

Abstract

The generation of common and stereochemically rich medium-sized benzo-fused sultams via complementary pairing of heretofore-unknown (o-fluoroaryl)sulfonyl aziridine building blocks with an array of amino alcohols/amines in a modular one-pot, sequential protocol using an aziridine ring opening and intramolecular nucleophilic aromatic substitution is reported. The strategy employs a variety of amino alcohols/amines and proceeds with 6 + 4/6 + 5 and 6 + 1 cycloetherification pathways in a highly chemo- and regioselective fashion to obtain skeletally and structurally diverse, polycyclic, 10- to 11- and 7-membered benzo-fused sultams for broad-scale screening.
Introduction
The development of efficient methods for the generation of medium- and large-sized heterocycles is an important facet of screening campaigns for facilitating drug discovery.1 In particular, medium and macrocyclic lactams1,2 constitute an important class of molecules in compound collections derived from target-oriented and diversity-oriented3 synthetic approaches. Their distinct properties, which include conformational constraint, reduced polarity, increased proteolytic stability, and potential for higher target binding and selectivity,4 are manifested in improved pharmacokinetics and pharmacodynamics,4 rendering them as attractive lead molecules for drug development.5 Taken collectively, these attributes have inspired production of natural product like6 medium-sized and macrocyclic ring systems that are stereochemically rich and enhanced in terms of their fraction of sp3 carbons,7 enabling efforts to address emerging difficult drug targets5,8 such as protein–protein interactions9 and epigenetic targets.10
Synthetic medium-sized (8–11 membered)11 and macrocyclic lactams have a rich biological profile and have been shown to exhibit broad activity in a variety of areas ranging from antitumor,2a antifungal,12 anthelmintic,13 neutral endopeptidase inhibitory,14 and hepatitis C virus protease inhibitory15 in drug discovery16 to insecticidal agents in agriculture (Figure 1).17 In contrast, their sulfonamide-based counterparts (amide surrogates),18 medium19 and macrocyclic sultams, are unnatural and less prevalent in the literature but have been found to exhibit antiproliferative,20 anti-HIV activity,21 inhibitory activity of trypsin-like serine protease Factor XIa involved in blood coagulation22 and, more recently, have been shown to be modulators of lysosomal acidification involved in critical cellular function (Figure 1).23 Despite advances in the field,24 methods to generate functionally rich, medium- to large-sized lactams and sultams remains a significant challenge.25
Figure 1.

Bioactive lactams and sultams.
We herein report a modular approach utilizing a heretofore-unknown class of sulfonamide building blocks, namely (o-fluoroaryl)sulfonyl aziridines, which react with amino alcohols via a process we term complementary pairing (CP), vide infra, with high chemo- and regioselectivity enabling access to 10- to 11-membered sultams. Overall, this one-pot protocol involves sequential aziridine ring opening by the amine component and intramolecular nucleophilic aromatic substitution (SNAr) via the alkoxy component. In addition, dual reactivity with primary amines facilitates access to 7-membered sultams. Taken collectively, the routes reported herein generate a diverse array of polycyclic, 10- to 11- and 7-membered benzo-fused sultam scaffolds.
Previously, our group has reported a strategy termed complementary ambiphile pairing (CAP)26,27 for the synthesis of skeletally diverse 7- and 8-membered benzo-fused sultams in a modular and efficient fashion. As shown in Figure 2A, CAP strategies unite a pair of ambiphilic compounds, possessing both electrophilic and nucleophilic components, in a synergistic complementary manner [(4 + 3) and (4 + 4) cyclizations]. In contrast, Yudin and co-workers have developed a number of elegant methods using aziridine aldehydes that contain a nucleophile and electrophile on the same molecule (Figure 2B) and which they term as amphoteric molecules.28
Figure 2.

Complementary ambiphile pairing and amphoteric molecules.
We have previously investigated and reported the use of o-haloaryl sulfonyl chlorides in a number of pairing strategies including Click, Click, Cyclize29 to generate a variety of bridged and benzo-fused sultams (Figure 3). Based on these studies, we sought to expand the scope to another unique class of bis-electrophiles, namely, heretofore unknown o-fluoroaryl-sulfonyl aziridines for use in complementary pairing to bis-nucleophilic counterparts, such as amino alcohols, as well as consecutive coupling with primary amines.30 We envisioned CP of activated sulfonyl aziridines (simple 6-atom bis-electrophilic synthon) via “chemo- and regioselective” ring opening by the amino component of the amino alcohol (bis-nucleophiles) and subsequent SNAr cyclization with the alcohol component to furnish unprecedented, functionally rich, medium-sized benzo-fused sultams in chemoselective “6 + 4” and “6 + 5” heterocyclization pathways. Moreover, the method can accommodate the use of (o-fluoroaryl)sulfonyl aziridines to generate 7-membered benzo-fused sultams via a “6 + 1” atom cyclization sequence where primary amines are utilized for sulfonyl aziridine ring opening and the resulting secondary amines cyclize via a subsequent SNAr reaction (Figure 3).
Figure 3.

Summary of CAP and CP routes to benzo-fused sultams.
Historically, intramolecular SNAr cyclizations have been utilized to access common-sized rings or macrocycles comprising 5-membered indoles and indolines,31 macrolactams such as complestatin (16-membered),32 vancomycin (16-membered and their modified derivatives),33 and cyclopeptide alkaloids.34 Reports of intramolecular heteroaryl cyclizations en route to sultams first surfaced in the 1990s, when Giannotti and co-workers reported Cu-catalyzed reactions on o-halobenzene-sulfonamides bearing amino side chains.35 In 2010, concurrent reports from several other laboratories detailed SNAr aryletherification protocols to 7- and 8-membered benzo-fused sultams.36 Use of intramolecular SNAr to access 10- and 11-membered sultams, however, to the best of our knowledge, is void in the literature, due to several challenging problems including methods of macrocyclization (cyclization vs oligomerization) and strain (distortion of standard bond angles, lengths and unfavorable transannular interactions) in the macrocyclic products.37
Results and Discussion
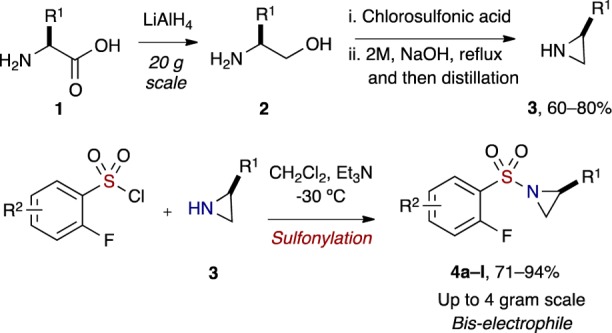
The titled investigation commenced with the preparation of chiral, nonracemic aziridines 3 via use of a mild Wenker synthesis38 from the respective amino alcohols, with all preparations occurring in good to excellent yields. Sulfonylation of aziridines with o-fluorobenzenesulfonyl chlorides and Et3N in CH2Cl2 at −30 °C furnished a variety of 1-((2-fluorophenyl)sulfonyl)aziridines in good yields (71–94%) (Scheme 1).
