

Significance
Extensive efforts have been devoted to revealing the cognitive and molecular bases of autism spectrum disorders. In this work, by using a reversal-learning paradigm that measures behavioral flexibility in Drosophila, we show that flies with mutations of five autism susceptibility genes—Fragile X mental retardation 1, Ubiquitin-protein ligase E3A, Neurexin-1, Neuroligin 4, and Tuberous sclerosis complex 1—displayed severe reversal-learning defects which were caused by the inability to forget the previously formed memory by the activation of Rac1 (Ras-related C3 botulinum toxin substrate 1). These findings indicate that Rac1-dependent forgetting is a functional converging point for multiple autism-risk genes.
Keywords: autism, forgetting, Rac1, behavioral flexibility, Drosophila
Abstract
The etiology of autism is so complicated because it involves the effects of variants of several hundred risk genes along with the contribution of environmental factors. Therefore, it has been challenging to identify the causal paths that lead to the core autistic symptoms such as social deficit, repetitive behaviors, and behavioral inflexibility. As an alternative approach, extensive efforts have been devoted to identifying the convergence of the targets and functions of the autism-risk genes to facilitate mapping out causal paths. In this study, we used a reversal-learning task to measure behavioral flexibility in Drosophila and determined the effects of loss-of-function mutations in multiple autism-risk gene homologs in flies. Mutations of five autism-risk genes with diversified molecular functions all led to a similar phenotype of behavioral inflexibility indicated by impaired reversal-learning. These reversal-learning defects resulted from the inability to forget or rather, specifically, to activate Rac1 (Ras-related C3 botulinum toxin substrate 1)-dependent forgetting. Thus, behavior-evoked activation of Rac1-dependent forgetting has a converging function for autism-risk genes.
Autism spectrum disorders are common polygenic neurodevelopmental disorders diagnosed by a cluster of core clinical syndromes characterized by impaired social interaction, communication deficits, and behavioral inflexibility (1–3). A large number of autism-risk genes have been identified through different genetic approaches (4–8). These candidate genes play highly diversified roles in synaptic organization, transmission, intracellular signaling pathways, and protein expression regulation, among others (9). How do variants of these risk genes lead to the core symptoms of autism? Are there causal paths that can determine the onset of autism? Answering these questions is difficult because the disorder is multifactorial in nature with multiple genetic variants and environmental factors contributing to the core symptoms (10–13). To face this challenge, extensive efforts have been made to identify the targeting and functional convergence of the autism-risk genes, such as those involved in translation regulation (14–16) and long-range connectivity among different brain regions (17–19). These targets and functions provide a better biological basis for elucidating the causal paths between risk genes and the disorders of autism.
The aim of the this study was to determine whether Rac1 (Ras-related C3 botulinum toxin substrate 1)-dependent forgetting is a functional converging point for autism-risk genes. This idea was inspired by two clues. First, inhibition of Rac1 activity in Drosophila leads to the failure to activate Rac1-dependent forgetting, resulting in behavioral inflexibility as assayed through a reversal-learning task (20). Behavioral inflexibility has been observed frequently in the clinical studies of autism patients (21, 22), and children with autism are reported to have impaired reversal-learning, although they can learn new behavioral patterns (23–26). Thus, the inability to evoke Rac1-dependent forgetting causes behavioral inflexibility in transgenic flies, and the inability to forget may cause behavioral inflexibility in autism patients. Second, a systems biology analysis concludes that Rac1 plays a critical role in the neuropathological events associated with autism based on gene-expression analysis of cerebellar samples from patients with autism (27). Thus, we were led to investigate whether Rac1-dependent forgetting is a converging function of homologs of autism-risk genes.
With a well-defined reversal-learning paradigm in fruit flies, we investigated the effects of five of the most promising or probable autism candidate genes: Fmr1 (Fragile X mental retardation 1), Ube3a (Ubiquitin-protein ligase E3A), Nrx-1 (Neurexin-1), Nlg4 (Neuroligin 4), and Tsc1 (Tuberous sclerosis complex 1) (28). Fmr1 encodes an RNA-binding protein that regulates translation and has been extensively characterized in Drosophila for its role in synaptic signaling and long-term memory formation (29, 30). Ube3a encodes a ubiquitin ligase that has functions in learning and memory, synapse development, and synaptic plasticity (31–34). Importantly, Drosophila Ube3a (dUbe3a) is highly homologous to human Ube3a (hUbe3a) (34). Nrx-1 and Nlg4 are involved in synapse formation, maturation, and plasticity (35–37). Tsc1 is a negative regulator in the mTORC1 pathway (38–42) which regulates cell growth and proliferation in flies (43). These five autism-risk genes play vital roles at the synapse to regulate cytoskeleton remodeling via interaction with Rac1 (44–48). Here we report that mutations of all these genes lead to reduced behavioral flexibility as shown by severe reversal-learning impairment, no matter whether learning is affected. This common behavioral phenotype results from the failure to activate Rac1-dependent forgetting.
Results
Mutation of Fmr1 Leads to Impaired Reversal-Learning Through an Inability to Activate Rac1-Dependent Forgetting.
First, we screened all mutants of the five susceptibility genes intended for this work and identified Fmr1 and Ube3a mutants with a phenotype that showed impaired reversal-learning without a learning defect. We began our analysis using Fmr1 mutants and then extended the findings to flies with mutations in Ube3a and other three genes. In this study, two Fmr1 mutant alleles (Fmr15-HA-1014 and Fmr1CB-0950-3) were used; flies with both mutations showed significantly reduced expression of FMRP (Fmr1-encoded protein) on Western blotting, with the protein being nearly undetectable in Fmr1CB-0950-3 flies (Fig. 1A). The aversive conditioning assay (Fig. 1D) (49) showed that the learning index was normal in flies with the Fmr15-HA-1014 mutation (the milder allele) or slightly lower in flies with the Fmr1CB-0950-3 mutation (the stronger allele) (Fig. 1B). In contrast, both mutants showed severe reversal-learning defects (Fig. 1C). These results are consistent with a previous study of Fmr1-knockout mice, which displayed mildly impaired spatial learning ability alongside a significant defect in complex reversal-learning (50).
