164F, were all clearly destabilized compared to hNaa60 WT (Figure S4C–F). Further, a higher initial fluorescence was seen for both hNaa60 D81A and I84A indicating that more hydrophobic areas are exposed to the solvent, most likely due to a destabilized β3–β4 loop.
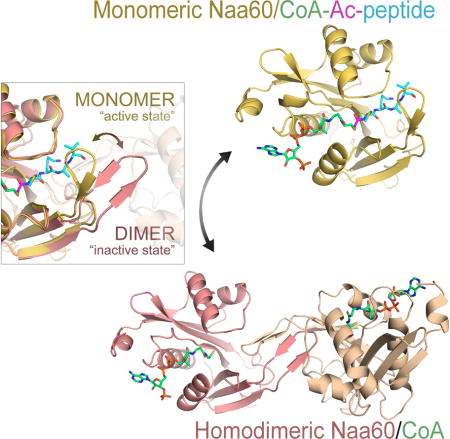
hNaa60 dimerizes through the hNaa60-specific β6–β7 loop in the absence of substrate peptide
In order to determine if hNaa60 undergoes any structural changes upon N-terminal peptide substrate binding, we determined the crystal structure of the hNaa60/CoA complex. We obtained crystals that formed in the P21 space group and determined the structure by molecular replacement using the protein portion of the SsNAT (PDB ID: 4LX9) as a search model. The crystals contained four hNaa60/CoA complexes per asymmetric unit and refined to a Rwork/Rfree of 0.2072/0.2547 with good geometry at 1.95 Å resolution (Table 1). Interestingly, in contrast to the monomeric hNaa60/CoA-Ac-MKAV7 complex, the hNaa60/CoA crystals show two symmetrical protein dimers, where dimer interactions are nucleated by the β6–β7 loops that form a four-stranded sheet that extends into the peptide-binding sites of both protomer of the dimer (Figure 6A). A superposition of the hNaa60/CoA-Ac-MKAV7 complex with a protomer of the hNaa60/CoA complex shows that the only significant structural change is a rotation of the β6–β7 loop in the hNaa60/CoA-Ac-MKAV7 by about 45° to accommodate the insertion of the β6–β7 loop from the opposing protomer of the hNaa60/CoA dimer (Figure 6B). The superimposition also reveals that the hNaa60 homodimer is incompatible with peptide binding by hNaa60. Specifically, the β6–β7 loop of the one protomer protrudes into the catalytic site of the second protomer of the dimer, leaving no room for a substrate peptide to bind (Figure 6C).
Figure 6. hNaa60 dimerization.
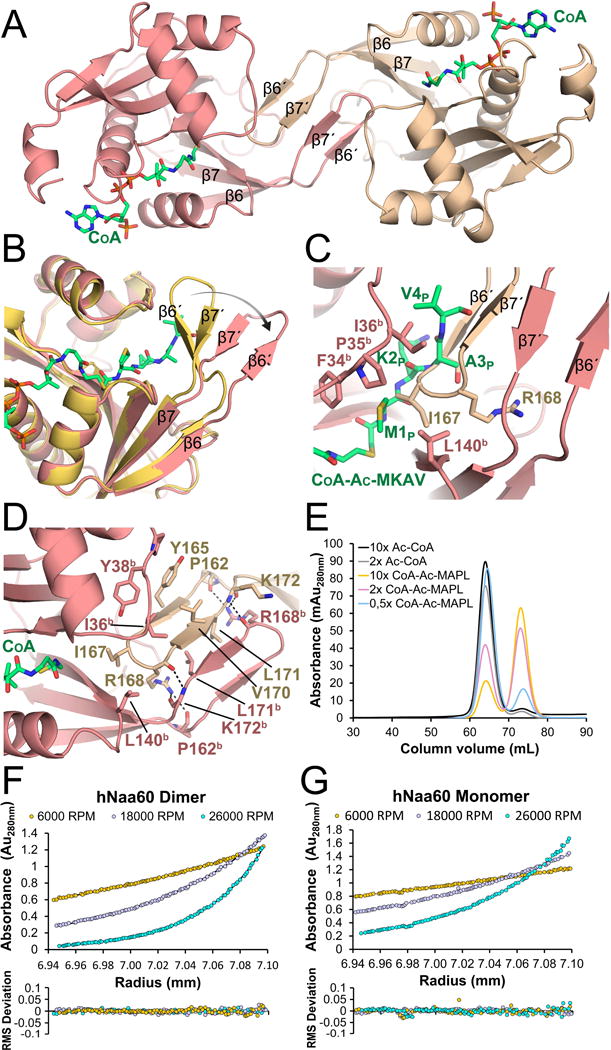
A) Crystal structure of the hNaa60-CoA homodimer complex. CoA is shown in green sticks. B) Superimposition of the β6–β7 loop in a monomeric state (shown in yellow) and in a dimeric state (shown in red). C) Superimposition of the hNaa60/CoA-Ac-MKAV7 structure (only showing CoA-Ac-MKAV7) with the hNaa60/CoA dimer. D) Contacts between the two hNaa60 protomers. The side-chain of K172b was omitted for clarity. E) Size exclusion chromatography of hNaa60 1–184 mixed with either 2× or 10× excess of Ac-CoA or 0.5×, 2× or 10× excess of the bisubstrate analogue CoA-Ac-MAPL7. The first peak corresponds to a hNaa60 homodimer and the second peak corresponds to a hNaa60 monomer. F) Sedimentation equilibrium analytical ultracentrifugation experiments of hNaa60 mixed with the bisubstrate analogue CoA-Ac-MAPL7, which eluted as a dimer. G) Sedimentation equilibrium analytical ultracentrifugation experiments of hNaa60 mixed with the bisubstrate analogue CoA-Ac-MAPL7 which eluted as a dimer. See also Figure S5.
