

Abstract
Background
In Mali, Plasmodium falciparum malaria is highly endemic and remains stable despite the implementation of various malaria control measures. Understanding P. falciparum population structure variations across the country could provide new insights to guide malaria control programmes. In this study, P. falciparum genetic diversity and population structure in regions of varying patterns of malaria transmission in Mali were analysed.
Methods
A total of 648 blood isolates adsorbed onto filter papers during population surveillance surveys (December 2012–March 2013, October 2013) in four distinct sites of Mali were screened for the presence of P. falciparum via quantitative PCR (qPCR). Multiple loci variable number of tandem repeats analysis (MLVA) using eight microsatellite markers was then performed on positive qPCR samples. Complete genotypes were then analysed for genetic diversity, genetic differentiation and linkage disequilibrium.
Results
Of 156 qPCR-positive samples, complete genotyping of 112 samples was achieved. The parasite populations displayed high genetic diversity (mean He = 0.77), which was consistent with a high level of malaria transmission in Mali. Genetic differentiation was low (FST < 0.02), even between sites located approximately 900 km apart, thereby illustrating marked gene flux amongst parasite populations. The lack of linkage disequilibrium further revealed an absence of local clonal expansion, which was corroborated by the genotype relationship results. In contrast to the stable genetic diversity level observed throughout the country, mean multiplicity of infection increased from north to south (from 1.4 to 2.06) and paralleled malaria transmission levels observed locally.
Conclusions
In Mali, the high level of genetic diversity and the pronounced gene flux amongst P. falciparum populations may represent an obstacle to control malaria. Indeed, results suggest that parasite populations are polymorphic enough to adapt to their host and to counteract interventions, such as anti-malarial vaccination. Additionally, the panmictic parasite population structure imply that resistance traits may disseminate freely from one area to another, making control measures performed at a local level ineffective.
Electronic supplementary material
The online version of this article (doi:10.1186/s12936-016-1397-0) contains supplementary material, which is available to authorized users.
Keywords: Malaria, Plasmodium falciparum, Mali, Genetic diversity, Epidemiology, MLVA
Background
Plasmodium falciparum, which is transmitted by mosquitoes of the genus Anopheles, is the most common and the deadliest human malaria parasite. Despite extensive malaria elimination efforts, 214 million cases of malaria and 438,000 deaths were estimated globally in 2015, which primarily affected the sub-Saharan African population and children under 5 years of age [1]. Control strategies include the distribution of long-lasting insecticide-treated bed nets (LLINs), improved diagnosis using malaria rapid diagnostic tests (RDTs) and wider availability of artemisinin-based combination therapy (ACT). Even if control strategies have been intensified during the past 10 years in Mali, P. falciparum malaria remains highly endemic; stable incidence of the disease has been reported in several malaria vaccine-testing sites, such as Bandiagara [2]. In contrast to the situation in Mali, recent studies have shown that in other countries of the Sahel region, such as Senegal [3–5], enhanced interventions have effectively reduced malaria transmission. The heterogeneous results of malaria control programmes highlight the complexity of malaria epidemiology and the necessity to adapt interventions to local epidemiological settings.
In Mali, from the Sahara Desert to the Sudano-Guinean savannah, throughout the Sahel region, the variety of malaria transmission pattern is characterized by a north to south increasing gradient [6]. Malaria transmission is highly seasonal and peaks during the rainy season, but it has been shown that transmission can continue late into the dry season [6–9]. Assessing P. falciparum genetic diversity may be useful to elucidate the mechanisms of transmission persistence and rebound. Such studies would provide insight into human reservoirs of the parasites, including symptomatic cases and those of asymptomatic carriage [3, 8, 10] and human migration patterns associated with parasite flux [11–15]. Additionally, a clear understanding of P. falciparum genetic diversity would shed light on the characteristics of malaria burden and the expected difficulties hindering malaria control. Indeed, it has been shown that P. falciparum genetic diversity is indicative of the ability of malaria parasites to adapt to their hosts by selection of advantageous traits, such as drug resistance and antigenic variability [16].
Plasmodium falciparum genetic diversity can be assessed by analysing genetic polymorphism of the merozoite surface proteins (msp-1 and msp-2), as previously published in Mali [8, 10, 17, 18]. These markers were useful to investigate genetic diversity, multiplicity of infections and parasite carriage. However, the msp-1 and msp-2 genes are under selective pressure and neutral markers such as microsatellites or single nucleotide polymorphism (SNPs) are better suited for population genetics assessment [19]. Highly polymorphic microsatellite markers have been widely used to study P. falciparum population genetics via multiple loci variable number of tandem repeats analysis (MLVA) [11–15, 20–23]. These studies provided insights into various P. falciparum population genetic features, including parasite migration and linkage disequilibrium. To date, microsatellite markers have never been assessed to study P. falciparum population genetics in Mali.
In this study, MLVA was performed on P. falciparum DNA extracted from blood samples collected from four Malian study sites displaying various malaria transmission patterns. The four study sites were located along a 900-km long north to south axis and included the city of Bamako.
Methods
Study sites
Blood samples were collected in Rharous (Timbuktu District), Bamako (Bamako District), Doneguebougou (Kati District), and Bougoula Hameau (Sikasso District) (Fig. 1). Each of the study sites represents a different pattern of malaria transmission as defined by previous Malian epidemiological reports [6, 24, 25]. According to these reports, malaria is hypo-endemic in the urban zone of Bamako, whereas the disease is sporadic with occasional epidemics in Rharous, which is located in the Sahara Desert. Malaria is hyper-endemic in Doneguebougou [a site located in the Sudano-Sahelian zone, where malaria is characterized by a short transmission season (3–4 months)] and Bougoula [which is located in the Sudano-Guinean zone, where the transmission season is longer (4–6 months)].
Fig. 1.

