

Abstract
The D14C variant of Pyrococcus furiosus ferredoxin provides an extraordinary framework to investigate a [3Fe-4S] cluster at two oxidation levels and compare the results to its physiologic [4Fe-4S] counterpart in the very same protein. Our spectroscopic and computational study reveals vibrational property changes related to electronic and structural aspects of both Fe-S clusters.
Graphical Abstract
A combined NRVS and DFT approach to characterize the redox-dependent vibrational properties of a [3Fe-4S]0/1+ cluster.
Iron-sulfur (Fe-S) clusters consist of iron atoms with bridging sulfides and play important roles in nature, e.g. in electron transport, as sensors, as catalytic heterometallic active sites, or have structural functions.1, 2 The thermostable ferredoxin (Fd) from Pyrococcus furiosus (Pf) functions as an electron donor for many oxidoreductases involved in carbohydrate and peptide fermentation. Reduced Fd supplies electrons for proton reduction by a membrane bound respiratory hydrogenase (MBH) that both produces molecular hydrogen (H2) and generates an ion gradient.3 The well-studied wild-type (WT) Pf Fd contains a single [4Fe-4S] cluster and represents an excellent framework for studying Fe-S clusters. The [4Fe-4S] coordination to the protein matrix via three cysteines (C11, C17, C56) and one aspartate (D14) side-chains is unusual in comparison to other proteins commonly utilizing four cysteines, and is found so far only in other ferredoxins and certain hydrogenases.4–6 Effects of this rare coordination with one aspartate have been scrutinized using Pf Fd site-directed mutagenesis. The redox potentials of the Pf Fd D14C and D14S mutants are respectively 58 and 133 mV more negative than the −368 mV of the WT protein.6, 7
Pf Fd D14C is sensitive to oxidative damage, which reversibly converts its [4Fe-4S] cluster into a [3Fe-4S] cluster with a 213 mV more positive redox potential (−155 mV) vs the WT protein.8 A 2.80 Å resolution X-ray diffraction (XRD) crystal structure of the ferricyanide-treated Pf Fd D14C reveals its [3Fe-4S] cofactor with the nearby Cys14 cysteine side chain facing away from the cluster (Fig. 1).8
Fig. 1.

[3Fe-4S] cluster of the ferricyanide-treated D14C ferredoxin from Pyrococcus furiosus. The cofactor inorganic core is shown in ball-and-stick representation. Sulfur and iron atoms are in yellow and brown, respectively. The cysteine at position 14 is facing away from the cluster. The presented crystal structure is based on chain A from PDB entry 3PNI.8
The best-known multi-cofactor enzymes hosting a [3Fe-4S] cluster are succinate dehydrogenase and periplasmic [NiFe] hydrogenases.9, 10 Their relatively high potential [3Fe-4S] clusters play important roles in electron transfer relays. In periplasmic hydrogenases, a [3Fe-4S]-to-[4Fe-4S] cluster conversion can be easily achieved by a single amino acid exchange from proline to cysteine at the missing fourth coordination site.11 This type of mutant-dependent cluster conversion (which is thus opposite to Pf Fd D14C) results in oxygen sensitivity of otherwise O2-tolerant Hyd1 hydrogenase from Escherichia coli.12 A detailed understanding of redox-dependent vibrational properties of [3Fe-4S] clusters would certainly be valuable; only the oxidized WT [3Fe-4S] Pf Fd state yielded useful resonance Raman (RR) spectra to date.13
Nuclear resonance vibrational spectroscopy (NRVS) is a relatively new synchrotron-based technique that selectively probes vibrational modes specific to Mössbauer-active nuclei.14, 15 Several [2Fe-2S] and [4Fe-4S] cluster systems as well as multi-cofactor enzymes have been so far characterized by 57Fe-NRVS.16–22 Density functional theory (DFT) has a long history of successful applications to Fe-S clusters, including those to Pf Fd and [3Fe-4S].19, 23–25 In this study, we use 57Fe-NRVS together with DFT to probe vibrational dynamics of the [3Fe-4S] cluster in Pf Fd D14C at two oxidation levels, and compare our results to those from an earlier account on the [4Fe-4S] variant from the same protein.19
The [4Fe-4S] cluster of Pf Fd D14C protein purified and reconstituted with 57Fe was converted by oxidation with ferricyanide and oxygen into a [3Fe-4S] cluster and the protein was subsequently treated with dithionite to obtain oxidized and fully reduced samples. The oxidation state and the conversion into the [3Fe-4S] cluster were confirmed by Mössbauer spectroscopy as expounded in the ESI†. The oxidized [3Fe-4S]1+ (S = 1/2) form of the cluster revealed doublets with isomer shift and quadrupole splitting corresponding to high-spin (S = 5/2) ferric (Fe3+) sites, and the reduced [3Fe-4S]0 (S = 2) form displayed hyperfine parameters corresponding to one ferric and two mixed-valent (Fe2.5+) ions (Table S1† and Fig. S1†). Our DFT results employing broken-symmetry (BS)26 concept (see ESI† discussion on spin coupling and Table S2†) are consistent with earlier spectroscopic indications on WT and mutant ferredoxins:27, 28 these are sites Fe1 and Fe2 that form the redox-active pair, coupled antiferromagnetically to Fe3 which remains ‘unique’ high-spin ferric in [3Fe-4S]0/1+ of Pf Fd D14C (see Fig. 1 and S2† for the Fe/S sites labeling).