Numerous contacts mediate hNaa60/CoA homodimerization, several of which participate in peptide substrate anchoring in the hNaa60 monomer (Figure 6D). L171 from each subunit make van der Waals contacts across the dimer. Van der Waals contacts are also mediated between Y165 and Y38b and P39b of the core (b designates the second protomer of the dimer), and I167 and V170 with L140b and I36b of the core, respectively. In addition, I36b of the core makes van der Waals interactions with the main chain of the beta-hairpin formed by residues S166-I167-R168-G169. R168 also hydrogen bonds to the main chain carbonyls of P162b and K172b of the core. Many of the residues in the β6–β7 loop that interact with the opposing subunit are specific for hNaa60 (Figure 1) and no other NAT structure determined to date shows this type of homodimerization (Liszczak et al., 2011; Liszczak et al., 2013; Liszczak and Marmorstein, 2013; Magin et al., 2015).
To investigate whether hNaa60 dimers are formed in solution, we performed size-exclusion chromatography and analytic ultracentrifugation (AUC). On a size exclusion chromatography column, hNaa60 1–184 with Ac-CoA elutes at a column volume corresponding to twice the theoretical size of the enzyme (Figure 6E). However, upon addition of 2 equivalents of the bisubstrate analogue CoA-Ac-MAPL7, more than 50% of the hNaa60 elutes as an apparent monomer from a size exclusion chromatography column. To further corroborate these findings, we performed AUC on the fractions of hNaa60 bound to Ac-CoA that eluted off the size exclusion column in an apparent dimer peak (Figure 6F), and on the fractions of hNaa60 bound to CoA-Ac-MAPL7, which eluted as an apparent monomer peak (Figure 6G). We performed the centrifugation at three different speeds and three different concentrations. The data fit extremely well to a hNaa60/CoA-Ac-MAPL7 monomer with a molecular weight of 22.9 kD, and a hNaa60/CoA dimer with a molecular weight of 39.9 kD, which are very close to the theoretical molecular weights of 22.6 kD and 43.6 kD, respectively. This data supports the peptide substrate dependent dimer to monomer transition of hNaa60 suggested by the crystal structures.
DISCUSSION
In this study, we have crystallized and biochemically characterized hNaa60, the only human NAT identified so far with distinct organellar localization and that primarily acetylates membrane proteins. As previously shown in vivo, hNaa60 has an organellar localization that is dependent on the C-terminal 60 amino acids (residues 182–242) (Aksnes et al., 2015b). The present study reveals that hNaa60 1–180 forms a GNAT fold that highly resembles the hNaa50 structure. Our studies also show that the C-terminus of hNaa60 is not necessary for its biochemical function. Secondary structure predictions suggest that the C-terminus of hNaa60 form two alpha-helices, which either facilitate the anchoring of hNaa60 to the membrane peripherally or form transmembrane helices (Aksnes et al., 2015b) (Figure S1).
In order to study the molecular basis for substrate specific acetylation by Naa60 we designed and synthesized two bisubstrate analogues, one of which was co-crystallized with Naa60 1–184. This approach was chosen as a result of difficulties in obtaining a crystal structure of a Naa60/CoA/peptide ternary complex. A similar approach was previously used in the crystallization of the p300/CBP transcriptional coactivator (Liu et al., 2008). Liu and colleagues prepared a bisubstrate analogue with CoA attached to the side chain of lysine, which both facilitated crystallization of this enzyme and enabled investigation of substrate-enzyme interactions and mechanism. Bisubstrate analogues have also been successfully used as enzyme-specific and potent NAT inhibitors (Foyn et al., 2013), and was also used in order to crystallize and study the SpNatA complex (Liszczak et al., 2013). The bisubstrate analogues presented here represent the first hNaa60 specific bisubstrate analogues and the second case where such analogues have been used as a tool for the investigation of amino acid substrate interactions in NAT enzymes.
The overall structure of hNaa60 aligns very well with other available NAT-structures and is very similar to the overall structure of hNaa50 (rmsd = 1.59 Å). Of particular interest to us was the active site of hNaa60 and whether there were any differences in its substrate peptide binding and catalytic mechanism as compared to hNaa50 and other NATs. Our data reveal that hNaa60 utilizes the same catalytic mechanism as hNaa50, and that all residues that are directly associated with catalysis are conserved between the two enzymes. Substrate peptide anchoring is also highly conserved, in addition to key residues involved in forming a hydrophobic pocket surrounding the M1 side chain of the substrate peptide. However, there are notable differences. In hNaa50, a second hydrophobic pocket is formed around substrate peptide residue 2p, enabling hNaa50 to efficiently acetylate substrates with a methionine in position 1p followed by a hydrophobic residue in position 2p. In comparison, the corresponding region of hNaa60 is more solvent exposed and has more room for substrates with larger charged side chains. Indeed, our enzymatic studies support this hypothesis. hNaa50 has a preference for substrates with a hydrophobic residue in the second position (MLGP24 and MVSM24), while hNaa60 has a preference for the substrates MAPL24, MVSM24, MKAV24 and MKQY24. Interestingly, although the catalytic efficiency towards the substrate polypeptide MKAV24 was very similar for hNaa50 and hNaa60, the catalytic efficiency towards another substrate peptide with a methionine in position 1p followed by a lysine in position 2p, MKQY24, was a 3-fold lower for hNaa50 compared to hNaa60. These findings suggest that although peptide anchoring in hNaa50 and hNaa60 mainly occurs through the first two residues of the substrate peptide, the amino acids in position 3p and 4p also may have a significant impact, and further that hNaa50 and hNaa60 differ in their amino acid preference in positions 3p and 4p. In this case, most likely the aspargine in position 3p of MKQY24 is pushing the substrate towards residues M75 and R62 of hNaa50 causing the peptide to bind improperly, while the more solvent exposed area of hNaa60 harbors enough room here to accommodate this peptide.
