

Abstract
PPARδ (peroxisome proliferator-activated receptor δ) mediates inflammation in response to lipid accumulation. Systemic administration of a PPARδ agonist can ameliorate atherosclerosis. Paradoxically, genetic deletion of PPARδ in hematopoietic cells led to a reduction of atherosclerosis in murine models, suggesting that downregulation of PPARδ expression in these cells may mitigate atherogenesis. To advance this finding forward to potential clinical translation through hematopoietic stem cell transplantation-based gene therapy, we employed a microRNA (miRNA) approach to knock down PPARδ expression in bone marrow cells followed by transplantation of the cells into LDLR −/− mice. We found that knockdown of PPARδ expression in the hematopoietic system caused a dramatic reduction in aortic atherosclerotic lesions. In macrophages, a key component in atherogenesis, knockdown of PPARδ led to decreased expression of multiple pro-inflammatory factors, including monocyte chemoattractant protein-1 (MCP-1), interleukin (IL)-1β and IL-6. Expression of CCR2, a receptor for MCP-1, was also decreased. The downregulation of pro-inflammatory factors is consistent with significant reduction of macrophage presence in the lesions, which may also be attributable to elevation of ABCA1 (ATP-binding cassette, subfamily A, member 1) and depression of adipocyte differentiate-related protein. Furthermore, the abundance of both MCP-1 and matrix metalloproteinase-9 proteins was reduced in plaque areas. Our results demonstrate that miRNA-mediated PPARδ knockdown in hematopoietic cells is able to ameliorate atherosclerosis.
INTRODUCTION
Peroxisome proliferator-activated receptors (PPARs α, γ and δ) function as ligand-dependent transcription factors that mediate a broad range of important metabolic functions. PPARs hetero-dimerize with the retinoid X receptor and then bind to specific regions on DNA of target genes to modulate their transcription.1 PPARs may act through mechanisms of transactivation, transrepression or corepression to regulate expression of target genes.2–4 For example, the PPARγ agonist thiazolidinedione induces adiponectin expression by transcriptional activation of its promoter,2,5 whereas PPARs downregulate expression of pro-inflammatory factors such as interleukin (IL)-6 and IL1-β via transrepression.2 PPAR agonists have emerged as important therapeutic agents that modulate plasma lipids and glucose homeostasis, and their metabolic benefits are expected to translate into reduced cardiovascular disease events.
Compared with other PPAR isoforms, PPARδ has been less studied. Recent data indicated that PPARδ has a beneficial role in both hypertriglyceridemia and insulin resistance.6–8 In addition, PPARδ suppresses inflammatory reactions initiated by macrophages via DNA-independent mechanisms—inhibiting nuclear factor-κB, activator protein-1, signal transducer and activator of transcription 3 and nuclear factor of activated T-cell signaling pathways6,9–14 or liberating B-cell lymphoma 6 protein to repress inflammatory gene expression on ligand binding or genetic deletion of PPARδ.15 Collectively, these studies suggest that PPARδ is becoming a potential therapeutic target for atherosclerosis, which is a result of both lipid accumulation and inflammatory reaction. PPARδ agonists may have an atheroprotective role by promoting endothelial cell survival,16,17 preventing endothelial cell dysfunction,18,19 upregulating ABCA1 (ATP-binding cassette, subfamily A, member 1) expression20 and inhibition of angiotensin II-mediated activation of the pro-atherogenic mitogen-activated protein kinases, p38 and extracellular signal-regulated kinase 1/2.21 Indeed, −/− mice treated with the PPARδ agonist GW0742X showed a marked attenuation of atherosclerosis, with a concomitant decrease in monocyte chemoattractant protein-1 (MCP-1) and intercellular vascular adhesion molecule-1.16,22 Surprisingly, Li et al.23 demonstrated that systematic administration of PPARδ agonist in −/− mice failed to inhibit lesion development and foam cell formation, undermining the efficacy of systemic application of PPARδ agonist in treatment of the disorder. On the other hand, transplantation of bone marrow cells with PPARδ genetic deletion leads to remarkable inhibition of aortic atherosclerotic lesion formation in animal model via inhibiting macrophage-dependent inflammatory reactions without affecting plasma lipid profiles.15 This finding suggests that genetic regulation of PPARδ expression in bone marrow cells, apart from systemic activation of PPARδ by agonists, may provide a new approach to reducing atherosclerosis. In this study, we hypothesized that PPARδ knockdown in hematopoietic cells, macrophages in particular, can protect against atherogenesis and provide a new potential gene therapy approach for atherosclerotic cardiovascular diseases. To test this hypothesis, bone marrow stem cells infected with lentiviruses expressing specific microRNA (miRNA) against PPARδ were transplanted into −/− mice. The effects of PPARδ knockdown in hematopoietic cells, in particular macrophages, on the formation of atherosclerosis were investigated.
RESULTS
Validation of miRNA-mediated PPARδ knockdown
Two sequences from PPARδ mRNA were selected as miRNA targets to direct construction of Lenti-PPARδ-miRNA vectors. The negative control plasmid Lenti-Ctl-miRNA was also constructed at the same time. Mouse myoblast C2C12 cells that highly express PPARδ were used initially to test the knockdown efficiency of these vectors. C2C12 cells were transfected with each construct and analyzed by western blotting. Compared with Lenti-Ctl-miRNA, the construct of Lenti-PPARδ-miRNA expressing sequence A (see Materials and Methods) knocked down target gene expression significantly (data not shown). Therefore, we use this construct to further validate its knockdown efficiency in the form of viral particles. When C2C12 cells were infected with Lenti-PPARδ-miRNA virus, the expression of PPARδ protein was reduced by more than 90% in comparison with Lenti-Ctl-miRNA virus (Figure 1a). To examine specificity of the miRNA-mediated PPARδ knockdown, bone marrow-derived macrophages (BMDM) were infected with the indicated viruses and expression of PPARδ, PPARα and PPARγ was determined by western blotting (Figure 1b). The results indicated that the PPARδ miRNA was able to efficiently knock down PPARδ expression, while having little effect on expression of either PPARα or PPARγ in BMDM. Real-time PCR analysis also showed that Lenti-PPARδ-miRNA virus-mediated PPARδ knockdown in BMDMs can reach about 80% at the transcriptional level (Figure 1c).