Scheme 1. Preparation of Aziridines and Sulfonylation To Provide (o-Fluoroaryl)sulfonyl Aziridines.
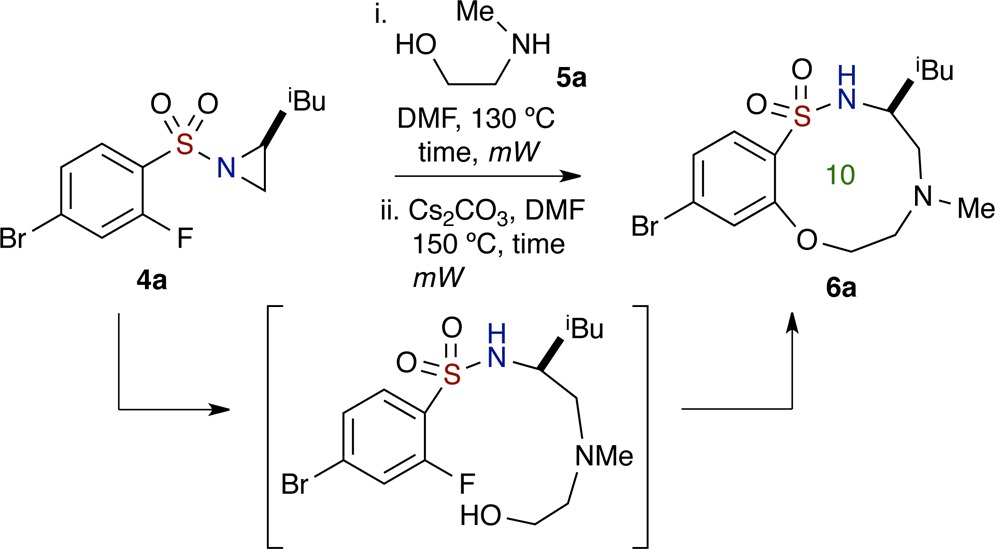
Studies on the one-pot, sequential process began with aziridinyl sulfonamide 4a (aziridine ring opening), which was reacted with N-methylethanolamine 5a (1.2 equiv) in DMF at 130 °C, using microwave (μW) irradiation for 30 min (Table 1, entry 1). The reaction was monitored by TLC, and upon disappearance of starting material, Cs2CO3 (2.5 equiv) was added to the crude mixture. The mixture was next subjected to 30 additional minutes of μW irradiation at 150 °C in order to facilitate the SNAr reaction and ultimately afford the desired benzo-oxathiadiazecine 1,1-dioxide 6a in moderate yield (43% over two reactions; 66% avg/reaction).
Table 1. Optimization of Reaction Conditions.
| entryc | conc (i–ii M) | time (i, ii min) | yielda (%) |
|---|---|---|---|
| 1 | 0.3 | 30, 30 | 43b |
| 2 | 0.3 | 30, 40 | 50b |
| 3 | 0.3–0.1 | 30, 40 | 58b |
| 4 | 0.3–0.08 | 30, 40 | 66b |
| 5 | 0.3–0.05 | 30, 40 | 34b,d |
Final isolated yield over two reactions after flash chromatography.
Aziridine opening: 1 (1.0 equiv) and 2 (1.05–1.3 equiv) in DMF at 130 °C. SNAr: Cs2CO3 (2.5 equiv) in DMF at 150 °C.
Reactions were monitored by TLC.
Reaction was run only once at 0.05.
With this result in hand, optimization of reaction conditions was carried out. Notably, it was found that solvent concentrations, reaction time, and temperature were key factors since the aziridine ring opening and SNAr reactions are inter- and intramolecular pathways, respectively (Table 1 and Scheme 2). In particular, increased reaction time and temperature were found to effect reaction decomposition. It should also be noted that the first reaction (intermolecular aziridine ring opening) was carried out under relatively high concentrations, while the subsequent intramolecular SNAr reaction requires dilute concentrations (Table 1, entries 3–5). Furthermore, it should also be noted that while aziridine ring opening proceeds at room temperature, the reaction took 5 days in order to go to completion, while utilization of μW irradiation allowed for completion of reaction in 30 min. Efforts to improve this reaction by screening other bases, for instance, CsF, K2CO3, K3PO4, DBU, and NaH, revealed that Cs2CO3 was optimal (see the Supporting Information for more data). After thorough investigation, the optimized conditions for this one-pot, sequential aziridine ring opening–SNAr protocol was achieved, whereby arylsulfonyl aziridine 4a and amino alcohol 5a were subjected to μW irradiation in DMF at 130 °C for 30 min and 150 °C for 40 min, respectively. This led to 10-membered sultam 6a in good yield (66% over two reactions; 81% avg/reaction) (Table 1, entry 4). The structure of sultam 6a was confirmed by X-ray crystallographic analysis (Figure 4). This set of optimized conditions was also utilized for the synthesis of 7-membered benzo-fused sultams, with some substrates having a shorter reaction time for SNAr cyclization.
Scheme 2. “6 + 4” and “6 + 5” Cyclization to Bi- and Tricyclic 10- and 11-Membered Sultams.

Figure 4.

X-ray structures of 6a, 6p, and 8b.