Maps of Mali showing four study sites and four malaria epidemiological patterns [6, 24]. Annual isohyets (mm) separate each climatic zone. The climatic zones from north to south are as follows: Saharian zone (malaria transmission is sporadic to epidemic), Sahelian and Sudano-Sahelian zones (hyper-endemic for malaria, short transmission season of 3–4 months), Sudano-Guinean zone (hyper-endemic for malaria, long transmission season of 4–6 months). In the urban area of Bamako, conditions are not favourable for malaria transmission (hypo-endemic malaria)
Study design
A total of 648 blood samples were collected by finger prick (~50 µL) and adsorbed onto filter paper (Whatman™ 3MM CHR). Dried blood spots on filter papers were transported and stored at ambient temperature at the Malaria Research and Training Centre (MRTC) Central Laboratory in Bamako, Mali. Along with blood sampling, epidemiological and clinical data were reported on a standardized form. Sample collection was performed at the end of the rainy season (October) and at the beginning of the dry season (January to March), which correspond to the highest malaria transmission periods [7] (Table 1). Samples were collected via active case detection, with the exception of Rharous, where passive case detection was employed due to political instability in this area (Table 1). Active case detection included both symptomatic and asymptomatic individuals for malaria, whereas passive case detection included only individuals presenting with a fever or reported fever to the community health centre staffed by the MRTC. Demographic and clinical features of the cohort are detailed in Table 1.
Table 1.
Demographic and clinical features of each cohort, four sites in Mali
| Rharous, n = 149 | Bamako, n = 249 | Doneguebougou, n = 100 | Bougoula, n = 150 | P value | |
|---|---|---|---|---|---|
| Malaria endemicity | Sporadic | Hypo-endemic | Hyper-endemic | Hyper-endemic | |
| Period of collection | December 2012–March 2013 | October 2013 | October 2013 | October 2013 | |
| Season | Dry (beginning) | Rainy | Rainy | Rainy | |
| Detection methoda | PCD | ACD | ACD | ACD | |
| Female (%) | 54/149 (64) | 132/249 (53) | 51/100 (51) | 96/150 (64) | 0.11 |
| Mean age ± SD | 23.82 ± 17.34 | 20.43 ± 18.10 | 16.68 ± 15.31 | 31.30 ± 25.26 | <0.0001 |
| Children (0–5 years old) (%) | 25/145 (17) | 49/249 (20) | 27/100 (27) | 39/148 (26) | |
| Children (6–18 years old) (%) | 42/145 (29) | 97/249 (39) | 41/100 (41) | 24/148 (16) | |
| Adults (>18 years old) (%) | 78/145 (54) | 103/249 (41) | 32/100 (32) | 85/148 (57) | <0.001 |
| Feverb (%) | 44/137 (32) | 38/249 (15) | 8/99 (8) | 18/149 (12) | <0.0001 |
a ACD active case detection; PCD passive case detection
b Fever axillary temperature ≥37.5 °C
Ethical approval
Ethical clearance was obtained from the Ethics Committee of the Faculty of Medicine of Bamako (No 2012/81/CE/FMPOS) and blood samples were obtained after individuals provided informed consent.
Plasmodium falciparum isolation and quantification
DNA extraction
Filter papers with dried blood spots were cut into two pieces of approximately 1 cm2 using scissors and were incubated for 48 h at room temperature in 800 µL EasyMAG lysis buffer (bioMérieux, Marcy l’Étoile, France). Nucleic acid extraction was then performed using a NucliSENS EasyMAG system (bioMérieux, Marcy l’Étoile, France) with an elution volume of 100 µL [26]. Screening for the presence of P. falciparum was performed using specific primers and probe targeting the 18S rRNA gene via qPCR with a LightCycler 480 PCR system (Roche Diagnostics, Meylan, France) as previously described [27]. Each experimental run included both a negative (no template) and a positive (plasmid of P. falciparum18S rRNA) control. Standard curves were generated from serial ten-fold dilutions of the plasmid for species-specific quantification of parasite density (number of parasites/μL of blood). It was assumed that each genome of P. falciparum has five copies of the 18S rRNA gene (as observed in the 3D7 genome) [28].
MLVA genotyping
Genotyping relied on the analysis of eight polymorphic microsatellite markers (Poly α, TA109, TA1, TA81, TA42, ARA2, PfPK2, Pfg377) distributed among five chromosomes of P. falciparum, as previously published [20, 29]. Microsatellites were amplified via a two-step, semi-nested PCR using fluorescent end-labelled primers as previously described [29, 30]. Primer sequences, chromosome location, as well as reaction and cycling conditions are detailed in Additional file 1. To determine repeat-length sizes, PCR products were analysed using an ABI 3130xl capillary sequencer (Applied Biosystems, Foster City, CA, USA). Microsatellite allele length was determined using internal size standards (GeneScan 500 LIZ Size Standard, Applied Biosystems) with the GeneMapper v4.0 software (Applied Biosystems). To differentiate allele peaks from stutter peak artefacts, multiple alleles per locus were scored if minor peaks were >33 % of the height of the major peak, corresponding to the predominant allele [20, 29]. Peaks with a fluorescence intensity <100 units were discarded.
Population genetics analysis
Only complete genotypes (presenting all eight loci) were included in the population genetics analysis (complete dataset).
Multiplicity of infection
Multiplicity of infection (MOI) is defined as the number of genetically distinct clones of P. falciparum (i.e., number of distinct parasite genomes) present in an individual. As P. falciparum is haploid in the human stage, multiple peaks or alleles correspond to an infection with multiple genotypes or strains (a polygenomic infection). In a given sample, MOI was scored as the maximum number of alleles observed when taking into account all analysed loci (n = 8). An average MOI was then calculated.
