

Abstract
While the etiology of rheumatoid arthritis (RA) remains to be fully elucidated, recent research has advanced the understanding of RA pathogenesis to the point where clinical trials for RA prevention are underway. The current paradigm for RA pathogenesis is that individuals progress through distinct preclinical stages prior to the onset of clinically apparent RA. These preclinical RA phases consist of genetic risk, local inflammation, presence of RA-related autoantibodies, asymptomatic systemic inflammation, and early non-specific symptoms prior to clinical seropositive RA. Epidemiologic studies have been important in forming hypotheses related to the biology occurring in preclinical RA. Specifically, studies associating cigarette smoking with overall RA risk as well as transitions between phases of preclinical RA were vital in helping to establish the lung as a potential important initiating site in the pathogenesis of seropositive RA. Herein, we review the epidemiology associating smoking with transitions in preclinical phases of RA as well as the recent literature supporting the lung as a critical site in RA pathogenesis.
Keywords: rheumatoid arthritis, lung, pulmonary, smoking, ACPA, pre-RA
Introduction
Rheumatoid arthritis (RA) causes an inflammatory polyarthritis affecting 1% of the population and is characterized by a chronic erosive arthropathy typically of the small joints [1 2]. RA is a serious public health problem that results in excess disability, morbidity, and mortality [3 4]. While the etiology of RA is currently not established, genetics, lifestyle factors, and biomarkers have been associated with risk of RA development in many research investigations over the past few decades [5]. The current paradigm for RA pathogenesis is that individuals with increased genetic susceptibility progress through preclinical stages prior to clinically apparent RA [6 7]. These preclinical RA stages include genetic risk, immune activation without symptoms (characterized by upregulation of cytokines and autoantibody production), arthralgias and other non-specific symptoms, and inflammatory arthritis prior to the development of classifiable RA [8]. Epidemiologic investigations have been helpful to guide basic and translational studies that aim to elucidate the biology of these pre-clinical RA transition stages.
Local loss of immune tolerance may trigger an inflammatory milieu and dysregulation of the adaptive immune system, causing systemic inflammation that eventually manifests as RA. Several anatomic sites have been posited as possible initiating sites for RA. Mucosal surfaces, in particular, are speculated to be the local sites of initial innate immune system dysregulation [9]. The anatomic sites that may be important in the development of RA include the lung, respiratory tract, periodontum, gut, bladder, and reproductive tract [9]. However, the lung and respiratory tract have the most evidence in support of being an initiating site for RA [10].
While much remains to be elucidated concerning the biologic mechanisms of RA pathogenesis, RA prevention strategies are actively being pursued. Epidemiologic studies have identified factors, such as genetics, family history, environment, and biomarkers that allow investigators to target those at highest risk for RA prevention [11 12]. The association of RA with potentially modifiable factors, in particular smoking and obesity, suggest that lifestyle interventions such as smoking cessation or weight loss may prevent or delay RA onset [13–16]. Since many effective targeted and non-targeted drugs for RA now exist, several clinical trials using pharmacologic interventions for RA prevention are currently underway among those at especially high risk for RA due to seropositivity or early symptoms [13]. Understanding the pathogenesis of RA remains crucial to the success, interpretation, and implementation of these ongoing as well as future RA prevention studies.
The aim of this article is to review the evidence supporting the lung and respiratory tract as an initiating site for RA pathogenesis, with a focus on how the epidemiology of smoking has informed investigations into the biology of pre-clinical seropositive RA pathogenesis in the lung.
Cigarette smoking, cessation, and overall risk for seropositive RA
Cigarette smoking is the best established lifestyle risk factor for RA and may provide the external trigger for those asymptomatic but at increased genetic risk for RA [17–24]. There is evidence that smoking is important throughout the preclinical phases of RA development (see Figure). The association of smoking is particularly strong for seropositive RA (defined as either rheumatoid factor [RF] or anti-cyclic citrullinated peptide [CCP] positivity). Smoking has a clear association with RA risk and may contribute up to 35% of the attributable risk for seropositive RA [25 26]. In a large meta-analysis that included 11 studies, 13,885 RA cases among a total of 593,576 individuals, current smokers had an odds ratio (OR) of 1.64 for seropositive RA compared to never smokers [17]. This effect of smoking for increased RA risk was more potent in men compared to women [17]. Current male smokers had an OR of 3.91 for seropositive RA compared to male non-smokers [17]. Both smoking status and intensity are associated with increased RA risk with clinically significant risk most apparent for those with >10 pack-years [27 28].
Figure.
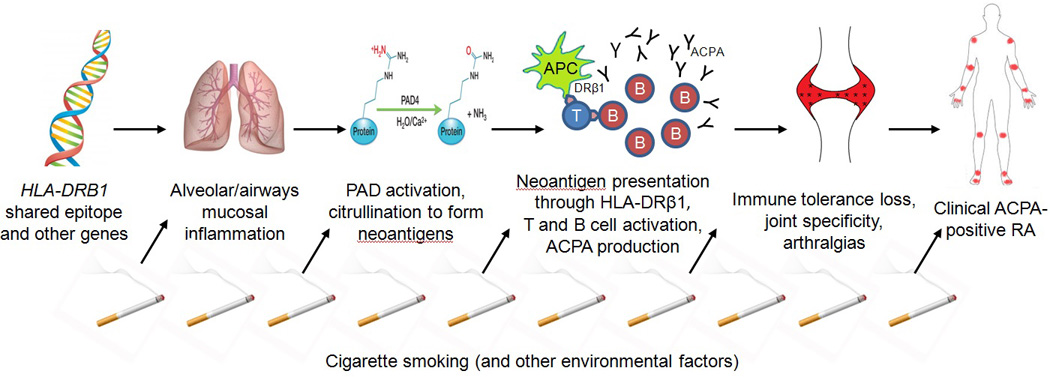
Schematic of proposed biologic mechanisms linking smoking and the lung to preclinical phases in the pathogenesis of anti-citrullinated protein antibody (ACPA)-positive rheumatoid arthritis (RA). An asymptomatic individual with high genetic risk, such as the HLA-DRB1 shared epitope, is exposed to environmental triggers such as cigarette smoking. This induces mucosal inflammation in the airways and alveoli of the lung. Activated enzymes, including peptidylarginine deiminase (PAD), stimulate citrullination of proteins at these sites to form neoantigens. Antigen presenting cells (APCs) in the innate immune system present these neoantigens through the HLA-DRβ1 protein to T cells that results in adaptive immune system dysregulation and leads to systemic inflammation. Stimulated B cells in the respiratory mucosa produce ACPA. Systemic inflammation leads to non-specific symptoms such as fatigue and arthralgias. Autoantibodies exhibit specificity to peripheral joints eventually leading to synovitis and the clinical diagnosis of ACPA-positive RA.
Studies suggest that smoking cessation may decrease RA risk [24 27 29]. Among women followed prospectively for 26 years in the Nurses’ Health Study, risk of RA for past smokers was similar to the RA risk of non-smokers ≥20 years after smoking cessation [27]. Similarly, in the Swedish Epidemiological Investigations of RA that compares incident RA cases to population-based controls, former smokers had similar RA risk compared to never smokers ≥20 years after cessation [24]. In the prospective Swedish Mammography Cohort, risk of RA was still significantly elevated 15 years after smoking cessation compared to never smokers [29]. While the risk for RA did not return to the baseline RA risk of never smokers in this study, risk steadily decreased over increasing duration of smoking cessation [29].