With the optimization conditions in hand, the substrate scope studies commenced with the synthesis of medium-sized, fused polycyclic and spirocyclic benzo-fused sultams using several secondary acyclic and cyclic amino alcohols 5a–e to yield the corresponding products 6a–p in average to good overall yields (Scheme 2). Notable applications include both (R)- and (S)-prolinol, racemic 2-piperidinemethanol, and 2-piperidine-ethanol to afford the 6,10,5-fused, 6,10,6-fused, and 6,11,6-fused tricyclic systems, respectively. During the investigation, it was determined that by increasing the reaction time for some substrates slightly higher yields were obtained. Thus, sultam 6b was generated in 70% yield over two reactions (84% avg/reaction) when the reaction time for SNAr reaction was extended to 50 min while maintaining all other reaction conditions (Scheme 2). In addition, two 10-membered benzo-fused sultams 6g and 6m were synthesized from sulfonamides derived from spiro-cyclohexyl aziridine in good yields. Finally, it is worth noting that a single diastereomer of the 6,11,6-fused tricyclic sultam 6p was synthesized, starting with racemic 2-piperidine-ethanol, suggesting only one diastereomer intermediate underwent cyclization reaction (or potentially aziridine ring opening). The relative stereochemistry of 6p was confirmed by X-ray crystallography (Figure 4, vide infra). Similar sultams 6n and 6o were also obtained as single diastereomers as observed by NMR.
A proposed plausible mechanism suggests that in all cases the secondary amino group reacts in a chemoselective fashion for the aziridine ring opening reaction, rendering the resulting tertiary amine incapable of executing the cyclization reaction (SNAr) and thus allowing the unprotected primary or secondary hydroxyl group to cyclize under basic conditions to provide the various benzo-oxathiadiazecine 1,1-dioxides. The resulting products have stereocenters on the core medium-sized rings, which consequently imparts “non-flatland” architecture.7
Next, we further investigated the scope of this one-pot, sequential procedure by using chiral, nonracemic, substituted secondary amino alcohols (Scheme 3). Commercially available derivatives of ephedrine, 7a–e, were subjected to the “Click” aziridine ring opening–SNAr reaction conditions, and to our delight, the secondary alcohols proceeded smoothly to afford medium-sized sultams (8a–f) in average to good yields over two reactions, albeit in lower yield for (1S,2S)-(+)-pseudoephedrine-derived 8c. Sultam 8b was confirmed by X-ray crystallography where the respective stereocenters (6R,7R) correspond to the structure as shown in Figure 4. In addition, in all cases studied, both primary and branched secondary hydroxyl groups were able to undergo SNAr cyclization to yield their respective sultams. Also, use of N-(methylamino) cyclohexyl methanol in the aforementioned method furnished the spiro-benzo-oxathiadiazecine-cyclohexane 1,1-dioxide 8f in 46% yield over two reactions (68% avg/reaction) (Scheme 3).
Scheme 3. Spiro- and Stereochemically Rich 10-Membered Sultams.

It is worth noting that the preferred conformations of these constrained structures are governed by stereoelectronic effects innate to sulfonamides, which place the nitrogen lone pair antiperiplanar to the S–Ar bond to maximize the σ* orbital delocalization and also bisect the O=S=O internuclear angle.39 The bisection of the O=S=O internuclear angle by the nitrogen lone pair has been confirmed in all X-ray crystallographic structures taken in this study (Figure 4). The consequence of this stereoelectronic effect/preferred rotomer of the Ar-SO2NR1R2 moiety is that it renders the core macrocyclic in a unique conformation, whereby the N–H bond points to the inner core of the macrocycle (see the circled highlighted area in 8b of Figure 4).40
With the aforementioned results in hand, the one-pot, sequential strategy was extended to several primary amines 9, whereby their ability to have dual reactivity facilitates access to 7-membered (common-sized) benzo-fused sultams in an overall “6 + 1” atom cyclization sequence involving consecutive aziridine ring opening and SNAr reaction (Scheme 4). The use of simple alkyl and aromatic amines containing different substituents furnished benzo-thiadiazepine 1,1-dioxides 10a–e in satisfactory yields (44–53% over two reactions, 67–73% avg/reaction). Amines with both cyclic and linear ether moieties were also employed successfully to provide 7-membered benzo-fused sultams 10f–i in moderate yields (46–56% over two reactions, 68–75% avg/reactions) (Scheme 4). The primary amines proceeded with aziridine ring opening, and the secondary amines generated from the first reaction were then cyclized to form cyclic sulfonamides.
Scheme 4. Substrate Scope of “6 + 1” Cyclization to 7-Membered Sultams.

Similarly, common-sized benzo-fused sultams 10j–p consisting of amines having hydroxyl motifs were generated with different (o-fluoroaryl)sulfonyl aziridines (cyclohexyl, iPr, and iBu) in albeit slightly lower yields (12–56% over two reactions, 35–75% avg/reaction). On the basis of the results, when primary amines with unprotected hydroxyl groups are used as the nucleophile, the resulting secondary amines from the aziridine ring opening reaction chemoselectively proceed to SNAr cyclization in preference to the free hydroxyl groups. A high degree of chemoselectivity was observed in the majority of cases, although in some cases formation of an unidentified side product during the SNAr reaction and some final product decomposition were seen.
A notable feature of these 7-membered sultams possessing a free N–H is their ability to undergo an additional facile Mitsunobu reaction to synthesize bridged [3.2.2] bicyclic benzo-fused sultams. Hence, sultam 10l was treated with Ph3P and DIAD in THF at room temperature, stirred overnight, and upon completion, provided ethanobenzothiadiazepine 1,1-dioxide 11a in 87% yield (Scheme 5). The structure of sultam 10m was confirmed by X-ray crystallography and shown to display an optimal positioning of the hydroxyl group in order to participate in facile intramolecular Mitsunobu alkylation to afford sultam 11b bearing a two-carbon bridgehead. Further demonstration of the intramolecular Mitsunobu reaction was realized in the production of the spiro-cyclohexyl-containing [3.2.2] bridged benzo-fused sultam 11c, albeit in a lower yield of 40%. The structure of 11c was confirmed by X-ray crystallography (Scheme 5). Sultam 10o, on the other hand, was unsuccessful in yielding the two-carbon bridged sultam after several attempts using similar reaction conditions. The recovery of starting material and several side products present in Mitsunobu reactions as well as excess reagents that were used in the reaction were collected.
Scheme 5. Utilization of the Mitsunobu Reaction To Access Bridged 7-Membered Sultams.