Previous NRVS data on Pf Fd D14C [3Fe-4S] samples suffered from inhomogeneity and were provided only in weighted-sum spectra.16 57Fe partial vibrational density of states (57Fe-PVDOS) for oxidized and reduced samples from the new NRVS measurements are presented in Fig. 2, A and B, respectively. The spectra are distantly similar to those from the Pf Fd D14C [4Fe-4S] variant (Fig. 1, C and D), nitrogenase Fe protein [4Fe-4S] cluster from Azotobacter vinelandii (Av2), and the [Fe4S4](SPh)4 model compound.18, 19, 21 In what follows below, the DFT-computed spectra (Fig. 2, A–M, Fig. S3† and S4†) aid in rationalization of the observed NRVS data.
Fig. 2.
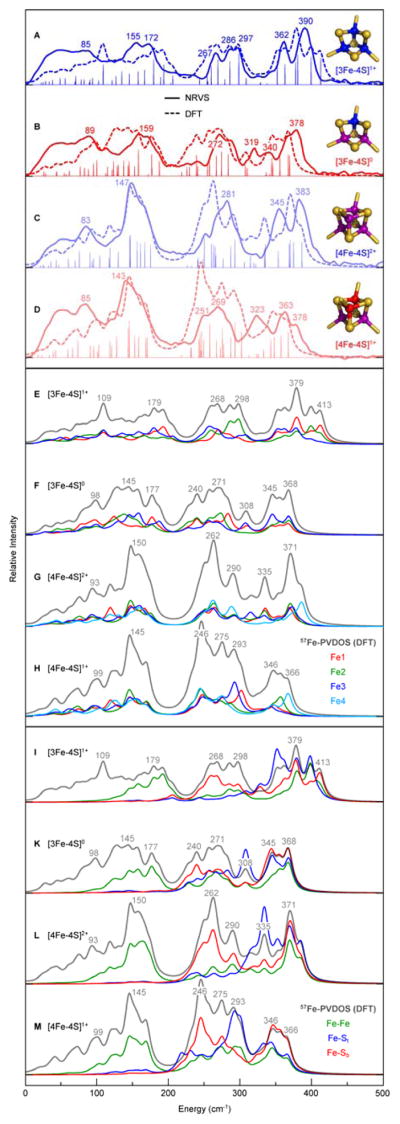
NRVS- and DFT-derived 57Fe-PVDOS and KED spectra for the 57Fe-enriched [3Fe-4S] and [4Fe-4S] variants of Pf Fd D14C at the reduced and oxidized levels. Top panel (A–D): overlay of the observed NRVS (solid line) and calculated DFT (broken line) spectra. The non-broadened intensities (and normal mode positions) from DFT are given in stick-style. On right, electronic structures of the Fe-S clusters are schematically shown, where Fe3+/2.5+/2+ are in blue/purple/red, respectively, and sulfide ligands are in yellow (see extended version of the top panel in Fig. S3†). Middle panel (EH): breakdown of the DFT spectra into contributions from individual Fe sites; Fe1/2/3/4 in red/green/blue/cyan, respectively. Bottom panel (I–M): DFT KED profiles for the Fe-bonding interactions; Fe–Fe/Fe–St/Fe–Sb lines are in green/blue/red, where St/b are terminal/bridging (cysteinate/inorganic) sulfides, respectively. Top to bottom in each panel: [3Fe-4S]1+ (A,E,I); [3Fe-4S]0 (B,F,K); [4Fe-4S]2+ (C,G,L); [4Fe-4S]1+ (D,H,M). The NRVS/DFT band positions are labeled in the top (A–D)/middle and bottom (E–M) panels, respectively. The data on the [4Fe-4S] variant have been in part published previously.19
In general, features range from ‘acoustic’ and torsional modes below 100 cm−1 to ‘breathing’ (Fe–Fe) and S–Fe–S bending modes around 100–200 cm−1, and to structurally-sensitive Fe–S stretching bands in the 200–420 cm−1 region. Stretching vibrational modes of the inorganic bridging sulfides (Fe–Sb) dominate the bands in the 200–300 cm−1 region, and stretching modes of the terminal cysteinate sulfurs (Fe–St) are mostly in the 300–420 cm−1 higher end of the spectra, as indicated by kinetic energy distribution (KED) analysis from DFT in Fig. 2, I–M (see ESI† for selected [3Fe-4S]0/1+ normal modes animated).18, 19, 29
