

A disease-specific, hybrid vector is developed for pneumococcal disease vaccine.
Keywords: Antigen delivery, vaccine, biomaterials, biological vector, pneumococcal disease
Abstract
The type and potency of an immune response provoked during vaccination will determine ultimate success in disease prevention. The basis for this response will be the design and implementation of antigen presentation to the immune system. Whereas direct antigen administration will elicit some form of immunological response, a more sophisticated approach would couple the antigen of interest to a vector capable of broad delivery formats and designed for heightened response. New antigens associated with pneumococcal disease virulence were used to test the delivery and adjuvant capabilities of a hybrid biological-biomaterial vector consisting of a bacterial core electrostatically coated with a cationic polymer. The hybrid design provides (i) passive and active targeting of antigen-presenting cells, (ii) natural and multicomponent adjuvant properties, (iii) dual intracellular delivery mechanisms, and (iv) a simple formulation mechanism. In addition, the hybrid format enables device-specific, or in situ, antigen production and consolidation via localization within the bacterial component of the vector. This capability eliminates the need for dedicated antigen production and purification before vaccination efforts while leveraging the aforementioned features of the overall delivery device. We present the first disease-specific utilization of the vector toward pneumococcal disease highlighted by improved immune responses and protective capabilities when tested against traditional vaccine formulations and a range of clinically relevant Streptococcus pneumoniae strains. More broadly, the results point to similar levels of success with other diseases that would benefit from the production, delivery, and efficacy capabilities offered by the hybrid vector.
INTRODUCTION
Vaccination has proven a powerful approach to infectious diseases predicated on the identification of antigen targets (1). Identifying these targets is one obstacle to vaccination success, with an equally important consideration being the effectiveness of antigen production and delivery. Starting from the base scenario of direct antigen administration, delivery technology can be designed to enhance and influence the associated immune response. For example, a protein antigen administered with an adjuvant such as alum would represent a classic vaccine formulation and would similarly be expected to initiate a predominantly humoral or antibody-based immune response, whereas delivery technology would offer the potential to provide a built-in adjuvant feature as well as variation in the antigen format and the corresponding immune response (2).
In particular, this study features a hybrid antigen delivery vector composed of biological and biomaterial components, each designed to facilitate, enhance, and direct the immune response process (Fig. 1) (3). The biological portion of the vector is a bacterial cell (nonpathogenic Escherichia coli) that, as a result of the microbial framework, has natural adjuvant properties (for example, cell wall containing lipopolysaccharide) and also allows the delivery of either protein or genetic antigens. A simple one-step electrostatic-based coating of the bacterial core with a poly(β-amino ester) (PBAE) covalently linked to mannose provides a combined delivery device with properties to assist antigen cargo cellular translocation (4). Namely, the size of the overall vector (~2 μm) allows passive targeting of phagocytic antigen-presenting cells (APCs) programmed to engulf such particles; in addition, the vector’s composition and the surface characteristics endowed by the mannosylated PBAE will engage APC receptors and enhance uptake upon vector administration (3, 4). The biomaterial and biological components can also be designed to facilitate endosomal escape and cytosolic trafficking after phagocytosis. Finally, the bacterial core of the vector allows antigen cargo to be delivered as protein, nucleic acid, or both.
Fig. 1. The hybrid biological-biomaterial vector.
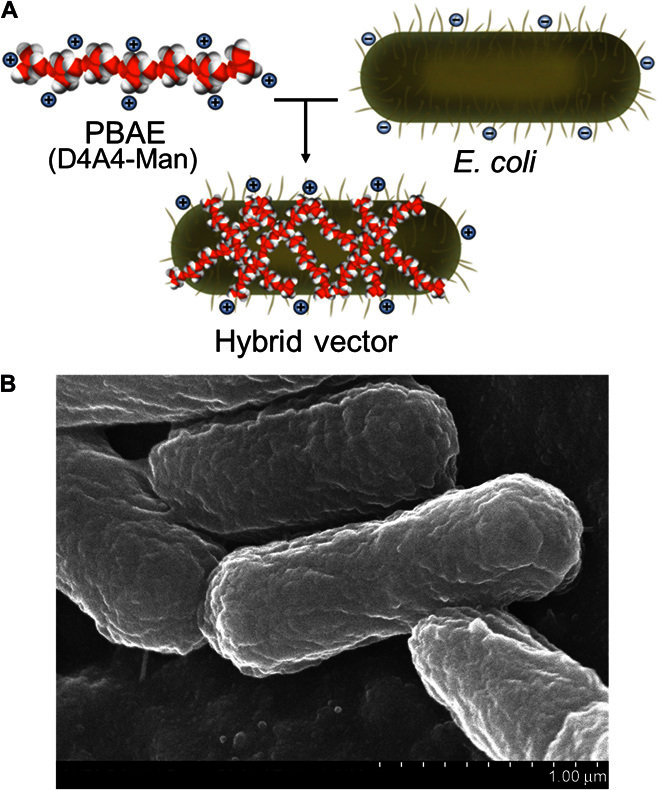
(A) Electrostatic interactions between a positively charged PBAE (D4A4-Man, in this case) and negatively charged E. coli bacteria result in the hybrid vector composed of both components contributing to the delivery of antigenic cargo within the E. coli core of vehicle. (B) Scanning electron microscopy image of the final vector.