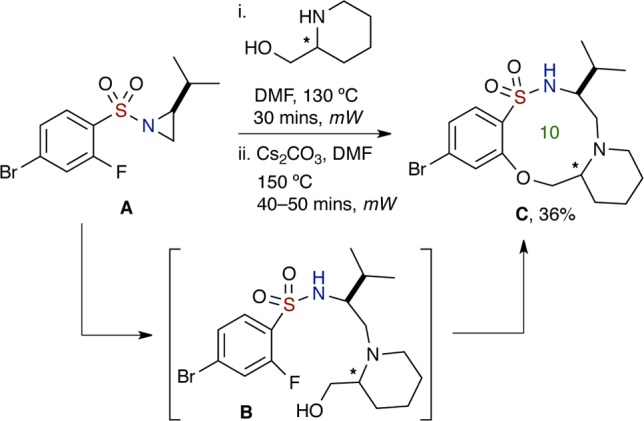
A key finding during the studies was the isolation of the intermediate, aziridine ring opened product, which was observable on 19F NMR (Scheme 6). A major difference between the intermediate and the final product is the presence of the fluorine atom, and use of 19F NMR provided a convenient way to monitor the progression of the SNAr reaction. In this experiment, aziridinyl-sulfonamide A was chosen and shown to contain a single resonance (triplet) in the 19F NMR spectrum (Figure 1a, Supporting Information). Reaction with racemic 2-piperidinemethanol furnished the ring opened intermediate B, which was detected as a single resonance (triplet) in the 19F NMR spectrum (Figure 1b, Supporting Information) but shifted marginally upfield due to the electronic changes within the sulfonamide. After SNAr reaction, the 19F NMR of the desired product C (6n) was obtained and showed complete disappearance of the fluorine resonance (Figure 1c, Supporting Information).41
Scheme 6. 19F NMR Studies: Comparison between Sulfonamide A, Ring-Opened B, and Product C.
See the 19F spectra in the Supporting Information.
Encouraged by these results, and in an effort to further highlight the efficiency of this modular approach, studies were focused toward the extension of the method using readily available chiral, nonracemic building blocks that were obtained in one step (Scheme 7). Hence, both (R)- and (S)-benzyl glycidyl ethers were subjected to “Click” epoxide ring opening30 with TBS-protected d-alaninol to furnish elaborate amino alcohols 16 and 17. The chiral, nonracemic building blocks were then utilized in the established aziridine ring opening–SNAr procedure to afford 10-membered sultams 18 and 19 with three stereocenters, along with pendant free hydroxy group, in moderate yields. In this regard, it should be noted that the TBS-ether protecting group was removed during the reaction, presumably by the displaced fluoride anion in the SNAr reaction, thus representing an overall one-pot, sequential aziridine ring opening–SNAr–desilylation protocol.
Scheme 7. “Click, Click, Click, Cyclize” to Stereochemically Rich, 10-Membered Sultams.

In summary, we have developed a one-pot CP strategy introducing (o-fluoroaryl)sulfonyl aziridine building blocks as versatile bis-electrophilic species for reaction with amino alcohols/amines for the preparation of common and medium-sized benzo-fused sultams containing up to three stereocenters. This approach was extended to the utilization of elaborate chiral, nonracemic building blocks as well as cyclic and spirocyclic amino alcohols to afford a diverse array of polycyclic scaffolds. Furthermore, the method is highly modular and adaptable for the preparation of sultam libraries in a one-pot, sequential manner. Work in this regard is underway and will be reported in due course.
Experimental Section
General Information
All air- and moisture-sensitive reactions were carried out in flame- or oven-dried glassware under argon atmosphere using standard gastight syringes, cannula, and septa. Stirring was achieved with oven-dried, magnetic stir bars. CH2Cl2 was purified by passage through the purification system employing activated Al2O3.42 Et3N was purified by passage over basic alumina and stored over KOH. Flash column chromatography was performed with SiO2. The crude mixture was also purified using an automated flash column chromatography system. Thin-layer chromatography was performed on silica gel plates. Deuterated solvents were purchased from commercial sources. 1H and 13C NMR spectra were recorded on a 400 MHz spectrometer as well as a 500 spectrometer operating at 500 MHz and 126 MHz, respectively. High-resolution mass spectrometry (HRMS) spectra were obtained on a TOF-MS operating on ESI. Microwave-assisted reactions were carried out in 1 dram vials utilizing a reaction heating block in an Anton Paar Synthos 3000 synthesizer and also Biotage Initiator both using an external calibrated external infrared (IR) sensor. All NMR peak assignments were assigned on the basis of both COSY and HSQC NMR methods.
General Procedure A: Preparation of (o-Fluoroaryl)sulfonyl Aziridines
To a round-bottom flask containing a solution of aziridine (2.2 mmol, 2.0 equiv) in dry CH2Cl2 (0.5 M) was added Et3N (2.2 mmol, 2.0 equiv). The reaction mixture was cooled to −40 °C and stirred for 10 min, and sulfonyl chloride (1.1 mmol, 1.0 equiv) was added to the reaction mixture in a dropwise fashion. The reaction was then stirred for 30 min after which conversion of starting material was monitored by TLC. Upon completion of the reaction, the mixture was warmed to rt and quenched with cold water (2.2 mL), and the layers were separated. The organic portion was washed with cold 10% aq HCl, and the resulting layers were separated. This partitioning was then repeated with cold water, cold satd NaHCO3, cold water again, and finally brine. The final organic layer was dried (Na2SO4) and concentrated under reduced pressure to afford the desired aziridinyl sulfonamide.
General Procedure B: One-Pot, Sequential (Aziridine Ring Opening and SNAr)
To a microwave vial containing a solution of sulfonamide (1.0 equiv) in DMF (0.3 M) was added amine/amino alcohol (1.05–1.2 equiv). The reaction vessel was capped and heated in the Biotage Initiator microwave at 130 °C for 30–40 min, after which conversion of starting material was monitored by TLC. To the crude mixture were added DMF (0.08 M) and Cs2CO3 (2.5 equiv), and the mixture underwent microwave irradiation again at 150 °C for 30–50 min. Water was added to the crude mixture, which was extracted with EtOAc (4×). The organic layer was separated, and the combined organic layers were washed with water and brine, dried (Na2SO4), and concentrated under reduced pressure to afford the crude product, which was purified by an automated flash column chromatography system.
General Procedure C: One-Pot, Sequential (Aziridine Ring Opening and SNAr)
To a microwave vial containing a solution of sulfonamide (1.0 equiv) in DMF (0.3 M) was added amine/amino alcohol (1.05–1.2 equiv). The reaction vessel was capped and heated in a Biotage Initiator microwave at 130 °C for 30–40 min, after which conversion of starting material was monitored by TLC. To the crude mixture was added Cs2CO3 (2.5 equiv), and the mixture underwent microwave irradiation again at 150 °C for 30–50 min. Water was added to the crude mixture, which was extracted with EtOAc (4×). The organic layer was separated, and the combined organic layers were washed with water and brine, dried (Na2SO4), and concentrated under reduced pressure to afford the crude product, which was purified by the automated flash column chromatography system.