In the mixed Fe–Sb/t stretching region, the strongest NRVS features of the [3Fe-4S]1+ Pf Fd D14C species are broad peaks at 362 and 390 cm−1 (Fig. 2, A). RR bands from 250 to 300 cm−1 of WT [3Fe-4S] Pf Fd13 overlap well with the NRVS peaks. The greatest contrasts between the NRVS and RR spectra are at 347 cm−1 with only a small NRVS signal and at 297 cm−1 with no detectable RR intensity, which can be explained with lack of Fe motion at 347 cm−1, and less change of polarizability at 297 cm−1. Upon reduction of Pf Fd with dithionite, a modified band pattern is observed for the spectral region between 240 and 420 cm−1 (Fig. 1, B), which is similar to the reduced [4Fe-4S] Pf Fd D14C (Fig. 1, D) and Av2 proteins.18, 19 For reduced [3Fe-4S] clusters, RR spectra have not been reported. The pronounced high-frequency maximum for the oxidized [3Fe-4S] D14C at 390 cm−1 downshifts to 378 cm−1 upon reduction, and the band at 362 cm−1 decreases in intensity; additionally, bands at 340 and 319 cm−1 appear.
NRVS of [3Fe-4S] revealed a drop of intensities of the ‘breathing’ and S–Fe–S bending bands in the 140–180 cm−1 region, otherwise very intensive in the [4Fe-4S] form (Fig. 2). Another remarkable feature of the [3Fe-4S] protein spectra is the split-band pattern of the stretching Fe–Sb modes region (267, 286 and 297 cm−1 bands for [3Fe-4S]1+) in comparison to the well-defined peaks at 269/281 cm−1 for the [4Fe-4S]1+/2+ counterpart. This type of 57Fe-PVDOS splitting has been also exposed for multi-cluster Desulfovibrio vulgaris Miyazaki F (Dv MF) [NiFe] hydrogenase with a [3Fe-4S] cluster in its electron relay.16 The dispersal of features in the [3Fe-4S] spectra can be explained by the loss of symmetry as an outcome of metal extraction from one of the [4Fe-4S] corners. The [4Fe-4S] core structure can be approximated as a Td point group or D2d, while the [3Fe-4S] structure is reminiscent of a lower symmetry group such as C3v, which would produce the observed breakdown in the pseudo-degeneracy of the bending and breathing modes.18, 19 High-resolution crystal structures of some [3Fe-4S]-containing ferredoxin indeed reveal distortions due to the absence of the fourth iron in contrast to [4Fe-4S] clusters, such as more open μ2S–Fe–μ2S (μ2S = Sb1/2/3, Fig. 1) conformation.30, 31 Consistently, our computational results predict the mean optimized Fe–μ3S (μ3S = Sb4) vs Fe–μ2S bonds longer by 0.07 Å (Table S3†).