Fig. 1.

The inability to forget underlies reduced reversal-learning in Fmr1 mutant flies. (A) Western blots for different genotypes show significantly reduced FMRP levels in two mutants, Fmr15-HA-1014 and Fmr1CB-0950-3. n = 3. (B) The immediate memory scores for learning ability. Note normal learning in Fmr15-HA-1014 flies and a slight defect in Fmr1CB-0950-3 flies. (C) Immediate memory scores for reversal-learning showing profound defects in both Fmr1 mutants. (D) Schematic representation of behavioral paradigms for reversal-learning and the third-odor test. (E) Immediate memory scores obtained through the third-odor test after reversal-learning training. The open bars represent the old memory (OCT vs. IA), and the solid bars represent the new memory (MCH vs. IA). The new memory was very similar to the simple learning as shown in B. The old memories were significantly enhanced in both mutants. Numerically, PI(reversal-learning) = PI(new memory) − PI(old memory), suggesting that impaired reversal-learning resulted from the inability to erase or forget the old memory. (F) Memory decay curves for the old memory. After reversal-learning training, enhancement of the old memory in Fmr15-HA-1014 flies was sustained for at least 6 h. n = 7–8 for all behavioral data. All data in this and following figures, unless otherwise indicated, are means ± SEM and were analyzed by ANOVA: *P < 0.05, **P < 0.01, ***P < 0.001; n.s., not significant, P > 0.05.
Because the reversal-learning paradigm used here consists of two opposite aversive conditionings, the reversal-learning performance index (PI) could be perceived as a combination or addition of two opposite memories, referred to as the “old memory,” which is associated with the first conditioning, and the “new memory,” which is associated with the second conditioning. Thus, the defect in reversal-learning could result from a decreased new memory (the inability to learn new behavioral patterns) or an increased old memory (the inability to forget the old memory). These two memories could be dissected through a third-odor test (Fig. 1D) (20). Our third-odor test assay using isopentyl acetate (IA) revealed that Fmr1 mutants had an enhanced old memory and an unaltered new memory (Fig. 1E). Furthermore, this enhancement was surprisingly persistent (Fig. 1F). Thus, the defective reversal-learning in Fmr1 mutants is the result of being unable to forget the old memory.
To determine whether forgetting is affected generally, we assayed time-based passive forgetting and interference-based forgetting in the Fmr15-HA-1014 mutant with normal learning performance. Indeed, memory decay was slower in the Fmr15-HA-1014 mutant (Fig. 2B). This slow-decay phenotype did not result from enhanced memory formation, because acquisition curves were completely normal under various training strengths (Fig. 2A). We also tested both Fmr1 mutants with an interference-based paradigm, which is also capable of inducing forgetting (Fig. 2C) (20). Again, interference-induced forgetting was blocked in both mutants (Fig. 2D). Thus once the memory has been formed in Fmr1 mutants, it is so solid that forgetting could not be induced by reversal-learning, time-based decay, or even interference.
Fig. 2.

Slower passive memory decay and weaker interference-induced forgetting in Fmr1 mutant flies. (A) Normal immediate memory acquisition in response to different training strengths with varied electric shock intensity and number of electric shock pulses in the Fmr15-HA-1014 mutant. (B) Passive memory decay curves. Passive decay was slower in the Fmr15-HA-1014 mutant. (C) Schematic representation of the interference paradigm. Flies were trained to learn the first odor pairing (OCT vs. MCH), followed immediately by a different odor pairing (EA vs. IA), which represented interference with the first training. The PI was measured immediately after the training procedure. (D) The earlier formed memory was seriously disrupted by a new memory in WT flies but not in the two Fmr1 mutants (n = 6–8 for all behavioral data presented).
Because the activation of the small G protein Rac1 is critical in regulating forgetting and reversal-learning as examined here (20), we compared the activation levels of Rac-GTP in WT and Fmr1 mutant flies immediately after reversal-learning. In the naive state, total Rac and basal activated Rac were similar in both fly types (Fig. 3A). However, after reversal-learning, but not after 2×learning training (Fig. S1), Rac1 activity was significantly evoked in control flies, but no such increase occurred in the Fmr1 mutant (Fig. 3A). Thus, the inability to activate Rac1 is responsible for the inflexibility in the reversal-learning paradigm in the Fmr15-HA-1014 mutant.
Fig. 3.

The inability to activate Rac1 in the Fmr15-HA-1014 mutant. (A) Western blots showing Rac1 activation in heads of naive (N) and reversal-learning (R)–trained flies. Reversal-learning activates Rac1 immediately in WT flies but not in the Fmr15-HA-1014 mutant. n = 3. (B) Rescue of the reversal-learning defect by acute overexpression of constitutively active Rac1(V12). The pan-neuronal Gene-Switch (GS) expression system was used to manipulate the expression of mutant Rac1(V12) acutely. In the RU486 feeding group (RU+), the reversal-learning defect of elav-GS/UAS-Drac1(V12),Fmr15-HA-1014 flies was partially rescued by acutely induced overexpression of Rac1(V12) (n = 6–8).
Fig. S1.

The inability to activate Rac after 2×learning training in WT and Fmr15-HA-1014 flies. Western blots show an increase in Rac activity by reversal-learning (R) rather than 2×learning (2×L) in WT flies. However, in Fmr15-HA-1014 flies, the Rac activity displays no alteration after either 2×learning or reversal-learning training.