General Procedure D: Mitsunobu Reaction
To a flame-dried round-bottom flask containing a solution of sultam (0.047 mmol, 1.0 equiv) in dry THF (0.05 M) was added triphenylphosphine (0.140 mmol, 3.0 equiv). The reaction mixture was stirred for 10 min, and diisopropyl azodicarboxylate (0.12 mmol, 2.5 equiv) was added to the mixture in a dropwise fashion. The reaction was then stirred overnight at rt, and conversion of starting material was monitored by TLC. The solvent was removed in vacuo to yield a yellow oil and was purified by an automated flash column chromatography system.
(S)-1-((4-Bromo-2-fluorophenyl)sulfonyl)-2-isopropylaziridine (4b)
According to general procedure A from 2-fluoro-4-bromosulfonyl chloride (1 g), 4b (854.4 mg, 72%) was isolated as a yellow oil: Rf = 0.60 (1:3 EtOAc/hexane); [α]D20 = −47.6 (c = 0.675, CHCl3); FTIR (thin film) 3094, 2962, 1589, 1472, 1398, 1333, 1167, 879, 735 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.88–7.76 (m, 1H, aromatic), 7.53–7.41 (m, 2H, aromatic), 2.80 (dd, J = 7.1, 1.1 Hz, 1H, NCHaHbCH), 2.72 (ddd, J = 7.3, 7.2, 4.8 Hz, 1H, NCHCH), 2.24 (d, J = 4.8 Hz, 1H, NCHaHbCH), 1.59–1.39 (m, 1H, CH3CHCH3), 0.96 (d, J = 6.8 Hz, 3H, CH3CHCH3), 0.91 (d, J = 6.7 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 159.1 (d, 1JC–F = 262.2 Hz), 131.4, 129.4 (d, 3JC–F = 9.0 Hz), 127.9 (d, 3JC–F = 3.8 Hz), 121.0 (d, 2JC–F = 24.5 Hz), 120.5 (d, 2JC–F = 24.2 Hz), 46.6, 33.8, 30.1, 19.5, 18.9; HRMS calcd for C11H13BrFNO2SH (M + H)+ 321.9913, found 321.9888 (TOF MS ES+).
(S)-1-((2,4-Difluorophenyl)sulfonyl)-2-isopropylaziridine (4d)
According to general procedure A from 2,4-difluorosulfonyl chloride (1 g), 4d (889.4 mg, 72%) was isolated as a yellow oil: Rf = 0.51 (1:3 EtOAc/hexane); [α]D20 = −5.0 (c = 1.43, CHCl3); FTIR (thin film) 3103, 2964, 1603, 1481, 1429, 1335, 1167, 854, 741, 727 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 8.10–7.85 (m, 1H, aromatic), 7.12–6.88 (m, 2H, aromatic), 2.80 (dd, J = 7.1, 1.1 Hz, 1H, NCHaHbCH), 2.70 (ddd, J = 7.3, 7.2, 4.6 Hz, 1H, NCHCH), 2.24 (d, J = 3.8 Hz, 1H, NCHaHbCH), 1.61–1.43 (m, 1H, CH3CHCH3), 0.96 (d, J = 6.8 Hz, 3H, CH3CHCH3), 0.90 (d, J = 6.7 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 166.3 (dd, J = 258.5, 11.4 Hz), 160.4 (dd, J = 260.8, 12.8 Hz), 132.4 (dd, J = 10.6, 1.6 Hz), 111.9 (dd, J = 22.0, 3.8 Hz), 105.8 (dd, J = 25.5, 25.4 Hz), 105.4 (dd, J = 26.0, 25.9 Hz), 46.5, 33.7, 30.1, 19.4, 18.9; HRMS calcd for C11H13F2NO2SH (M + H)+ 262.0713, found 262.0720 (TOF MS ES+).
(S)-10-Bromo-3-isobutyl-5-methyl-2,3,4,5,6,7-hexahydrobenzo[b][1,4,5,8]oxathiadiazecine 1,1-Dioxide (6a)
According to the reaction protocol described in general procedure B from 4a (49.8 mg), compound 6a (66%, 38.7 mg) was isolated after chromatography as a white solid: mp 192–195 °C; Rf = 0.44 (2:1 EtOAc/hexane); [α]D20 = +167.5 (c = 1.02, CHCl3); FTIR (thin film) 3267, 3090, 2966, 1576, 1558, 1462, 1400, 1323, 1165, 1059, 824, 781, 748, 725 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.85 (d, J = 8.3 Hz, 1H, aromatic), 7.34–7.28 (m, 2H, aromatic), 7.09 (s, 1H, NH), 4.41–4.31 (m, 2H, OCH2CH2), 2.73 (dddd, J = 9.2, 9.0, 4.7, 4.6 Hz, 1H, NHCHCH2N), 2.66 (dd, J = 12.6, 4.9 Hz, 1H, NHCHCHaHbN), 2.58 (ddd, J = 14.8, 10.8, 3.3 Hz, 1H, NCHaHbCH2O), 2.46 (s, 3H, NCH3), 2.34 (ddd, J = 15.2, 1.7, 1.6 Hz, 1H, NCHaHbCH2O), 2.24 (dd, J = 12.6, 10.5 Hz, 1H, NHCHCHaHbN), 1.85 (ddd, J = 13.7, 9.4, 4.2 Hz, 1H, NHCHCHaHb), 1.62–1.49 (m, 1H, CH3CHCH3), 1.33 (ddd, J = 13.8, 8.8, 5.0 Hz, 1H, NHCHCHaHb), 0.86 (d, J = 6.6 Hz, 3H, CH3CHCH3), 0.73 (d, J = 6.6 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 155.1, 132.8, 129.3, 128.4, 125.7, 120.6, 69.7, 60.0, 53.2, 49.8, 44.3, 43.1, 24.4, 23.6, 21.9; HRMS calcd for C15H23BrN2O3SH (M + H)+ 391.0691, found 391.0670 (TOF MS ES+).