Measure of genetic diversity
Because of the frequent occurrence of polygenomic infections, only major peaks were considered to assemble genotypes for genetic diversity analysis, as previously described [11, 12, 15, 20, 21]. The robustness of the genotyping assay was tested using two datasets: (1) built with major peaks only (first dataset); and, (2) built with minor peaks only (second dataset). The dataset including the major peaks only was used, as no significant difference in results has been observed. Allelic diversity is usually measured using expected heterozygosity (He). As P. falciparum is haploid in the human host, the terms ‘genetic diversity’ and ‘Nei’s unbiased genetic diversity index’ were employed instead of ‘expected heterozygosity (He)’. Nei’s unbiased genetic diversity index was calculated using ARLEQUIN v.3.5 [31] with the following formula:
in which n = number of samples analysed and p = frequency of each allele at a given locus [32]. For a haploid organism, Nei’s genetic diversity index can be considered as a measure of the probability to randomly draw a pair of different alleles from an allelic pool. Potential values range from 0 (no diversity, 100 % similarity between alleles) to 1 (maximal diversity, 100 % of the alleles are different). Allelic richness, which is an index derived from the normalization of the number of alleles with respect to sampling size, was calculated for each site using FSTAT v.2.9.3.2 [33].
Population genetic structure
The complete dataset, including genotypes from the four study sites, was analysed to calculate Wright’s F-statistics (FST), a measure of genetic differentiation between populations, using ARLEQUIN v 3.5 [31].
Linkage disequilibrium
The overall multi-locus linkage disequilibrium (LD) was assessed using the LIAN v.3.7 web interface [34]. This genetic phenomenon follows recurrent recombination between alleles at distinct loci instead of a random association of alleles. LIAN compares the variance in the number of alleles shared between all pairs of genotypes with the variance expected under a random association hypothesis and calculates the Standard Index of Association (IAS), a quantitative measure of LD. Values range from 0 (no loci in LD) to 1 (all loci in LD).
Genotype analysis
A minimum spanning tree was constructed based on the complete dataset to investigate genetic connections between genotypes. The chosen plug-in and the cluster analysis tool were implemented in BioNumerics v7.1 Software (Applied Maths, Ghent, Belgium).
Statistical analysis
Parasite densities of the four sites were compared using a negative binomial regression model (Genmod procedure). The influence of categorical variables including gender, age category (up to 5 years, 6–18 years, >18 years) and fever on the occurrence of polygenomic infections was assessed via Chi squared test (Chi2). Plasmodium falciparum MOI association with sex, age group, study site, and parasite density was tested via univariate unconditional logistic regression analysis (Logistic procedure). Multivariate unconditional logistic regression analysis with stepwise selection was performed to select the most parsimonious, best-fitting model of P. falciparum MOI. The analyses were performed using two-sided tests with a P < 0.05 significance level using SAS v9.2 statistical software (SAS Institute Inc, Cary, NC, USA).
Results
PCR results
The P. falciparum qPCR was positive in 156 of 648 tested samples (see study flow chart on Fig. 2). Positivity rate of P. falciparum qPCR differed significantly between the different locations, ranging from 9 % in Bamako to 44 % in Doneguebougou (P < 0.0001, Table 2). Positive qPCR samples were genotyped until 30 complete genotypes (i.e., successfully genotyped for all eight tested loci) were obtained for each study site. However, in Bamako, only 22 complete genotypes were obtained overall due to low malaria incidence. Of 125 genotyped samples, 112 genotypes were complete (Fig. 2). The results of the P. falciparum qPCR and estimated parasite density for each study site are detailed in Table 2. The microsatellite dataset is included in Additional file 2.
Fig. 2.
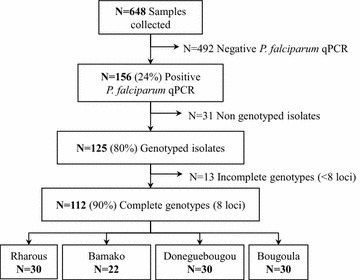
Study flow chart of samples collected from the four sites in Mali. Positive qPCR samples were genotyped until 30 complete genotypes (successfully genotyped for all tested loci) were obtained for each study site. In the site of Bamako, only 22 complete genotypes were obtained due to relatively low case numbers
Table 2.
Plasmodium falciparum qPCR results, estimated parasite density and polygenomic infections, four sites in Mali
| Rharous | Bamako | Doneguebougou | Bougoula | P value | |
|---|---|---|---|---|---|
| Total | |||||
| Positive qPCR for P. falciparum (%) | 44/149 (30) | 23/249 (9) | 44/100 (44) | 45/149 (30) | <0.0001 |
| Polygenomic infectionsd (%) | 11/30 (37) | 9/22 (41) | 21/30 (70) | 25/30 (83) | <0.0001 |
| Mean MOIc | 1.4e | 1.5f | 1.9 | 2.06 | <0.001 |
| Mean estimated parasitaemia ± SDb (parasites/µL blood) | 4397 ± 8127 | 5348 ± 5209 | 2942 ± 5863 | 8755 ± 24,395 | 0.061 |
| Individuals with fevera | |||||
| Positive qPCR for P. falciparum (%) | 22/44 (50) | 13/38 (34) | 6/8 (75) | 12/18 (66) | 0.051 |