If the failure to activate Rac1 contributes to impaired reversal-learning, one would expect to see better reversal-learning performance associated with more Rac1-GTP. To test this possibility, constitutively active Rac1(V12) and dominant-negative Rac1(N17) were expressed under the control of an inducible pan-neuronal driver, elav (Embryonic lethal abnormal vision) in the Gene-Switch (GS) system (elav-GS) (51). Rac1(V12) expression acutely induced by feeding adult flies with the drug RU486 was confirmed by Western blotting (Fig. S2). We showed that reversal-learning was rescued by acute overexpression of Rac1(V12) in the Fmr1 mutant [elav-GS/UAS-Drac1(V12);Fmr15-HA-1014] (Fig. 3B). However, further inhibition of Rac1 activity through the expression of Rac1(N17) in the Fmr1 mutant [elav-GS/UAS-Drac1(N17);Fmr15-HA-1014] had no further effect on reversal-learning. In summary, the failure to activate Rac1 contributes to the phenotype of being unable to forget in the Fmr1 mutant, and that inability in turn leads to behavioral inflexibility.
Fig. S2.
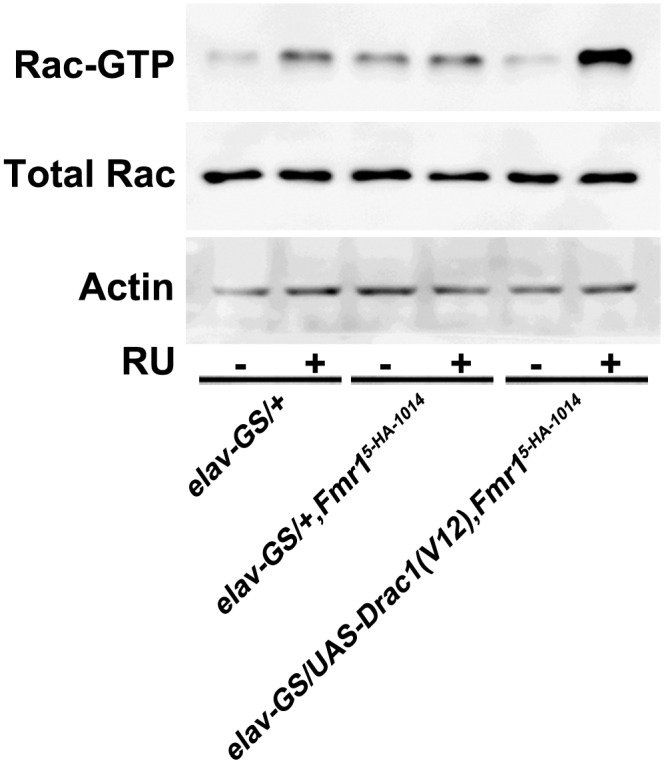
Acute overexpression of constitutively active Rac1(V12) by the GS system. Western blots show induced Rac1 activity in elav-GS/UAS-Drac1(V12),Fmr15-HA-1014 flies after RU486 feeding.
Acute Down-Regulation of Fmr1 Expression Leads to Behavioral Inflexibility.
One argument for the above phenotypes is that Fmr1 mutants have a developmental defect, because the Fmr1 gene is known to play a vital role in regulating neuronal elaboration and the differentiation of dendritic spines (52, 53). To rule out this possibility, we used heat-shock–induced acute expression of Fmr1-RNAi to down-regulate FMRP. Western blotting confirmed that heat-shock induction of UAS-Fmr1-RNAi was effective, because FMRP was significantly reduced in hs-Gal4/UAS-Fmr1-RNAi27484 transgenic flies 3 h after the heat-shock treatment (Fig. 4A). Consistent with the phenotypes of Fmr1 mutants, these RNAi flies displayed a reversal-learning defect 3 h after heat shock (Fig. 4B), and this reversal-learning defect also resulted from the inability to forget the old memory (Fig. 4C). In other words, within a short time window of less than 3 h, reduced Fmr1 expression could result in a defect in forgetting, although the phenotype is milder than that in the mutants. Thus, mutations of the Fmr1 gene can make an acute physiological contribution to the regulation of forgetting.
Fig. 4.

Acute knockdown of Fmr1 expression leads to diminished forgetting. (A) Western blots and statistical analysis confirm that the level of FMRP is reduced by acutely heat-shock–induced expression of Fmr1-RNAi. n = 3. (B) Reduced reversal-learning in transgenic flies with down-regulation of FMRP expression. (C) Enhanced old memory in transgenic flies subjected to reversal-learning training. Acute down-regulation of FMRP expression led to enhanced old memory in the third-odor test after reversal-learning training (n = 6–8 for all behavioral data presented).
Disruption of Ube3a Impairs Rac1-Dependent Forgetting.
The Ube3a mutant flies exhibited normal learning ability (Fig. 5A) but significantly impaired reversal-learning (Fig. 5B). The third-odor test revealed that the reversal-learning phenotype in these flies also resulted from the inability to forget old memory although the ability to learn new behavior patterns was normal (Fig. 5C). Also, spatiotemporal down-regulation of Ube3a expression by elav-GS induced phenotypes identical to those in the Ube3aEP3214 mutant (Fig. 5 D–F). Consistently, the inability to forget was associated with the failure in reversal-learning–induced activation of Rac1 (Fig. 5G). Thus, the Ube3a mutant also showed behavioral inflexibility that resulted from the failure in the activation of Rac1-dependent forgetting.
Fig. 5.