(S)-10-Bromo-3-isopropyl-5-methyl-2,3,4,5,6,7-hexahydrobenzo[b][1,4,5,8]oxathiadiazecine-1,1-Dioxide (6b)
According to the reaction protocol described in general procedure B from 4b (50.0 mg), compound 6b (70%, 41.0 mg) was isolated after chromatography as a white solid: mp 175–180 °C; Rf = 0.45 (2:1 EtOAc/hexane); [α]D20 = +170.4 (c = 0.545, CHCl3); FTIR (thin film) 3263, 3099, 2968, 1578, 1560, 1452, 1371, 1323, 1163 1057, 820, 737 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.84 (d, J = 8.4 Hz, 1H, aromatic), 7.36–7.29 (m, 2H, aromatic), 7.05 (s, 1H, NH), 4.43–4.27 (m, 2H, OCH2CH2), 2.68 (ddd, J = 11.2, 4.6, 4.5 Hz, 1H, NHCHCH2N), 2.60 (ddd, J = 14.5, 10.6, 3.5 Hz, 1H, NHCHCHaHbN), 2.49 (dd, J = 12.7, 5.1 Hz, 1H, NCHaHbCH2O), 2.46 (s, 3H, NCH3), 2.38–2.23 (m, 3H, NHCHCHaHbN, NCHaHbCH2O, CH3CHCH3), 0.98 (d, J = 6.9 Hz, 3H, CH3CHCH3), 0.79 (d, J = 7.3 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 155.2, 132.9, 129.2, 128.3, 125.7, 120.7, 69.7, 55.6, 53.7, 53.1, 44.3, 28.9, 18.3, 15.6; HRMS calcd for C14H21BrN2O3SH (M + H)+ 377.0535, found 377.0495 (TOF MS ES+).
(S)-10-Fluoro-3-isobutyl-5-methyl-2,3,4,5,6,7-hexahydrobenzo[b][1,4,5,8]oxathiadiazecine 1,1-Dioxide (6c)
According to the reaction protocol described in general procedure C from 4c (52.0 mg), compound 6c (43%, 26.8 mg) was isolated after chromatography as a white solid: mp 145–148 °C; Rf = 0.33 (2:1 EtOAc/hexane); [α]D20 = +148.7 (c = 0.87, CHCl3); FTIR (thin film) 3265, 2962, 1603, 1587, 1458, 1350, 1323, 1163, 1057, 818, 785, 733, 704 cm–1; 1H NMR (400 MHz, CDCl3) δ ppm 7.99 (dd, J = 8.6, 6.4 Hz, 1H, aromatic), 7.07 (s, 1H, NH), 6.92–6.78 (m, 2H, aromatic), 4.46–4.19 (m, 2H, OCH2CH2), 2.70 (dddd, J = 9.0, 8.8, 4.6, 4.4 Hz, 1H, NHCHCH2N), 2.65 (dd, J = 12.4, 4.9 Hz, 1H, NHCHCHaHbN), 2.58 (ddd, J = 14.9, 8.9, 5.5 Hz, 1H, NCHaHbCH2O), 2.46 (s, 3H, NCH3), 2.34 (ddd, J = 15.1, 1.8, 1.7 Hz, 1H, NCHaHbCH2O), 2.23 (dd, J = 12.4, 10.5 Hz, 1H, NHCHCHaHbN), 1.85 (ddd, J = 13.7, 9.5, 3.9 Hz, 1H, NHCHCHaHb), 1.61–1.52 (m, 1H, CH3CHCH3), 1.33 (ddd, J = 13.8, 8.6, 5.0 Hz, 1H, NHCHCHaHb), 0.86 (d, J = 6.6 Hz, 3H, CH3CHCH3), 0.72 (d, J = 6.6 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 166.3 (d, 1JC–F = 254.8 Hz), 156.3 (d, 3JC–F = 10.5 Hz), 133.7 (d, 3JC–F = 10.8 Hz), 126.2 (d, 4JC–F = 3.2 Hz), 109.7 (d, 2JC–F = 22.1 Hz), 104.9 (d, 2JC–F = 25.0 Hz), 69.7, 60.0, 53.2, 49.9, 44.4, 43.1, 24.4, 23.6, 21.8; HRMS calcd for C15H23FN2O3SH (M + H)+ 331.1492, found 331.1519 (TOF MS ES+).
(S)-10-Fluoro-3-isopropyl-5-methyl-2,3,4,5,6,7-hexahydrobenzo[b][1,4,5,8]oxathiadiazecine 1,1-Dioxide (6d)
According to the reaction protocol described in general procedure C from 4d (94.2 mg), compound 6d (40%, 44.6 mg) was isolated after chromatography as a white solid: mp 183–188 °C; Rf = 0.30 (2:1 EtOAc/hexane); [α]D20 = +128.8 (c = 0.745, CHCl3); FTIR (thin film) 3257, 2974, 1603, 1587, 1470, 1448, 1373, 1323, 1163, 1067, 818, 777, 756, 729 cm–1; 1H NMR (400 MHz, CDCl3) δ ppm 7.99 (dd, J = 8.6, 6.4 Hz, 1H, aromatic), 7.04 (s, 1H, NH), 6.93–6.73 (m, 2H, aromatic), 4.43–4.25 (m, 2H, OCH2CH2), 2.71–2.56 (m, 2H, NHCHCH2N, NHCHCHaHbN), 2.50 (dd, J = 12.8, 5.0 Hz, 1H, NCHaHbCH2O), 2.47 (s, 3H, NCH3), 2.37–2.29 (m, 3H, NHCHCHaHbN, NCHaHbCH2O, CH3CHCH3), 0.99 (d, J = 6.9 Hz, 3H, CH3CHCH3), 0.80 (d, J = 7.2 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 166.3 (d, 1JC–F = 254.7 Hz), 156.4 (d, 3JC–F = 10.6 Hz), 133.7 (d, 3JC–F = 10.8 Hz), 126.2 (d, 4JC–F = 3.4 Hz), 109.7 (d, 2JC–F = 22.1 Hz), 105.1 (d, 2JC–F = 25.0 Hz), 69.7, 55.7, 53.8, 53.1, 44.4, 28.9, 18.4, 15.6; HRMS calcd for C14H21FN2O3SH (M + H)+ 317.1335, found 317.1320 (TOF MS ES+).