While smoking is clearly associated with seropositive RA, this association is attenuated or null for seronegative RA [17 28 30]. Some studies suggest that seronegative RA may be a heterogeneous collection of diseases without a common pathogenesis or distinct risk factors [31]. A recent study aimed to define a homogeneous population of seronegative RA patients by removing patients with detectable RA-related autoantibodies on sensitive research-only assays for anti-citrullinated protein antibodies (ACPA) as well as patients with HLA-B27 where spondyloarthropathy may have masqueraded as seronegative RA [32]. Using these methods, distinct genetic differences were detected between patients with seropositive and seronegative RA implying separate pathogenesis for each disease phenotype. However, most studies rely on the clinical classification of seronegative RA which has inherent clinical heterogeneity. For this reason, the pathogenesis of seropositive RA, particularly ACPA-positive RA, is best developed and is therefore the focus of this article. We refer to ACPA as the general presence of any research or commercial assay for anti-citrullinated protein antibodies, while we refer to CCP (and particular assays, such as CCP2, CCP3, or CCP3.1) as the specific commercial assay.
Cigarette smoking and transitions between preclinical phases of RA
Smoking has been associated with increased risk for progression among preclinical RA transition states (Table 1). Among patients with arthralgias and detectable serum IgM-RF or CCP2, smoking may hasten the emergence of classifiable RA [33]. However, when evaluating all patients with arthralgias, there is no clear association of smoking with progression to detectable inflammatory arthritis on physical examination [34]. Among first-degree relatives without RA in the Studies for the Etiology of RA, smoking >10 pack-years, in addition to current smoking status, was associated with two-fold increased risk of developing inflammatory joint signs [35]. Most of these unaffected relatives were asymptomatic and further away from RA development compared to those with seropositive arthralgias, so the particular timing of smoking in relation to preclinical RA stages is likely important.
Table 1.
Selected epidemiologic studies investigating smoking in RA preclinical phase transitions.
| First author year [reference] |
Preclinical RA phase |
Study Population Number of subjects |
Study design |
Summary | Comments |
|---|---|---|---|---|---|
| Voigt, 1994 [22] | Overall RA risk |
King County, WA US women N=349 RA cases and 1,457 controls |
Case-control | Current smoking: OR 1.33 (95%CI 1.00–1.77) Ever smoking: OR 1.31 (95%CI 1.07–1.60) Compared to non-smoking |
|
| Criswell, 2002 [18] | Overall RA risk |
Iowa Women’s Health Study US women N=31,336 |
Prospective cohort |
Current smoking: RR 2.0 (95%CI 1.3–2.9) for RA Quit smoking ≤10 years: RR 1.8 (95%CI 1.1–3.1) for RA Quit smoking >10 years: RR 0.9 (95%CI 0.5–2.6) for RA Compared to non-smoking |
|
| Costenbader, 2006 [27] | Overall RA risk |
Nurses’ Health Study US women N=103,818 |
Prospective cohort |
Current smoking: RR 1.43 (95%CI 1.16–1.75) for RA Past smoking: RR 1.47 (95%CI 1.23–1.76) for RA Current smoking: RR 1.58 (95%CI 1.21–2.06) for RF+RA Compared to non-smoking |
|
| Sugiyama, 2010 [17] | Overall RA risk |
11 case-control studies (N=4,764 RA cases and 13,647 controls) and 5 cohort studies (9,121 cases from 566,044 subjects) |
Meta- analysis of case-control and cohort studies |
All participants: Ever smokers: OR 1.40 (95%CI 1.25–1.58) for RA Ever smokers: OR 1.66 (95%CI 1.42–195) for RF+ RA Among men: Ever smokers: OR 1.89 (95%CI 1.56–2.28) for RA Ever smokers: OR 3.02 (95%CI 2.35–3.88) for RF+ RA Compared to non-smoking |
|
| Di Giuseppe, 2013 [29] | Overall RA risk |
Swedish Mammography Cohort Swedish women N=34,101 |
Prospective cohort |
Current smoking: RR 2.20 (95%CI 1.58–3.04) for RA Quit smoking ≥15 years: RR 1.99 (95%CI 1.23−.320) for RA Compared to non-smoking |
|
| Bhatia, 2007 [38] | Genetic susceptibility to serum RA-related antibody risk |
Studies of the Etiology of RA US women N=304 |
Cross- sectional |
≥20 pack-years smoking: HR 56.38 (95%CI 4.31- 736.98) for RF positivity Compared to non-smoking |
|
| Kokkonen, 2015 [39] | Genetic susceptibility to serum RA-related antibody risk |
Medical Biobank of Northern Sweden Sweden N=370 pre-RA cases, N=203 RA cases, N=585 controls |
Case-control | Among pre-RA cases within 10.5 years of diagnosis: Ever smoking and shared epitope positivity: OR 3.96 (1.87–8.39) for CCP2 positivity Compared to non-smoking, shared epitope negative |
|
| Kim, 2015 [65] | Genetic susceptibility to RA risk |
Nurses’ Health Studies, Epidemiological Investigations of RA, Korean RA Cohort US women, Sweden, Korea N=229 RA cases and N=360 controls; N=1654 RA cases and N=1934 controls; N=1390 cases and N=735 controls |
Case-control | >10 pack-years and high genetic risk score: OR ranging from 4.42 to 61.49 for seropositive RA Compared to 0–10 pack- years and low genetic risk score |
|
| Sparks, 2015 [35] | Genetic susceptibility to inflammatory arthritis risk |
Studies of the Etiology of RA / US N=966 |
Prospective cohort |
Current smoking: OR 2.12 (95%CI 1.33–3.38) for inflammatory joint signs Smoking >10 pack-years: OR 1.89 (95%CI 1.26–2.82) for inflammatory joint signs Compared to non-smoking |
|
| De Hair, 2013 [33] | Seropositive arthralgias to RA risk |
Seropositive Arthralgia Cohort Netherlands N=55 patients with seropositivity and arthralgias but no synovitis |
Prospective cohort |
Current smoking: HR 9.6 (95%CI 1.3–73.0) for RA Compared to non-smoking |
|
ACPA, anti-citrullinated protein antibodies; CCP, cyclic citrullinated peptide; CI, confidence interval; HR, hazard ratio; OR, odds ratio; RA, rheumatoid arthritis; RF, rheumatoid factor; RR, relative risk; US, United States.
Smoking may also be important in the development of RA-related autoantibodies. Patients with chronic obstructive pulmonary disease (COPD) without RA have increased levels of CCP and RF compared to controls. Smoking is associated with development of RA-related autoantibodies (CCP, ACPA, and RF) in patients that later develop RA [36 37] and in those at high genetic risk [38]. A Swedish study analyzed banked samples prior to the onset of RA patients and examined the HLA shared epitope, CCP, particular ACPA targeting specific antigens, and smoking. Smoking was associated with presence of ACPA antibodies closer to disease onset. Ever smokers with the shared epitope had a 4-fold increased odds of being CCP2 positive as well as other ACPA within 10.5 years of RA diagnosis compared to never smokers without the shared epitope [39]. Since the presence of circulating RA-related autoantibodies is associated with very elevated risk for RA, smoking may propagate the generation of RF, CCP, and other ACPA [40 41].
Finally, other inhaled irritants such as silica, air pollution, and textile dust may also increase the risk for RA, particularly seropositive RA [42–46]. While the epidemiology of these environmental factors is less developed compared to smoking, the associations of these other environmental factors emphasize that the lung, and potentially other mucosal sites, may provide the common link and micro-environment important for a variety of biologic processes that ultimately present clinically as RA.
HLA–smoking statistical interaction provides clues to the biology of transitions of preclinical RA
The HLA shared epitope, present in the Major Histocompatability Complex Class II on chromosome 6, is the strongest genetic risk factor for RA, among many other known genetic risk factors [47–49]. Shared epitope alleles are the site of transcription for specific amino acid haplotypes important in antigen processing and presentation and thus important in immune function [50]. Many epidemiologic studies have detected a statistical interaction between the shared epitope and smoking [51–58]. This finding provided background for the hypothesis that smoking may induce autoantigens interacting with the innate system that stimulates the adaptive immune system and eventually leads to joint specificity through molecular mimicry of autoantibodies and, finally, clinical symptoms of RA [59–64].