Figure 1.

Validate miRNA-mediated PPARδ knockdown. (a) C2C12 cells were infected with the lentiviruses expressing control and PPARδ miRNA (sequence A). After 48 h, the cells were collected and expression of endogenous PPARδ was determined using western blot analysis. (b) Specific PPARδ knockdown. BMDM were infected with the indicated miRNA and the expression levels of PPAR δ, α and γ were determined using western blot analysis (In a and b, the numbers indicate the relative expression of the indicated proteins after normalization to β-actin). (c) The expression of endogenous PPARδ in BMDM infected with indicated viruses was determined by real-time PCR. The mRNA levels in BMDM infected with Lenti-PPARδ-miRNA were normalized to those in BMDM infected with Lenti-Ctl-miRNA (**P<0.01).
PPARδ knockdown in bone marrow hematopoietic cells ameliorates atherosclerosis
To investigate the effects of PPARδ knockdown on the development of atherosclerosis, donor bone marrow cells enriched for hematopoietic stem cells were infected with either Lenti-PPARδ-miRNA or Lenti-Ctl-miRNA virus and then transplanted into irradiated female −/− mice. Beginning 4 weeks post transplant, the recipient mice were fed with an atherogenic diet for 8 weeks and then euthanized for tissue collections. Mice transplanted with Lenti-PPARδ-miRNA-modified bone marrow cells showed an average 35% reduction of atherosclerotic lesions at the aortic root as revealed by oil red O staining, compared with mice transplanted with Lenti-Ctl-miRNA (Figures 2a and b). Moreover, the intensity of CD68 immunostaining in atherosclerotic lesions showed a significant reduction in the recipients of Lenti-PPARδ-miRNA-transduced bone marrow cells versus those in the recipients of Lenti-Ctl-miRNA-transduced bone marrow cells (Figures 2c and d), suggesting a decreased content of macrophages in atherosclerotic lesions. As PPARδ has a regulatory role in metabolism in vivo, the body weights were recorded during the entire experimental period. There was no significant weight difference between the two groups of mice (data not shown) nor was there a significant difference in food consumption, suggesting that hematopoietic PPARδ knockdown was generally well tolerated under these conditions.
Figure 2.

miRNA-mediated PPARδ knockdown in hematopoietic stem cells ameliorates the development of atherosclerosis in −/− mice. Lethally irradiated −/− mice were randomly divided into two groups (12 mice each) and transplanted with bone marrow cells infected with the indicated viruses. The mice were fed with an atherogenic diet for 8 weeks beginning 4 weeks post bone marrow transplantation and then killed. The lesion size in the aorta root was examined using oil red O staining as described in Materials and Methods. (a) Representative images of oil red O staining of the aortic root. (b) Statistical analysis of the atherosclerotic lesion sizes in two groups (**P<0.01). (c) Immunostaining with anti-CD68 antibody to show the content of macrophages in lesion areas. (d) Statistical analysis of the CD68-positive staining of two groups (**P<0.01).
PPARδ knockdown has little effect on plasma lipid levels, but alters expression of ABCA1 and adipocyte differentiate related protein in macrophages
As the action of PPARδ agonists on lipid metabolism in vivo has been controversial,23–25 total cholesterol and triglycerides were measured in plasma of both the Lenti-PPARδ-miRNA and Lenti-Ctl-miRNA recipient mice. No significant difference was observed between the two groups in amounts of either plasma cholesterol or triglycerides (Figure 3a). ABCA1 mediates the efflux of cholesterol and phospholipids from cells to lipid-poor apolipoproteins (apo-A1 and apoE) to form nascent high-density lipoproteins. As ABCA1 is responsible for reverse cholesterol transport from macrophages in aortic lesion sites and adipocyte differentiate-related protein enhances foam cell formation,26 we compared the expression of these two genes in peritoneal macrophages from the two groups using quantitative PCR. Peritoneal macrophages with PPARδ knockdown displayed significantly increased ABCA1 expression and decreased adipocyte differentiate-related protein expression (Figure 3b), implying that PPARδ knockdown may inhibit foam cell formation in vivo and thus suppress the process of atherogenesis.
Figure 3.

miRNA-mediated PPARδ knockdown has no effect on plasma lipid levels, but alters expression of ABCA1 and adipocyte differentiate related protein (ADRP) in macrophages. (a) Plasma total cholesterol and triglycerides measured by enzymatic assay. (b) ABCA1 and ADRP mRNA expression in peritoneal macrophages elicited from recipient mice transplanted with bone marrow cells transduced with either Lenti-Ctl-miRNA (open bars) or Lenti-PPARδ-miRNA (solid bars) viruses (*P<0.05, **P<0.01). Total RNA was isolated and the relative mRNA levels of PPARδ, ABCA1 and ADRP were determined by real-time PCR.
PPARδ knockdown decreases expression of MCP-1 and MMP-9 in atherosclerotic lesions, as well as macrophages
The migration of monocytes into the arterial subendothelial space is a critical event in atherogenesis and MCP-1 is a potent mediator of this process. Interestingly, either genetic depletion or pharmacological activation of PPARδ in macrophages leads to inhibition of MCP-1 promoter activity through derepression or activation, respectively, of PPARδ target genes, as well as mitigation of atherosclerosis.15,22 Thus, we inferred that the levels of MCP-1 might be reduced in aorta root lesion area of the recipients transplanted with Lenti-PPARδ-miRNA bone marrow cells. As expected, immunostaining against MCP-1 showed deceased expression in the atherosclerotic plaques of mice receiving Lenti-PPARδ-miRNA compared with those of mice receiving Lenti-Ctl-miRNA (Figure 4a). Matrix metalloproteinases (MMPs) are involved in atherosclerosis by remodeling the vasculature and promoting vulnerable plaque development. The expression of MMP-9 was decreased in atherosclerotic plaques in mice receiving Lenti-PPARδ-miRNA (Figure 4a). Similarly, the expression levels of both MCP-1 and MMP-9 were decreased significantly in peritoneal macrophages of mice receiving Lenti-PPARδ-miRNA-transduced bone marrow cells compared with those of control mice receiving Lenti-Ctl-miRNA-transduced bone marrow cells (Figure 4b), consistent with the reported findings in PPARδ-deficient macrophages.15
Figure 4.