This last feature also provides an opportunity to directly generate and maintain the required antigen in lieu of a dedicated bioprocess to produce genetic or protein formats. Thus, the hybrid vector provides the added capability of “in situ” antigen production that can be coupled to the delivery features of the device. Combined, the vector offers a broad array of engineering opportunities to improve vaccine production (via rapid and scalable component generation, in situ antigen provision, and simple hybrid formulation) and potency (via the innate design and diverse engineering tools, that is, polymer chemistry and molecular biology).
This premise is the basis for the following results. Within the framework of pneumococcal disease, new antigens were generated and delivered via the hybrid vector. We obtained directed, broad, and potent results as assessed within disease challenge assays against a range of clinically relevant Streptococcus pneumoniae strains. Beyond the protection data collected for this particular disease type, the vector format offers opportunities for other maladies (such as cancer or viral-based infectious disease) similarly expected to require an advanced delivery device capable of supporting a similarly advanced immune response.
RESULTS
Hybrid vector assessment with model and novel antigen delivery
The data presented in this study marks the first application of the hybrid vector, and the utility of the device is assessed in the context of pneumococcal disease, which afflicts millions worldwide annually, especially resource-limited children and the elderly (5–7). Initial experiments were dedicated to optimizing and characterizing the use of the hybrid vector when incorporating a model protein antigen for pneumococcal disease, that is, pneumococcal surface protein A (PspA) (8, 9); the results demonstrate improved outcomes when compared to traditional vaccine formulations containing either alum or complete Freund’s adjuvant (CFA; Fig. 2, A to C). Specifically, both time to death extension and antibody responses are improved when using the hybrid vector to deliver PspA. These results were consistent across three different administration routes (intraperitoneal, subcutaneous, and intranasal), implying a generality and flexibility toward different means of vaccination. The hybrid vector allowed for a reduced amount of PspA to be delivered (5 to 15 μg of PspA with traditional adjuvants versus ~0.007 to 0.7 μg of PspA with the hybrid vector), with the resulting improvements to immune response outcomes indicating the effectiveness of the vector in antigen delivery and dose sparing. There was also a balanced antibody response (Fig. 2D) with a transition over time to enhanced immunoglobulin G (IgG) content for intraperitoneal and subcutaneous administrations and IgG and IgA content for intranasal administration.
Fig. 2. Comparative assessment of vaccine outcomes for the hybrid vector across different formulations and administration routes.
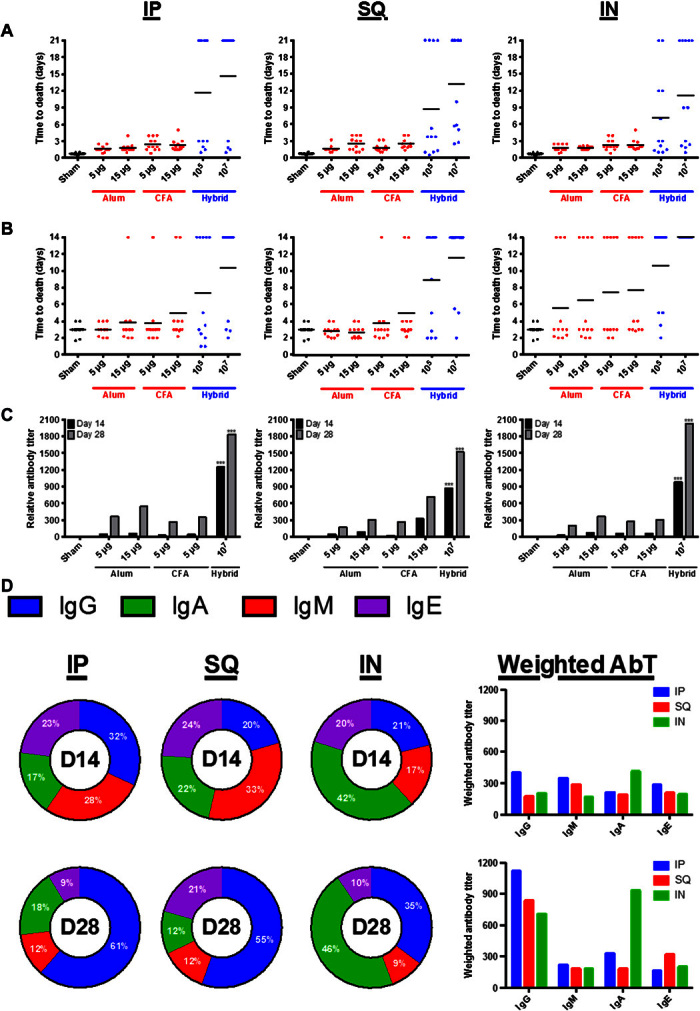
(A to C) The PspA protein antigen at two dose levels (5 and 15 μg) was formulated with either alum or CFA and compared to the hybrid vector housing PspA (via expression plasmid) at two dose levels (105 and 107) across intraperitoneal (IP), subcutaneous (SQ), and intranasal (IN) administration routes using sepsis (A) or pneumonia (B) mouse vaccination models that were challenged with S. pneumoniae strain D39. (C) Similarly, anti-PspA antibody titers are compared. ***P < 0.001, relative to controls on associated days. (D) Antibody distributions upon vaccination with the hybrid vector containing PspA are provided at days 14 and 28 across administration routes. The x axes for all plots represent PspA antigen delivered as either protein or within the hybrid vector; 105 and 107 hybrid vectors equate to ~0.007 and 0.7 μg of PspA, respectively. AbT, antibody titer.