Specific amino acid positions within the peptide binding groove of the HLA-DRβ1 protein are associated with especially high risk of seropositive RA [50]. These sites also have statistical interactions with cigarette smoking for risk of seropositive, especially ACPA-positive, RA [51–57]. In a trans-ethnic study using data from the Nurses’ Health Studies in the US, the Swedish Epidemiological Investigations of RA, and the Korean RA Cohort Study, heavy smoking (defined as >10 pack-years) interacted with HLA-DRβ1 positions 11 and 13 for increased risk for seropositive RA [65]. Specific amino acid haplotypes conferred increased risk of seropositive RA (RF or CCP in the Nurses’ Health Study; CCP in both the Swedish and Korean studies) [65]. For example, presence of valine at HLA-DRβ1 position 11 or histidine at position 13 significantly increased seropositive RA risk while presence of serine at HLA-DRβ1 positions 11 or 13 significantly decreased RA risk [65]. These results suggest a physical interaction of a neoantigen, specifically induced by smoking, with the HLA-DRβ1 protein. However, the neoantigen(s) produced by this process are yet to be definitively identified.
Alveolar and airway inflammation in preclinical RA
While pulmonary involvement of patients with RA is well known, particularly interstitial lung disease (ILD) as an extra-articular RA disease manifestation, data suggest that lung inflammation can precede RA diagnosis [66 67]. While rates vary depending on RA duration, smoking exposure, medication history, and sensitivity of the testing modality, 8–36% of RA patients have pulmonary function tests suggesting airways obstruction, 60–80% of RA patients have airways disease on high-resolution computed tomography (CT) scans, and parenchymal disease is present in up to 79% of RA patients [10].
In a cross-sectional study of newly diagnosed RA patients, Wilsher and colleagues evaluated risk factors for lung imaging abnormalities on 60 patients within one year of RA diagnosis [68]. They found that bronchial wall thickening was present in 50% of patients, bronchiectasis in 35%, ground glass opacities in 18%, and reticular changes in 12% [68]. Additionally, CCP and RF correlated with decreasing diffusion capacity to carbon monoxide, suggesting significant alveolar damage in patients with early seropositive RA [68]. Similarly, Fischer and colleagues described significant disease burden in patients with respiratory symptoms who had CCP positivity without RA [69]. On high-resolution CT scans of the chest, 54% had isolated airways disease, 14% had ILD alone, and 26% had airways disease and ILD [69]. A subset who had bronchial biopsies performed showed significant airways inflammation [69].
A study in Sweden compared imaging findings on high-resolution CT scans of the chest among 105 untreated patients with early RA to healthy controls [70]. Parenchymal lung abnormalities were present in 63% of CCP-positive RA patients compared to only 37% of CCP-negative patients and 30% of controls, suggesting that lung inflammation is particularly relevant to ACPA-positive RA [70]. Airway changes on CT were detected more frequently in RA patients (66%) compared to controls (42%), suggesting that local airways inflammation is already present in patients recently diagnosed with RA [70]. Another study by the same investigators examined bronchial biopsy in similar patients with untreated early RA and found significant local inflammation in the lung at the alveoli and airways [71]. Adaptive immune cells (B cells, T cells, and plasma cells) were much more likely to be present and activated in CCP-positive patients compared to the CCP-negative RA patients or controls in both bronchial biopsies as well as bronchioalveolar lavage [71].
These pulmonary abnormalities may have been due to factors other than preclinical RA disease processes. In particular, cigarette smoking may cause many of these abnormalities. However, similar airways changes are also seen in patients with RA who were non-smokers suggesting that smoking alone does not cause these abnormalities. When adjusted for smoking, differences remained between ACPA-positive RA, ACPA-negative RA, and controls. Since patients in these studies had very early RA and were untreated, it is less likely that other clinical factors such as decreased physical activity or medication use explain these differences.
Demoruelle and colleagues investigated pulmonary function and imaging among subjects with seropositivity (positive CCP2 or CCP3.1 +/− ≥2 RF isotypes) without inflammatory arthritis [72]. This group at very elevated risk of RA was compared to autoantibody-negative controls as well as patients with early RA. Seropositive subjects showed significant burden or pulmonary abnormalities compared to controls [72]. Of those who were seropositive, 76% had airways disease compared to only 33% of controls. Fifty percent of seropositive cases without RA had bronchial thickening compared to only 13% of seronegative controls; 69% of seropositive cases had air trapping compared to 7% of seronegative controls [72]. Early RA patients had more bronchial wall thickening and parenchymal abnormalities but were otherwise similar to seropositive cases without RA. Smoking was a matching factor and similar trends were seen when analyzing only never smokers.
Together, these studies in early RA and subjects with seropositivity without RA are compelling to emphasize the inflammation that occurs in airways and alveoli prior to disease. Further, many of these patients with RA had no evidence of ILD on biopsy or imaging arguing that these abnormalities are important for ACPA-positive RA and not only patients with RA who develop ILD.
Citrullination of antigens in respiratory mucosa
While studies demonstrate excess local inflammation in the alveoli and airways in preclinical RA, the biological processes occurring here are challenging to study since these individuals are difficult to identify and are otherwise often healthy. Citrullination is a process by which arginine residues on proteins, such as fibrinogen and enolase, are converted to citrulline by the enzyme peptidylarginine deiminase (PAD) [73]. Autoantibodies to citrullinated protein antigens are highly specific to RA and presence of these in preclinical phases of RA greatly increases the risk of progression to clinical RA. Therefore, citrullination has been postulated to be one of the biologic processes occurring locally in the lung that may initiate RA pathogenesis. Citrullination may be the important first step in the loss of immune tolerance, forming self-antigens that are then presented to T cells that eventually result in activated B cells producing ACPA [74 75].
Reynisdottir and colleagues analyzed fluid from bronchoalveolar lavage and found increased citrullination of proteins in CCP2-positive early untreated RA patients compared to CCP2-negative RA and healthy controls [71]. Ytterberg and colleagues performed proteomic screening of bronchial and synovial biopsies of RA patients to find proteins in common at both sites that might link the lung and joints [76]. They found that two citrullinated vimentin peptides were identified in the majority of these patients at both anatomic sites, perhaps providing a link between initial breakdown of the innate immune system locally and the lung and inflammation in the joints after adaptive immune system dysregulation [76].
While these findings are provocative, they require replication and would likely not explain the full spectrum of ACPA-positive RA pathogenesis. Smoking may be the extrinsic factor that causes local mucosal inflammation in the lung, upregulating PAD in susceptible hosts, leading to citrullination of proteins that form neoantigens that stimulate the immune system and induce autoimmunity. However, there may be other parallel biologic processes that also lead to RA. These might include a collection of neoantigens (instead of a single instigator), inflammation at other mucosal sites (in particular the gut and periodontum) or genetic susceptibility factors that are currently undiscovered.
The lung as a site of initial RA-related autoantibody formation
While the particular immunologic mechanisms occurring locally in the lung prior to RA development remains to be fully elucidated, data support the notion that autoantibody development is initiated specifically in the lung (Table 2). Willis and colleagues investigated individuals at high risk of RA due to family history or detectable serum antibodies [77]. They evaluated a variety of RA-related autoantibodies in the serum and induced sputum of seronegative relatives, seropositive relatives, and early RA patients. They found that 65% of seropositive at-risk relatives had evidence for at least one RA-related autoantibody in the sputum (CCP2, CCP3, CCP3.1, IgG-RF, IgA-RF, or IgM-RF) compared to 86% of early RA subjects, and only 35% of seronegative at-risk relatives [77]. In addition, most of those with detectable serum RA-related autoantibodies had the same autoantibody detectable in the sputum [77]. The authors conclude that these results provide evidence that autoantibodies are initially locally produced in the lung prior to epitope spreading and subsequent detection in the serum. Some argued that sputum may have been contaminated from oral secretions in this study, especially relevant since the periodontum is another mucosal surface that may be important in RA pathogenesis [78]. In particular, the bacterium Propionibacterium gingivalis synthesizes a type of PAD that may induce local citrullination in the gingiva [79 80].