PPARδ knockdown represses expression of MCP-1 and MMP-9. (a) The expression of MCP-1 and MMP-9 in sections of mouse aortic roots were examined by immunostaining using antibodies against MCP-1 (upper panels) and MMP-9 (lower panels) as indicated. The arrows indicate the expression of the proteins. (b) The relative expression of MCP-1 and MMP-9 at the transcriptional level in peritoneal macrophages elicited from recipient mice transplanted with bone marrow cells transduced with either Lenti-Ctl-miRNA (open bars) or Lenti-PPARδ-miRNA (solid bars) viruses were determined by real-time PCR (*P<0.05).
PPARδ knockdown represses expression of proinflammatory factors in macrophages
Among proinflammatory factors, both tumor necrosis factor-α (TNFα) and IL-6 have critical roles for atherogenesis.27 Therefore, we examined TNFα and IL-6 levels in plasma and BMDMs. Although the levels of TNFα and IL-6 in plasma showed no difference between the two groups (data not shown), the expression of IL-6 at the transcript level was decreased in peritoneal macrophages of Lenti-PPARδ-miRNA mouse group (Figure 5a). PPARδ knockdown in BMDMs also led to reduced expression of IL-6 and IL-1β with or without PPARδ agonist. The agonist administration further reduced the expression levels of both factors under the condition of PPARδ downregulation (Figures 5b and c), suggesting further liberation of B-cell lymphoma 6 protein under this condition.11,15,28,29
Figure 5.

PPARδ knockdown represses expression of IL-6 and IL-1β in macrophages. The relative expression of IL-6 in elicited peritoneal macrophages (a) as well as IL-6 and IL-1β transcripts in BMDM treated with GW 501516 was determined using quantitative PCR (b and c, respectively) as indicated (open bars: Lenti-Ctl-miRNA, solid bars: Lenti-PPARδ-miRNA). BMDMs were subjected to incubation in Dulbecco’s modified Eagle’s medium supplemented with 10% lipoprotein-deficient serum (LPDS) overnight, followed by GW501516 (0.1 μM) treatment for 24 h. (*P<0.05 and **P<0.01).
PPARδ knockdown represses CCR2 expression in BMDMs
Interaction between MCP-1 and its receptor CCR2 promotes recruitment of macrophages to the arterial wall with lipid accumulation and atherogenesis. A previous study demonstrated that genetic deletion of CCR2 leads to impaired macrophage recruitment into the arterial wall, despite lipid accumulation, and blocks the development of atherosclerosis in animal model.30 Therefore, we asked whether PPARδ knockdown inhibits CCR2 expression in BMDMs. As shown in Figure 6, PPARδ knockdown in BMDMs reduced the expression of CCR2 regardless of the presence of the PPARδ agonist. Nevertheless, the agonist enhanced PPARδ knockdown-mediated inhibition of CCR2 expression in BMDMs (Figure 6).
Figure 6.
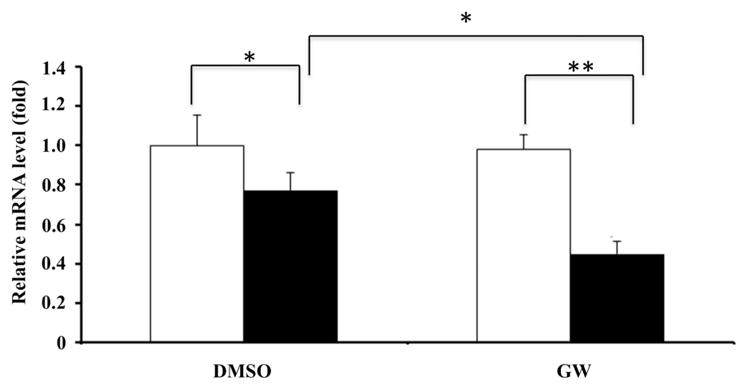
PPARδ knockdown represses expression of CCR2 in BMDM. BMDM infected with the indicated viruses expressing miRNA (open bars: Lenti-Ctl-miRNA, solid bars: Lenti-PPARδ-miRNA) were subjected to incubation in Dulbecco’s modified Eagle’s medium supplemented with 10% lipoprotein-deficient serum (LPDS) overnight, followed by dimethyl sulfoxide (DMSO; vehicle) or GW501516 (0.1 μM) treatment for 24 h. The cells were collected for total RNA isolation. The relative mRNA levels of CCR2 in BMDM were determined by real-time PCR (*P<0.05 and **P<0.01).
DISCUSSION
PPARδ agonists have been emerging as potential agents for treatment of atherosclerotic diseases. Studies demonstrated that PPARδ agonists, including GW1516, GW0742, GW0742X and L-165041, are able to repress atherogenesis in animal models through modulating lipid metabolism, macrophage-mediated inflammation, vascular smooth muscle cell proliferation and migration, and attachment of adhesion molecules on vascular endothelial cells.16,22,31,32 As PPARδ is highly expressed in various tissues, including the colon, small intestine, liver, heart, spleen, skeletal muscle, lung, brain and thymus,33,34 systemic PPARδ activation by its agonists may cause unpredictable adverse effects. In addition, recent studies showed that PPARδ is strongly expressed in certain cancer cells, although its potential role in carcinogenesis remains controversial.35–42 Moreover, the PPARδ agonist GW50156 induces activation of proinflammatory macrophages (M1) but not anti-inflammatory macrophages (M2).43 Finally, the existence of PPARδ genetic polymorphisms complicates the consideration of PPARδ agonists as anti-atherosclerosis agents; for example, the +294T/C polymorphism of the PPARδ gene associates with risk of coronary artery disease in normolipidemic Tunisians.44 Taken together, concerns about the efficacy and safety of systemic PPARδ agonist administration for treatment of atherosclerosis prompted us to seek a new therapeutic approach to this disease process.