Dosing levels of the hybrid vector containing PspA were also assessed and optimized across administration routes and in response to S. pneumoniae sepsis challenge using a D39 strain (fig. S1). D39 was selected because of its notable virulence profile and the fact that it is one of the harshest preclinical challenge strains. Increasing the challenge inocula of S. pneumoniae can be countered with increasing hybrid vector vaccination doses (fig. S1A); however, particularly aggressive challenge levels [106 S. pneumoniae colony-forming units (CFU)] require a vaccination dose of ≥1010 hybrid devices to mediate full protection. Because of the bacterial content of the hybrid vector, it is important to assess these enhanced dose levels for associated toxicity. Across administration routes, toxicity was not observed until ≥1013 hybrid vectors were administered (fig. S1B). However, the design of the hybrid vector allows both the chemical and biological aspects of the device to extend dosing level safety. Namely, the vector’s polymer coating and the inclusion of a lysis E (LyE) gene provide alternative mechanisms of attenuation (3, 10). In the case of LyE, the membrane perforative activity of this protein affords safety of up to 1020 dose levels for the resulting hybrid device (fig. S1B). Given the added tissue sensitivity associated with intranasal administration, histology was performed with 1013 hybrid devices. This administration dose was selected because it was the highest delivery load with no associated toxicity. In support of the mouse subject viability tests in fig. S1B, no tissue damage was observed via histological analysis as compared to the untreated control (fig. S2).
The hybrid vector was then used to screen several antigen candidates selected due to enhanced expression during S. pneumoniae virulence transition during the course of pneumococcal disease progression (table S1) (11). As such, the antigens would serve as the basis of a “smart” vaccine capable of directing a subsequent immune response to only a virulent subset of S. pneumoniae cells (Fig. 3A). Figure 3B presents the degree of protection provided by protein antigen candidates StkP, DexB, GlpO, and PncO relative to PspA, with each individually delivered via the hybrid vector. Although individual protection varied by antigen, the design parameters of the hybrid vector allowed consolidated testing of all the candidates. Specifically, a dual expression plasmid system (pCJ10 and pLF; tables S2 and S3 and fig. S3) was used to enable the vector-based in situ production of the antigen products while leveraging the delivery capabilities of the hybrid device. Using the two-plasmid system, we provided complete protection against the S. pneumoniae challenge strain D39 in both sepsis and pneumonia mouse models (Fig. 3, C and D).
Fig. 3. Directed pneumococcal disease antigen assessment via the hybrid vector.
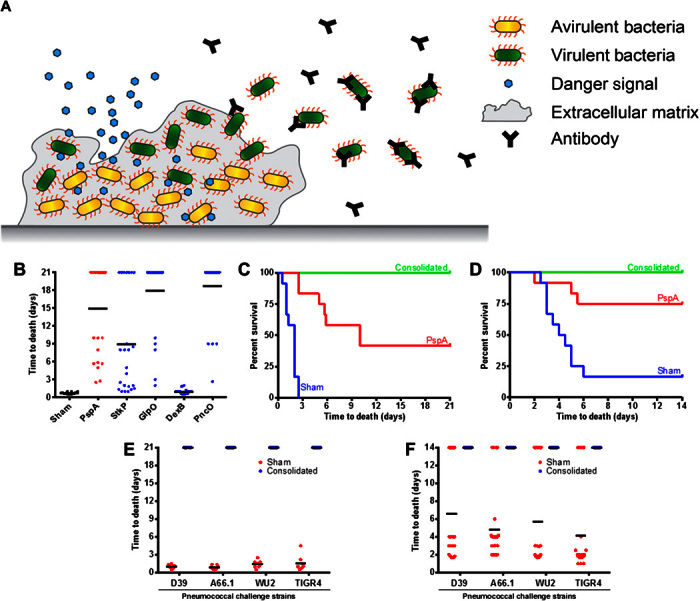
(A) Asymptomatic S. pneumoniae biofilm carriage is established in the nasopharynx and can be triggered (via signals such as viral infection) for virulent cellular release and dissemination characterized by extended tissue burden and disease. The antigens delivered with the hybrid vector were chosen to elicit a directed immune response to only the virulent subpopulation of S. pneumoniae. (B to D) Vaccine screening of individual virulent-specific antigens (x axis) (B) before consolidating the antigens to plasmids within the hybrid vector tested within sepsis (C) and pneumonia (D) disease challenge protection mouse model assays against the virulent S. pneumoniae strain D39. (E and F) Vaccination was extended to test other clinically relevant S. pneumoniae strains within sepsis (E) and pneumonia (F) challenge protection mouse models.
To demonstrate the potency and versatility of the hybrid vector two-plasmid system, we varied challenge levels and strains of S. pneumoniae within vaccine protection assays. Challenge with strain D39 was tested up to 1010 CFU (fig. S4), with strong protection (>80% survival) observed for the 107 level and varying degrees of protection provided beyond this level across administration routes. Protection conferred on these advanced challenge levels is considered impressive and highlights the combined adjuvant, delivery, and antigen consolidation capabilities of the hybrid vector. Furthermore, as supported by the data of fig. S1, even better levels of protection would be expected with increased doses of the hybrid vector, readily possible as a result of engineering the degree of vector attenuation. Finally, to further assess the protective capabilities of the antigens delivered with the hybrid vector, we tested vaccination against an extended panel of S. pneumoniae strains representing diverse serotypes notably difficult to protect against using current vaccine formulations; complete protection and reduced bacterial burden were observed (Fig. 3, E and F, and fig. S5).