Table 2.
Selected epidemiologic and translational studies investigating the lung in RA preclinical phase transitions.
| First author year [reference] |
Preclinical RA phase |
Study Population Number of subjects |
Study design | Summary | Comments |
|---|---|---|---|---|---|
| Reynisdottir, 2014 [70] | Alveolar/airways mucosal inflammation, citrullination to form neoantigens, pulmonary RA- related autoantibodies |
Lung Investigation in Early RA Sweden N=105 untreated early RA patients, N=43 healthy controls |
Translational | 63% of ACPA- positive RA patients had parenchymal lung abnormalities, compared with only 37% of ACPA- negative RA patients and 30% of healthy controls (P<0.05) Airway changes detected were more frequent in RA patients than in healthy controls (66% versus 42%; P<0.05) |
|
| Reynisdottir, 2015 [71] | Alveolar/airways mucosal inflammation |
Lung Investigation in Early RA Sweden N=24 untreated early RA patients, N=79 healthy controls |
Translational | Lymphocyte infiltration was more frequently found in ACPA-positive patients (50%) as compared with ACPA-negative patients (17%) and controls (13%) BAL samples of patients with ACPA- positive, but not ACPA-negative, RA had significantly higher relative numbers of lymphocytes and expressed higher levels of activation markers compared with controls |
|
| Wilsher, 2012 [68] | Alveolar/airways mucosal inflammation |
New Zealand N=60 early RA patients |
Cross-sectional | 30% of early RA patients had respiratory symptoms 50% had bronchial wall thickening CCP and RF positivity was associated with decreased DLCO |
|
| Willis, 2013 [77] | Asymptomatic pulmonary RA- related autoantibodies |
Studies of the Etiology of RA US N=49 cases |
Case-control | One or more RA- related antibodies (CCP2, CCP3, CCP3.1, IgG-RF, IgA-RF, and IgM- RF) was detected in the sputum of 39% of at-risk seronegative cases |
|
| Demoruelle, 2012 [72] | Alveolar/airways mucosal inflammation, Asymptomatic serum RA- related autoantibodies |
Studies of the Etiology of RA US N=42 autoantibody- positive cases, N=15 autoantibody- negative controls, N=12 seropositive patients with early RA |
Case-control | 76% of seropositive controls without RA had any airways disease compared to 33% of seronegative controls (P=0.005) 50% of seropositive controls without RA had bronchial thickening compared to 13% of seronegative controls (P=0.015) 69% of seropositive controls without RA had air trapping compared to 7% of seronegative controls (P<0.001) |
|
| Janssen, 2015 [81] | Asymptomatic serum RA- related autoantibodies |
Netherlands N=86 RA patients, N=114 periodontitis patients, N=80 with bronchiectasis, N=41 cystic fibrosis patients, N=36 healthy controls |
Translational | CCP seropositivity was significantly associated with RA, bronchiectasis, and cystic fibrosis |
|
| Fischer, 2012 [69] | Alveolar/airways mucosal inflammation, asymptomatic serum RA- related autoantibodies |
US N=74 patients with respiratory symptoms but no RA |
Retrospective cohort | On high-resolution computed tomography scans, 54% had isolated airways disease, 14% had isolated interstitial lung disease (ILD), 26% had airways disease and ILD, and 7% had pulmonary fibrosis and emphysema |
|
| Ytterberg, 2015 [76] | Citrullination to form neoantigens, immune tolerance loss, joint specificity |
Epidemiological Investigations of RA Sweden N=393 patients with RA, N=152 healthy controls, N=236 disease controls |
Translational | Five peptides were shared between synovial and bronchial biopsy specimens in RA patients Two citrullinated vimentin (cit-vim) peptides were detected in the majority of synovial and lung tissue biopsies |
|
| Nannini, 2013 [94] | Clinical RA | Mayo Clinic US N=594 RA patients, N=596 non-RA patients |
Prospective cohort | Significantly more RA patients (9.6%) developed obstructive lung disease than non- RA patients (6.2%), HR 1.54 (95%CI 1.01–2.34) |
|
| Sparks, 2015 [3] | Preclinical to Clinical RA |
Nurses’ Health Study US N=119,209 |
Prospective cohort | RA was associated with increased respiratory mortality (HR 2.06, 95%CI 1.51–2.80) Seropositive RA associated with nearly 3-fold increased respiratory mortality (2.67, 95%CI 1.89– 3.77) Compared to non- RA |
|
ACPA, anti-citrullinated protein antibodies; BAL, bronchoalveolar lavage; CCP, cyclic citrullinated peptide; CI, confidence interval; DLCO, Diffusing capacity of the lungs for carbon monoxide; ILD, interstitial lung disease; HR, hazard ratio; RA, rheumatoid arthritis; RF, rheumatoid factor; RR, relative risk; US, United States.
Reynisodottir and colleagues also studied whether RA-related autoantibodies were present locally in the lung of newly-diagnosed untreated RA patients by obtaining specimens from bronchoalveolar lavage [70]. They found that CCP-positive RA patients had higher levels of ACPA autoantibodies in the bronchoalveolar lavage samples than in the serum, suggesting that the autoantibody production may have started in the lung [70]. These findings also correlated to patients with structural lung disease on high-resolution CT scans of the chest [70]. Only one CCP-negative RA patient and no healthy controls had detectable ACPA in the lung [70]. Since the fluid was collected by bronchoalveolar lavage, it was unlikely that oral flora contaminated these specimens. While preclinical cases were unavailable in this study, the RA patients were all untreated and were recently diagnosed arguing that these changes likely preceded the clinical onset of RA.
A study by Janssen and colleagues evaluated whether other diseases involving inflammation of the mucosal surfaces might have detectable RA-related autoantibodies in the serum. They tested CCP2 in the sera of patients with periodontitis, bronchiectasis, and cystic fibrosis as well as patients with RA and healthy controls [81]. CCP2 seropositivity was associated with RA, bronchiectasis, and cystic fibrosis but not periodontitis [81]. The authors conclude that this provides further evidence that local inflammation of the mucosa in the lung may provide conditions necessary for citrullination and ACPA production. Studies of seropositive RA-ILD without joint involvement and the previously described study of patients with pulmonary complaints and CCP positivity without RA argue that there is a group of patients on the spectrum of RA where clinical pulmonary disease precedes or is more severe than joint symptoms [69 82–85].
Future directions
While much progress has been made in elucidating the pathogenesis of RA, further work is needed in nearly every phase of preclinical RA. While the shared epitope is the strongest genetic risk factor for RA, currently described genetic factors only explain 18% of the genetic variance for RA [49 86]. Other potential RA susceptibility factors such as epigenetics, the microbiome, and metabolomics may be useful in identifying high-risk individuals and further bridge genetics with environmental factors for RA development [6 87]. Other lifestyle factors, particularly metabolic factors such as diet and obesity, may be important in the development of RA [88–90]. Future epidemiologic studies of interactions of smoking with other lifestyle factors or biomarkers may offer novel hypotheses for the biology behind preclinical RA phase transitions. While smoking is key in RA etiology, many non-smokers develop RA so studying other environmental factors may provide important alternate hypotheses for RA. Studies concerning the microbiome and periodontitis have already offered other hypotheses for RA development [80 87].