As monocytes derived from bone marrow cells contribute to macrophage or foam cell accumulation in atherosclerotic plaque, we developed a novel means to achieve specific regulation of PPARδ expression in bone marrow hematopoietic cells and evaluated its impact on the development of atherosclerosis in animal models. In this study, we employed miRNA technology to knock down the expression of PPARδ in hematopoietic stem cell-containing bone marrow cells that were then transplanted into atherosclerosisprone −/− mice. We demonstrated that in the −/− mouse model, PPARδ knockdown in hematopoietic blood cells: (i) reduced atherosclerotic development by 35% compared with control group (Figures 2a and b), without an influence on the plasma lipid profile (Figure 3a); (ii) impaired macrophage recruitment to atherosclerotic lesions (Figure 2c); (iii) led to decreased expression of proinflammtory factors and adhesion molecules (Figures 4 and 5); and (iv) increased ABCA1 expression in peritoneal and BMDMs (Figure 3b). These changes occurred without significant alternations in either food consumption or body weight of the recipient animals. The decrease in macrophage recruitment and reduction of proinflammatory factors in atherosclerosis lesions may reinforce each other to attenuate the progression of atherogenesis when PPARδ expression is down-regulated in hematopoietic cells. The decreased content of macrophages in atherosclerosis lesions may be due to PPARδ knockdown-mediated inhibition of expression of MCP-1 and CCR2 in macrophages (Figures 4a and 6).30,45 We observed that the PPARδ knockdown group exhibited reduced MMP-9 expression compared with the control group using immunohistochemical assessment of atherosclerotic lesions (Figure 4a, lower panel), which may impede remodeling of vasculature and thus mitigate atherosclerotic progression. Repression of IL-6 and IL-1β expression in macrophages (Figure 5) would also contribute to inhibition of local inflammation and amelioration of vascular endothelial cell dysfunction due to lipid accumulation.46,47
In summary, our research suggests a potential new therapeutic approach to atherosclerosis based on miRNA-mediated PPARδ knockdown in hematopoietic blood cells, including macrophages. This therapy would overcome unwanted side effects associated with the pan-action of PPARδ agonists administrated systemically. The existence of PPARδ polymorphisms48–50 further emphasizes this consideration. Recent advances in basic and clinical investigations on lentiviral hematopoietic stem cell gene therapy encourage its extension to cardiovascular disorders.
MATERIALS AND METHODS
Generation of construct and virus preparation
Using Invitrogen miRNA design program, two PPARδ mRNA target sites were selected and their miRNA oligo sequences designed as follows: first PPARδ miRNA sequence: top strand: tgctgttctggatcttgcagatccgagttttggccactgactgactcggatctaagatccagaa, bottom strand: cctgttctggatcttagatccgagtcagtcagtggccaaaactcggatctgcaagatccagaa (sequence A); second PPARδ miRNA sequence, top strand: tgctgttctttagccactgcatcatcgttttggccactgactgacgatgatgctggctaaagaa, bottom strand: cctgttctttagccagcatcatcgtcagtcagtggccaaaacgatgatgcagtggctaaagaa (sequence B). The top and bottom strands were annealed followed by cloning into pcDNA6.2-GW/EmGFP-miroRNA vector using BP Clonase II Enzyme Mix (Carlsbad, CA, USA). The resulting pcDNA6.2-GW/EmGFP-PPARδ miRNA plasmids and the scrambled miRNA expression plasmid (provided by Invitrogen, Carlsbad, CA, USA) were reacted with pDONR221 donor vector and pLenti6/V5-DEST vector using LR Clonase II enzyme Mix (Carlsbad, CA, USA), to produce a lentiviral expression clone, that is, pLenti6/EmGFP-PPARδ-miRNA or pLenti6/EmGFP-Ctl-miroRNA (all vectors were provided by Invitrogen). The clones were further identified by sequence analysis.
Viruses were prepared as previously described.51 Briefly, plasmid Lenti-PPARδ-miRNA or Lenti-Ctl-miRNA (short for pLenti6/EmGFP-PPARδ-miRNA or pLenti6/EmGFP-Ctl-miroRNA, respectively) with packaging plasmids pMDLg/p.RRE, pRSV-Rev and pMD.G were co-transfected into 293T cells in the ratio of 10:6:1.5:2.5. The supernatants containing viruses were collected at 36 and 72 h post transfection, respectively. The supernatants were filtered through 0.45-μm pore size filters. For in-vitro infection use, the supernatants were concentrated 20-fold by high-speed centrifugation at 56 000 g for 90 min and the viral pellets were resuspended in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum or macrophage growth medium depending on downstream experiments. For bone marrow cell transplantation use, the supernatants were concentrated 1000-fold by 3 rounds of high-speed centrifugation at 56 000 g for 90 min and finally at 72 000 g for 90 min. Viral pellets were resuspended in StemPro-34 SFM (Invitrogen) and stored at −80 °C for downstream use.
Cell culture
293T and C2C12 cells were purchased from ATCC (Manassas, VA, USA) and identified for free of mycoplasma. Both cell lines were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. BMDMs were prepared as follows. Mouse bone marrow cells were flushed from the femora and tibia followed by adding red blood cell lysis buffer to remove erythrocytes. The cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, 20% horse serum and 20% L929 cell conditioned medium for 7 days. The formation of BMDMs was validated by examination of CD11b expression. The procedure for preparation of BMDMs infected with lentiviruses expressing miRNA is as follows. The bone marrow cells were maintained in specific macrophage growth medium at the density of 2.5 × 106 per ml (5 × 106 per well) for 3 days and then non-adherent cells were removed; the adherent cells were thrice rinsed with phosphate-buffered saline (PBS) followed by addition of 20-fold concentrated viruses for infection. After 24 h, the media containing viruses were removed and the cells were thrice washed with PBS before addition of fresh macrophage medium. The cells were continued to incubate at 37 °C until day 7 for deriving into macrophages. BMDMs were subjected to incubation with Dulbecco’s modified Eagle’s medium supplemented with 10% lipoprotein-deficient serum overnight before PPARδ agonist treatment. Elicited peritoneal macrophages were obtained 4 days after intraperitoneal injection of a 3.0-ml volume of thioglycolate. Briefly, peritoneal macrophage cells were collected by lavage with 5 ml of ice-cold RPMI supplemented with 2 mmol l−1 L-alanyl-L-glutamin, 1% (V/V) non-essential amino acids and penicillin/streptomycin (100 IU ml−1 and 100 μg ml−1), respectively. The cells were incubated for 4 h and non-adherent cells were removed by washing with medium. The adherent cells were collected as peritoneal macrophages for downstream analysis.