Directed protection and expanded strain coverage
The directed nature of the vaccination strategy was tested first by comparing vaccinated and nonvaccinated mice subjects challenged with avirulent and virulent S. pneumoniae strains (Fig. 4A). In these experiments, S. pneumoniae strain EF3030 was used because of planktonic (or broth cultured) cells demonstrating an avirulent phenotype, whereas those cells released from in vitro or in vivo biofilms are virulent and cause disease (12, 13). The avirulent strains are cleared either with or without vaccination, whereas only vaccinated mice are capable of demonstrating virulent bacterial clearance over time. This effect is further tested in the context of S. pneumoniae colonization followed by the addition of an in vivo virulence trigger (that is, administration of IAV). Designed to more closely mimic the common clinical setting where influenza spurs pneumonia development (13–15), bacterial dissemination, as indicated by the results, is significantly reduced over time, in line with the theme of the immune response targeting only virulent populations, whereas complete protection is maintained with the consolidated set of antigens tested (Fig. 4, B to D).
Fig. 4. Directed and extended protection using the hybrid vector.

(A) Assessment of bacterial burden was conducted across anatomical sites for unimmunized (filled circles) and immunized (using the consolidated antigens; open circles) mice challenged with avirulent (planktonic; red) or virulent (biofilm-released; blue) S. pneumoniae strain EF3030. (B to D) Site-specific bacterial burden and protection were also tested over time for mice colonized with the S. pneumoniae strain EF3030 and triggered for virulence progression using influenza A virus (IAV) inoculation. Dotted lines represent limit of detection for bacterial counts. ***P < 0.001. (E) Protection assessment (using a mouse sepsis challenge model) was then extended to 10 additional clinically relevant S. pneumoniae strains.
The consolidated aspect of the vaccine formulation also offers extended protection. Namely, the conserved nature of the individual antigens (table S1), when presented in combination, provides the potential to cover a broad range of S. pneumoniae strains (table S4). Challenge assays were therefore tested with a series of 10 additional S. pneumoniae strains, with enhanced protection observed in all cases and complete protection provided in six cases (Fig. 4E).
Predicting protection potential
Although the data presented support the broad protection potential of the consolidated antigens generated and delivered within the hybrid vector, the mutational potential of individual and combined antigens was predicted to further assess widespread and extended utilization of the vaccination strategy (fig. S6). The results emphasize the cumulative resistance of the combined antigen vaccination strategy to mutation. Together, using five antigens specific to virulence provides ample coverage and resistance to strain evasion. Thus, antigenic drift (as a result of innate S. pneumoniae mutation) and the associated threat to vaccine efficacy are minimized through consolidation.
DISCUSSION
The hybrid vector was designed at one level to assist the intracellular delivery of antigen cargo to APCs. As a combination of biomaterial and biological components, each of which was chosen because of individual use as delivery vehicles, the combined vector allows synergistic delivery mechanisms and disparate engineering tools to influence the process of antigen transport. Thus, the steps associated with APC targeting, uptake, activation, and intracellular antigen release have been designed through a combination of the two components of the hybrid device. Highlighting the dual engineering capabilities of the hybrid vector are the polymer chemistry steps used to conjugate a mannose ligand to the PBAE shell of the device, which aids in APC targeting and vector uptake. Similarly, expression and localization of the LyE protein to the bacterial component of the hybrid vector safely extend the dose limits of the device.
Here, however, a different aspect of the vector is also highlighted, namely, the option for in situ antigen generation. The biological core of the vector allows the molecular biology tools associated with a facile microbe like E. coli to be used in both antigen consolidation and internal expression before delivery. This capability makes the normal requirement of separate bioprocesses dedicated to antigen generation and purification unnecessary and allows a “one-pot” production and delivery device for vaccine applications. Future opportunities include a greatly extended capability for antigen diversity and valency by further leveraging the molecular biology and recombinant tools available to E. coli, including the option of extensive operons capable of coordinately expressing >20 genes (16). Similarly, robust bioreactor protocols and one-step polymer synthesis and vector formation support the prospect of a scalable and rapid route to vaccine products in response to emerging pandemic infectious disease demands.
The hybrid vector design also offers options associated with stable storage and widespread distribution; however, features related to production and access are targets of future research. Here, the results illustrate the first directed application of the vector, tested in the context of pneumococcal disease. The hybrid vector demonstrated improved immune response metrics using a model protein antigen (PspA) when compared to traditional vaccine formulations with alum and CFA. Once the molecular biology tools of the vector were used to consolidate newly identified antigens, protection was provided across a range of clinically relevant S. pneumoniae strains and disease models.
The antigens selected for consolidation to the hybrid vector are based on those up-regulated during virulent transition; thus, the objective is to target the disease-causing subset from a greater population of colonizing and asymptomatic S. pneumoniae cells. This capability was observed across different pneumococci strains, administration routes, and in vivo conditions, including disease onset triggered by influenza. Furthermore, the effectiveness of the antigens delivered with the hybrid vector resulted in protection extended to 10 additional S. pneumoniae strains. Although not all levels of protection were the same, complete protection was observed in 11 of 15 cases, emphasizing the broad coverage potential of the consolidated antigens tested in this study. Owing to the tools associated with the hybrid vector, even broader coverage can be expected upon the consolidation of additional virulence-associated antigens into the vector design. This feature, coupled with the conserved nature of the antigens identified and tested (fig. S6 and table S1), offers an excellent prospect of combating antigenic drift and the resulting reduction in vaccine efficacy. These concerns currently plague various vaccines associated with this and other diseases (17). Thus, the conserved nature of the antigens selected and consolidation options provided through the hybrid vector offer a solution to this issue.