| Polygenomic infectionsd (%) | 9/16 (56) | 5/13 (38) | 3/5 (60) | 6/8 (75) | <0.0001 |
| Mean MOIc | 1.62 | 1.46 | 1.6 | 1.75 | 0.72 |
| Mean estimated parasitaemia ± SDb (parasites/µL blood) | 7335 ± 10639 | 6916 ± 5934 | 9063 ± 12,318 | 5731 ± 9192 | 0.86 |
| Apyretic individualsa | |||||
| Positive qPCR for P. falciparum (%) | 20/93 (21) | 10/211 (5) | 38/91 (42) | 32/131 (24) | <0.0001 |
| Polygenomic infectionsd (%) | 2/14 (14) | 4/9 (44) | 18/25 (72) | 19/22 (86) | <0.0001 |
| Mean MOIc | 1.14 | 1.55 | 1.96 | 2.18 | <0.0001 |
| Mean estimated parasitaemia ± SDb (parasites/µL blood) | 1368 ± 2066 | 3311 ± 3348 | 1976 ± 3523 | 10148 ± 28,407 | <0.01 |
Differences in the denominator are due to missing data (n = 14 missing axillary temperature data)
Statistically significant results are indicated in italic
aFever: axillary temperature ≥37.5 °C and apyretic: axillary temperature <37.5 °C
bEstimation of parasite densities: [(copy number of the gene in 1 µL of DNA) × (100/125)]. Assuming that each genome of P. falciparum has five copies of the 18S rRNA gene [28], that blood volume spotted onto filter papers was 25 µL (half filter paper) and that extracted DNA was eluted into 100 µL
cMOI was calculated based on complete genotypes
MOI calculated in each site = ∑(maximum number alleles for a given sample) ÷ ∑(genotyped samples)
dPolygenomic infections are defined as individuals infected by ≥2 distinct parasite genotypes
eStatistically significant compared with Doneguebougou and Bougoula
fStatistically significant compared with Bougoula
Multiplicity of infection (MOI)
The proportion of polygenomic infections and mean MOI values per study site are shown in Table 2. Mean proportion of polygenomic infections was 58.9 % of the total cohort. The mean MOI significantly differed according to the study site (P < 0.001). Mean MOI values ranged from 1.4 (Rharous) to 2.06 (Bougoula) and were intermediate in Bamako (1.5). Similarly, polygenomic infection prevalence significantly differed based on the study sites (P < 0.0001). The lowest polygenomic infection rate was observed in Rharous (37 %), while the highest rate was noted in Bougoula (83 %). Univariate analysis revealed no significant effect of gender, age, fever, and parasite density on the occurrence of P. falciparum polygenomic infections (Table 3). In contrast, univariate analysis confirmed that the risk of polygenomic infection was significantly associated with study site (P < 0.001). Accordingly, the risk was significantly lower in Rharous (P < 0.01) and Bamako (P < 0.05) when compared with Bougoula.
Table 3.
Univariate logistic analysis of Plasmodium falciparum polygenomic infections
| Proportion of monogenomic infections (%) | Proportion of polygenomic infections (%) | OR | 95 % CI | P value | |
|---|---|---|---|---|---|
| Female | 24/57 (42) | 33/57 (58) | 0.917 | [0.432–1.947] | 0.82 |
| Age group | 0.19 | ||||
| Children (0–5 years old) | 6/23 (26) | 17/23 (74) | 3.051 | [0.921–10.114] | 0.083 |
| Children (6–18 years old) | 26/62 (42) | 36/62 (58) | 1.491 | [0.601–3.697] | 0.69 |
| Adults (>18 years old) | 14/27 (52) | 13/27 (48) | 1 | – | – |
| Fever (axillary temp ≥37.5 °C) | 19/42 (45) | 23/42 (55) | 0.760 | [0.350–1.650] | 0.49 |
| Ln (parasite density) | – | – | 1.063 | [0.823–1.373] | 0.64 |
| Site | <0.001 | ||||
| Rharous | 19/30 (63) | 11/30 (37) | 0.116 | [0.034–0.390] | <0.01 |
| Bamako | 13/22 (59) | 9/22 (41) | 0.138 | [0.038–0.499] | <0.05 |
| Doneguebougou | 9/30 (30) | 21/30 (70) | 0.467 | [0.135–1.609] | 0.19 |
| Bougoula | 5/30 (17) | 25/30 (83) | 1 | – | Reference |
Statistically significant results are indicated in italics
Genetic diversity
Genetic diversity values (He) for each study site are shown in Table 4. Genetic diversity values were also high between the different study sites (He = 0.76–0.79; mean He ± SD = 0.77 ± 0.16).
Table 4.
Genetic diversity of Plasmodium falciparum at the four study sites in Mali
| Locus | Rharous, n = 30 | Bougoula, n = 30 | Doneguebougou, n = 30 | Bamako, n = 22 | ||||
|---|---|---|---|---|---|---|---|---|
| A | He | A | He | A | He | A | He | |
| PolyA | 13.51 | 0.92 | 13.45 | 0.91 | 14.37 | 0.91 | 9.00 | 0.84 |
| TA109 | 8.72 | 0.85 | 9.66 | 0.86 | 7.86 | 0.85 | 12.00 | 0.93 |
| Pfpk2 | 10.58 | 0.88 | 9.72 | 0.87 | 6.86 | 0.80 | 7.00 | 0.85 |
| ARA2 | 6.85 | 0.71 | 5.92 | 0.75 | 8.78 | 0.85 | 7.00 | 0.82 |
| Pfg377 | 3.00 | 0.54 | 4.92 | 0.68 | 4.86 | 0.63 | 4.00 | 0.60 |
| TA42 | 3.86 | 0.43 | 3.86 | 0.35 | 4.79 | 0.56 | 4.00 | 0.46 |
| TA81 | 8.72 | 0.83 | 7.72 | 0.78 | 6.79 | 0.79 | 7.00 | 0.73 |
| TA1 | 11.52 | 0.88 | 9.79 | 0.89 | 8.92 | 0.90 | 9.00 | 0.86 |
| All loci | ||||||||
| Mean | 8.34 | 0.76 | 8.13 | 0.76 | 7.90 | 0.79 | 7.37 | 0.76 |
| SD | 3.64 | 0.17 | 3.14 | 0.18 | 3.04 | 0.12 | 2.66 | 0.15 |
A Allelic richness based on minimum sample size of 22 individuals (Bamako)
He Nei’s unbiased genetic diversity index
Population structure between geographic sites
Table 5 shows the pairwise FST between the four study sites, which reveals an almost complete panmixia with very low FST values (FST = 0.0011–0.018). Pairwise FST was statistically significant only between Bamako and Rharous (P = 0.03).
Table 5.