Despite the large overall structural similarity between hNaa50 and hNaa60, there are several features that are specific for hNaa60. A prominent difference maps to the β3–β4 loop, which compared to other human NATs is extended by 10–15 residues in hNaa60 (Figure 1). The elongated loop interacts with residues in β-strands β5, β6 and β7, and point mutation of key residues in this loop resulted in a slightly increased catalytic efficiency of the enzyme. The exact function of this loop is still unclear, and interestingly we show that loss of the interaction between the β3–β4 loop and β-strands β5, β6 and β7 not only results in altered acetyltransferase activity, but also cause a substantial drop in protein stability with a melting point that is 5–10 °C lower than wild-type hNaa60.
A second difference between Naa50 and Naa60 maps to the slightly elongated β6–β7 loop of Naa60. This loop mediates dimerization of hNaa60 in the absence of substrate peptide, a feature that, to our knowledge, has not been seen for any other NAT. The β6–β7 loop of each protomer of the dimer extends into the peptide-binding site of the second protomer of the dimer and is stabilized by hydrogen bond and van der Waals interactions. As the hNaa60 dimerization is mediated by the β6–β7 loop of both protomers extending into the active site of each other, it seems very unlikely that hNaa60 can be catalytically active in its dimeric form. However, our size exclusion chromatography and AUC data shows that hNaa60 can dynamically shift between a monomeric and dimeric state. This is also reflected in enzymatic assays; although hNaa60 is purified as a homodimer, this dimer is catalytically active. The substrate peptide therefore likely competes with the β6–β7 loop of the other hNaa60 protomer for binding in the active site. This could be a way to regulate substrate-specificity as the affinity for a potential substrate may have to be higher than the affinity for the other protomer of the dimer. In vivo hNaa60 specifically associates with the Golgi membrane through the C-terminal 60 amino acids (Naa60 182–242). In order for the dimerization of hNaa60 to occur in vivo each hNaa60 monomer has to associate to the membrane in a way that enables homodimerization. Consistent with this, in the hNaa60 homodimer the C-terminal end of both hNaa60 1–184 monomers are pointing in the same direction, supporting that dimerization is compatible with hNaa60 attachment to organellar membranes (Figure S5).
The majority of proteins that are Nt-acetylated are modified co-translationally as they emerge from the ribosomal exit tunnel and several NATs have previously been shown to associate with the ribosome (Arnesen et al., 2005a; Gautschi et al., 2003; Polevoda et al., 2008; Starheim et al., 2008; Starheim et al., 2009). As Naa60 was shown to localize to the cytosolic side of Golgi membranes, and as the bulk of all detected Naa60 is associated with membranes (Aksnes et al., 2015b) it is likely that Naa60 does not directly associate with ribosomes but rather acts post-translationally acetylating transmembrane proteins with N-termini facing the cytosol. Naa60 has yet to be studied in the presence of membranes in vitro, but based on the current data we suspect that membrane interaction will have limited effect on Naa60 biochemical activity and specificity per se. Rather, it may anchor Naa60 in close proximity to its transmembrane substrates. The exact mechanism for Naa60 anchoring to cell membranes also remains unknown and the functional implications of the Naa60-dimerization in terms of functionality and membrane anchoring will be a topic of future studies.
Several reports over the last decade have either described NATs as lysine acetyltransferases or have reported roles for NATs in lysine acetylation. Most of these reports describe Naa10 and the KAT-activity of this enzyme (Lim et al., 2006; Yoon et al., 2014), but there are also reports describing Naa40 (Liu et al., 2009), Naa50 (Chu et al., 2011; Evjenth et al., 2009) and Naa60 (Yang et al., 2011) as lysine acetyltransferases. Recently, depletion of several NAT subunits were shown to affect H3 and H4 lysine acetylation levels in drosophila cells either directly or indirectly (Feller et al., 2015) and Naa50 was picked up in a screen for HAT enzymes with a photoclickable CoA-Lys bisubstrate analogue (Montgomery et al., 2014). The role of NATs in lysine acetylation is however controversial (Arnesen et al., 2005b; Friedmann and Marmorstein, 2013) and recently we showed that Naa10 has high in vitro NAT activity, but no detectable in vitro KAT-activity towards several of the earlier reported Naa10 KAT-substrates (Magin et al., 2016). It has previously also shown that although Naa50 was able to acetylate lysine side chains to some extent, Naa50 had approximately a 1000 fold higher NAT activity compared to KAT activity in vitro (Evjenth et al., 2009). With the emergence of experimental crystal structures of NATs, it is becoming evident that these enzymes predominantly are NATs with N-terminal acetylation as their main role. Even though NATs share the same overall GNAT fold as several KAT enzymes, a key difference between NATs and KATs is the β6–β6 loop which in NATs hangs over the peptide binding site creating a tunnel that accommodates substrate N-termini but not lysine side chains (Liszczak et al., 2011; Liszczak et al., 2013; Magin et al., 2015). This is also the case for Naa60, which despite having a slightly more solvent exposed peptide binding site would not be able to accommodate lysine side chains in its closed state. Although Naa60 previously was described as a histone acetyltransferase (Yang et al., 2011), the Naa60 crystal structure reported here together with recent data described elsewhere (Aksnes et al., 2015b; Van Damme et al., 2011b) strongly suggest that Naa60 is predominantly a NAT with the same globular fold and similar catalytic mechanism as all other NAT enzymes characterized thus far.