Immunoblotting
The cells were thrice washed with PBS and then collected by centrifugation. The cell pellets were resuspended in lysis buffer containing 25 mM Tris-HCl pH 7.4, 0.5 mM EDTA, 0.5 mM EGTA, 0.05% Triton X-100, 10 mM β-mercaptoethanol plus protease inhibitors. The cells were disrupted by ultrasonication. The lysates were collected by centrifugation and loading buffer was added. The samples were boiled for 5 min and then resolved by 12% SDS-polyacrylmide gel electrophoresis. The proteins were blotted onto nitrocellulose membrane. The expression levels of PPARδ, PPARα and PPARγ were measured using anti-PPARδ, PPARα and PPARγ-specific antibodies (sc-7197, sc-9000 and sc-7196, Santa Cruz Biotechnology, Santa Cruz, CA, USA) at a dilution of 1:500, respectively. The immunoblotting was performed in three independent experiments.
Quantitative PCR
Total RNA was isolated using TRIzol reagent (Invitrogen) and reverse transcribed using iScript cDNA synthesis Kit (Bio-Rad Laboratory, Hercules, CA, USA). The synthesized cDNA was used as a template for quantitative PCR analysis by SYBR green method. The primer sequences for quantitative PCR are listed in Table 1. Real-time PCR analysis was performed in triplicate with three independent experiments.
Table 1.
Primer sequences for real-time PCR
| Genes | Access number | Primer sequences (5–3′) | Temperature (°C) | Product size |
|---|---|---|---|---|
| β-Actin | NM_007393 | F: TTGCTGACAGGATGCAGAAGGAGA | 59.9 | 159 bp |
| R: ACTCCTGCT TGC TGATCCACATCT | 60.0 | |||
| PPARδ | NM_011145 | F: GCCCAAGTTCGAGTTTGCTGTCAA | 59.9 | 161 bp |
| R: ATTCTAGAGCCCGCAGAATGGTGT | 60.0 | |||
| ABCA1 | AF287263.1 | F: CCCTGCTACACTGGTCATTATC | 61.0 | 103 bp |
| R: CCCATACAGCAAGAGCAGAA | 60.9 | |||
| CCR2 | MMU56819 | F: AATGAGAAGAAGAGGCACAGGGCT | 60.0 | 161 bp |
| R: ATGGCCTGGTCTAAGTGCTTGTCA | 60.2 | |||
| MCP-1 | NM_011333 | F: AGCAGGTGTCCCAAAGAAGCTGTA | 60.1 | 176 bp |
| R: TGAAGACCTTAGGGCAGATGCAGT | 60.2 | |||
| MMP-9 | NM_013599 | F: CCATGGCAAATTCTTCTGGCGTGT | 60.1 | 162 bp |
| R: TAGAGACTTGCACTGCACGGTTGA; | 60.0 | |||
| IL6 | DQ788722 | F:ATCCAGTTGCCTTCTTGGGACTGA | 60.0 | 134 bp |
| R:TAAGCCTCCGACTTGTGAAGTGGT | 59.8 | |||
| TNFα | Y00467 | F:AGCCGTGACTGTAATTGCCCTACA | 60.1 | 115 bp |
| R:TTTAGGCCTCCGCAAAGAGATGGA | 60.0 | |||
| ADRP | M93275 | F:ACCTTGTGTCCTCCGCTTATGTCA | 59.8 | 170 bp |
| R:GGCATAGGTATTGGCAACCGCAAT | 59.9 |
Bone marrow cell transduction and transplantation
C57BL/6J mice were injected intravenously with 5-fluoruracil (110 mg kg−1) 4 days in advance before collecting bone marrow cells. Bone marrow cells were maintained in Stempro medium containing 6 ng ml−1 of mIL-3, 10 ng ml−1 of hIL-6, 10 ng ml−1 of mIL-1α and 100 ng ml−1 of mouse stem cell factor overnight.52 Before bone marrow transplantation, the cells were exposed to concentrated viruses containing the above factors in retro-nectin-coated 24-well plates for 5 h. Eight-week-old female LDL receptor-deficient mice (on C57BL/6J background, purchased from Jackson Laboratory, Bar Harbor, ME, USA) were irradiated (950 cGy) with a cesium gamma source and then divided randomly into two groups (16 mice per group) with comparable body weight. Lenti-PPARδ-miRNA- or Lenti-Ctl-miRNA-transduced hematopoietic stem cell-enriched bone marrow cells (8 × 106) were transplanted by tail vein injection. All studies were performed with the approval of the University of Texas Health Science Center at San Antonio Institutional Animal Care and Use Committee.
Development of atherosclerosis and lesion analysis
Allowing donor marrow cells to repopulate for 4 weeks with chow, the recipient animals were then fed with atherogenic diet containing 15.8% fat and 1.25% cholesterol (Harlan Teklad diet 94059, Madison, WI, USA) ad libitum for 8 weeks and killed for atherosclerotic lesion analysis. The mice were anesthetized during operation. Atherosclerotic lesions were analyzed by oil red O staining. The mean lesion area for each animal was determined in five sections (10 μm per section) taken every 40 μm through the aortic valve with 300 μm span length using NIH ImageJ software (NIH, Bethesda, MD, USA). Based on our and other’s previous studies, 12 mice per group were used for assessment of atherosclerosis formation. Quantification of atherosclerotic lesion size was assessed by two blinded investigators following the same criteria.