Matrix of between study site pairwise F ST
| Rharous | Bougoula | Doneguebougou | |
|---|---|---|---|
| Bougoula | 0.0011 | – | – |
| Doneguebougou | 0.0065 | 0.0072 | – |
| Bamako | 0.018* | 0.011 | 0.0026 |
* P value = 0.03
Linkage disequilibrium (LD)
Global multi-locus LD analysis of genotypes showed an absence of significant LD among alleles of P. falciparum genotypes in the different study sites. The IAS values and associated P values are shown in Table 6.
Table 6.
Multilocus linkage disequilibrium analysis of Plasmodium falciparum genotypes
| I AS | P value | |
|---|---|---|
| Rharous | −0.0015 | 0.54 |
| Bougoula | 0.0065 | 0.25 |
| Doneguebougou | 0.0005 | 0.48 |
| Bamako | −0.0071 | 0.72 |
Genotype analysis
Of the 112 complete genotypes, 100 % were unique genotypes as they were found only in a single sample. Most genotypes differed by two, three or more than three loci variants of the microsatellite markers. No major clustering of genotypes was detected between and within study sites as shown in Fig. 3.
Fig. 3.
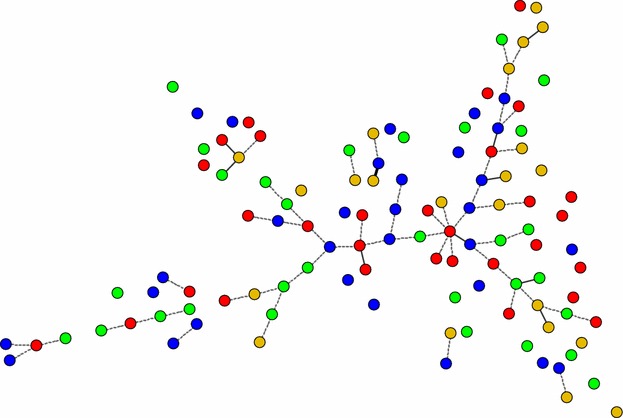
Minimum spanning tree showing the genetic relationship between 112 Plasmodium falciparum genotypes. Each genotype was unique. The thick solid lines indicate that the connected genotypes differ by only one locus of microsatellite marker, i.e., single-locus variant. Similarly, the thin solid lines represent double-loci variants and the dotted lines indicate triple-loci variants. Therefore, unlinked genotypes differ by more than three loci variants of the tested microsatellite markers. Isolates are coloured-coded according to geographic location in the dendrogram as follow: Rharous (red), Bougoula (blue), Doneguebougou (green), Bamako (gold). The distance between genotypes in the diagram does not reflect any relationship with genetic distance between genotypes
Discussion
This work represents the most extensive investigation of genetic diversity of P. falciparum populations in Mali to date and the first study to employ MLVA genotyping of P. falciparum in this country. Plasmodium falciparum isolates from Mali displayed high genetic diversity (mean He = 0.77) and low genetic differentiation (FST < 0.02). The lack of LD further revealed an absence of local clonal expansion, which was corroborated by the genotype relationship results. These findings highlight the marked gene flux between parasite populations, including within sites of lower transmission rates (Rharous and Bamako) and between sites located 900 km from each other (Rharous and Bougoula). In contrast with the lack of variability between genetic diversity levels observed in the country, mean MOI and proportions of polygenomic infection showed a north to south increasing gradient paralleling local malaria transmission levels [6].
Comparable levels of genetic diversity (He = 0.72–0.8) have already been observed in multi-locus microsatellite studies of P. falciparum in African regions highly endemic for malaria [11, 12, 20, 21]. This African pattern differs dramatically to those observed in hypo-endemic Latin American countries where genetic diversity is low (He = 0.30–0.40) [20, 23, 35] and Southeast Asia, where genetic diversity is intermediate (He = 0.51–0.65), which is probably due to more advanced malaria control progress in these areas [13, 20]. Indeed, the lower genetic diversity levels in Latin America and Southeast Asia, compared with Africa, might stem from a reduction in parasite population size following a decline in malaria transmission rates [20, 36].
Genetic diversity metrics have been useful to assess malaria control interventions and to highlight the disparities in malaria control programme results in endemic African countries [5, 11, 12, 15, 20, 21, 37]. The present study confirms the high genetic diversity previously reported in analyses of genetic polymorphism of the merozoite surface proteins (msp-1 and msp-2) [8, 10, 17, 18] and further indicates an absence of local clonal expansion in Mali. In Sudan, it has also been reported that P. falciparum genetic diversity remained high despite bolstered malaria control efforts [37]. In contrast, declining rates of genetic diversity and oligo-clonal expansion of parasite populations have been reported during a longitudinal study of P. falciparum microsatellite markers in Djibouti between 1998 and 2009 [15]. This setting was compatible with pre-elimination objectives. Comparable results of P. falciparum clonality have also been observed in Senegal (Thies city) during a longitudinal study using a set of 24 SNPs molecular barcode [5]. Clonal transmission of malaria has been detected after enhanced interventions, thereby revealing a dramatic change in the population structure. Such differences in P. falciparum population genetics in African countries suggest that in Mali, malaria control interventions still exert a limited impact on P. falciparum populations and malaria transmission.
Findings highlight pronounced gene flux between parasite populations. As illustrated in Rharous (northern Mali), the association between a high proportion of monogenetic infections (63 %) and high genetic diversity (He = 0.76) suggest that genetically diverse parasite strains have been imported from highly endemic regions via extensive human migration events. A similar trend was observed in the capital city of Bamako, thereby suggesting that many malaria cases are being imported from rural areas. These hypotheses are also supported by the lack of genetic differentiation between the different study sites. In Nigeria [12], a lack of genetic differentiation between P. falciparum populations located approximately 200 km apart was depicted as the marker of substantial parasite flux between the two locations. In the present study, a similar pattern was observed between study sites located up to 900 km apart (Rharous and Bougoula). A possible explanation could be the serious political crisis that occurred in 2012 and 2013 in Mali, which led to significant deterioration of security in the northern regions as well as displacement of large populations to neighbouring countries and the southern areas of Mali. Importantly, these findings imply that the effectiveness of malaria control efforts may be impaired if all malaria foci are not tackled simultaneously. High parasite flux from one site to another may also favour the diffusion of anti-malarial drug-resistant strains.