Countering the capabilities offered by the hybrid design is the potential for toxicity, given the bacterial nature of the vector’s core and the cationic charge of the polymer coating. However, an attempt to mitigate this issue was made in selecting the starting components, both of which were chosen because of nontoxic or nonpathogenic aspects. As demonstrated, the final vector provides attenuation through cellular engineering of the bacterial core and via the process of polymer coating. The vector has not provoked any toxicity upon administration unless heightened doses are used. Yet, even under enhanced dose levels, it is possible to negate toxic effects due to the biological attenuation afforded by the LyE gene, providing another level of engineering design in applying the hybrid vector.
In summary, this study presents the first disease-specific application of a hybrid biological-biomaterial vector designed to enhance the cellular elements of an immune response upon administration. Consolidating virulent-specific antigens to the hybrid device also offers a smart vaccine with built-in antigen production or an in situ means of cargo generation. Combined, the vector offers engineering elements that range from process-level scalability and distribution to cellular-level immune response tunability, with the potential to be directed at a number of challenging vaccine opportunities beyond pneumococcal disease.
MATERIALS AND METHODS
Experimental design
A hybrid biological-biomaterial vector was tested in the delivery of virulent-specific antigens targeting pneumococcal disease progression. Success was assessed by mouse model immune response outcomes that included balanced antibody profiles, directed targeting of virulent S. pneumoniae cells, reduced bacterial dissemination and tissue burden, and bacterial challenge protection. The hybrid vector allowed innate (or in situ) maintenance, consolidation, and production of the antigens in addition to other features that span vaccine production and enhance delivery capability.
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH). The protocols were approved by the Institutional Animal Care and Use Committee at the University at Buffalo, Buffalo, NY. All bacterial inoculations and treatments were performed under conditions designed to minimize any potential suffering of the animals.
Materials
Bacterial and cell culture media and reagents were purchased from Fisher Chemical and Sigma-Aldrich. Chemically defined bacterial growth medium (CDM) was obtained from JRH Biosciences. Sheep blood was purchased from Hemostat Laboratories. Monomers were purchased from Sigma-Aldrich and TCI. Acetone [high-performance liquid chromatography (HPLC)–grade], chloroform (HPLC-grade), N-hexadecane (99%), N,N′-dimethylformamide (HPLC-grade), and dimethyl sulfoxide (DMSO) (≥99.7%) were purchased from Fisher Chemical. Phosphate-buffered saline (PBS) was purchased from Life Technologies. All remaining chemicals and reagents were purchased from Sigma-Aldrich.
Polymer synthesis
The polymer used in this study, D4A4-Man, was synthesized in a four-step reaction as previously described (fig. S7) (3, 4, 18, 19). Briefly, in step 1, allyl-α-d-mannopyranoside (ADM) was synthesized by dissolving 3 g of d(+)-mannose and 18 mg of p-toluenesulfonyl chloride in allyl alcohol (20 ml) at 90°C under reflux for 24 hours. The resulting reaction solution was concentrated by vacuum distillation at 35°C. Next, in step 2, acrylate-terminated D4A4 was synthesized in DMSO at a 1:1.2 amine/diacrylate molar ratio for 5 days at 60°C with continuous stirring at 1000 rpm. Acrylate-terminated polymers were purified by dialysis followed by evaporation under vacuum. Dialysis procedures were conducted against acetone using molecular porous membrane tubing (Spectra/Por Dialysis Membrane, Spectrum Laboratories Inc.), with an approximate molecular weight cutoff of 3500 daltons. In step 3, acrylate-terminated D4A4 was reacted with excess ethylenediamine to amine-cap the terminal ends (D4A4-EDA). Specifically, acrylate-terminated D4A4 was dissolved in DMSO (167 mg/ml) and reacted with 5 M ethylenediamine (in DMSO) at room temperature for 24 hours. D4A4-EDA was purified by dialysis followed by evaporation under vacuum. In the last step, D4A4-EDA was reacted with ADM at a 1:2 molar ratio in DMSO at 90°C for 24 hours and then purified via dialysis. Structure and purity of D4A4-Man were confirmed using 1H nuclear magnetic resonance spectroscopy (fig. S7).
Bacterial strains and plasmid generation
Antigen genes were amplified from the genomic DNA of S. pneumoniae strains using primers summarized in table S2. Each polymerase chain reaction (PCR) product was cloned into pET expression plasmids using the flanking restriction sites indicated (designed within the primers). Constructs were verified by colony PCR and restriction digest analysis and were then chemically transformed into the BL21(DE3) E. coli cell line (Novagen). Resulting single-colony transformants were cultured in 3 ml of lysogeny broth (LB) at 37°C with shaking before 15% glycerol stock storage at −80°C. Plasmid selection antibiotics were used as needed during bacterial culture within LB medium. Expression was confirmed by 3 to 5 ml of LB cultures started from glycerol stocks and grown at 37°C with shaking until an OD600 of 0.4 was achieved. After which, cultures were induced with isopropyl-β-d-thiogalactopyranoside (IPTG) (1 mM) for 1 hour. Upon collection via centrifugation, cells were washed twice with PBS and resuspended in 25 mM tris-HCl before boiling with loading dye and expression analysis or confirmation via SDS–polyacrylamide gel electrophoresis (SDS-PAGE). Plasmid pUAB055 was used to express and clone the pspA gene (20).