(S)-11-Chloro-3-isopropyl-5-methyl-2,3,4,5,6,7-hexahydrobenzo[b][1,4,5,8]oxathiadiazecine 1,1-Dioxide (6e)
According to the reaction protocol described in general procedure B from 4e (75.0 mg), compound 6e (51%, 45.8 mg) was isolated after chromatography as a yellow oil: Rf = 0.32 (1:1 EtOAc/hexane); [α]D20 = +89.0 (c = 0.125, CHCl3); FTIR (neat) 3149, 2962, 1587, 1469, 1371, 1307, 1222, 1161, 1107, 1060, 835, 821, 729 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.95 (d, J = 2.7 Hz, 1H, aromatic), 7.50 (dd, J = 8.8, 2.7 Hz, 1H, aromatic), 7.14 (s, 1H, NH), 7.12 (d, J = 8.9 Hz, 1H, aromatic), 4.39–4.29 (m, 2H, OCH2CH2), 2.75–2.69 (m, 1H, NHCHCH2N), 2.56 (ddd, J = 14.5, 10.5, 3.6 Hz, 1H, NCHaHbCH2O), 2.48 (dd, J = 12.6, 5.1 Hz, 1H, NCHaHbCH2O), 2.45 (s, 3H, NCH3), 2.38–2.25 (m, 3H, NHCHCHaHb, NHCHCHaHbN, CH3CHCH3), 0.98 (d, J = 6.9 Hz, 3H, CH3CHCH3), 0.81 (d, J = 7.2 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 153.2, 134.1, 131.6, 131.3, 127.5, 118.7, 69.7, 55.4, 53.6, 53.0, 44.2, 28.9, 18.3, 15.5; HRMS calcd for C14H21ClN2O3SH (M + H)+ 333.1040, found 333.1022 (TOF MS ES+).
(S)-3-((S)-sec-Butyl)-11-chloro-5-methyl-2,3,4,5,6,7-hexahydrobenzo[b][1,4,5,8]oxathiadiazecine 1,1-Dioxide (6f)
According to the reaction protocol described in general procedure B from 4f (78.7 mg), compound 6f (45%, 42.1 mg) was isolated after chromatography as a white solid: mp 138–142 °C; Rf = 0.31 (1:1 EtOAc/hexane); [α]D20 = −93.2 (c = 0.125, CHCl3); FTIR (neat) 2962, 1588, 1467, 1371, 1159, 1060, 831, 819, 750 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.95 (d, J = 2.7 Hz, 1H, aromatic), 7.50 (dd, J = 8.8, 2.7 Hz, 1H, aromatic), 7.11 (d, J = 8.8 Hz, 1H, aromatic), 7.05 (s, 1H, NH), 4.44–4.24 (m, 2H, OCH2CH2), 2.83–2.72 (m, 1H, NHCHCH2N), 2.58 (ddd, J = 14.5, 10.6, 3.5 Hz, 1H, NCHaHbCH2O), 2.50 (dd, J = 12.6, 5.1 Hz, 1H, NCHaHbCH2O), 2.44 (s, 3H, NCH3), 2.35–2.25 (m, 2H, NHCHCHaHbN, NHCHCHaHb), 2.01–1.92 (m, 1H, CH3CHCHaHbCH3), 1.92–1.82 (m, 1H, CH3CHCHaHbCH3), 1.03–0.95 (m, 1H, CH3CHCHaHbCH3), 0.95–0.90 (m, 3H, CH3CHCH2CH3), 0.80 (d, J = 7.1 Hz, 3H, CH3CHCH2CH3); 13C NMR (126 MHz, CDCl3) δ ppm 153.3, 134.1, 131.6, 131.3, 127.5, 118.6, 69.6, 55.7, 54.6, 52.8, 44.2, 36.0, 23.0, 15.3, 12.4; HRMS calcd for C15H23ClN2O3SH (M + H)+ 347.1196, found 347.1200 (TOF MS ES+).
12-Fluoro-5-methyl-4,5,6,7-tetrahydro-2H-spiro[benzo[b][1,4,5,8]oxathiadiazecine-3,1′-cyclohexane] 1,1-Dioxide (6g)
According to the reaction protocol described in general procedure B from 4g (77.7 mg), compound 6g (46%, 42.6 mg) was isolated after chromatography as a brownish oil: Rf = 0.35 (1:1 EtOAc/hexane); FTIR (neat) 3245, 2956, 2931, 1591, 1488, 1458, 1319, 1153, 1062, 891, 821, 705 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.47 (ddd, J = 8.2, 8.2, 5.9 Hz, 1H, aromatic), 7.13 (ddd, J = 8.2, 1.1, 1.0 Hz, 1H, aromatic), 6.98 (ddd, J = 10.5, 8.4, 1.1 Hz, 1H, aromatic), 6.55 (s, 1H, NH), 5.61–5.56 (m, 1H, OCHaHbCH2), 3.79 (dd, J = 5.1, 4.9 Hz, 1H, OCHaHbCH2), 3.38 (d, J = 5.8 Hz, 2H, NCH2CH2O), 3.25–3.09 (m, 2H, NHCCH2N), 2.82 (s, 3H, NCH3), 1.99–1.87 (m, 4H, cyclohexyl), 1.70–1.44 (m, 6H, cyclohexyl); 13C NMR (126 MHz, CDCl3) δ ppm 160.3(d, 1JC–F = 257.2 Hz), 154.5, 133.5 (d, 3JC–F = 11.2 Hz), 123.8 (d, 3JC–F = 10.2 Hz), 119.5 (d, 4JC–F = 3.2 Hz), 113.4 (d, 2JC–F = 24.5 Hz), 59.5, 58.9, 50.1, 42.7, 26.4 (2C), 25.0, 22.4, 22.1 (2C); HRMS calcd for C16H23FN2O3SH (M + H)+ 343.1492, found 343.1485 (TOF MS ES+).
(6S,14aR)-11-Bromo-6-isobutyl-1,2,3,5,6,7,14,14a-octahydrobenzo[b]pyrrolo[1,2-h][1,4,5,8]oxathiadiazecine 8,8-Dioxide (6h)
According to the reaction protocol described in general procedure C from 4a (81.8 mg), compound 6h (57%, 57.7 mg) was isolated after chromatography as a colorless oil: Rf = 0.34 (1:1 EtOAc/hexane); [α]D20 = −48.1 (c = 1.805, CHCl3); FTIR (thin film) 3275, 2955, 1578, 1466, 1317, 1159, 1063, 852, 733, 702 cm–1; 1H NMR (400 MHz, CDCl3) δ ppm 7.93–7.71 (m, 1H, aromatic), 7.26–7.18 (m, 2H, aromatic), 5.69 (s, 1H, NH), 4.48 (dd, J = 11.5, 3.3 Hz, 1H, OCHaHbCHN), 3.90 (dd, J = 11.4, 11.3 Hz, 1H, OCHaHbCHN), 3.40–3.26 (m, 1H, NHCHCH2N), 3.17 (ddt, J = 12.0, 8.1, 3.8 Hz, 1H, NCHCH2O), 3.14–3.03 (m, 1H, NCHaHbCH2CH2), 2.50–2.38 (m, 3H, NHCHCH2N, NCHaHbCH2CH2), 2.01–1.90 (m, 1H, NCH2CH2CHaHb), 1.88–1.77 (m, 3H, NCH2CH2CH2, CH3CHCH3), 1.67 (ddd, J = 14.3, 8.4, 6.2 Hz, 1H, NHCHCHaHb), 1.40 (td, J = 11.4, 4.6 Hz, 1H, NCH2CH2CHaHb), 1.10 (ddd, J = 14.0, 7.8, 6.2 Hz, 1H, NHCHCHaHb), 0.90 (dd, J = 6.9, 6.8 Hz, 6H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 156.0, 131.1, 130.8, 128.0, 124.5, 118.2, 74.0, 62.8, 61.4, 56.7, 55.5, 40.9, 27.4, 24.7, 24.7, 22.7, 22.3; HRMS calcd for C17H25BrN2O3SH (M + H)+ 417.0848, found 417.0836 (TOF MS ES+).