EXPERIMENTAL PROCEDURES
hNaa60 expression and protein purification
A C-terminal truncation construct coding for His-SUMO-hNaa60 1–184 was expressed in Rosetta (DE3) pLys competent cells over night at 17°C. Cells were lysed in lysis buffer ((25 mM Tris-HCl (pH 8.0), 1 M NaCl, 10 mM β-mercaptoethanol and 10 μg/ml phenylmethanesulfonylfluoride) by sonication, cell lysate centrifuged at 17000 g for 30 minutes and overexpressed His-tagged proteins were isolated by passing the supernatant over nickel resin. The resin was washed with wash buffer (25 mM Tris-HCl (pH 8.0), 250 mM NaCl, 20 mM Imidazole, 10 mM β-mercaptoethanol) and bound proteins eluted by slowly increasing the imidazole concentration to 300 mM. The His-SUMO tag was removed by adding SUMO protease (Ulp-1) to the eluted protein while the protein was dialyzed into a buffer without imidazole (25 mM Tris-HCl (pH 8.0), 250 mM NaCl, 10 mM β-mercaptoethanol). The His-SUMO tag was removed from the sample by reverse nickel purification, the flow through collected and dialyzed into size exclusion buffer (25 mM HEPES (pH 7.5), 300 mM NaCl, 1 mM DTT). The dialyzed sample was concentrated to a final volume of 1 mL and loaded on a HiLoad Superdex 200 size exclusion chromatography column. Fractions containing hNaa60 1–184 were collected and used for protein crystallization. For a thorough description of protein expression and protein purification for all experiments, see supplemental experimental procedures.
Synthesis of bisubstrate analogues
A detailed description of the synthesis of bisubstrate analogues can be found in the supplemental experimental procedures. The structure, theoretical molecular weight and experimental molecular weight can be found in Table S3.
Crystallization and data collection
The hNaa60/CoA structure was obtained by mixing 18 mg/ml purified hNaa60 1–184 with 1:3:5 molar ratio of hNaa60:CoA:MAPL7 peptide (although electron density corresponding to the MAPL7 peptide was not observed in the final structure). Crystals were obtained by hanging-drop vapor diffusion at 20°C for 1–3 days against a reservoir solution of 3% Tascimate, 0.1 M Bistris pH 6.5 and 16% PEG 3350. For the hNaa60/CoA-Ac-MKAV7 bisubstrate analogue structure, the N- and C-terminal truncation variant hNaa60 5–184 was purified, concentrated to 13 mg/ml and mixed with the bisubstrate analogue CoA-Ac-MKAV7 at a 1:3 molar ratio. Crystals were obtained by hanging-drop vapor diffusion at 20°C over 3 months against a reservoir solution of 4% Tascimate pH 4.0 and 12% PEG 3500. hNaa60:CoA crystals were transferred to a series of cryoprotectant solutions containing well solution supplemented with 300 mM NaCl and 5%, 10% and 25% glycerol prior to flash freezing in liquid nitrogen. hNaa60/CoA-Ac-MKAV7 crystals were transferred into a cryoprotectant containing it’s reservoir solution supplemented with 20% glycerol. Both data sets were collected at beamline X29A at the National Synchrotron Light source (Brookhaven National Laboratory) and were processed using HKL2000 (Otwinowski and Minor, 1997).
Structure determination and refinement
The binary hNaa60/CoA complex and hNaa60/CoA-Ac-MKAV7 complex was determined by molecular replacement using a structure of SsNAT prepared with CHAINSAW (Stein, 2008) without solvent molecules or cofactors as a search model. Molecular replacement was performed with Phaser in the CCP4 suite. Initial rounds of manual model building were performed with Coot (Emsley and Cowtan, 2004), and all subsequent rounds of refinement were performed using Phenix refine and Coot interchangeably. For both hNaa60 complexes, the cofactor/bisubstrate analogue was built into F0 – Fc electron density maps. Refinement statistics are shown in Table 1.
In vitro acetylation assays
A colorimetric acetylation assay was used to measure the catalytic activity of hNaa60 variants (Thompson et al., 2004). Purified enzymes were mixed with substrate peptides and Ac-CoA in acetylation buffer (50 mM HEPES pH 7.5, 100 mM NaCl, and 2 mM EDTA), and reactions were quenched with quenching buffer (3.2 M guanidinium-HCl, 100 mM sodium phosphate dibasic pH 6.8). To measure CoA production, DTNB (2 mM final, dissolved in 100 mM sodium phosphate dibasic pH 6.8 and 10 mM EDTA) was added to the quenched reaction and the absorbance at 412 nm was measured. Background absorbance was determined in negative controls, and subtracted from the absorbance determined in each individual reaction. Thiophenolate production was quantified assuming ɛ = 13.7 × 103 M−1 cm−1. Product formation was measured in the linear phase of the reaction, and kinetic constants were determined by GraphPad Prism. Peptide sequences and a full description of experiments determining IC50 values, Kiapp and modality of the two inhibitors can be found in the supplemental experimental procedures.