While structural abnormalities in the alveoli and airways of individuals with preclinical RA are well documented, less is known about the immunologic changes occurring locally in these patients. Most studies investigating this have used early RA patients and inferred that these changes occurred prior to clinical onset [70 71]. Since ACPA-positive RA patients often develop seropositivity within five years before clinical onset, these transition states have not been able to be studied directly given the difficulty in identifying these patients. Since many are otherwise healthy, the societal benefit of lung biopsy or even bronchoscopy may not outweigh the risk to the individual subjects. However, if the particular citrullinated neoantigen(s) formed during these processes were identified, targeted approaches to RA prevention would likely have much more potential for success and may also have importance in identifying targets for treatment of early and established RA. Lastly, while much progress has been made concerning the natural history of RA using banked specimens of patients who later developed RA, there are only a few longitudinal studies of at risk subjects available (typically unaffected first-degree relatives or those with arthralgias or undifferentiated inflammatory arthritis) [5 11 41 74 91]. Larger longitudinal studies with longer follow-up of individuals at increased risk are necessary to understand the natural history, but recruitment and retention are major challenges in this otherwise healthy population.
Preclinical disease processes may have clinical implications for patients with established RA. Several groups have recently identified respiratory mortality as a major contributor to the excess mortality of RA patients, particularly those with seropositive RA [3 92 93]. Emerging data suggest that this increased risk may be due to obstructive lung diseases and that RA patients are at particularly increased risk for asthma and COPD [3 94–96]. Since airways disease is particularly common in preclinical RA, these lesions may predispose RA patients to later develop clinical obstructive disease, beyond the effect of smoking. Preclinical pulmonary processes may also further subset patients with RA who later develop ILD which has known excess mortality in RA [97]. Identifying RA patients early in the development of ILD may similarly have major prevention or treatment implications and many of these processes likely are initiated in the preclinical RA phases [98].
Finally, understanding the pathogenesis of RA may have public health implications beyond RA. Studies of excess CVD outcomes among RA patients have impacted the understanding of how chronic inflammation affects the biology of CVD and has had broad public health significance beyond RA [99–103]. Similar to this framework, inflammation and autoimmunity may also be the common link between RA and increased respiratory disease burden and mortality. Bronchiolar immune tolerance loss with resultant systemic inflammation may bridge RA pathogenesis to clinical outcomes [93]. Evaluating the effect of RA on respiratory outcomes therefore has importance not only for RA patients, but may establish the roles of systemic inflammation and autoimmunity on respiratory outcomes beyond RA, which may have broad biologic, clinical, and public health implications.
Conclusions
The understanding of the pathogenesis of RA has progressed over the past two decades through both epidemiologic and translational studies. Specifically, epidemiologic investigations of smoking and risk of RA have informed the paradigm for RA development and generated novel hypotheses that have been tested in translational studies to further understand the biology of RA pathogenesis. The association of smoking with RA led investigators to initially consider the lung as a site of RA pathogenesis. Later studies associated smoking with progression to inflammatory arthritis or classifiable RA in high-risk populations. The particular association of smoking with seropositive RA, particularly CCP-positive RA, led investigators to consider citrullination as a biologic process central to RA pathogenesis. The identification of structural lung abnormalities and local autoantibody production in the lungs add to this hypothesis. Finally, the HLA shared epitope-smoking, occurring specifically in the peptide binding groove of HLA-DRβ1 offer the possibility of a physical interaction of a neoantigen induced by smoking and the immune cells where dysregulation may be initiated.
Acknowledgments
Funding: Dr. Sparks was supported by the Rheumatology Research Foundation Scientist Development Award and the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Numbers K23 AR069688 and L30 AR066953. Dr. Karlson was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Numbers R01 AR049880, K24 AR052403, and P60 AR047782. The funders had no role in the preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard University, its affiliated academic health care centers, or the National Institutes of Health.
Footnotes
Conflicts of Interest: None
Human and Animal Rights and Informed Consent: This article does not contain any primary data concerning studies with animals or human subjects.
Contributor Information
Jeffrey A. Sparks, Email: jasparks@partners.org, Brigham and Women’s Hospital, 75 Francis Street, Boston, MA 20115, USA.
Elizabeth W. Karlson, Email: ekarlson@partners.org, Brigham and Women’s Hospital, 75 Francis Street, Boston, MA 20115, USA.
References
- 1.Kirwan JR. Links between radiological change, disability, and pathology in rheumatoid arthritis. J Rheumatol. 2001;28(4):881–886. [PubMed] [Google Scholar]
- 2.Crowson CS, Matteson EL, Myasoedova E, et al. The lifetime risk of adult-onset rheumatoid arthritis and other inflammatory autoimmune rheumatic diseases. Arthritis Rheum. 2011;63(3):633–639. doi: 10.1002/art.30155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sparks JA, Chang SC, Liao KP, et al. Rheumatoid arthritis and mortality among women during 36 years of prospective follow-up: Results from the Nurses' Health Study. Arthritis Care Res (Hoboken) 2015 doi: 10.1002/acr.22752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dadoun S, Zeboulon-Ktorza N, Combescure C, et al. Mortality in rheumatoid arthritis over the last fifty years: systematic review and meta-analysis. Joint Bone Spine. 2013;80(1):29–33. doi: 10.1016/j.jbspin.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 5.van Steenbergen HW, Huizinga TW, van der Helm-van Mil AH. The preclinical phase of rheumatoid arthritis: what is acknowledged and what needs to be assessed? Arthritis Rheum. 2013;65(9):2219–2232. doi: 10.1002/art.38013. [DOI] [PubMed] [Google Scholar]