The analysis of the relationship between genotypes further revealed that each P. falciparum genotype was unique, with an absence of population clustering, even in the remote low-transmission site of Rharous. Human migration events might play a role in this phenomenon by boosting genetic diversity amongst P. falciparum populations. The absence of local clonal expansion was further corroborated by the lack of LD, which revealed a panmixia parasite population. Nevertheless, other markers, such as SNPs, might be more prone to show clonal expansion of parasites [19]. For instance, in Senegal, which is adjacent to Mali, a local clonal expansion of P. falciparum populations has been observed in Thies city using SNP molecular barcode [3, 5]. Therefore, it would be interesting to complete this study, especially in the hypo-endemic site of Rharous, to determine to which extent the absence of local clonal expansion can be confirmed using SNP markers.
The lowest levels of MOI and proportion of polygenomic infections were found in the areas exhibiting low malaria transmission, namely the urban site of Bamako and the Sahelo-Saharan site of Rharous. In contrast, the highest levels were found in the Sudano-Guinean site of Bougoula, where malaria transmission is almost perennial. A similar trend has been observed in two Malawian districts characterized by different transmission settings [38]. Although the association between prevalence of polygenomic infections and age group has been shown [10, 38], a longitudinal study in Mali [10] found that the increase in prevalence of polygenomic infections affected all age groups during the peak malaria transmission season, which is concordant with the results of the present study.
A limitation of this study may be that the human cohorts of each study site were not totally comparable. Indeed, a passive sampling procedure was used in Rharous due to security issues that precluded active case detection (Table 1). This explains why the proportion of febrile malaria cases in Rharous was significantly higher than in the other sites (P < 0.0001). Nevertheless, the significantly lower proportion of polygenomic infections observed in Rharous is probably not due to differences in case detection, as the univariate analysis revealed no association between proportion of polygenomic infections and fever, patient sex, age group, or estimated parasitaemia. Variations in proportion of polygenomic infections correlated with the study site only (P < 0.001). Additionally, disparities between malaria cohorts, such as fever prevalence, seem not to have impacted genetic diversity level, as it was similar in the different study sites. Likewise, this observation was further supported by a comparable high level of allelic richness.
Conclusions
Overall, these results reveal that P. falciparum genetic diversity level is particularly high in Mali. Such diversity may constitute an obstacle to control malaria as genetic polymorphism may facilitate parasite populations to adapt to their host and counteract certain interventions, such as anti-malarial vaccination. The pronounced parasite gene flux mediated by human migration events likely represents another hindrance for malaria control. Indeed, high parasite flux from one site to another could favour the diffusion of parasite strains, including those resistant to anti-malarial drugs. Therefore, these findings argue that malaria control measures should be intensified in Mali and that all malaria foci must be tackled simultaneously to reduce P. falciparum genetic diversity. As the measures currently in place (ACT and LLINs) seem to have a limited impact on P. falciparum populations, malaria control programmes aiming at reducing both malaria burden and parasite populations are greatly warranted.
Authors’ contributions
SD, SK, BF, AC, MAT, and OKD designed the study and collected the field samples and data; CN processed the samples; CN, FJ, TM, CL, SM, SR, and RP analysed the data; CN, SD, SM, OKD, and RP wrote the draft paper. All authors read and approved the final version. All authors have read and approved the final manuscript.
Acknowledgements
We would like to express our gratitude to the study population and volunteers as well as the MRTC field and laboratory staff. We would like to acknowledge Anne Cécile Normand and APPLIED MATHS for use of the BioNumerics software platform.
Availability of data and materials
The dataset supporting the conclusions of this article is included within the article and the additional files.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
This manuscript does not contain any individual person’s data that need a published consent.
Ethics approval and consent to participate
Ethical clearance was obtained from the Ethics Committee of the Faculty of Medicine of Bamako (No 2012/81/CE/FMPOS) and blood samples were obtained after individuals provided informed consent.
Funding
The fieldwork was supported by the WANECAM/EDTCP, the FMERIEUX Grant and WAMI/MRTC projects. Sample genotyping was supported by the Parasitology and Mycology Laboratory, La Timone Hospital, Marseilles, France.
Abbreviations
- RDT
rapid diagnostic test
- MLVA
multiple loci variable number of tandem repeats analysis
- SNP
single nucleotide polymorphism
- MRTC
Malaria Research and Training Centre
- ACT
artemisinin-based combination therapy
- LLIN
long-lasting insecticide-treated bed net
- MOI
multiplicity of infection
- LD
linkage disequilibrium
Additional files
10.1186/s12936-016-1397-0 Primer sequences of eight Plasmodium falciparum microsatellite loci, chromosome location and PCR reaction conditions. As previously described [29], primers listed third are internal to the other two and are end-labelled (*). PCR master mixes were comprised of 12 µL of Light Cycler 480 Probes Master (Roche Diagnostics, Meylan, France), 0.5 µL of each primer at 10 µM (Eurogentec, Seraing, Belgium), (Applied Biosystems, Foster, USA) and 1 µL of DNA. Thermocycling was performed in a 96-well T3 thermocycler (Biometra, Goettingen, Germany). For the first PCR reaction, the PCR programme was as follows: denaturation at 94 °C for 2 min, 1 cycle; (denaturation at 94 °C for 30 s, annealing at 42 °C for 30 s, annealing at 40 °C for 30 s, final elongation at 65 °C for 40 s), 25 cycles; extension time of 2 min at 65 °C, 1 cycle. For the second PCR reaction, the PCR programme was as follows: denaturation at 94 °C for 2 min, 1 cycle; (denaturation at 94 °C for 20 s, annealing at 45 °C for 30 s, final elongation at 65 °C for 30 s), 25 cycles; extension time of 2 min at 65 °C, 1 cycle.