Analytical Ultracentrifugation
Sedimentation equilibrium analytical ultracentrifugation experiments were performed at 4 °C with absorbance optics at 280 nm using a Beckman Optima XL-I analytical ultracentrifuge. We employed the use of a 4-hole rotor containing six-channel centerpieces with quartz windows, spinning at 6000, 18000 and 26000 rpm. hNaa60 was mixed with a five molar excess of either acetyl-CoA or CoA-Ac-MKAV7 and run on a superdex 75 gel filtration column. Protein samples were analyzed at A280 = 0.7, 0.5, and 0.3 in gel filtration buffer (25 mM HEPES pH 7.5, 300 mM NaCl, 1 mM DTT). Data for each speed were collected in quadruplicate. The viscosity of the samples were estimated using Sedenterp (http://sednterp.unh.edu/) and the most representative runs were included to calculate a theoretical molecular weight using the program HeteroAnalysis (http://www.biotech.uconn.edu/auf/?i=aufftp)
Supplementary Material
Highlights.
Crystal structure of Naa60, a unique membrane-associated N-terminal acetyltransferase
Naa60-specific loops are important for enzyme stability and substrate binding
Naa60 β6–β7 loop mediates dimer to monomer transition for substrate-specific binding
A novel CoA-peptide bisubstrate Naa60-inhibitor was developed and co-crystallized
Acknowledgments
We thank John Domsic and Adam Olia for help with obtaining crystal data sets at the synchrotron at Brookhaven National Labs, and for help with Molecular Replacement; and Aurora Martinez, Helene J. Bustad and Jarl Underhaug for providing infrastructure and help with performing and analyzing results from differential scanning analysis of wild-type hNaa60 and mutants. We also thank Jan Bjørnstad at Statistics Norway for deriving a function to determine the standard deviations of Ki. These studies were supported by grants from the Norwegian Cancer Society (to T.A.), The Bergen Research Foundation BFS (to T.A.), the Research Council of Norway (grant 230865 to T.A), the Western Norway Regional Health Authority (to T.A.), and the National Institutes of Health (grants R01 GM060293 to R.M. and T32 GM071339 to R.S.M).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
S.I.S and R.S.M designed and performed experiments described in the manuscript. H.F. designed, synthesized and tested bisubstrate analogues. S.I.S prepared figures and wrote the manuscript. B.E.H designed and supervised synthesis of bisubstrate analogues. R.M and T.A designed and supervised experiments by S.I.S and R.M. All authors read, contributed and approved the submitted manuscript.
References
- Aksnes H, Hole K, Arnesen T. Molecular, cellular, and physiological significance of N-terminal acetylation. International review of cell and molecular biology. 2015a;316:267–305. doi: 10.1016/bs.ircmb.2015.01.001. [DOI] [PubMed] [Google Scholar]
- Aksnes H, Van Damme P, Goris M, Starheim KK, Marie M, Stove SI, Hoel C, Kalvik TV, Hole K, Glomnes N, et al. An organellar nalpha-acetyltransferase, naa60, acetylates cytosolic N termini of transmembrane proteins and maintains Golgi integrity. Cell reports. 2015b;10:1362–1374. doi: 10.1016/j.celrep.2015.01.053. [DOI] [PubMed] [Google Scholar]
- Arnesen T, Anderson D, Baldersheim C, Lanotte M, Varhaug JE, Lillehaug JR. Identification and characterization of the human ARD1-NATH protein acetyltransferase complex. Biochem J. 2005a;386:433–443. doi: 10.1042/BJ20041071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnesen T, Kong X, Evjenth R, Gromyko D, Varhaug JE, Lin Z, Sang N, Caro J, Lillehaug JR. Interaction between HIF-1 alpha (ODD) and hARD1 does not induce acetylation and destabilization of HIF-1 alpha. FEBS Lett. 2005b;579:6428–6432. doi: 10.1016/j.febslet.2005.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnesen T, Van Damme P, Polevoda B, Helsens K, Evjenth R, Colaert N, Varhaug JE, Vandekerckhove J, Lillehaug JR, Sherman F, et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc Natl Acad Sci U S A. 2009;106:8157–8162. doi: 10.1073/pnas.0901931106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnia R, Panic B, Whyte JR, Munro S. Targeting of the Arf-like GTPase Arl3p to the Golgi requires N-terminal acetylation and the membrane protein Sys1p. Nat Cell Biol. 2004;6:405–413. doi: 10.1038/ncb1120. [DOI] [PubMed] [Google Scholar]
- Bienvenut WV, Sumpton D, Martinez A, Lilla S, Espagne C, Meinnel T, Giglione C. Comparative large scale characterization of plant versus mammal proteins reveals similar and idiosyncratic N-alpha-acetylation features. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.M111.015131. M111 015131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustad HJ, Skjaerven L, Ying M, Halskau O, Baumann A, Rodriguez-Larrea D, Costas M, Underhaug J, Sanchez-Ruiz JM, Martinez A. The peripheral binding of 14-3-3gamma to membranes involves isoform-specific histidine residues. PloS one. 2012;7:e49671. doi: 10.1371/journal.pone.0049671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey JP, Stove SI, McGorrian C, Galvin J, Blenski M, Dunne A, Ennis S, Brett F, King MD, Arnesen T, et al. NAA10 mutation causing a novel intellectual disability syndrome with Long QT due to N-terminal acetyltransferase impairment. Scientific reports. 2015;5:16022. doi: 10.1038/srep16022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CW, Hou F, Zhang J, Phu L, Loktev AV, Kirkpatrick DS, Jackson PK, Zhao Y, Zou H. A novel acetylation of beta-tubulin by San modulates microtubule polymerization via down-regulating tubulin incorporation. Mol Biol Cell. 2011;22:448–456. doi: 10.1091/mbc.E10-03-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland RA, Dr. William McDowell Mastin Fund . Enzymes: a practical introduction to structure, mechanism, and data analysis. 2nd. New York: J. Wiley-VCH; 2000. [Google Scholar]