Lipid determination
The mice were fasted overnight before blood collection for plasma lipid analysis. Plasma cholesterol and triglycerides were determined by enzymatic assay following the supplier’s instructions (Thermo DMA, Arlington, TX, USA, and Thermo Trace Ltd, Melbourne, VIC, Australia).
Determination of TNFα and IL-6 in plasma
The levels of TNFα and IL-6 in plasma were determined using enzyme-linked immunosorbent assay method following the manufacturer’s description (OptEIA Sets, Pharmingen, San Diego, CA, USA).
Immunohistochemistry
Standard avidin–biotin complex method was employed to localize CD68, MCP-1 and MMP-9 in 10-μm-thick frozen sections of the aorta. Briefly, sections dried at room temperature for 30 min were washed in PBS and treated with 0.3% hydrogen peroxide to inhibit endogenous peroxidase activity. The sections were washed again in PBS for 15 min and incubated in 1% bovine serum albumin in PBS containing 0.3% Triton X-100 (blocking solution). Sections were then incubated with rabbit polyclonal antibodies against CD68, MCP-1 and MMP-9 (Santa Cruz Biotechnology) at 1:50 dilution overnight at 4 °C. Sections were rinsed in PBS for 10 min and incubated with biotinylated goat anti-rabbit secondary antibody for 40 min, followed by avidin–biotin complex (Vectastain ABC kit, Vector Laboratories, Burlingame, CA, USA). 3-Amino-9-ethyl carbazole (Sigma-Aldrich, St Louis, MO, USA) was used as chromogen to visualize the reaction product; reddish brown precipitate indicated the presence of CD68, MCP-1 and MMP-9 in the tissue sections. Sections were washed in distilled water and mounted in glycerol gelatin. Optical densities of the CD68+ cells per area were measured using NIH ImageJ software (NIH).
Statistical analysis
All numerical results are expressed as means ± s.d. derived from three independent experiments, unless otherwise stated. Statistical analyses were performed after establishment of similarity of the variance between groups using one-way analysis of variance (GraphPad Prism 5.03, GraphPad Software Inc., La Jolla, CA, USA) and statistically significant differences were established as P<0.05.
Acknowledgments
We thank Xiaoling Xue and Qiong Zhang for excellent laboratory assistance. This study was supported by a Merit Review grant from the Research Division of the Department of Veterans Affairs and by a grant from William and Ella Owens Medical Research Foundation awarded to SL. RAC is supported by Clinical and Translational Science Award TR001120 from the National Institutes of Health. This study was supported by a grant from the US Veterans Health Administration.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Berger J, Moller DE. The mechanisms of action of PPARs. Ann Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 2.Ricote M, Glass CK. PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta. 2007;1771:926–935. doi: 10.1016/j.bbalip.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schnegg CI, Kooshki M, Hsu FC, Sui G, Robbins ME. PPARdelta prevents radiation-induced proinflammatory responses in microglia via transrepression of NF-kappaB and inhibition of the PKCalpha/MEK1/2/ERK1/2/AP-1 pathway. Free Radic Biol Med. 2012;52:1734–1743. doi: 10.1016/j.freeradbiomed.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serrano-Marco L, Barroso E, El KI, Palomer X, Michalik L, Wahli W, et al. The peroxisome proliferator-activated receptor (PPAR) beta/delta agonist GW501516 inhibits IL-6-induced signal transducer and activator of transcription 3 (STAT3) activation and insulin resistance in human liver cells. Diabetologia. 2012;55:743–751. doi: 10.1007/s00125-011-2401-4. [DOI] [PubMed] [Google Scholar]
- 5.Iwaki M, Matsuda M, Maeda N, Funahashi T, Matsuzawa Y, Makishima M, et al. Induction of adiponectin, a fat-derived antidiabetic and antiatherogenic factor, by nuclear receptors. Diabetes. 2003;52:1655–1663. doi: 10.2337/diabetes.52.7.1655. [DOI] [PubMed] [Google Scholar]
- 6.Barish GD, Narkar VA, Evans RM. PPAR delta: a dagger in the heart of the metabolic syndrome. J Clin Invest. 2006;116:590–597. doi: 10.1172/JCI27955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benetti E, Patel NS, Collino M. The role of PPARbeta/delta in the management of metabolic syndrome and its associated cardiovascular complications. Endocr Metab Immune Disord Drug Targets. 2011;11:273–284. doi: 10.2174/187153011797881175. [DOI] [PubMed] [Google Scholar]
- 8.Berger JP, Akiyama TE, Meinke PT. PPARs: therapeutic targets for metabolic disease. Trends Pharmacol Sci. 2005;26:244–251. doi: 10.1016/j.tips.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 9.Barish GD, Atkins AR, Downes M, Olson P, Chong LW, Nelson M, et al. PPARdelta regulates multiple proinflammatory pathways to suppress atherosclerosis. Proc Natl Acad Sci USA. 2008;105:4271–4276. doi: 10.1073/pnas.0711875105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coll T, Barroso E, Alvarez-Guardia D, Serrano L, Salvado L, Merlos M, et al. The role of peroxisome proliferator-activated receptor beta/delta on the inflammatory basis of metabolic disease. PPAR Res. 2010;2010:1–11. doi: 10.1155/2010/368467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding G, Cheng L, Qin Q, Frontin S, Yang Q. PPARdelta modulates lipopolysaccharide-induced TNFalpha inflammation signaling in cultured cardio-myocytes. J Mol Cell Cardiol. 2006;40:821–828. doi: 10.1016/j.yjmcc.2006.03.422. [DOI] [PubMed] [Google Scholar]