Breakdown of the 57Fe-PVDOS spectra from DFT into contributions from individual Fe sites offers an alternative rationalization of the [3Fe-4S] vs [4Fe-4S] spectral differences, see Fig. 2, E–H. The major collective bands of the inorganic core calculated for [4Fe-4S]1+/2+ at 145/150 (Fe–Fe modes) and 246/262 (Fe–Sb modes) cm−1 respectively, result from consolidated displacements of the four Fe sites at these vibrational energies, while in the case of [3Fe-4S]0/1+ the corresponding individual bands of the three metal sites are distributed and therefore lack their cooperative effect. Notably, in both cluster types the degree of Fe vibrational cooperation is lower among the Fe–St modes above 300 cm−1, which represent interactions outside the inorganic core.
Consistently at both the oxidized [3Fe-4S]1+/[4Fe-4S]2+ and reduced [3Fe-4S]0/[4Fe-4S]1+ levels, prominent 57Fe-PVDOS bands of the [3Fe-4S] cluster display a blue shift within ~20 cm−1 when compared to the counterpart bands of the [4Fe-4S] form of Pf Fd D14C (Fig. 2, A vs C and B vs D). A similar effect has been observed for the [3Fe-4S] vs [4Fe-4S] derivatives of the WT Fd in a previous RR study.13 Comparable in their magnitude, band shifts as well apply to the [3Fe-4S]0/1+ and [4Fe-4S]1+/2+ redox couples of the same cluster type (Fig. 2, A vs B and C vs D). The latter observation is in line with a systematic 10–30 cm−1 stepwise decrease of Fe S stretching frequencies upon each one-electron reduction to the [4Fe-4S] cluster from Av2 Fe protein.18 Mean oxidation level per Fe site, which averages to +3/2.6/2.5/2.25 respectively for the [3Fe-4S]1+/[3Fe-4S]0/[4Fe-4S]2+/[4Fe-4S]1+ species, is a qualitative parameter which rationalizes the trends displayed in Fig. 2: upon gradual decrease of the mean Fe charge, the red shift enhances in both the experimental and calculated vibrational spectra. This tendency is mapped by the reduction-dependent expansion of both clusters,19, 32, 33 as reflected by elongation of the mean DFT-optimized Fe–Fe/St/Sb internuclear distances in the [3Fe-4S]1+/0 and [4Fe-4S]2+/1+ pairs (Table S3†). The mean Fe–Fe distance of 2.61/2.68/2.68/2.70 Å respectively for the [3Fe-4S]1+/[3Fe-4S]0/[4Fe-4S]2+/[4Fe-4S]1+ species is found to be the most relevant structural parameter which follows the above-mentioned general trend in all four Pf Fd D14C 57Fe-PVDOS spectra.
The redox-dependent structural shift of the [3Fe-4S] cluster is markedly superior to that of the [4Fe-4S] cluster (by +0.07/0.10/0.08 Å for [3Fe-4S] vs +0.02/0.05/0.01 Å for [4Fe-4S] in terms of the mean Fe–Fe/St/Sb distances, respectively, see Table S3†), which can be readily accepted in view of less efficient electron delocalization in the smaller cluster. This structural shift has a tight connection with reorganization energy λ required to relax the structure and its environment following the redox event. In semi-classical Marcus theory of electron transfer, the activation barrier for electron transfer ΔG‡ is predicted to depend on λ and the driving force ΔGo (difference in reduction potentials) by the relationship ΔG‡ = (λ + ΔGo)2/4λ.34 Our computed inner-sphere contribution to reorganization energy λi (see ESI† discussion on reorganization energy, Table S2†, and earlier theoretical works19, 25, 35–38) of the [3Fe-4S]0/1+ redox couple is 111.3 kJ/mol, which is about 6 times higher than the 18.8 kJ/mol value19 obtained for the [4Fe-4S]1+/2+ variant within the same Pf Fd D14C protein framework. The significantly higher reorganization energy of [3Fe-4S] explains its less frequent occurrence in electron-transfer proteins as compared to [4Fe-4S]. Interestingly, Asp (D14) coordination to [4Fe-4S] in the native Pf Fd WT protein has been indicated to increase the [4Fe-4S]1+/2+ reorganization energy λ by 80 kJ/mol, as compared to that of conventional Cys-only ligated [4Fe-4S] in the Pf Fd D14C mutant.39 In the native protein, a unique capability of aspartate to switch between mono- and bidentate coordination modes to [4Fe-4S] (“carboxylate shift”)24 was proposed to allow for conformational control over the electron transfer, in expense of its retarded rate.39