- De Angelis J, Gastel J, Klein DC, Cole PA. Kinetic analysis of the catalytic mechanism of serotonin N-acetyltransferase (EC 2.3.1.87) J Biol Chem. 1998;273:3045–3050. doi: 10.1074/jbc.273.5.3045. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica. Section D, Biological crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Esmailpour T, Riazifar H, Liu LN, Donkervoort S, Huang VH, Madaan S, Shoucri BM, Busch A, Wu J, Towbin A, et al. A splice donor mutation in NAA10 results in the dysregulation of the retinoic acid signalling pathway and causes Lenz microphthalmia syndrome. J Med Genet. 2014;51:185–196. doi: 10.1136/jmedgenet-2013-101660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evjenth R, Hole K, Karlsen OA, Ziegler M, Arnesen T, Lillehaug JR. Human Naa50p (Nat5/San) displays both protein N alpha- and N epsilon-acetyltransferase activity. J Biol Chem. 2009;284:31122–31129. doi: 10.1074/jbc.M109.001347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evjenth RH, Brenner AK, Thompson PR, Arnesen T, Froystein NA, Lillehaug JR. Human protein N-terminal acetyltransferase hNaa50p (hNAT5/hSAN) follows ordered sequential catalytic mechanism: combined kinetic and NMR study. J Biol Chem. 2012;287:10081–10088. doi: 10.1074/jbc.M111.326587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller C, Forne I, Imhof A, Becker PB. Global and specific responses of the histone acetylome to systematic perturbation. Mol Cell. 2015;57:559–571. doi: 10.1016/j.molcel.2014.12.008. [DOI] [PubMed] [Google Scholar]
- Forte GM, Pool MR, Stirling CJ. N-terminal acetylation inhibits protein targeting to the endoplasmic reticulum. PLoS biology. 2011;9:e1001073. doi: 10.1371/journal.pbio.1001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foyn H, Jones JE, Lewallen D, Narawane R, Varhaug JE, Thompson PR, Arnesen T. Design, synthesis, and kinetic characterization of protein N-terminal acetyltransferase inhibitors. ACS chemical biology. 2013;8:1121–1127. doi: 10.1021/cb400136s. [DOI] [PubMed] [Google Scholar]
- Friedmann DR, Marmorstein R. Structure and mechanism of non-histone protein acetyltransferase enzymes. FEBS J. 2013;280:5570–5581. doi: 10.1111/febs.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautschi M, Just S, Mun A, Ross S, Rucknagel P, Dubaquie Y, Ehrenhofer-Murray A, Rospert S. The yeast N(alpha)-acetyltransferase NatA is quantitatively anchored to the ribosome and interacts with nascent polypeptides. Mol Cell Biol. 2003;23:7403–7414. doi: 10.1128/MCB.23.20.7403-7414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hole K, Van Damme P, Dalva M, Aksnes H, Glomnes N, Varhaug JE, Lillehaug JR, Gevaert K, Arnesen T. The human N-alpha-acetyltransferase 40 (hNaa40p/hNatD) is conserved from yeast and N-terminally acetylates histones H2A and H4. PloS one. 2011;6:e24713. doi: 10.1371/journal.pone.0024713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes WM, Mannakee BK, Gutenkunst RN, Serio TR. Loss of aminoterminal acetylation suppresses a prion phenotype by modulating global protein folding. Nature communications. 2014;5:4383. doi: 10.1038/ncomms5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou F, Chu CW, Kong X, Yokomori K, Zou H. The acetyltransferase activity of San stabilizes the mitotic cohesin at the centromeres in a shugoshin-independent manner. J Cell Biol. 2007;177:587–597. doi: 10.1083/jcb.200701043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang CS, Shemorry A, Varshavsky A. N-terminal acetylation of cellular proteins creates specific degradation signals. Science. 2010;327:973–977. doi: 10.1126/science.1183147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvik TV, Arnesen T. Protein N-terminal acetyltransferases in cancer. Oncogene. 2013;32:269–276. doi: 10.1038/onc.2012.82. [DOI] [PubMed] [Google Scholar]
- Lim JH, Park JW, Chun YS. Human arrest defective 1 acetylates and activates beta-catenin, promoting lung cancer cell proliferation. Cancer Res. 2006;66:10677–10682. doi: 10.1158/0008-5472.CAN-06-3171. [DOI] [PubMed] [Google Scholar]