10.1186/s12936-016-1397-0 Microsatellite data of the genotyped samples. The allele length (in base pairs) for each Plasmodium falciparum tested loci (Poly α, TA109, TA1, TA81, TA42, ARA2, PfPK2, Pfg377) is provided. The empty cells of the Table correspond to negative loci.
Footnotes
Cécile Nabet and Safiatou Doumbo contributed equally to this work
Contributor Information
Cécile Nabet, Email: cecile.nabet@ap-hm.fr.
Safiatou Doumbo, Email: sdoumbo@icermali.org.
Fakhri Jeddi, Email: fakhri.jeddi@ap-hm.fr.
Salimata Konaté, Email: sakonate@icermali.org.
Tommaso Manciulli, Email: tommaso.manciulli01@universitadipavia.it.
Bakary Fofana, Email: bfofana@icermali.org.
Coralie L’Ollivier, Email: coralie.lollivier@ap-hm.fr.
Aminata Camara, Email: micamara111@yahoo.fr.
Sandra Moore, Email: sandy.moore17@gmail.com.
Stéphane Ranque, Email: stephane.ranque@ap-hm.fr.
Mahamadou A. Théra, Email: mthera@icermali.org
Ogobara K. Doumbo, Email: okd@icermali.org
Renaud Piarroux, Email: renaud.piarroux@ap-hm.fr.
References
- 1.WHO. World Malaria Report 2015. Geneva: World Health Organization; 2015. http://www.who.int/malaria/publications/world-malaria-report-2015/report/en/.
- 2.Coulibaly D, Travassos MA, Kone AK, Tolo Y, Laurens MB, Traore K, et al. Stable malaria incidence despite scaling up control strategies in a malaria vaccine-testing site in Mali. Malar J. 2014;13:374. doi: 10.1186/1475-2875-13-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daniels RF, Schaffner SF, Wenger EA, Proctor JL, Chang H-H, Wong W, et al. Modeling malaria genomics reveals transmission decline and rebound in Senegal. Proc Natl Acad Sci USA. 2015;112:7067–7072. doi: 10.1073/pnas.1505691112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trape J-F, Tall A, Sokhna C, Ly AB, Diagne N, Ndiath O, et al. The rise and fall of malaria in a West African rural community, Dielmo, Senegal, from 1990 to 2012: a 22 year longitudinal study. Lancet Infect Dis. 2014;14:476–488. doi: 10.1016/S1473-3099(14)70712-1. [DOI] [PubMed] [Google Scholar]
- 5.Daniels R, Chang HH, Séne PD, Park DC, Neafsey DE, Schaffner SF, et al. Genetic surveillance detects both clonal and epidemic transmission of malaria following enhanced intervention in Senegal. PLoS One. 2013;8:e60780. doi: 10.1371/journal.pone.0060780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.PNLP, MRTC and INFORM. An epidemiological profile of malaria in Mali. A report prepared for the Ministry of Health, Mali, the Roll Back Malaria Partnership and the Department for International Development, UK. 2014. http://www.inform-malaria.org/wp-content/uploads/2015/03/Mali-Malaria-Epi-Profile-Report_030315.pdf Accessed 13 Apr 2016.
- 7.Coulibaly D, Rebaudet S, Travassos M, Tolo Y, Laurens M, Kone AK, et al. Spatio-temporal analysis of malaria within a transmission season in Bandiagara, Mali. Malar J. 2013;12:82. doi: 10.1186/1475-2875-12-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krogstad DJ, Koita OA, Diallo M, Gerone JL, Poudiougou B, Diakité M, et al. Molecular incidence and clearance of Plasmodium falciparum infection. Malar J. 2015;14:415. doi: 10.1186/s12936-015-0941-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sogoba N, Doumbia S, Vounatsou P, Bagayoko MM, Dolo G, Traore SF, et al. Malaria transmission dynamics in Niono, Mali: the effect of the irrigation systems. Acta Trop. 2007;101:232–240. doi: 10.1016/j.actatropica.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 10.Sonden K, Doumbo S, Hammar U, Vafa Homann M, Ongoiba A, Traore B, et al. Asymptomatic multiclonal Plasmodium falciparum infections carried through the dry season predict protection against subsequent clinical malaria. J Infect Dis. 2015;212:608–616. doi: 10.1093/infdis/jiv088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bogreau H, Renaud F, Bouchiba H, Durand P, Assi S, Henry M, et al. Genetic diversity and structure of African Plasmodium falciparum populations in urban and rural areas. Am J Trop Med Hyg. 2006;74:953–959. [PubMed] [Google Scholar]
- 12.Oyebola MK, Idowu ET, Nyang H, Olukosi YA, Otubanjo OA, Nwakanma DC, et al. Microsatellite markers reveal low levels of population sub-structuring of Plasmodium falciparum in south western Nigeria. Malar J. 2014;13:493. doi: 10.1186/1475-2875-13-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pumpaibool T, Arnathau C, Durand P, Kanchanakhan N, Siripoon N, Suegorn A, et al. Genetic diversity and population structure of Plasmodium falciparum in Thailand, a low transmission country. Malar J. 2009;8:155. doi: 10.1186/1475-2875-8-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schultz L, Wapling J, Mueller I, Ntsuke PO, Senn N, Nale J, et al. Multilocus haplotypes reveal variable levels of diversity and population structure of Plasmodium falciparum in Papua New Guinea, a region of intense perennial transmission. Malar J. 2010;9:336. doi: 10.1186/1475-2875-9-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khaireh BA, Assefa A, Guessod HH, Basco LK, Khaireh MA, Pascual A, et al. Population genetics analysis during the elimination process of Plasmodium falciparum in Djibouti. Malar J. 2013;12:201. doi: 10.1186/1475-2875-12-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghansah A, Amenga-Etego L, Amambua-Ngwa A, Andagalu B, Apinjoh T, Bouyou-Akotet M, et al. Monitoring parasite diversity for malaria elimination in sub-Saharan Africa. Science. 