Disrupted expression of Ube3a produces the inability to forget. (A) Immediate memory scores for learning ability show normal learning in the Ube3aEP3214 mutant. (B) Immediate memory scores for reversal-learning show a profound defect in the Ube3aEP3214 mutant. (C) Immediate memory scores obtained through the third-odor test after reversal-learning training. The new memory was normal in the Ube3aEP3214 mutant, whereas the old memory was significantly enhanced in mutant flies. (D) Normal learning ability in transgenic flies with acute down-regulation of Ube3a by the elav-GS system. (E) Reduced reversal-learning in transgenic flies with down-regulation of Ube3a by the elav-GS system. (F) Enhanced old memory in transgenic flies subjected to reversal-learning training. Acute down-regulation of Ube3a expression led to enhanced old memory in the third-odor test after reversal-learning training. n = 6–8 for all behavioral data. (G) The inability to activate Rac1 in the Ube3aEP3214 mutant. Rac-GTP was significantly activated in WT flies, whereas there was little activation in the Ube3aEP3214 mutant (n = 3).
Impairment of Rac1-Dependent Active Forgetting in Three Other Autism-Susceptibility Gene Mutants.
The above findings made us highly interested in knowing whether the observed behavioral inflexibility, resulting from the inability to activate Rac1-dependent forgetting, is a general phenotype for autism-risk genes. We chose three additional autism-risk genes that are functionally unrelated to Fmr1 and Ube3a: Nrx-1, Nlg4, and Tsc1. All mutants used in this study were well characterized (31, 54–56) and were confirmed through PCR and DNA sequencing (Fig. S3).
Fig. S3.

Verification of autism-susceptibility gene mutations. (A–C) PCR primers were designed for amplification near the upstream and downstream of the P-element insertion point for Ube3aEP3214 (A) Nrx-1HP35068 (B), and Nlg4MB03367 (C) mutant flies. Primers for Ube3aEP3214 are: sense (S): GCCACCGTTCATCTTCGC, antisense (A): ATCCCTTGGAGTTTCACAATCA; primers for Nrx-1HP35068 are S: CCAAACCCATAGCACCAA, A: ATACGCACTCGGACAAGG; and primers for Nlg4MB03367 are S: TCAACAGAACGGTTTCCC, A: TGTGGTGTCCTTGTAGCG. (D) DNA sequencing for testing the mutation in Tsc1R460X from 460 CGA (Arg) to TGA (stop). Primers for Tsc1R460X sequencing are S: AACGAGATTGCCACGACC, A: CTGTAGCTTGCCATACGATT.
To our surprise, all three mutants had severe reversal-learning impairments (Fig. 6B) with mild learning defects (Fig. 6A). Using the third-odor test, we found that the Nlg4 and Tsc1 mutant flies displayed significantly enhanced old memory; the Nrx-1 mutant flies also showed an enhanced old memory, but the difference between the Nrx-1 mutants and WT flies was not statistically significant (Fig. 6C). Also, spatiotemporal down-regulation of Nrx-1 and Tsc1 expression by elav-GS induced normal learning (Fig. 6D) but reduced reversal-learning (Fig. 6E) and unforgettable old memory (Fig. 6F). Furthermore, Rac1 could not be activated through reversal training in any of the mutants (Fig. 6G). Thus, the behavioral inflexibility caused by impaired Rac1-dependent forgetting appears to be a common phenotype for autism-risk genes.
Fig. 6.

Impairment of Rac1-dependent forgetting results in behavioral inflexibility in mutants of autism candidate genes. (A) Flies with mutations of the autism candidate genes Nrx-1, Nlg4, and Tsc1 displayed mild learning defects. (B) Flies with mutations of these autism candidate genes displayed severe reversal-learning deficiencies. (C) The old memories were significantly enhanced in the Nlg4MB03367 and Tsc1R460X mutant flies. (D) Transgenic flies with acute down-regulation of Nrx-1 and Tsc1 by the elav-GS system had normal learning ability. (E) Reversal-learning was reduced in transgenic flies with down-regulation of Nrx-1 and Tsc1 by the elav-GS system. (F) Old memory was enhanced in transgenic flies with down-regulation of Nrx-1 and Tsc1 by the elav-GS system. n = 6–8 for all behavioral data. (G) The inability to activate Rac1 after reversal-learning in flies with mutations in the autism candidate genes. N, naive; R, reversal-learning training (n = 3).
Fmr1, Ube3a, Nrx-1, and Tsc1 Function in the Mushroom Body to Regulate Forgetting.
Considering the vital roles of the mushroom body in learning, memory, and forgetting (20, 57, 58), we asked whether the autism-risk genes we studied were involved in regulating forgetting in the mushroom body. After acute knockdown of Fmr1, Ube3a, Nrx-1, and Tsc1 expression in the mushroom body (59), normal learning ability, significant reversal-learning defects, and unforgettable old memory were observed (Fig. 7). The involvement of the mushroom body in regulating forgetting was confirmed further by knockdown of Fmr1 with the OK107 driver (Fig. S4). Thus, the autism-susceptibility genes Fmr1, Ube3a, Nrx-1, and Tsc1 are critical for regulating forgetting in Drosophila mushroom body.
Fig. 7.

Forgetting cannot be produced by acute down-regulation of Fmr1, Ube3a, Nrx-1, and Tsc1 expression in the mushroom body. (A) Transgenic flies with acute down-regulation of Fmr1, Ube3a, Nrx-1, and Tsc1 in the mushroom body by the MB-GS system have normal learning ability. (B) Transgenic flies with acute down-regulation of Fmr1, Ube3a, Nrx-1, and Tsc1 in the mushroom body have reduced reversal-learning ability. (C) Old memory is enhanced in transgenic flies with acute down-regulation of Fmr1, Ube3a, Nrx-1, and Tsc1 in mushroom body neurons (n = 6 for all behavioral data presented).
Fig. S4.

Down-regulation of Fmr1 expression in the mushroom body leads to diminished forgetting. UAS-Fmr1-RNAi34944 flies were crossed with WT flies (+) and the MB Gal4, OK107. (A) No effect on learning was found when Fmr1 was down-regulated in the mushroom body. (B) Reversal-learning was reduced in UAS-Fmr1-RNAi34944/+;OK107/+ flies with down-regulation of FMRP in the mushroom body. (C) Down-regulation of FMRP expression led to enhanced old memory in transgenic flies subjected to reversal-learning training.