- Liszczak G, Arnesen T, Marmorstein R. Structure of a ternary Naa50p (NAT5/SAN) N-terminal acetyltransferase complex reveals the molecular basis for substrate-specific acetylation. J Biol Chem. 2011;286:37002–37010. doi: 10.1074/jbc.M111.282863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszczak G, Goldberg JM, Foyn H, Petersson EJ, Arnesen T, Marmorstein R. Molecular basis for N-terminal acetylation by the heterodimeric NatA complex. Nature structural & molecular biology. 2013;20:1098–1105. doi: 10.1038/nsmb.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszczak G, Marmorstein R. Implications for the evolution of eukaryotic aminoterminal acetyltransferase (NAT) enzymes from the structure of an archaeal ortholog. Proc Natl Acad Sci U S A. 2013;110:14652–14657. doi: 10.1073/pnas.1310365110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang L, Zhao K, Thompson PR, Hwang Y, Marmorstein R, Cole PA. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature. 2008;451:846–850. doi: 10.1038/nature06546. [DOI] [PubMed] [Google Scholar]
- Liu Z, Liu Y, Wang H, Ge X, Jin Q, Ding G, Hu Y, Zhou B, Chen Z, Ge X, et al. Patt1, a novel protein acetyltransferase that is highly expressed in liver and downregulated in hepatocellular carcinoma, enhances apoptosis of hepatoma cells. Int J Biochem Cell Biol. 2009;41:2528–2537. doi: 10.1016/j.biocel.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Magin RS, Liszczak GP, Marmorstein R. The molecular basis for histone H4-and H2A-specific amino-terminal acetylation by NatD. Structure. 2015;23:332–341. doi: 10.1016/j.str.2014.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magin RS, March ZM, Marmorstein R. The N-terminal Acetyltransferase Naa10/ARD1 Does Not Acetylate Lysine Residues. J Biol Chem. 2016;291:5270–5277. doi: 10.1074/jbc.M115.709428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery DC, Sorum AW, Meier JL. Chemoproteomic profiling of lysine acetyltransferases highlights an expanded landscape of catalytic acetylation. Journal of the American Chemical Society. 2014;136:8669–8676. doi: 10.1021/ja502372j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen JR, Kayne PS, Moerschell RP, Tsunasawa S, Gribskov M, Colavito-Shepanski M, Grunstein M, Sherman F, Sternglanz R. Identification and characterization of genes and mutants for an N-terminal acetyltransferase from yeast. EMBO J. 1989;8:2067–2075. doi: 10.1002/j.1460-2075.1989.tb03615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myklebust LM, Van Damme P, Stove SI, Dorfel MJ, Abboud A, Kalvik TV, Grauffel C, Jonckheere V, Wu Y, Swensen J, et al. Biochemical and cellular analysis of Ogden syndrome reveals downstream Nt-acetylation defects. Hum Mol Genet. 2015;24:1956–1976. doi: 10.1093/hmg/ddu611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Park EC, Szostak JW. ARD1 and NAT1 proteins form a complex that has N-terminal acetyltransferase activity. EMBO J. 1992;11:2087–2093. doi: 10.1002/j.1460-2075.1992.tb05267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimenta-Marques A, Tostoes R, Marty T, Barbosa V, Lehmann R, Martinho RG. Differential requirements of a mitotic acetyltransferase in somatic and germ line cells. Dev Biol. 2008;323:197–206. doi: 10.1016/j.ydbio.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polevoda B, Brown S, Cardillo TS, Rigby S, Sherman F. Yeast N(alpha)-terminal acetyltransferases are associated with ribosomes. J Cell Biochem. 2008;103:492–508. doi: 10.1002/jcb.21418. [DOI] [PubMed] [Google Scholar]
- Polevoda B, Cardillo TS, Doyle TC, Bedi GS, Sherman F. Nat3p and Mdm20p are required for function of yeast NatB Nalpha-terminal acetyltransferase and of actin and tropomyosin. J Biol Chem. 2003;278:30686–30697. doi: 10.1074/jbc.M304690200. [DOI] [PubMed] [Google Scholar]
- Polevoda B, Norbeck J, Takakura H, Blomberg A, Sherman F. Identification and specificities of N-terminal acetyltransferases from Saccharomyces cerevisiae. EMBO J. 1999;18:6155–6168. doi: 10.1093/emboj/18.21.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polevoda B, Sherman F. NatC Nalpha-terminal acetyltransferase of yeast contains three subunits, Mak3p, Mak10p, and Mak31p. J Biol Chem. 2001;276:20154–20159. doi: 10.1074/jbc.M011440200. [DOI] [PubMed] [Google Scholar]
- Popp B, Stove SI, Endele S, Myklebust LM, Hoyer J, Sticht H, Azzarello-Burri S, Rauch A, Arnesen T, Reis A. De novo missense mutations in the NAA10 gene cause severe non-syndromic developmental delay in males and females. European journal of human genetics: EJHG. 2015;23:602–609. doi: 10.1038/ejhg.2014.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathore OS, Faustino A, Prudencio P, Van Damme P, Cox CJ, Martinho RG. Absence of N-terminal acetyltransferase diversification during evolution of eukaryotic organisms. Scientific reports. 2016;6:21304. doi: 10.1038/srep21304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rope AF, Wang K, Evjenth R, Xing J, Johnston JJ, Swensen JJ, Johnson WE, Moore B, Huff CD, Bird LM, et al. Using VAAST to identify an X-linked disorder resulting in lethality in male infants due to N-terminal acetyltransferase deficiency. Am J Hum Genet. 2011;89:28–43. doi: 10.1016/j.ajhg.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunier C, Stove SI, Popp B, Gerard B, Blenski M, AhMew N, de Bie C, Goldenberg P, Isidor B, Keren B, et al. Expanding the Phenotype Associated with NAA10 Related N-terminal Acetylation Deficiency. Hum Mutat. 2016 doi: 10.1002/humu.23001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DC, Monda JK, Bennett EJ, Harper JW, Schulman BA. N-Terminal Acetylation Acts as an Avidity Enhancer Within an Interconnected Multiprotein Complex. Science. 2011;334:674–678. doi: 10.1126/science.1209307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setty SR, Strochlic TI, Tong AH, Boone C, Burd CG. Golgi targeting of ARF-like GTPase Arl3p requires its Nalpha-acetylation and the integral membrane protein Sys1p. Nat Cell Biol. 2004;6:414–419. doi: 10.1038/ncb1121. [DOI] [PubMed] [Google Scholar]