- 6.Sparks JA, Costenbader KH. Genetics, environment, and gene-environment interactions in the development of systemic rheumatic diseases. Rheum Dis Clin North Am. 2014;40(4):637–657. doi: 10.1016/j.rdc.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karlson EW, Deane K. Environmental and gene-environment interactions and risk of rheumatoid arthritis. Rheum Dis Clin North Am. 2012;38(2):405–426. doi: 10.1016/j.rdc.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerlag DM, Raza K, van Baarsen LG, et al. EULAR recommendations for terminology and research in individuals at risk of rheumatoid arthritis: report from the Study Group for Risk Factors for Rheumatoid Arthritis. Ann Rheum Dis. 2012;71(5):638–641. doi: 10.1136/annrheumdis-2011-200990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Demoruelle MK, Deane KD, Holers VM. When and where does inflammation begin in rheumatoid arthritis? Curr Opin Rheumatol. 2014;26(1):64–71. doi: 10.1097/BOR.0000000000000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demoruelle MK, Solomon JJ, Fischer A, et al. The lung may play a role in the pathogenesis of rheumatoid arthritis. Int J Clin Rheumtol. 2014;9(3):295–309. doi: 10.2217/ijr.14.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karlson EW, van Schaardenburg D, van der Helm-van Mil AH. Strategies to predict rheumatoid arthritis development in at-risk populations. Rheumatology (Oxford) 2014 doi: 10.1093/rheumatology/keu287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sparks JA, Chen CY, Jiang X, et al. Improved performance of epidemiologic and genetic risk models for rheumatoid arthritis serologic phenotypes using family history. Ann Rheum Dis. 2015;74(8):1522–1529. doi: 10.1136/annrheumdis-2013-205009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deane KD. Can rheumatoid arthritis be prevented? Best Pract Res Clin Rheumatol. 2013;27(4):467–485. doi: 10.1016/j.berh.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lahiri M, Morgan C, Symmons DP, et al. Modifiable risk factors for RA: prevention, better than cure? Rheumatology (Oxford) 2012;51(3):499–512. doi: 10.1093/rheumatology/ker299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sparks JA, Iversen MD, Miller Kroouze R, et al. Personalized Risk Estimator for Rheumatoid Arthritis (PRE-RA) Family Study: rationale and design for a randomized controlled trial evaluating rheumatoid arthritis risk education to first-degree relatives. Contemp Clin Trials. 2014;39(1):145–157. doi: 10.1016/j.cct.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerlag DM, Norris JM, Tak PP. RA: from risk factors and pathogenesis to prevention: Towards prevention of autoantibody-positive rheumatoid arthritis: from lifestyle modification to preventive treatment. Rheumatology (Oxford) 2015 doi: 10.1093/rheumatology/kev347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugiyama D, Nishimura K, Tamaki K, et al. Impact of smoking as a risk factor for developing rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis. 2010;69(1):70–81. doi: 10.1136/ard.2008.096487. [DOI] [PubMed] [Google Scholar]
- 18.Criswell LA, Merlino LA, Cerhan JR, et al. Cigarette smoking and the risk of rheumatoid arthritis among postmenopausal women: results from the Iowa Women's Health Study. Am J Med. 2002;112(6):465–471. doi: 10.1016/s0002-9343(02)01051-3. [DOI] [PubMed] [Google Scholar]
- 19.Heliovaara M, Aho K, Aromaa A, et al. Smoking and risk of rheumatoid arthritis. J Rheumatol. 1993;20(11):1830–1835. [PubMed] [Google Scholar]
- 20.Hazes JM, Dijkmans BA, Vandenbroucke JP, et al. Lifestyle and the risk of rheumatoid arthritis: cigarette smoking and alcohol consumption. Ann Rheum Dis. 1990;49(12):980–982. doi: 10.1136/ard.49.12.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vessey MP, Villard-Mackintosh L, Yeates D. Oral contraceptives, cigarette smoking and other factors in relation to arthritis. Contraception. 1987;35(5):457–464. doi: 10.1016/0010-7824(87)90082-5. [DOI] [PubMed] [Google Scholar]
- 22.Voigt LF, Koepsell TD, Nelson JL, et al. Smoking, obesity, alcohol consumption, and the risk of rheumatoid arthritis. Epidemiology. 1994;5(5):525–532. [PubMed] [Google Scholar]
- 23.Symmons DP, Bankhead CR, Harrison BJ, et al. Blood transfusion, smoking, and obesity as risk factors for the development of rheumatoid arthritis: results from a primary care-based incident case-control study in Norfolk, England. Arthritis Rheum. 1997;40(11):1955–1961. doi: 10.1002/art.1780401106. [DOI] [PubMed] [Google Scholar]
- 24.Stolt P, Bengtsson C, Nordmark B, et al. Quantification of the influence of cigarette smoking on rheumatoid arthritis: results from a population based case-control study, using incident cases. Ann Rheum Dis. 2003;62(9):835–841. doi: 10.1136/ard.62.9.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kallberg H, Ding B, Padyukov L, et al. Smoking is a major preventable risk factor for rheumatoid arthritis: estimations of risks after various exposures to cigarette smoke. Ann Rheum Dis. 2011;70(3):508–511. doi: 10.1136/ard.2009.120899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sparks JA, Chen CY, Hiraki LT, et al. Contributions of familial rheumatoid arthritis or lupus and environmental factors to risk of rheumatoid arthritis in women: a prospective cohort study. Arthritis Care Res (Hoboken) 2014;66(10):1438–1446. doi: 10.1002/acr.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Costenbader KH, Feskanich D, Mandl LA, et al. Smoking intensity, duration, and cessation, and the risk of rheumatoid arthritis in women. Am J Med. 2006;119(6):503. doi: 10.1016/j.amjmed.2005.09.053. e1–9. [DOI] [PubMed] [Google Scholar]
- 28.Di Giuseppe D, Discacciati A, Orsini N, et al. Cigarette smoking and risk of rheumatoid arthritis: a dose-response meta-analysis. Arthritis Res Ther. 2014;16(2):R61. doi: 10.1186/ar4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Di Giuseppe D, Orsini N, Alfredsson L, et al. Cigarette smoking and smoking cessation in relation to risk of rheumatoid arthritis in women. Arthritis Res Ther. 2013;15(2):R56. doi: 10.1186/ar4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yahya A, Bengtsson C, Lai TC, et al. Smoking is associated with an increased risk of developing ACPA-positive but not ACPA-negative rheumatoid arthritis in Asian populations: evidence from the Malaysian MyEIRA case-control study. Mod Rheumatol. 2012;22(4):524–531. doi: 10.1007/s10165-011-0544-2. [DOI] [PubMed] [Google Scholar]
- 31.Pedersen M, Jacobsen S, Klarlund M, et al. Environmental risk factors differ between rheumatoid arthritis with and without auto-antibodies against cyclic citrullinated peptides. Arthritis Res Ther. 2006;8(4):R133. doi: 10.1186/ar2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han B, Diogo D, Eyre S, et al. Fine Mapping Seronegative and Seropositive Rheumatoid Arthritis to Shared and Distinct HLA Alleles by Adjusting for the Effects of Heterogeneity. Am J Hum Genet. 2014;94(4):522–532. doi: 10.1016/j.ajhg.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Hair MJ, Landewe RB, van de Sande MG, et al. Smoking and overweight determine the likelihood of developing rheumatoid arthritis. Ann Rheum Dis. 2013;72(10):1654–1658. doi: 10.1136/annrheumdis-2012-202254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Steenbergen HW, Mangnus L, Reijnierse M, et al. Clinical factors, anticitrullinated peptide antibodies and MRI-detected subclinical inflammation in relation to progression from clinically suspect arthralgia to arthritis. Ann Rheum Dis. 2015 doi: 10.1136/annrheumdis-2015-208138. [DOI] [PubMed] [Google Scholar]
- 35.Sparks JA, Chang SC, Deane KD, et al. Genetic, environmental, and serologic risk factors for inflammatory joint signs among first-degree relatives without rheumatoid arthritis in a prospective cohort [abstract] [Accessed December 12, 2015];Arthritis Rheumatol. 2015 67(suppl 10) http://acrabstracts.org/abstract/genetic-environmental-and-serologic-risk-factors-for-inflammatory-joint-signs-among-first-degree-relatives-without-rheumatoid-arthritis-in-a-prospective-cohort/ [Google Scholar]
- 36.Hensvold AH, Magnusson PK, Joshua V, et al. Environmental and genetic factors in the development of anticitrullinated protein antibodies (ACPAs) and ACPA-positive rheumatoid arthritis: an epidemiological investigation in twins. Ann Rheum Dis. 2015;74(2):375–380. doi: 10.1136/annrheumdis-2013-203947. [DOI] [PubMed] [Google Scholar]
- 37.Mahdi H, Fisher BA, Kallberg H, et al. Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritis. Nat Genet. 2009;41(12):1319–1324. doi: 10.1038/ng.480. [DOI] [PubMed] [Google Scholar]