Discussion
This study investigates whether forgetting-dependent behavioral inflexibility is a common phenotype for autism-risk genes and whether behavior-dependent activation of Rac1 is the convergent molecular target for these genes. To minimize genetic and behavioral variations, flies for all genotypes had the same isogenic background, and behavioral assays were performed using the well-established balanced protocol (Methods). We used aversive conditioning and its associated reversal-learning task to investigate the functions of five autism-susceptibility genes: Fmr1, Ube3a, Nrx-1, Nlg4, and Tsc1. Four major findings emerge from the current study. First, mutation and RNAi-induced knockdown of these five autism-risk genes result in strong reversal-learning defects independent of learning ability. Second, these reversal-learning phenotypes are all caused by the inability to forget the old memory. Third, all the autism-risk gene mutants studied failed to trigger the behavior-dependent activation of Rac1, a reported regulator of forgetting. Fourth, the effects of these genes are confined within the mushroom body, in which Rac1-dependent forgetting is involved. These results suggest that the inability to activate Rac1-dependent forgetting is a converging mechanism for multiple autism-risk genes.
It is interesting that all five of the autism-risk genes with diverse functions funnel to a deficit in Rac1 activation. These five autism-risk genes have been reported to be involved in the Rac1 signaling pathway. The cytoplasmic FMRP-interacting protein (CYFIP) directly links Rac1 and FMRP to modulate cytoskeleton remodeling (45); Tsc1 functionally regulates Rac1 activity (46, 60); Ube3a promotes Rho-GEF Pbl degradation via ubiquitination to affect Rac1 activation (47); and upon synaptic activation Rho-GEF Kal-7 disassembles from the Nrx-1/Nlg4/DISC1 complex to modulate the Rac1 pathway (48). Several other autism-risk genes, such as Nlg1, Nrx-4, P-Rex1, and Shank-3, have also been reported to participate in the Rac1-signaling pathway (61–64). In addition, when the gene–environment interactions of 122 genes and 191 factors in the autistic context were analyzed by systems biology, Rac1 was predicted to be a converging node that genetically links to the neurobiology of autism (27, 65). Taken together, these findings indicate that Rac1 is a functional converging site for autism-risk genes.
Although autism is considered to be a developmental disorder, emerging evidence points to the postdevelopmental effects of autism-risk genes in adults (66–68). In this study, acute down-regulation of these five autism-risk genes at the adult stage led to impaired behavioral flexibility with reduced reversal-learning and resistant old memory. Thus, all five autism-risk genes in our study are physiologically involved in regulating behavioral flexibility.
In addition to behavioral flexibility, the autism genes in our study participate in some other cognitive processes such as learning and long-term memory (29, 33, 69, 70). For instance, as a general translational repressor, Fmr1 interacts with Staufen and the RNAi pathway to regulate long-term memory formation. However, Rac1-dependent forgetting has been reported to be independent of protein synthesis and long-term memory (20). These findings indicate that Fmr1 plays a dual role in regulating long-term memory and forgetting. Taken together, the data presented here indicate that the inability to forget is a cognitive mechanism in autism.
Materials and Methods
Drosophila Stock.
Flies were raised at the room temperature of 23 °C, with 70% relative humidity. The following strains were obtained from the Bloomington Drosophila Stock Center: Fmr1CB-0950-3 (no. 123391), Fmr15-HA-1014 (no. 125009), Ube3aEP3214 (no. 17099), Nrx-1HP35068 (no. 21977), Nlg4MB03367 (no. 23608), UAS-Fmr1-RNAi (no. 27484), UAS-Drac1(V12) (no. 6291), UAS-Drac1(N17) (no. 6292), UAS-Ube3a-RNAi (no. 31972), UAS-Nrx1-RNAi (no. 27502), and UAS-Tsc1-RNAi (no. 31314). OK107 and hs-GAL4 are laboratory stocks. elav-GS and mushroom body (MB)-GS were kindly provided by Ronald L. Davis, The Scripps Research Institute, Jupiter, FL. UAS-Fmr1-RNAi (no. 34933) was kindly provided by Jianquan Ni, Tsinghua Fly Center, Beijing. Tsc1R460X was kindly provided by Rongwen Xi, National Institute of Biological Sciences, Beijing. All flies with autism-susceptibility gene mutations were backcrossed with the w1118 flies (stock in our laboratory) for five generations.
Heat-Shock Regimen.
When the hs-GAL4 and UAS-Fmr1-RNAi strains were crossed, flies were raised at 23 °C. Flies for our studies were collected 2 to 4 d after eclosion. Before the behavioral and Western-blotting experiments, the heat-induced group was transferred to a 37 °C incubator for 40 min and then was placed at 23 °C for 3 h to induce the expression of RNAi. The uninduced group was kept at 23 °C.
Drug Feeding.
The procedures were described previously (71). For RU486 feeding (RU+), flies were fed with 500 μM RU486 (mifepristone; J&K Chemicals) dissolved in control solution.
Pavlovian Olfactory Associative Aversive Learning.
During one training session, about 100 flies were loaded in a training tube and then were sequentially exposed to two odors, 3-octanol (OCT) (1.5 × 10−3 dilution in heavy mineral oil; Fluka ) and 4-methylcyclohexanol (MCH) (1.0 × 10−3 dilution in heavy mineral oil; Fluka), for 60 s each, with 45 s of fresh air between the two odor presentations. Foot shock (60 V, 12 1.5-s pulses) was presented during the presentation of the first odor [positive conditioned stimulus (CS+)], but not during the second [negative conditioned stimulus (CS−)]. To test the learning (immediate memory) index, the flies were transferred to a T-maze and allowed to choose between the CS+ and the CS− arms for 60 s. The PI was calculated from the distribution of the number of flies in the two arms of the T-maze, according to the equation (CS− − CS+)/(CS− + CS+) × 100. To eliminate any odor-preference bias, two reciprocal groups of flies were trained using OCT and MCH as the CS+ respectively. Each PI (n = 1) is the average of PI (OCT) and PI (MCH). A PI of 0 suggests no learning, whereas a PI of 100 suggests perfect learning.