To use the two-plasmid system, pCJ10 was used in conjunction with a plasmid containing pspA, dexB, stkP, and glpO. The consolidated plasmid was assembled by subcloning each respective gene into a pACYCDuet-1 plasmid (fig. S3 and table S3). To construct pLF, the restriction enzymes or sites were used for the corresponding multiple cloning sites within pACYCDuet-1. Plasmids were chemically transformed into the E. coli strain BL21(DE3) (as the basis for hybrid device preparation), and resulting transformants were prepared and stored as glycerol stocks.
Hybrid device preparation
Bacterial and hybrid vectors were prepared from bacterial cultures inoculated at 2% (v/v) from overnight starter cultures. Following incubation at 37°C with shaking until OD600 of 0.4 to 0.5, samples were induced with 0.1 mM IPTG at 30°C for 3 hours. Bacterial vectors were then washed once and standardized to OD600 of 0.5 in PBS, whereas bacteria to be used in hybrid vector formation were washed once and standardized to OD600 of 1.0 in 25 mM NaOAc (pH 5.15). Polymer was dissolved in chloroform, desiccated, and resuspended at 1 mg/ml in 25 mM NaOAc (pH 5.15) before it was added in an equal volume to bacteria and mixed by vortexing on a setting of 5 (Analog Vortex Mixer, Fisher Scientific) for 10 s. Polymer/bacteria self-assembly was allowed to continue for 15 min, and devices were then diluted in PBS. Scanning electron microscopy of the hybrid vectors was performed as described previously (3).
Hybrid vaccine immunization
Outbred 6-week-old female CD-1 mice (Harlan Laboratories) were used in immunization experiments. Mice were immunized by intraperitoneal injection (200 μl), subcutaneous injection (200 μl), and intranasal aspiration (40 μl) using isoflurane; unless indicated otherwise, 107 hybrid vectors were used in vaccination experiments. Immunization of controls included sham (PBS; all samples were prepared in PBS as the background solution), PspA plus alum, and PspA plus CFA, which was replaced with incomplete Freund’s adjuvant during booster immunizations (adjuvants were added per the manufacturer’s instructions). If not indicated otherwise, mice were immunized via subcutaneous injection. After 14 days, mice were boosted with the same formulations, except in the case of the CFA adjuvant as described previously. At days 14 and 21, samples were collected by retro-orbital bleeding and clarified by centrifugation to collect serum. SDS-PAGE expression analysis and densitometry assessment (ImageJ; https://imagej.nih.gov/ij/) were completed (relative to PspA standards and subtracting the expression profile from a background sample) to quantify PspA levels per dose of hybrid vector, resulting in ~0.007 and 0.7 μg of PspA delivered with 105 and 107 hybrid vectors, respectively.
S. pneumoniae bacterial preparation and biofilm release
S. pneumoniae strains used in this study are listed in table S4 and were initially grown on Todd-Hewitt agar plates supplemented with 0.5% yeast extract and 5% sheep blood and incubated overnight at 37°C. Single colonies were used to inoculate 5 ml of Todd-Hewitt broth containing 0.5% yeast extract and incubated at 37°C to an OD600 of 0.6. At this point, S. pneumoniae strains used for challenge studies were washed once with and resuspended in PBS. NCI-H292 epithelial cells were cultured in RPMI 1640 medium in T75 flasks at 37°C and 5% CO2. After reaching 100% confluency, H292 cells were prefixed in 4% buffered paraformaldehyde at 34°C for 48 hours followed by three washes with PBS. CDM-grown pneumococci were then seeded onto fixed H292 cells, with change of media occurring every 12 hours. Formed biofilms were exposed to heat (38.5°C) for 4 hours, and released cells were then collected by centrifugation, washed and resuspended in PBS, and quantified by OD600 measurement. Strains presented in table S4 are a mix of mouse-passaged strains, which directly confer virulence on culture and administration, and clinical isolates, which usually do not cause disease upon direct administration and require heat release from the in vitro biofilm model (previously discussed in detail) to render a virulent phenotype.
Challenge models
To induce sepsis or pneumonia, mice were administered intraperitoneally or intranasally (with isoflurane), respectively, with 1 × 104 to 1 × 1010 CFU of pneumococci strains (table S4). To induce colonization, mice were administered 1 × 106 CFU, intranasally, without isoflurane. To mimic influenza-induced pneumonia, pneumococci colonization was followed by intranasal inoculation with 40 plaque-forming units of IAV. The mouse-adapted IAV strain A/PR/8/34 (H1N1) (ATCC VR-95) was used, and titers were determined by plaque assays. Mice were monitored every 4 hours for signs of morbidity (huddling, ruffled fur, lethargy, and abdominal surface temperature). Mice found to be moribund were euthanized via CO2 asphyxiation and cervical dislocation.
Tissue bacterial count
At predefined time points (24 and 48 hours after infection for intraperitoneal and intranasal challenges, respectively) or upon becoming moribund, mice were euthanized (as described previously), and a combination of nasopharynx tissue, nasopharyngeal lavage fluid, lung, liver, spleen, and blood samples was collected and assessed for bacterial burden, as described by Tyx et al. (21). Briefly, tissue and organ samples, lavage fluid, and blood were homogenized (on a setting of 10 for 30 s or until homogenized completely; Tissue-Tearor, BioSpec Products Inc.) to ensure dissociation of bacterial aggregates and then were serially diluted on tryptic soy and 5% blood agar plates before enumeration.