- Shemorry A, Hwang CS, Varshavsky A. Control of protein quality and stoichiometries by N-terminal acetylation and the N-end rule pathway. Mol Cell. 2013;50:540–551. doi: 10.1016/j.molcel.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song OK, Wang X, Waterborg JH, Sternglanz R. An Nalpha-acetyltransferase responsible for acetylation of the N-terminal residues of histones H4 and H2A. J Biol Chem. 2003;278:38109–38112. doi: 10.1074/jbc.C300355200. [DOI] [PubMed] [Google Scholar]
- Starheim KK, Arnesen T, Gromyko D, Ryningen A, Varhaug JE, Lillehaug JR. Identification of the human N(alpha)-acetyltransferase complex B (hNatB): a complex important for cell-cycle progression. Biochem J. 2008;415:325–331. doi: 10.1042/BJ20080658. [DOI] [PubMed] [Google Scholar]
- Starheim KK, Gromyko D, Evjenth R, Ryningen A, Varhaug JE, Lillehaug JR, Arnesen T. Knockdown of human N alpha-terminal acetyltransferase complex C leads to p53-dependent apoptosis and aberrant human Arl8b localization. Mol Cell Biol. 2009;29:3569–3581. doi: 10.1128/MCB.01909-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein N. CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J Appl Crystallogr. 2008;41:641–643. [Google Scholar]
- Tercero JC, Wickner RB. MAK3 encodes an N-acetyltransferase whose modification of the L-A gag NH2 terminus is necessary for virus particle assembly. J Biol Chem. 1992;267:20277–20281. [PubMed] [Google Scholar]
- Thompson PR, Kurooka H, Nakatani Y, Cole PA. Transcriptional coactivator protein p300. Kinetic characterization of its histone acetyltransferase activity. J Biol Chem. 2001;276:33721–33729. doi: 10.1074/jbc.M104736200. [DOI] [PubMed] [Google Scholar]
- Thompson PR, Wang D, Wang L, Fulco M, Pediconi N, Zhang D, An W, Ge Q, Roeder RG, Wong J, et al. Regulation of the p300 HAT domain via a novel activation loop. Nature structural & molecular biology. 2004;11:308–315. doi: 10.1038/nsmb740. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Evjenth R, Foyn H, Demeyer K, De Bock PJ, Lillehaug JR, Vandekerckhove J, Arnesen T, Gevaert K. Proteome-derived Peptide Libraries Allow Detailed Analysis of the Substrate Specificities of N{alpha}-acetyltransferases and Point to hNaa10p as the Post-translational Actin N{alpha}-acetyltransferase. Mol Cell Proteomics. 2011a;10 doi: 10.1074/mcp.M110.004580. M110 004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Hole K, Gevaert K, Arnesen T. N-terminal acetylome analysis reveals the specificity of Naa50 (Nat5) and suggests a kinetic competition between N-terminal acetyltransferases and methionine aminopeptidases. Proteomics. 2015;15:2436–2446. doi: 10.1002/pmic.201400575. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Hole K, Pimenta-Marques A, Helsens K, Vandekerckhove J, Martinho RG, Gevaert K, Arnesen T. NatF contributes to an evolutionary shift in protein N-terminal acetylation and is important for normal chromosome segregation. PLoS genetics. 2011b;7:e1002169. doi: 10.1371/journal.pgen.1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Lasa M, Polevoda B, Gazquez C, Elosegui-Artola A, Kim DS, De Juan-Pardo E, Demeyer K, Hole K, Larrea E, et al. N-terminal acetylome analyses and functional insights of the N-terminal acetyltransferase NatB. Proc Natl Acad Sci U S A. 2012;109:12449–12454. doi: 10.1073/pnas.1210303109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetting MW, SdC LP, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys. 2005;433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Williams BC, Garrett-Engele CM, Li Z, Williams EV, Rosenman ED, Goldberg ML. Two putative acetyltransferases, san and deco, are required for establishing sister chromatid cohesion in Drosophila. Curr Biol. 2003;13:2025–2036. doi: 10.1016/j.cub.2003.11.018. [DOI] [PubMed] [Google Scholar]
- Yang X, Yu W, Shi L, Sun L, Liang J, Yi X, Li Q, Zhang Y, Yang F, Han X, et al. HAT4, a Golgi apparatus-anchored B-type histone acetyltransferase, acetylates free histone H4 and facilitates chromatin assembly. Mol Cell. 2011;44:39–50. doi: 10.1016/j.molcel.2011.07.032. [DOI] [PubMed] [Google Scholar]
- Yoon H, Kim HL, Chun YS, Shin DH, Lee KH, Shin CS, Lee DY, Kim HH, Lee ZH, Ryoo HM, et al. NAA10 controls osteoblast differentiation and bone formation as a feedback regulator of Runx2. Nature communications. 2014;5:5176. doi: 10.1038/ncomms6176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.