In context of the discussion above (and as well in absence of high-resolution XRD structures of the [3Fe-4S] cluster hosted specifically by Pf Fd D14C), it is remarkable to note that 1.4 Å resolution determinations of the [3Fe-4S]0/1+ structures from Azotobacter vinelandii Fd (Av FdI) displayed deviations within experimental error only (see ESI† discussion on structural aspects and Table S3†).30, 31 The reduced [3Fe-4S]0 form has been proposed to be a subject to an entatic state40 forced by the Av FdI protein in order to facilitate electron transfer. Our DFT calculation on the reduced [3Fe-4S]0 state using the equilibrium [3Fe-4S]1+ structure shows that stabilization of the entatic state should be backed by energetic input of as much as 14 kcal/mol from the protein (Table S2†). Further, 57Fe-PVDOS spectra computed for the entatic state produces high-intensity extensions in the 400–460 cm−1 region (Fig. S3†, B), which are not consistent with the NRVS experiment. Collectively, this indicates that the entatic state of the [3Fe-4S] cluster core is not realized in Pf Fd D14C.
Conclusions
In conclusion, our results highlight the power of combined application of NRVS and DFT to understanding of Fe-S clusters.18, 19, 41 The gradual red shift of vibrational bands, both upon cluster reduction and the [3Fe-4S]-to-[4Fe-4S] variant transition in the very same Pf Fd D14C protein, is explained by steadily increasing ferrous character of the inorganic core; this trend is well mapped by expansion of the Fe–Fe internuclear distances. The reduction-dependent expansion of the [3Fe-4S] cluster is markedly superior to that of the [4Fe-4S] cluster, which ultimately translates to higher reorganization energy of [3Fe-4S] and rationalizes its less frequent occurrence in electron-transfer proteins. The dispersal and splitting of the NRVS bands caused by conversion from [4Fe-4S] to [3Fe-4S] are related to electronic and structural aspects of both cluster types, in particular to the symmetry loss in the smaller cluster with one open corner. A new approach in breakdown of the collective DFT-computed NRVS spectra into contributions from individual 57Fe nuclei shows that the bands dispersal results from inhomogeneity of the metal site properties in [3Fe-4S], which is an alternative indication for a less efficient electron delocalization, as compared to [4Fe-4S]. To our best knowledge, this is the first comprehensive NRVS analysis of a [3Fe-4S] cluster in protein, and pioneering vibrational study of its reduced [3Fe-4S]0 form, which therefore completes the list of typical Fe-S clusters. An extension of the present approach is expected to shed light on structural and functional properties of electron relays and active sites of enzymes with multiple Fe-S cofactors. Applicable examples are membrane-bound O2-tolerant [NiFe] hydrogenase with its unique [4Fe-3S] proximal cluster,10, 42 and CO dehydrogenase/acetyl-CoA synthase hosting a heterometallic NiFe-S cluster.43
Supplementary Material
Acknowledgments
This work was supported through the Cluster of Excellence “Unifying Concepts in Catalysis” initiative of DFG (to L.L. and V.P.) and NIH grant number GM-65440 (to S.P.C. and L.B.G.). Preparation of ferredoxin samples was supported by the U.S. Department of Energy, Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences (DE-FG05-95ER20175 to M.W.W.A.). NRVS experiments were performed at BL09XU of SPring8 with approval of JASRI (Proposal No. 2014B1032).
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures, spectral data, computational chemistry details, cartesian coordinates of the optimized structures, animated vibrational modes as GIF files. See DOI: 10.1039/x0xx00000x
Contributor Information
Lars Lauterbach, Email: lars.lauterbach@tu-berlin.de.
Vladimir Pelmenschikov, Email: pelmentschikov@tu-berlin.de.