2014;345:1297–1298. doi: 10.1126/science.1259423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bereczky S, Dolo A, Maiga B, Hayano M, Granath F, Montgomery SM, et al. Spleen enlargement and genetic diversity of Plasmodium falciparum infection in two ethnic groups with different malaria susceptibility in Mali, West Africa. Trans R Soc Trop Med Hyg. 2006;100:248–257. doi: 10.1016/j.trstmh.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 18.Auburn S, Campino S, Miotto O, Djimde AA, Zongo I, Manske M, et al. Characterization of within-host Plasmodium falciparum diversity using next-generation sequence data. PLoS One. 2012;7:e32891. doi: 10.1371/journal.pone.0032891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su X-Z, Jiang H, Yi M, Mu J, Stephens RM. Large-scale genotyping and genetic mapping in Plasmodium parasites. Korean J Parasitol. 2009;47:83–91. doi: 10.3347/kjp.2009.47.2.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson TJ, Haubold B, Williams JT, Estrada-Franco JG, Richardson L, Mollinedo R, et al. Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol Biol Evol. 2000;17:1467–1482. doi: 10.1093/oxfordjournals.molbev.a026247. [DOI] [PubMed] [Google Scholar]
- 21.Mobegi VA, Loua KM, Ahouidi AD, Satoguina J, Nwakanma DC, Amambua-Ngwa A, et al. Population genetic structure of Plasmodium falciparum across a region of diverse endemicity in West Africa. Malar J. 2012;11:223. doi: 10.1186/1475-2875-11-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nkhoma SC, Nair S, Al-Saai S, Ashley E, McGready R, Phyo AP, et al. Population genetic correlates of declining transmission in a human pathogen. Mol Ecol. 2013;22:273–285. doi: 10.1111/mec.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Larrañaga N, Mejía RE, Hormaza JI, Montoya A, Soto A, Fontecha GA. Genetic structure of Plasmodium falciparum populations across the Honduras-Nicaragua border. Malar J. 2013;12:354. doi: 10.1186/1475-2875-12-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doumbo OK. Epidémiologie du paludisme au Mali, étude de la chloroquinorésistance, essai de stratégie de contrôle basée sur l’utilisation des rideaux impregnés de perméthrine associée au traitement systématique des accès fébriles. Thèse de Doctorat: Université de Montpellier II; 1992. [Google Scholar]
- 25.Ceesay SJ, Bojang KA, Nwakanma D, Conway DJ, Koita OA, Doumbia SO, et al. Sahel, savana, riverine and urban malaria in West Africa: similar control policies with different outcomes. Acta Trop. 2012;121:166–174. doi: 10.1016/j.actatropica.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boom R, Sol CJ, Salimans MM, Jansen CL, Wertheim-van Dillen PM, Van der Noordaa J. Rapid and simple method for purification of nucleic acids. J Clin Microbiol. 1990;28:495–503. doi: 10.1128/jcm.28.3.495-503.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rougemont M, Van Saanen M, Sahli R, Hinrikson HP, Bille J, Jaton K. Detection of four Plasmodium species in blood from humans by 18S rRNA gene subunit-based and species-specific real-time PCR assays. J Clin Microbiol. 2004;42:5636–5643. doi: 10.1128/JCM.42.12.5636-5643.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mercereau-Puijalon O, Barale J-C, Bischoff E. Three multigene families in Plasmodium parasites: facts and questions. Int J Parasitol. 2002;32:1323–1344. doi: 10.1016/S0020-7519(02)00111-X. [DOI] [PubMed] [Google Scholar]
- 29.Anderson TJ, Su XZ, Bockarie M, Lagog M, Day KP. Twelve microsatellite markers for characterization of Plasmodium falciparum from finger-prick blood samples. Parasitology. 1999;119:113–125. doi: 10.1017/S0031182099004552. [DOI] [PubMed] [Google Scholar]
- 30.Su XZ, Ferdig MT. Microsatellite analysis in Plasmodium falciparum. Methods Mol Med. 2002;72:131–136. doi: 10.1385/1-59259-271-6:131. [DOI] [PubMed] [Google Scholar]
- 31.Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 32.Nei M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics. 1978;89:583–590. doi: 10.1093/genetics/89.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goudet J. FSTAT (version 2.9.3.2), a program to estimate and test population genetic parameters. http://www2.unil.ch/popgen/softwares/fstat.htm (2012). Accessed 13 April 2016.
- 34.Haubold B, Hudson RR. Linkage analysis, Version 3.7. Program for testing the null hypothesis of linkage equilibrium for multilocus data.http://guanine.evolbio.mpg.de/cgi-bin/lian/lian.cgi.pl/query. Accessed 13 April 2016.
- 35.Chenet SM, Schneider KA, Villegas L, Escalante AA. Local population structure of Plasmodium: impact on malaria control and elimination. Malar J. 2012;11:412. doi: 10.1186/1475-2875-11-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Escalante AA, Ferreira MU, Vinetz JM, Volkman SK, Cui L, Gamboa D, et al. Malaria molecular epidemiology: lessons from the International Centers of Excellence for Malaria Research Network. Am J Trop Med Hyg. 2015;93(Suppl 3):79–86. doi: 10.4269/ajtmh.15-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bakhiet AM, Abdel-Muhsin AM, Elzaki SE, Al-Hashami Z, Albarwani HS, Al Qamashoui BA, et al. Plasmodium falciparum population structure in Sudan post artemisinin-based combination therapy. Acta Trop. 2015;148:97–104. doi: 10.1016/j.actatropica.2015.04.013. [DOI] [PubMed] [Google Scholar]
- 38.Bruce MC, Macheso A, McConnachie A, Molyneux ME. Comparative population structure of Plasmodium malariae and Plasmodium falciparum under different transmission settings in Malawi. Malar J. 2011;10:38. doi: 10.1186/1475-2875-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The dataset supporting the conclusions of this article is included within the article and the additional files.