Reversal-Learning and Third-Odor Test.
In the reversal-learning paradigm, following a regular learning session, flies were retrained immediately by another session in which the conditioned stimulus–unconditioned stimulus contingency was reversed. In the third-odor test, after reversal-learning training (note that OCT was paired with shock in the initial session), flies were required to choose between OCT and a previously unexposed odor, IA (7.0 × 10−3 dilution; Avocado Research Chemicals) to measure their old memory and to choose between MCH and IA (1.0 × 10−3 dilution) to measure their new memory. In the end, reciprocal groups were averaged to get n = 1. Before the behavioral test, untrained flies were used to balance the odor preference, and the score of the untrained group was subtracted from that of the trained group to yield a final PI.
Interference Learning.
After a regular learning session (OCT/MCH), trained flies immediately received another learning session with a novel pair of odors: ethyl acetate (EA) (2.0 × 10−3 dilution; Alfa Aesar) and IA (2.0 × 10−3 dilution; Avocado Research Chemicals) as CS+ and CS− (referred to as “interference+”), respectively. The control group (“interference−”) received only the initial learning session. After the interference training session, flies were required to choose between OCT and MCH to measure their previous memory.
Rac1 Activity Assay and Western Blotting.
The heads of approximately 400 flies were isolated to detect the relative levels of Rac1-GTP by PBD pull-down assay (no. 17-441; Millipore) according to the manufacturer’s procedure. The anti-human Rac1 monoclonal antibody (1:3,000 dilution; BD Transduction Laboratories) was used to detect total and activated Rac1 levels. For the detection of FMRP levels, the heads of 50 flies were homogenized in 100 μL of radioimmunoprecipitation assay lysis buffer (no. P0013; Beyotime). The anti-FMRP antibody (no. 05-1512; Millipore) was diluted 1:3,000.
Statistics.
The statistical results are shown as means ± SEM and were analyzed by ANOVA [*P < 0.05; **P < 0.01; ***P < 0.001; n.s., nonsignificant (P > 0.05)].
Acknowledgments
We thank Ronald L. Davis, Jianquan Ni, Rongwen Xi, the Bloomington Stock Center, and the Tsinghua Fly Center for fly stocks and Yichun Shuai, Qian Li, Cheng Huang, Weiwei Ma, Zhiyong Xie, and Xuchen Zhang for their help. This work was supported by National Basic Research Project Grant 2013cb835100 (973 Program) from the Ministry of Science and Technology of China (to Y.Z.), National Science Foundation of China Grant 91332207 (to Y.Z.), Tsinghua University Initiative Scientific Research Program Grant 20111080956 (to Y.Z.), a grant of Yunnan Provincial Zhongyi Expert Workstation (2014IC046), and the Tsinghua-Peking Joint Center for Life Sciences.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1602152113/-/DCSupplemental.
References
- 1.Hill EL. Evaluating the theory of executive dysfunction in autism. Dev Rev. 2004;24(2):189–233. [Google Scholar]
- 2.Rajendran G, Mitchell P. Cognitive theories of autism. Dev Rev. 2007;27(2):224–260. [Google Scholar]
- 3.Dajani DR, Uddin LQ. Demystifying cognitive flexibility: Implications for clinical and developmental neuroscience. Trends Neurosci. 2015;38(9):571–578. doi: 10.1016/j.tins.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12(5):363–376. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Itsara A, et al. De novo rates and selection of large copy number variation. Genome Res. 2010;20(11):1469–1481. doi: 10.1101/gr.107680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iossifov I, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74(2):285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neale BM, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485(7397):242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Roak BJ, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43(6):585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandler WM, Sebat J. From de novo mutations to personalized therapeutic interventions in autism. Annu Rev Med. 2015;66:487–507. doi: 10.1146/annurev-med-091113-024550. [DOI] [PubMed] [Google Scholar]
- 10.Phillips M, Pozzo-Miller L. Dendritic spine dysgenesis in autism related disorders. Neurosci Lett. 2015;601:30–40. doi: 10.1016/j.neulet.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsai NP, et al. Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell. 2012;151(7):1581–1594. doi: 10.1016/j.cell.2012.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim YS, Leventhal BL. Genetic epidemiology and insights into interactive genetic and environmental effects in autism spectrum disorders. Biol Psychiatry. 2015;77(1):66–74. doi: 10.1016/j.biopsych.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalkbrenner AE, Schmidt RJ, Penlesky AC. Environmental chemical exposures and autism spectrum disorders: A review of the epidemiological evidence. Curr Probl Pediatr Adolesc Health Care. 2014;44(10):277–318. doi: 10.1016/j.cppeds.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gkogkas CG, et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature. 2013;493(7432):371–377. doi: 10.1038/nature11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ebert DH, Greenberg ME. Activity-dependent neuronal signalling and autism spectrum disorder. Nature. 2013;493(7432):327–337. doi: 10.1038/nature11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelleher RJ, 3rd, Bear MF. The autistic neuron: Troubled translation? Cell. 2008;135(3):401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 17.Just MA, Keller TA, Malave VL, Kana RK, Varma S. Autism as a neural systems disorder: A theory of frontal-posterior underconnectivity. Neurosci Biobehav Rev. 2012;36(4):1292–1313. doi: 10.1016/j.neubiorev.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maximo JO, Cadena EJ, Kana RK. The implications of brain connectivity in the neuropsychology of autism. Neuropsychol Rev. 2014;24(1):16–31. doi: 10.1007/s11065-014-9250-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Mello AM, Stoodley CJ. Cerebro-cerebellar circuits in autism spectrum disorder. Front Neurosci. 2015;9:408. doi: 10.3389/fnins.2015.00408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shuai Y, et al. Forgetting is regulated through Rac activity in Drosophila. Cell. 2010;140(4):579–589. doi: 10.1016/j.cell.2009.12.044. [DOI] [PubMed] [Google Scholar]