- 38.Bhatia SS, Majka DS, Kittelson JM, et al. Rheumatoid factor seropositivity is inversely associated with oral contraceptive use in women without rheumatoid arthritis. Ann Rheum Dis. 2007;66(2):267–269. doi: 10.1136/ard.2006.060004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kokkonen H, Brink M, Hansson M, et al. Associations of antibodies against citrullinated peptides with human leukocyte antigen-shared epitope and smoking prior to the development of rheumatoid arthritis. Arthritis Res Ther. 2015;17:125. doi: 10.1186/s13075-015-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rantapaa-Dahlqvist S, de Jong BA, Berglin E, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48(10):2741–2749. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- 41.Arkema EV, Goldstein BL, Robinson W, et al. Anti-citrullinated peptide autoantibodies, human leukocyte antigen shared epitope and risk of future rheumatoid arthritis: a nested case-control study. Arthritis Res Ther. 2013;15(5):R159. doi: 10.1186/ar4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hart JE, Laden F, Puett RC, et al. Exposure to traffic pollution and increased risk of rheumatoid arthritis. Environ Health Perspect. 2009;117(7):1065–1069. doi: 10.1289/ehp.0800503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hart JE, Kallberg H, Laden F, et al. Ambient air pollution exposures and risk of rheumatoid arthritis: results from the Swedish EIRA case-control study. Ann Rheum Dis. 2013;72(6):888–894. doi: 10.1136/annrheumdis-2012-201587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stolt P, Kallberg H, Lundberg I, et al. Silica exposure is associated with increased risk of developing rheumatoid arthritis: results from the Swedish EIRA study. Ann Rheum Dis. 2005;64(4):582–586. doi: 10.1136/ard.2004.022053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stolt P, Yahya A, Bengtsson C, et al. Silica exposure among male current smokers is associated with a high risk of developing ACPA-positive rheumatoid arthritis. Ann Rheum Dis. 2010;69(6):1072–1076. doi: 10.1136/ard.2009.114694. [DOI] [PubMed] [Google Scholar]
- 46.Too CL, Muhamad NA, Ilar A, et al. Occupational exposure to textile dust increases the risk of rheumatoid arthritis: results from a Malaysian population-based case-control study. Ann Rheum Dis. 2015 doi: 10.1136/annrheumdis-2015-208278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fernando MM, Stevens CR, Walsh EC, et al. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4(4):e1000024. doi: 10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30(11):1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 49.Okada Y, Wu D, Trynka G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506(7488):376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raychaudhuri S, Sandor C, Stahl EA, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. 2012;44(3):291–296. doi: 10.1038/ng.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karlson EW, Chang SC, Cui J, et al. Gene-environment interaction between HLA-DRB1 shared epitope and heavy cigarette smoking in predicting incident rheumatoid arthritis. Ann Rheum Dis. 2010;69(1):54–60. doi: 10.1136/ard.2008.102962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lundstrom E, Kallberg H, Alfredsson L, et al. Gene-environment interaction between the DRB1 shared epitope and smoking in the risk of anti-citrullinated protein antibody-positive rheumatoid arthritis: all alleles are important. Arthritis Rheum. 2009;60(6):1597–1603. doi: 10.1002/art.24572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karlson EW, Ding B, Keenan BT, et al. Association of environmental and genetic factors and gene-environment interactions with risk of developing rheumatoid arthritis. Arthritis Care Res (Hoboken) 2013;65(7):1147–1156. doi: 10.1002/acr.22005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Criswell LA, Saag KG, Mikuls TR, et al. Smoking interacts with genetic risk factors in the development of rheumatoid arthritis among older Caucasian women. Ann Rheum Dis. 2006;65(9):1163–1167. doi: 10.1136/ard.2005.049676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kallberg H, Padyukov L, Plenge RM, et al. Gene-gene and gene-environment interactions involving HLA-DRB1, PTPN22, and smoking in two subsets of rheumatoid arthritis. Am J Hum Genet. 2007;80(5):867–875. doi: 10.1086/516736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lundberg K, Bengtsson C, Kharlamova N, et al. Genetic and environmental determinants for disease risk in subsets of rheumatoid arthritis defined by the anticitrullinated protein/peptide antibody fine specificity profile. Ann Rheum Dis. 2013;72(5):652–658. doi: 10.1136/annrheumdis-2012-201484. [DOI] [PubMed] [Google Scholar]
- 57.Jiang X, Kallberg H, Chen Z, et al. An Immunochip-based interaction study of contrasting interaction effects with smoking in ACPA-positive versus ACPA-negative rheumatoid arthritis. Rheumatology (Oxford) 2016;55(1):149–155. doi: 10.1093/rheumatology/kev285. [DOI] [PubMed] [Google Scholar]
- 58.Too CL, Yahya A, Murad S, et al. Smoking interacts with HLA-DRB1 shared epitope in the development of anti-citrullinated protein antibody-positive rheumatoid arthritis: results from the Malaysian Epidemiological Investigation of Rheumatoid Arthritis (MyEIRA) Arthritis Res Ther. 2012;14(2):R89. doi: 10.1186/ar3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klareskog L, Stolt P, Lundberg K, et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006;54(1):38–46. doi: 10.1002/art.21575. [DOI] [PubMed] [Google Scholar]
- 60.Klareskog L, Ronnelid J, Lundberg K, et al. Immunity to citrullinated proteins in rheumatoid arthritis. Annu Rev Immunol. 2008;26:651–675. doi: 10.1146/annurev.immunol.26.021607.090244. [DOI] [PubMed] [Google Scholar]
- 61.Lee HS, Irigoyen P, Kern M, et al. Interaction between smoking, the shared epitope, and anti-cyclic citrullinated peptide: a mixed picture in three large North American rheumatoid arthritis cohorts. Arthritis Rheum. 2007;56(6):1745–1753. doi: 10.1002/art.22703. [DOI] [PubMed] [Google Scholar]
- 62.Padyukov L, Silva C, Stolt P, et al. A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum. 2004;50(10):3085–3092. doi: 10.1002/art.20553. [DOI] [PubMed] [Google Scholar]
- 63.Bos WH, Wolbink GJ, Boers M, et al. Arthritis development in patients with arthralgia is strongly associated with anti-citrullinated protein antibody status: a prospective cohort study. Ann Rheum Dis. 2010;69(3):490–494. doi: 10.1136/ard.2008.105759. [DOI] [PubMed] [Google Scholar]
- 64.El-Gabalawy HS, Robinson DB, Hart D, et al. Immunogenetic risks of anti-cyclical citrullinated peptide antibodies in a North American Native population with rheumatoid arthritis and their first-degree relatives. J Rheumatol. 2009;36(6):1130–1135. doi: 10.3899/jrheum.080855. [DOI] [PubMed] [Google Scholar]
- 65.Kim K, Jiang X, Cui J, et al. Interactions between amino acid-defined major histocompatibility complex class II variants and smoking in seropositive rheumatoid arthritis. Arthritis Rheumatol. 2015;67(10):2611–2623. doi: 10.1002/art.39228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chatzidionisyou A, Catrina AI. The lung in rheumatoid arthritis, cause or consequence? Curr Opin Rheumatol. 2016;28(1):76–82. doi: 10.1097/BOR.0000000000000238. [DOI] [PubMed] [Google Scholar]
- 67.Catrina AI, Ytterberg AJ, Reynisdottir G, et al. Lungs, joints and immunity against citrullinated proteins in rheumatoid arthritis. Nat Rev Rheumatol. 2014;10(11):645–653. doi: 10.1038/nrrheum.2014.115. [DOI] [PubMed] [Google Scholar]
- 68.Wilsher M, Voight L, Milne D, et al. Prevalence of airway and parenchymal abnormalities in newly diagnosed rheumatoid arthritis. Respir Med. 2012;106(10):1441–1446. doi: 10.1016/j.rmed.2012.06.020. [DOI] [PubMed] [Google Scholar]
- 69.Fischer A, Solomon JJ, du Bois RM, et al. Lung disease with anti-CCP antibodies but not rheumatoid arthritis or connective tissue disease. Respir Med. 2012;106(7):1040–1047. doi: 10.1016/j.rmed.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reynisdottir G, Karimi R, Joshua V, et al. Structural changes and antibody enrichment in the lungs are early features of anti-citrullinated protein antibody-positive rheumatoid arthritis. Arthritis Rheumatol. 2014;66(1):31–39. doi: 10.1002/art.38201. [DOI] [PubMed] [Google Scholar]