Notes and references
- 1.Beinert H, Holm RH, Munck E. Science. 1997;277:653–659. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 2.Lill R. Nature. 2009;460:831–838. doi: 10.1038/nature08301. [DOI] [PubMed] [Google Scholar]
- 3.McTernan PM, Chandrayan SK, Wu CH, Vaccaro BJ, Lancaster WA, Yang Q, Fu D, Hura GL, Tainer JA, Adams MW. J Biol Chem. 2014;289:19364–19372. doi: 10.1074/jbc.M114.567255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schäfer C, Friedrich B, Lenz O. Appl Environ Microbiol. 2013;79:5137–5145. doi: 10.1128/AEM.01576-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vitt S, Ma K, Warkentin E, Moll J, Pierik AJ, Shima S, Ermler U. J Mol Biol. 2014;426:2813–2826. doi: 10.1016/j.jmb.2014.05.024. [DOI] [PubMed] [Google Scholar]
- 6.Zhou ZH, Adams MW. Biochemistry. 1997;36:10892–10900. doi: 10.1021/bi9708141. [DOI] [PubMed] [Google Scholar]
- 7.Brereton PS, Verhagen MF, Zhou ZH, Adams MW. Biochemistry. 1998;37:7351–7362. doi: 10.1021/bi972864b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Løvgreen MN, Martic M, Windahl MS, Christensen HE, Harris P. J Biol Inorg Chem. 2011;16:763–775. doi: 10.1007/s00775-011-0778-7. [DOI] [PubMed] [Google Scholar]
- 9.Yankovskaya V, Horsefield R, Tornroth S, Luna-Chavez C, Miyoshi H, Leger C, Byrne B, Cecchini G, Iwata S. Science. 2003;299:700–704. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- 10.Fritsch J, Scheerer P, Frielingsdorf S, Kroschinsky S, Friedrich B, Lenz O, Spahn CM. Nature. 2011;479:249–252. doi: 10.1038/nature10505. [DOI] [PubMed] [Google Scholar]
- 11.Rousset M, Montet Y, Guigliarelli B, Forget N, Asso M, Bertrand P, Fontecilla-Camps JC, Hatchikian EC. Proc Natl Acad Sci U S A. 1998;95:11625–11630. doi: 10.1073/pnas.95.20.11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans RM, Armstrong FA. Methods Mol Biol. 2014;1122:73–94. doi: 10.1007/978-1-62703-794-5_6. [DOI] [PubMed] [Google Scholar]
- 13.Conover RC, Kowal AT, Fu W, Park JB, Aono S, Adams MWW, Johnson MK. J Biol Chem. 1990;265:8533–8541. [PubMed] [Google Scholar]
- 14.Sturhahn W. J Phys Condens Matter. 2004;16:S497–S530. [Google Scholar]
- 15.Scheidt WR, Durbin SM, Sage JT. J Inorg Biochem. 2005;99:60–71. doi: 10.1016/j.jinorgbio.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Kamali S, Wang H, Mitra D, Ogata H, Lubitz W, Manor BC, Rauchfuss TB, Byrne D, Bonnefoy V, Jenney FE, Jr, Adams MW, Yoda Y, Alp E, Zhao J, Cramer SP. Angew Chem Int Ed. 2013;52:724–728. doi: 10.1002/anie.201204616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lauterbach L, Wang HX, Horch M, Gee LB, Yoda Y, Tanaka Y, Zebger I, Lenz O, Cramer SP. Chem Sci. 2015;6:1055–1060. doi: 10.1039/c4sc02982h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitra D, George SJ, Guo Y, Kamali S, Keable S, Peters JW, Pelmenschikov V, Case DA, Cramer SP. J Am Chem Soc. 2013;135:2530–2543. doi: 10.1021/ja307027n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitra D, Pelmenschikov V, Guo Y, Case DA, Wang H, Dong W, Tan ML, Ichiye T, Jenney FE, Adams MW, Yoda Y, Zhao J, Cramer SP. Biochemistry. 2011;50:5220–5235. doi: 10.1021/bi200046p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogata H, Kramer T, Wang HX, Schilter D, Pelmenschikov V, van Gastel M, Neese F, Rauchfuss TB, Gee LB, Scott AD, Yoda Y, Tanaka Y, Lubitz W, Cramer SP. Nat Commun. 2015;6 doi: 10.1038/ncomms8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao Y, Koutmos M, Case DA, Coucouvanis D, Wang H, Cramer SP. Dalton Trans. 2006;18:2192–2201. doi: 10.1039/b513331a. [DOI] [PubMed] [Google Scholar]