- 21.Uddin LQ, et al. Brain state differentiation and behavioral inflexibility in autism. Cereb Cortex. 2015;25(12):4740–4747. doi: 10.1093/cercor/bhu161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D’Cruz AM, et al. Reduced behavioral flexibility in autism spectrum disorders. Neuropsychology. 2013;27(2):152–160. doi: 10.1037/a0031721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merchán-Naranjo J, et al. Executive function is affected in autism spectrum disorder, but does not correlate with intelligence. Rev Psiquiatr Salud Ment. 2016;9(1):39–50. doi: 10.1016/j.rpsm.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 24.Corbett BA, Constantine LJ, Hendren R, Rocke D, Ozonoff S. Examining executive functioning in children with autism spectrum disorder, attention deficit hyperactivity disorder and typical development. Psychiatry Res. 2009;166(2-3):210–222. doi: 10.1016/j.psychres.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geurts HM, Corbett B, Solomon M. The paradox of cognitive flexibility in autism. Trends Cogn Sci. 2009;13(2):74–82. doi: 10.1016/j.tics.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaland N, Smith L, Mortensen EL. Brief report: Cognitive flexibility and focused attention in children and adolescents with Asperger syndrome or high-functioning autism as measured on the computerized version of the Wisconsin Card Sorting Test. J Autism Dev Disord. 2008;38(6):1161–1165. doi: 10.1007/s10803-007-0474-1. [DOI] [PubMed] [Google Scholar]
- 27.Zeidán-Chuliá F, et al. Exploring the multifactorial nature of autism through computational systems biology: Calcium and the Rho GTPase RAC1 under the spotlight. Neuromolecular Med. 2013;15(2):364–383. doi: 10.1007/s12017-013-8224-3. [DOI] [PubMed] [Google Scholar]
- 28.Abrahams BS, Geschwind DH. Advances in autism genetics: On the threshold of a new neurobiology. Nat Rev Genet. 2008;9(5):341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bolduc FV, Bell K, Cox H, Broadie KS, Tully T. Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat Neurosci. 2008;11(10):1143–1145. doi: 10.1038/nn.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedman SH, Dani N, Rushton E, Broadie K. Fragile X mental retardation protein regulates trans-synaptic signaling in Drosophila. Dis Model Mech. 2013;6(6):1400–1413. doi: 10.1242/dmm.012229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu Y, et al. A Drosophila model for Angelman syndrome. Proc Natl Acad Sci USA. 2008;105(34):12399–12404. doi: 10.1073/pnas.0805291105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greer PL, et al. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell. 2010;140(5):704–716. doi: 10.1016/j.cell.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun J, et al. UBE3A regulates synaptic plasticity and learning and memory by controlling SK2 channel endocytosis. Cell Reports. 2015;12(3):449–461. doi: 10.1016/j.celrep.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu Y, et al. The Drosophila homologue of the Angelman syndrome ubiquitin ligase regulates the formation of terminal dendritic branches. Hum Mol Genet. 2009;18(3):454–462. doi: 10.1093/hmg/ddn373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh SK, et al. Astrocytes assemble thalamocortical synapses by bridging NRX1α and NL1 via Hevin. Cell. 2016;164(1-2):183–196. doi: 10.1016/j.cell.2015.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larkin A, et al. Neurexin-1 regulates sleep and synaptic plasticity in Drosophila melanogaster. Eur J Neurosci. 2015;42(7):2455–2466. doi: 10.1111/ejn.13023. [DOI] [PubMed] [Google Scholar]
- 37.Li Y, et al. Drosophila neuroligin 4 regulates sleep through modulating GABA transmission. J Neurosci. 2013;33(39):15545–15554. doi: 10.1523/JNEUROSCI.0819-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hatton DD, et al. Autistic behavior in children with fragile X syndrome: Prevalence, stability, and the impact of FMRP. Am J Med Genet A. 2006;140A(17):1804–1813. doi: 10.1002/ajmg.a.31286. [DOI] [PubMed] [Google Scholar]
- 39.Nurmi EL, et al. Linkage disequilibrium at the Angelman syndrome gene UBE3A in autism families. Genomics. 2001;77(1-2):105–113. doi: 10.1006/geno.2001.6617. [DOI] [PubMed] [Google Scholar]
- 40.Kim HG, et al. Disruption of neurexin 1 associated with autism spectrum disorder. Am J Hum Genet. 2008;82(1):199–207. doi: 10.1016/j.ajhg.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jamain S, et al. Paris Autism Research International Sibpair Study Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34(1):27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baker P, Piven J, Sato Y. Autism and tuberous sclerosis complex: Prevalence and clinical features. J Autism Dev Disord. 1998;28(4):279–285. doi: 10.1023/a:1026004501631. [DOI] [PubMed] [Google Scholar]
- 43.Tapon N, Ito N, Dickson BJ, Treisman JE, Hariharan IK. The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell. 2001;105(3):345–355. doi: 10.1016/s0092-8674(01)00332-4. [DOI] [PubMed] [Google Scholar]