- 12.Planavila A, Laguna JC, Vazquez-Carrera M. Nuclear factor-kappaB activation leads to down-regulation of fatty acid oxidation during cardiac hypertrophy. J Biol Chem. 2005;280:17464–17471. doi: 10.1074/jbc.M414220200. [DOI] [PubMed] [Google Scholar]
- 13.Serrano-Marco L, Rodriguez-Calvo R, El K, Palomer I, Michalik X, Wahli L, et al. Activation of peroxisome proliferator-activated receptor-beta/-delta (PPAR-beta/-delta) ameliorates insulin signaling and reduces SOCS3 levels by inhibiting STAT3 in interleukin-6-stimulated adipocytes. Diabetes. 2011;60:1990–1999. doi: 10.2337/db10-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zingarelli B, Piraino G, Hake PW, O’Connor M, Denenberg A, Fan H, et al. Peroxisome proliferator-activated receptor {delta} regulates inflammation via NF-{kappa}B signaling in polymicrobial sepsis. Am J Pathol. 2010;177:1834–1847. doi: 10.2353/ajpath.2010.091010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee CH, Chawla A, Urbiztondo N, Liao D, Boisvert WA, Evans RM. Transcriptional repression of atherogenic inflammation: modulation by PPARδ. Science. 2003;302:453–457. doi: 10.1126/science.1087344. [DOI] [PubMed] [Google Scholar]
- 16.Bojic LA, Burke AC, Chhoker SS, Telford DE, Sutherland BG, Edwards JY, et al. Peroxisome proliferator-activated receptor delta agonist GW1516 attenuates diet-induced aortic inflammation, insulin resistance, and atherosclerosis in low-density lipoprotein receptor knockout mice. Arterioscler Thromb Vasc Biol. 2014;34:52–60. doi: 10.1161/ATVBAHA.113.301830. [DOI] [PubMed] [Google Scholar]
- 17.Brunelli L, Cieslik KA, Alcorn JL, Vatta M, Baldini A. Peroxisome proliferator-activated receptor-δ upregulates 14-3-3 epsilon in human endothelial cells via CCAAT/enhancer binding protein-beta. Circ Res. 2007;100:e59–e71. doi: 10.1161/01.RES.0000260805.99076.22. [DOI] [PubMed] [Google Scholar]
- 18.d’Uscio LV, Das P, Santhanam AVR, He T, Younkin SG, Katusic ZS. Activation of PPARδ prevents endothelial dysfunction induced by overexpression of amyloid-β precursor protein. Cardiovasc Res. 2012;96:504–512. doi: 10.1093/cvr/cvs266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quintela AM, Jiménez R, Gómez-Guzmán M, Zarzuelo MaJ, Galindo P, Sánchez M, et al. Activation of peroxisome proliferator-activated receptor-β/-δ (PPARβ/δ) prevents endothelial dysfunction in type 1 diabetic rats. Free Radic Biol Med. 2012;53:730–741. doi: 10.1016/j.freeradbiomed.2012.05.045. [DOI] [PubMed] [Google Scholar]
- 20.Sprecher DL, Massien C, Pearce G, Billin AN, Perlstein I, Willson TM, et al. Triglyceride:high-density lipoprotein cholesterol effects in healthy subjects administered a peroxisome proliferator activated receptor delta agonist. Arterioscler Thromb Vasc Biol. 2007;27:359–365. doi: 10.1161/01.ATV.0000252790.70572.0c. [DOI] [PubMed] [Google Scholar]
- 21.Takata Y, Liu J, Yin F, Collins AR, Lyon CJ, Lee CH, et al. PPARdelta-mediated antiinflammatory mechanisms inhibit angiotensin II-accelerated atherosclerosis. Proc Natl Acad Sci USA. 2008;105:4277–4282. doi: 10.1073/pnas.0708647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graham TL, Mookherjee C, Suckling KE, Palmer CN, Patel L. The PPARdelta agonist GW0742X reduces atherosclerosis in LDLR(−/−) mice. Atherosclerosis. 2005;181:29–37. doi: 10.1016/j.atherosclerosis.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 23.Li AC, Binder CJ, Gutierrez A, Brown KK, Plotkin CR, Pattison JW, et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J Clin Invest. 2004;114:1564–1576. doi: 10.1172/JCI18730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reilly SM, Lee CH. PPAR delta as a therapeutic target in metabolic disease. FEBS Lett. 2008;582:26–31. doi: 10.1016/j.febslet.2007.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takahashi S, Tanaka T, Kodama T, Sakai J. Peroxisome proliferator-activated receptor delta (PPARdelta), a novel target site for drug discovery in metabolic syndrome. Pharmacol Res. 2006;53:501–507. doi: 10.1016/j.phrs.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 26.Imamura M, Inoguchi T, Ikuyama S, Taniguchi S, Kobayashi K, Nakashima N, et al. ADRP stimulates lipid accumulation and lipid droplet formation in murine fibroblasts. Am J Physiol Endocrinol Metab. 2002;283:E775–E783. doi: 10.1152/ajpendo.00040.2002. [DOI] [PubMed] [Google Scholar]
- 27.Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovasc Res. 2008;79:360–376. doi: 10.1093/cvr/cvn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan Y, Wang Y, Tang Z, Zhang H, Qin X, Zhu Y, et al. Suppression of pro-inflammatory adhesion molecules by PPAR-delta in human vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:315–321. doi: 10.1161/ATVBAHA.107.149815. [DOI] [PubMed] [Google Scholar]
- 29.Li Z, Wang X, Yu RY, Ding BB, Yu JJ, Dai XM, et al. BCL-6 negatively regulates expression of the NF-kappaB1 p105/p50 subunit. J Immunol. 2005;174:205–214. doi: 10.4049/jimmunol.174.1.205. [DOI] [PubMed] [Google Scholar]
- 30.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 31.Di PR, Esposito E, Mazzon E, Paterniti I, Galuppo M, Cuzzocrea S. GW0742, a selective PPAR-beta/delta agonist, contributes to the resolution of inflammation after gut ischemia/reperfusion injury. J Leukoc Biol. 2010;88:291–301. doi: 10.1189/jlb.0110053. [DOI] [PubMed] [Google Scholar]