- 71.Reynisdottir G, Olsen H, Joshua V, et al. Signs of immune activation and local inflammation are present in the bronchial tissue of patients with untreated early rheumatoid arthritis. Ann Rheum Dis. 2015 doi: 10.1136/annrheumdis-2015-208216. 10.1136/annrheumdis-2015-208216. [DOI] [PubMed] [Google Scholar]
- 72.Demoruelle MK, Weisman MH, Simonian PL, et al. Brief report: airways abnormalities and rheumatoid arthritis-related autoantibodies in subjects without arthritis: early injury or initiating site of autoimmunity? Arthritis Rheum. 2012;64(6):1756–1761. doi: 10.1002/art.34344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pruijn GJ. Citrullination and carbamylation in the pathophysiology of rheumatoid arthritis. Front Immunol. 2015;6:192. doi: 10.3389/fimmu.2015.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sokolove J, Bromberg R, Deane KD, et al. Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PLoS One. 2012;7(5):e35296. doi: 10.1371/journal.pone.0035296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dwivedi N, Radic M. Citrullination of autoantigens implicates NETosis in the induction of autoimmunity. Ann Rheum Dis. 2014;73(3):483–491. doi: 10.1136/annrheumdis-2013-203844. [DOI] [PubMed] [Google Scholar]
- 76.Ytterberg AJ, Joshua V, Reynisdottir G, et al. Shared immunological targets in the lungs and joints of patients with rheumatoid arthritis: identification and validation. Ann Rheum Dis. 2015;74(9):1772–1777. doi: 10.1136/annrheumdis-2013-204912. [DOI] [PubMed] [Google Scholar]
- 77.Willis VC, Demoruelle MK, Derber LA, et al. Sputum autoantibodies in patients with established rheumatoid arthritis and subjects at risk of future clinically apparent disease. Arthritis Rheum. 2013;65(10):2545–2554. doi: 10.1002/art.38066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lundberg K, Wegner N, Yucel-Lindberg T, et al. Periodontitis in RA-the citrullinated enolase connection. Nat Rev Rheumatol. 2010;6(12):727–730. doi: 10.1038/nrrheum.2010.139. [DOI] [PubMed] [Google Scholar]
- 79.Wegner N, Wait R, Sroka A, et al. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and alpha-enolase: implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010;62(9):2662–2672. doi: 10.1002/art.27552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mikuls TR, Thiele GM, Deane KD, et al. Porphyromonas gingivalis and disease-related autoantibodies in individuals at increased risk of rheumatoid arthritis. Arthritis Rheum. 2012;64(11):3522–3530. doi: 10.1002/art.34595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Janssen KM, de Smit MJ, Brouwer E, et al. Rheumatoid arthritis-associated autoantibodies in non-rheumatoid arthritis patients with mucosal inflammation: a case-control study. Arthritis Res Ther. 2015;17:174. doi: 10.1186/s13075-015-0690-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang T, Zheng XJ, Liang BM, et al. Clinical features of rheumatoid arthritis-associated interstitial lung disease. Sci Rep. 2015;5:14897. doi: 10.1038/srep14897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yunt ZX, Solomon JJ. Lung disease in rheumatoid arthritis. Rheum Dis Clin North Am. 2015;41(2):225–236. doi: 10.1016/j.rdc.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen J, Shi Y, Wang X, et al. Asymptomatic preclinical rheumatoid arthritis-associated interstitial lung disease. Clin Dev Immunol. 2013;2013:406927. doi: 10.1155/2013/406927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gochuico BR, Avila NA, Chow CK, et al. Progressive preclinical interstitial lung disease in rheumatoid arthritis. Arch Intern Med. 2008;168(2):159–166. doi: 10.1001/archinternmed.2007.59. [DOI] [PubMed] [Google Scholar]
- 86.Viatte S, Plant D, Raychaudhuri S. Genetics and epigenetics of rheumatoid arthritis. Nat Rev Rheumatol. 2013;9(3):141–153. doi: 10.1038/nrrheum.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Scher JU, Sczesnak A, Longman RS, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2:e01202. doi: 10.7554/eLife.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lu B, Solomon DH, Costenbader KH, et al. Alcohol consumption and risk of incident rheumatoid arthritis in women: a prospective study. Arthritis Rheumatol. 2014;66(8):1998–2005. doi: 10.1002/art.38634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qin B, Yang M, Fu H, et al. Body mass index and the risk of rheumatoid arthritis: a systematic review and dose-response meta-analysis. Arthritis Res Ther. 2015;17(1):86. doi: 10.1186/s13075-015-0601-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Di Giuseppe D, Wallin A, Bottai M, et al. Long-term intake of dietary long-chain n-3 polyunsaturated fatty acids and risk of rheumatoid arthritis: a prospective cohort study of women. Ann Rheum Dis. 2014;73(11):1949–1953. doi: 10.1136/annrheumdis-2013-203338. [DOI] [PubMed] [Google Scholar]
- 91.Deane KD, Norris JM, Holers VM. Preclinical rheumatoid arthritis: identification, evaluation, and future directions for investigation. Rheum Dis Clin North Am. 2010;36(2):213–241. doi: 10.1016/j.rdc.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.England BR, Sayles H, Michaud K, et al. Cause-specific mMortality in US veteran men with rheumatoid arthritis. Arthritis Care Res (Hoboken) 2015 [Google Scholar]
- 93.Gonzalez A, Icen M, Kremers HM, et al. Mortality trends in rheumatoid arthritis: the role of rheumatoid factor. J Rheumatol. 2008;35(6):1009–1014. [PMC free article] [PubMed] [Google Scholar]
- 94.Nannini C, Medina-Velasquez YF, Achenbach SJ, et al. Incidence and mortality of obstructive lung disease in rheumatoid arthritis: a population-based study. Arthritis Care Res (Hoboken) 2013;65(8):1243–1250. doi: 10.1002/acr.21986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shen TC, Lin CL, Chen CH, et al. Increased risk of chronic obstructive pulmonary disease in patients with rheumatoid arthritis: a population-based cohort study. QJM. 2014;107(7):537–543. doi: 10.1093/qjmed/hcu027. [DOI] [PubMed] [Google Scholar]
- 96.Shen TC, Lin CL, Wei CC, et al. The risk of asthma in rheumatoid arthritis: a population-based cohort study. QJM. 2014;107(6):435–442. doi: 10.1093/qjmed/hcu008. [DOI] [PubMed] [Google Scholar]
- 97.Bongartz T, Nannini C, Medina-Velasquez YF, et al. Incidence and mortality of interstitial lung disease in rheumatoid arthritis: a population-based study. Arthritis Rheum. 2010;62(6):1583–1591. doi: 10.1002/art.27405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Doyle TJ, Patel AS, Hatabu H, et al. Detection of Rheumatoid Arthritis-Interstitial Lung Disease is Enhanced by Serum Biomarkers. Am J Respir Crit Care Med. 2015 doi: 10.1164/rccm.201411-1950OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Skeoch S, Bruce IN. Atherosclerosis in rheumatoid arthritis: is it all about inflammation? Nat Rev Rheumatol. 2015;11(7):390–400. doi: 10.1038/nrrheum.2015.40. [DOI] [PubMed] [Google Scholar]
- 100.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473(7347):317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 101.Ridker PM, Luscher TF. Anti-inflammatory therapies for cardiovascular disease. Eur Heart J. 2014;35(27):1782–1791. doi: 10.1093/eurheartj/ehu203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Everett BM, Pradhan AD, Solomon DH, et al. Rationale and design of the Cardiovascular Inflammation Reduction Trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J. 2013;166(2):199–207. doi: 10.1016/j.ahj.2013.03.018. e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ridker PM, Thuren T, Zalewski A, et al. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) Am Heart J. 2011;162(4):597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]