- 44.Peça J, Ting J, Feng G. SnapShot: Autism and the synapse. Cell. 2011;147(3):706, 706.e1. doi: 10.1016/j.cell.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 45.Abekhoukh S, Bardoni B. CYFIP family proteins between autism and intellectual disability: Links with Fragile X syndrome. Front Cell Neurosci. 2014;8:81. doi: 10.3389/fncel.2014.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohsawa M, et al. TSC1 controls distribution of actin fibers through its effect on function of Rho family of small GTPases and regulates cell migration and polarity. PLoS One. 2013;8(1):e54503. doi: 10.1371/journal.pone.0054503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reiter LT, Seagroves TN, Bowers M, Bier E. Expression of the Rho-GEF Pbl/ECT2 is regulated by the UBE3A E3 ubiquitin ligase. Hum Mol Genet. 2006;15(18):2825–2835. doi: 10.1093/hmg/ddl225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Owczarek S, Bang ML, Berezin V. Neurexin-Neuroligin synaptic complex regulates schizophrenia-related DISC1/Kal-7/Rac1 “Signalosome”. Neural Plast. 2015;2015:167308. doi: 10.1155/2015/167308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tully T, Quinn WG. Classical conditioning and retention in normal and mutant Drosophila melanogaster. J Comp Physiol A Neuroethol Sens Neural Behav Physiol. 1985;157(2):263–277. doi: 10.1007/BF01350033. [DOI] [PubMed] [Google Scholar]
- 50.D’Hooge R, et al. Mildly impaired water maze performance in male Fmr1 knockout mice. Neuroscience. 1997;76(2):367–376. doi: 10.1016/s0306-4522(96)00224-2. [DOI] [PubMed] [Google Scholar]
- 51.Nicholson L, et al. Spatial and temporal control of gene expression in Drosophila using the inducible GeneSwitch GAL4 system. I. Screen for larval nervous system drivers. Genetics. 2008;178(1):215–234. doi: 10.1534/genetics.107.081968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci. 2001;21(14):5139–5146. doi: 10.1523/JNEUROSCI.21-14-05139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pan L, Zhang YQ, Woodruff E, Broadie K. The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr Biol. 2004;14(20):1863–1870. doi: 10.1016/j.cub.2004.09.085. [DOI] [PubMed] [Google Scholar]
- 54.Quan Z, Sun P, Lin G, Xi R. TSC1/2 regulates intestinal stem cell maintenance and lineage differentiation through Rheb-TORC1-S6K but independently of nutritional status or Notch regulation. J Cell Sci. 2013;126(Pt 17):3884–3892. doi: 10.1242/jcs.125294. [DOI] [PubMed] [Google Scholar]
- 55.Staudt N, et al. Gain-of-function screen for genes that affect Drosophila muscle pattern formation. PLoS Genet. 2005;1(4):e55. doi: 10.1371/journal.pgen.0010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bellen HJ, et al. The BDGP gene disruption project: Single transposon insertions associated with 40% of Drosophila genes. Genetics. 2004;167(2):761–781. doi: 10.1534/genetics.104.026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heisenberg M, Borst A, Wagner S, Byers D. Drosophila mushroom body mutants are deficient in olfactory learning. J Neurogenet. 1985;2(1):1–30. doi: 10.3109/01677068509100140. [DOI] [PubMed] [Google Scholar]
- 58.Heisenberg M. Mushroom body memoir: From maps to models. Nat Rev Neurosci. 2003;4(4):266–275. doi: 10.1038/nrn1074. [DOI] [PubMed] [Google Scholar]
- 59.Mao Z, Roman G, Zong L, Davis RL. Pharmacogenetic rescue in time and space of the rutabaga memory impairment by using Gene-Switch. Proc Natl Acad Sci USA. 2004;101(1):198–203. doi: 10.1073/pnas.0306128101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goncharova E, Goncharov D, Noonan D, Krymskaya VP. TSC2 modulates actin cytoskeleton and focal adhesion through TSC1-binding domain and the Rac1 GTPase. J Cell Biol. 2004;167(6):1171–1182. doi: 10.1083/jcb.200405130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Newell-Litwa KA. Breaking down to build up: Neuroligin’s C-terminal domain strengthens the synapse. J Cell Biol. 2016;212(4):375–377. doi: 10.1083/jcb.201601081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mack NA, Georgiou M. The interdependence of the Rho GTPases and apicobasal cell polarity. Small GTPases. 2014;5(2):10. doi: 10.4161/21541248.2014.973768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duffney LJ, et al. Autism-like deficits in Shank3-deficient mice are rescued by targeting actin regulators. Cell Reports. 2015;11(9):1400–1413. doi: 10.1016/j.celrep.2015.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li J, et al. Synaptic P-Rex1 signaling regulates hippocampal long-term depression and autism-like social behavior. Proc Natl Acad Sci USA. 2015;112(50):E6964–E6972. doi: 10.1073/pnas.1512913112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zeidán-Chuliá F, Salmina AB, Noda M, Verkhratsky A. Rho GTPase RAC1 at the molecular interface between genetic and environmental factors of autism spectrum disorders. Neuromolecular Med. 2015;17(4):333–334. doi: 10.1007/s12017-015-8366-6. [DOI] [PubMed] [Google Scholar]
- 66.Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315(5815):1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mei Y, et al. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature. 2016;530(7591):481–484. doi: 10.1038/nature16971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Clement JP, et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell. 2012;151(4):709–723. doi: 10.1016/j.cell.2012.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zeng X, et al. Neurexin-1 is required for synapse formation and larvae associative learning in Drosophila. FEBS Lett. 2007;581(13):2509–2516. doi: 10.1016/j.febslet.2007.04.068. [DOI] [PubMed] [Google Scholar]
- 70.Choi YB, et al. Neurexin-neuroligin transsynaptic interaction mediates learning-related synaptic remodeling and long-term facilitation in aplysia. Neuron. 2011;70(3):468–481. doi: 10.1016/j.neuron.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang C, et al. A permissive role of mushroom body α/β core neurons in long-term memory consolidation in Drosophila. Curr Biol. 2012;22(21):1981–1989. doi: 10.1016/j.cub.2012.08.048. [DOI] [PubMed] [Google Scholar]