- 32.Lim HJ, Park JH, Lee S, Choi HE, Lee KS, Park HY. PPARdelta ligand L-165041 ameliorates Western diet-induced hepatic lipid accumulation and inflammation in LDLR−/− mice. Eur J Pharmacol. 2009;622:45–51. doi: 10.1016/j.ejphar.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 33.Girroir EE, Hollingshead HE, He P, Zhu B, Perdew GH, Peters JM. Quantitative expression patterns of peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) protein in mice. Biochem Biophys Res Commun. 2008;371:456–461. doi: 10.1016/j.bbrc.2008.04.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Higashiyama H, Billin AN, Okamoto Y, Kinoshita M, Asano S. Expression profiling of peroxisome proliferator-activated receptor-delta (PPAR-delta) in mouse tissues using tissue microarray. Histochem Cell Biol. 2007;127:485–494. doi: 10.1007/s00418-007-0279-5. [DOI] [PubMed] [Google Scholar]
- 35.Gupta RA, Tan J, Krause WF, Geraci MW, Willson TM, Dey SK, et al. Prostacyclin-mediated activation of peroxisome proliferator-activated receptor delta in colorectal cancer. Proc Natl Acad Sci USA. 2000;97:13275–13280. doi: 10.1073/pnas.97.24.13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pedchenko TV, Gonzalez AL, Wang D, DuBois RN, Massion PP. Peroxisome proliferator-activated receptor beta/delta expression and activation in lung cancer. Am J Respir Cell Mol Biol. 2008;39:689–696. doi: 10.1165/rcmb.2007-0426OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scholtysek C, Katzenbeisser J, Fu H, Uderhardt S, Ipseiz N, Stoll C, et al. PPARbeta/delta governs Wnt signaling and bone turnover. Nat Med. 2013;19:608–613. doi: 10.1038/nm.3146. [DOI] [PubMed] [Google Scholar]
- 38.Takayama O, Yamamoto H, Damdinsuren B, Sugita Y, Ngan CY, Xu X, et al. Expression of PPARdelta in multistage carcinogenesis of the colorectum: implications of malignant cancer morphology. Br J Cancer. 2006;95:889–895. doi: 10.1038/sj.bjc.6603343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wagner KD, Benchetrit M, Bianchini L, Michiels JF, Wagner N. Peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) is highly expressed in liposarcoma and promotes migration and proliferation. J Pathol. 2011;224:575–588. doi: 10.1002/path.2910. [DOI] [PubMed] [Google Scholar]
- 40.Yang L, Yu YY, Zhou ZG, Luo HZ, Zhou B, Tian C, et al. Quantification of PPARdelta mRNA by real-time RT-PCR in rectal cancer tissues. Sichuan Da Xue Xue Bao Yi Xue Ban. 2007;38:78–80. [PubMed] [Google Scholar]
- 41.Yang L, Zhang H, Zhou ZG, Yan H, Adell G, Sun XF. Biological function and prognostic significance of peroxisome proliferator-activated receptor delta in rectal cancer. Clin Cancer Res. 2011;17:3760–3770. doi: 10.1158/1078-0432.CCR-10-2779. [DOI] [PubMed] [Google Scholar]
- 42.Zeng L, Geng Y, Tretiakova M, Yu X, Sicinski P, Kroll TG. Peroxisome proliferator-activated receptor-delta induces cell proliferation by a cyclin E1-dependent mechanism and is up-regulated in thyroid tumors. Cancer Res. 2008;68:6578–6586. doi: 10.1158/0008-5472.CAN-08-0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thulin P, Wei T, Werngren O, Cheung L, Fisher RM, Grander D, et al. MicroRNA-9 regulates the expression of peroxisome proliferator-activated receptor delta in human monocytes during the inflammatory response. Int J Mol Med. 2013;31:1003–1010. doi: 10.3892/ijmm.2013.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jguirim-Souissi I, Jelassi A, Hrira Y, Najah M, Slimani A, Addad F, et al. +294T/C polymorphism in the PPAR-delta gene is associated with risk of coronary artery disease in normolipidemic Tunisians. Genet Mol Res. 2010;9:1326–1333. doi: 10.4238/vol9-3gmr831. [DOI] [PubMed] [Google Scholar]
- 45.Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. 2004;95:858–866. doi: 10.1161/01.RES.0000146672.10582.17. [DOI] [PubMed] [Google Scholar]
- 46.Fan J, Watanabe T. Inflammatory reactions in the pathogenesis of atherosclerosis. J Atheroscler Thromb. 2003;10:63–71. doi: 10.5551/jat.10.63. [DOI] [PubMed] [Google Scholar]
- 47.Norata GD, Grigore L, Raselli S, Redaelli L, Hamsten A, Maggi F, et al. Post-prandial endothelial dysfunction in hypertriglyceridemic subjects: molecular mechanisms and gene expression studies. Atherosclerosis. 2007;193:321–327. doi: 10.1016/j.atherosclerosis.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 48.Aberle J, Hopfer I, Beil FU, Seedorf U. Association of peroxisome proliferator-activated receptor delta +294T/C with body mass index and interaction with peroxisome proliferator-activated receptor alpha L162V. Int J Obes (Lond) 2006;30:1709–1713. doi: 10.1038/sj.ijo.0803345. [DOI] [PubMed] [Google Scholar]
- 49.Burch LR, Donnelly LA, Doney AS, Brady J, Tommasi AM, Whitley AL, et al. Peroxisome proliferator-activated receptor-delta genotype influences metabolic phenotype and may influence lipid response to statin therapy in humans: a genetics of diabetes audit and research Tayside study. J Clin Endocrinol Metab. 2010;95:1830–1837. doi: 10.1210/jc.2009-1201. [DOI] [PubMed] [Google Scholar]
- 50.Gu SJ, Guo ZR, Zhou ZY, Hu XS, Wu M. PPAR alpha and PPAR gamma polymorphisms as risk factors for dyslipidemia in a Chinese Han population. Lipids Health Dis. 2014;13:23. doi: 10.1186/1476-511X-13-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He W, Qiang M, Ma W, Valente AJ, Quinones MP, Wang W, et al. Development of a synthetic promoter for macrophage gene therapy. Hum Gene Ther. 2006;17:949–959. doi: 10.1089/hum.2006.17.949. [DOI] [PubMed] [Google Scholar]
- 52.Pawliuk R, Westerman KA, Fabry ME, Payen E, Tighe R, Bouhassira EE, et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science. 2001;294:2368–2371. doi: 10.1126/science.1065806. [DOI] [PubMed] [Google Scholar]