- 22.Xiao Y, Tan ML, Ichiye T, Wang H, Guo Y, Smith MC, Meyer J, Sturhahn W, Alp EE, Zhao J, Yoda Y, Cramer SP. Biochemistry. 2008;47:6612–6627. doi: 10.1021/bi701433m. [DOI] [PubMed] [Google Scholar]
- 23.Dey A, Glaser T, Moura JJG, Holm RH, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2004;126:16868–16878. doi: 10.1021/ja0466208. [DOI] [PubMed] [Google Scholar]
- 24.Jensen KP, Ooi BL, Christensen HEM. Inorg Chem. 2007;46:8710–8716. doi: 10.1021/ic7009836. [DOI] [PubMed] [Google Scholar]
- 25.Jensen KP. J Inorg Biochem. 2008;102:87–100. doi: 10.1016/j.jinorgbio.2007.07.025. [DOI] [PubMed] [Google Scholar]
- 26.Noodleman L, Case DA. Adv Inorg Chem. 1992;38:423–470. [Google Scholar]
- 27.Duderstadt RE, Staples CR, Brereton PS, Adams MWW, Johnson MK. Biochemistry. 1999;38:10585–10593. doi: 10.1021/bi990670l. [DOI] [PubMed] [Google Scholar]
- 28.Donaire A, Gorst CM, Zhou ZH, Adams MWW, Lamar GN. J Am Chem Soc. 1994;116:6841–6849. [Google Scholar]
- 29.Johnson MK, Czernuszewicz RS, Spiro TG, Fee JA, Sweeney WV. J Am Chem Soc. 1983;105:6671–6678. [Google Scholar]
- 30.Schipke CG, Goodin DB, McRee DE, Stout CD. Biochemistry. 1999;38:8228–8239. doi: 10.1021/bi983008i. [DOI] [PubMed] [Google Scholar]
- 31.Stout CD, Stura EA, McRee DE. J Mol Biol. 1998;278:629–639. doi: 10.1006/jmbi.1998.1732. [DOI] [PubMed] [Google Scholar]
- 32.Collins DJ, Zhou H-C. Encyclopedia of Inorganic and Bioinorganic Chemistry. John Wiley & Sons, Ltd; 2011. [Google Scholar]
- 33.Scott TA, Berlinguette CP, Holm RH, Zhou HC. Proc Natl Acad Sci U S A. 2005;102:9741–9744. doi: 10.1073/pnas.0504258102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcus RA, Sutin N. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 35.Olsson MHM, Ryde U, Roos BO. Protein Sci. 1998;7:2659–2668. doi: 10.1002/pro.5560071220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sigfridsson E, Olsson MHM, Ryde U. Inorg Chem. 2001;40:2509–2519. doi: 10.1021/ic000752u. [DOI] [PubMed] [Google Scholar]
- 37.Jensen KP, Ooi BL, Christensen HEM. J Phys Chem A. 2008;112:12829–12841. doi: 10.1021/jp8014782. [DOI] [PubMed] [Google Scholar]
- 38.Pelmenschikov V, Kaupp M. J Am Chem Soc. 2013;135:11809–11823. doi: 10.1021/ja402159u. [DOI] [PubMed] [Google Scholar]
- 39.Calzolai L, Zhou ZH, Adams MWW, LaMar GN. J Am Chem Soc. 1996;118:2513–2514. [Google Scholar]
- 40.Vallee BL, Williams RJP. Proc Natl Acad Sci U S A. 1968;59:498–505. doi: 10.1073/pnas.59.2.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scott AD, Pelmenschikov V, Guo YS, Yan LF, Wang HX, George SJ, Dapper CH, Newton WE, Yoda Y, Tanaka Y, Cramer SP. J Am Chem Soc. 2014;136:15942–15954. doi: 10.1021/ja505720m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shomura Y, Yoon KS, Nishihara H, Higuchi Y. Nature. 2011;479:253–U143. doi: 10.1038/nature10504. [DOI] [PubMed] [Google Scholar]
- 43.Can M, Armstrong FA, Ragsdale SW. Chem Rev. 2014;114:4149–4174. doi: 10.1021/cr400461p. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.