Histologic analysis
Following induction of isoflurane anesthesia, a midline abdominal incision was made, and the abdominal aorta was transected to exsanguinate and euthanize the animal. A midline incision was continued through the thoracic cavity and neck to allow injection of 5 ml of normal saline into the right ventricle of the heart to flush the pulmonary vasculature of residual blood. A luer lock–hubbed 20-gauge × ½-inch cannula was inserted into the trachea and secured in place with a suture. The lungs were fixed by insufflation with 10% neutral buffered formalin at 20 cm H2O for 24 hours at room temperature. The lung lobes were removed and embedded in paraffin, and 5-μm tissue slices were prepared and stained with hematoxylin and eosin by standard histopathology techniques (22). Histology images were acquired with an Aperio ScanScope CS slide scanner (Leica Biosystems) using a 40× objective lens and subsequently analyzed using Aperio ImageScope software version 12.3 (Leica Biosystems).
Antibody analysis
To characterize antibody titers associated with delivery optimization, an enzyme-linked immunosorbent assay was performed by coating a 96-well Costar high-binding polystyrene plate with PspA (10 μg/ml) in tris-buffered saline (TBS) at 4°C overnight. The plate was blocked with 3% bovine serum albumin in TBS–Tween 20 (TBS-T) for 1 hour at 22°C. Sera was diluted into TBS-T in ratios of 1:1000, 1:5000, 1:7500, and 1:10,000 and added to the plate. The plates were then incubated at 37°C with mild agitation for 3 hours. The secondary antibody [anti-mouse IgG, IgA, IgM (H + L), IgE, highly X-adsorbed (biotin)] was added to the wells in a 1:1000 ratio and agitated for 2 hours. Streptavidin was added to each well in a 1:1000 ratio and allowed to shake for 30 min. The signal was developed with p-nitrophenylphosphate, and the reaction was quenched using 0.75 M NaOH. The signal was detected using a plate reader spectrophotometer at an absorbance of 405 nm.
Statistical analysis
Column comparisons were analyzed for statistical significance using a two-tailed Student’s t test for unpaired data. Multivariance analysis was done using one-way analysis of variance (ANOVA) that was corrected using the Bartlett’s variance test, and for multiple comparisons, the Bonferroni multiple-comparison test was used. For both tests, a P value of 0.05 was considered significant. Statistical analysis was performed using the GraphPad Prism software (version 6.0h.283; GraphPad Software Inc.). All data resulted from animal experiments using 6 to 12 subjects per group except histology analysis, which used two mouse subjects per group.
Supplementary Material
Acknowledgments
We thank D. Briles (University of Alabama at Birmingham) for providing S. pneumoniae strains and pUAB055. Funding: The authors recognize support from NIH awards AI088485 and AI117309 (to B.A.P.) and a SUNY-Buffalo Schomburg fellowship (to C.H.J.). Author contributions: C.H.J. and B.A.P. designed the study, analyzed the data, and wrote the manuscript; Y.L. and C.H.J. conducted challenge assays; L.F. cloned the antigens into a consolidated plasmid; M.K.A. cloned and prepared the LyE plasmid; M.C. prepared all polymer samples; M.B. analyzed antibody titers; B.A.D. and P.K. conducted histological assessment and provided assistance with in vivo IAV administration; R.J.S. and S.T.A. conducted the hybrid vector PspA expression quantification analysis; A.P.H. provided technical assistance with vaccine models; and A.H. completed the antigenic drift probability model. Competing interests: Several authors are associated with Abcombi Biosciences, a company focused on vaccine design. Patent applications have been filed on items related to the enclosed study. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/7/e1600264/DC1
fig. S1. Dosing and toxicity assessment of hybrid and bacterial vectors.
fig. S2. Histological intranasal toxicity evaluation of hybrid devices.
fig. S3. The pLF consolidation design and organization.
fig. S4. Challenge levels of D39.
fig. S5. Bacterial burden assessed for hybrid vector vaccination with the consolidated antigens against pneumococcal challenge strains that included D39, A66.1, WU2, and TIGR4 in either a sepsis or pneumonia model.
fig. S6. Probability of mutations occurring in S. pneumoniae genes (glpO, pncO, dexB, and stkP) through cellular division.
fig. S7. Synthetic scheme for PBAE D4A4-Man.
table S1. DexB, GlpO, StkP, and PncO antigen description and analysis.
table S2. Antigen cloning summary.
table S3. Consolidated plasmid (pLF) cloning summary.
table S4. S. pneumoniae strains used in the current study.
Reference (23)
REFERENCES AND NOTES
- 1.Plotkin S. A., Vaccines: Past, present and future. Nat. Med. 11, S5–S11 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones C. H., Chen C.-K., Ravikrishnan A., Rane S., Pfeifer B. A., Overcoming nonviral gene delivery barriers: Perspective and future. Mol. Pharm. 10, 4082–4098 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones C. H., Ravikrishnan A., Chen M., Reddinger R., Ahmadi M. K., Rane S., Hakansson A. P., Pfeifer B. A., Hybrid biosynthetic gene therapy vector development and dual engineering capacity. Proc. Natl. Acad. Sci. U.S.A. 111, 12360–12365 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones C. H., Chen M., Ravikrishnan A., Reddinger R., Zhang G., Hakansson A. P., Pfeifer B. A., Mannosylated poly(beta-amino esters) for targeted antigen presenting cell immune modulation. Biomaterials 37, 333–344 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bakaletz L. O., Bacterial biofilms in the upper airway - evidence for role in pathology and implications for treatment of otitis media. Paediatr. Respir. Rev. 13, 154–159 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hausdorff W. P., Hoet B., Adegbola R. A., Predicting the impact of new pneumococcal conjugate vaccines: Serotype composition is not enough. Expert Rev. Vaccines 14, 413–428 (2015). [DOI] [PubMed] [Google Scholar]
- 7.O’Brien K. L., Wolfson L. J., Watt J. P., Henkle E., Deloria-Knoll M., McCall N., Lee E., Mulholland K., Levine O. S., Cherian T.; Hib and Pneumococcal Global Burden of Disease Study Team , Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: Global estimates. Lancet 374, 893–902 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Roche H., Håkansson A., Hollingshead S. K., Briles D. E., Regions of PspA/EF3296 best able to elicit protection against Streptococcus pneumoniae in a murine infection model. Infect. Immun. 71, 1033–1041 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roche H., Ren B., McDaniel L. S., Håkansson A., Briles D. E., Relative roles of genetic background and variation in PspA in the ability of antibodies to PspA to protect against capsular type 3 and 4 strains of Streptococcus pneumoniae. Infect. Immun. 71, 4498–4505 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung T.-C., Jones C. H., Gollakota A., Ahmadi M. K., Rane S., Zhang G., Pfeifer B. A., Improved Escherichia coli bactofection and cytotoxicity by heterologous expression of bacteriophage ΦX174 lysis gene E. Mol. Pharm. 12, 1691–1700 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pettigrew M. M., Marks L. R., Kong Y., Gent J. F., Roche-Hakansson H., Hakansson A. P., Dynamic changes in the Streptococcus pneumoniae transcriptome during transition from biofilm formation to invasive disease upon influenza a virus infection. Infect. Immun. 82, 4607–4619 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marks L. R., Parameswaran G. I., Hakansson A. P., Pneumococcal interactions with epithelial cells are crucial for optimal biofilm formation and colonization in vitro and in vivo. Infect. Immun. 80, 2744–2760 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marks L. R., Davidson B. A., Knight P. R., Hakansson A. P., Interkingdom signaling induces Streptococcus pneumoniae biofilm dispersion and transition from asymptomatic colonization to disease. mBio 4, e00438-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chonmaitree T., Howie V. M., Truant A. L., Presence of respiratory viruses in middle ear fluids and nasal wash specimens from children with acute otitis media. Pediatrics 77, 698–702 (1986). [PubMed] [Google Scholar]
- 15.Pettigrew M. M., Gent J. F., Pyles R. B., Miller A. L., Nokso-Koivisto J., Chonmaitree T., Viral-bacterial interactions and risk of acute otitis media complicating upper respiratory tract infection. J. Clin. Microbiol. 49, 3750–3755 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H., Wang Y., Wu J., Skalina K., Pfeifer B. A., Complete biosynthesis of erythromycin A and designed analogs using E. coli as a heterologous host. Chem. Biol. 17, 1232–1240 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Rappuoli R., Bridging the knowledge gaps in vaccine design. Nat. Biotechnol. 25, 1361–1366 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Jones C. H., Gollakota A., Chen M., Chung T.-C., Ravikrishnan A., Zhang G., Pfeifer B. A., Influence of molecular weight upon mannosylated bio-synthetic hybrids for targeted antigen presenting cell gene delivery. Biomaterials 58, 103–111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones C. H., Chen M., Gollakota A., Ravikrishnan A., Zhang G., Lin S., Tan M., Cheng C., Lin H., Pfeifer B. A., Structure–function assessment of mannosylated poly(β-amino esters) upon targeted antigen presenting cell gene delivery. Biomacromolecules 16, 1534–1541 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Briles D. E., Ades E., Paton J. C., Sampson J. S., Carlone G M., Hueber R. C., Virolainen A., Swiatlo E., Hollingshead S. K., Intranasal immunization of mice with a mixture of the pneumococcal proteins PsaA and PspA is highly protective against nasopharyngeal carriage of Streptococcus pneumoniae. Infect. Immun. 68, 796–800 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tyx R. E., Roche-Hakansson H., Hakansson A. P., Role of dihydrolipoamide dehydrogenase in regulation of raffinose transport in Streptococcus pneumoniae. J. Bacteriol. 193, 3512–3524 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davidson B. A., Vethanayagam R. R., Grimm M. J., Mullan B. A., Raghavendran K., Blackwell T. S., Freeman M. L., Ayyasamy V., Singh K. K., Sporn M. B., Itagaki K., Hauser C. J., Knight P. R., Segal B. H., NADPH oxidase and Nrf2 regulate gastric aspiration–induced inflammation and acute lung injury. J. Immunol. 190, 1714–1724 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stevens K. E., Sebert M. E., Frequent beneficial mutations during single-colony serial transfer of Streptococcus pneumoniae. PLOS Genet. 7, e1002232 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/7/e1600264/DC1
fig. S1. Dosing and toxicity assessment of hybrid and bacterial vectors.
fig. S2. Histological intranasal toxicity evaluation of hybrid devices.
fig. S3. The pLF consolidation design and organization.
fig. S4. Challenge levels of D39.
fig. S5. Bacterial burden assessed for hybrid vector vaccination with the consolidated antigens against pneumococcal challenge strains that included D39, A66.1, WU2, and TIGR4 in either a sepsis or pneumonia model.
fig. S6. Probability of mutations occurring in S. pneumoniae genes (glpO, pncO, dexB, and stkP) through cellular division.
fig. S7. Synthetic scheme for PBAE D4A4-Man.
table S1. DexB, GlpO, StkP, and PncO antigen description and analysis.
table S2. Antigen cloning summary.
table S3. Consolidated plasmid (pLF) cloning summary.
table S4. S. pneumoniae strains used in the current study.
Reference (23)