(6S,14aR)-11-Fluoro-6-isopropyl-1,2,3,5,6,7,14,14a-octahydrobenzo[b]pyrrolo[1,2-h][1,4,5,8]oxathiadiazecine 8,8-Dioxide (6i)
According to the reaction protocol described in general procedure C from 4d (54.3 mg), compound 6i (37%, 26.1 mg) was isolated after chromatography as a semiwhite sticky oil: Rf = 0.28 (1:1 EtOAc/hexane); [α]D20 = −6.6 (c = 1.33, CHCl3); FTIR (thin film) 3300, 2961, 1603, 1587, 1468, 1387, 1323, 1157, 1070, 839 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.99–7.76 (m, 1H, aromatic), 6.82–6.64 (m, 2H, aromatic), 4.84 (d, J = 6.9 Hz, 1H, NH), 4.45 (dd, J = 11.6, 3.1 Hz, 1H, OCHaHbCHN) 3.99 (dd, J = 11.2, 11.1 Hz, 1H, OCHaHbCHN), 3.32 (ddd, J = 11.3, 5.9, 5.7 Hz, 1H, NHCHCH2N), 3.08 (dddd, J = 11.2, 8.7, 5.8, 3.1 Hz, 1H, NCHCH2O), 2.97 (ddd, J = 9.6, 6.2, 4.5 Hz, 1H, NCHaHbCH2CH2), 2.71 (dd, J = 14.4, 5.6 Hz, 1H, NHCHCHaHbN), 2.56–2.43 (m, 2H, NCHaHbCH2CH2, NHCHCHaHbN), 2.00–1.85 (m, 2H, CH3CHCH3, NCH2CH2CHaHb), 1.79–1.69 (m, 2H, NCH2CH2CH2), 1.45–1.31 (m, 1H, NCH2CH2CHaHb), 0.97 (dd, J = 6.9, 4.4 Hz, 6H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 166.0 (d, 1JC–F = 253.8 Hz), 157.5 (d, 3JC–F = 10.8 Hz), 132.0 (d, 3JC–F = 10.9 Hz), 126.7 (d, 4JC–F = 3.2 Hz), 107.9 (d, 2JC–F = 22.2 Hz), 102.6 (d, 2JC–F = 25.6 Hz), 74.0, 64.9, 62.8, 59.4, 57.5, 31.3, 27.2, 23.8, 18.7, 18.2; HRMS calcd for C16H23FN2O3SH (M + H)+ 343.1492, found 343.1492 (TOF MS ES+).
(6S,14aR)-6-((S)-sec-Butyl)-10-chloro-1,2,3,5,6,7,14,14a-octahydrobenzo[b]pyrrolo[1,2-h][1,4,5,8]oxathiadiazecine 8,8-Dioxide (6j)
According to the reaction protocol described in general procedure B from 4f (78.8 mg), compound 6j (54%, 54.4 mg) was isolated after chromatography as a yellow oil: Rf = 0.37 (1:1 EtOAc/hexane); [α]D20 = −68.4 (c = 0.125, CHCl3); FTIR (neat) 2962, 2875, 1598, 1467, 1407, 1380, 1338, 1163, 1064, 786, 761 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.91 (d, J = 2.7 Hz, 1H, aromatic), 7.46 (dd, J = 8.8, 2.7 Hz, 1H, aromatic), 7.06 (d, J = 8.8 Hz, 1H, aromatic), 6.85 (d, J = 5.7 Hz, 1H, NH), 4.35 (dd, J = 11.8, 3.2 Hz, 1H, OCHaHbCHN), 3.90 (t, J = 11.6 Hz, 1H, OCHaHbCHN), 3.20–2.95 (m, 2H, NHCHCH2N, NCHCH2O), 2.92–2.78 (m, 1H, NCHaHbCH2CH2), 2.63–2.50 (m, 2H, NHCHCHaHbN, NCHaHbCH2CH2), 2.41 (dd, J = 13.5, 5.1 Hz, 1H, NHCHCHaHbN), 2.02–1.86 (m, 1H, NCH2CHaHbCH2), 1.86–1.75 (m, 2H, NCH2CHaHbCH2, NCH2CHaHbCHaHb), 1.76–1.62 (m, 2H, NCH2CHaHbCHaHb, CH3CHCHaHbCH3), 1.40–1.29 (m, 1H, CH3CHCHaHbCH3), 1.11–0.97 (m, 1H, CH3CHCHaHbCH3), 0.95 (d, J = 6.8 Hz, 3H, CH3CHCH2CH3), 0.90 (t, J = 7.3 Hz, 3H, CH3CHCH2CH3); 13C NMR (126 MHz, CDCl3) δ ppm 153.1, 133.6, 131.6, 130.3, 126.7, 116.4, 73.5, 61.9, 58.0, 57.2, 55.7, 37.0, 27.0, 25.4, 24.2, 15.8, 11.6; HRMS calcd for C17H25ClN2O3SH (M + H)+ 373.1353, found 373.1334 (TOF MS ES+).
(6S,14aR)-6-((S)-sec-Butyl)-9-fluoro-1,2,3,5,6,7,14,14a-octahydrobenzo[b]pyrrolo[1,2-h][1,4,5,8]oxathiadiazecine 8,8-Dioxide (6k)
According to the reaction protocol described in general procedure B from 4h (74.4 mg), compound 6k (57%, 54.9 mg) was isolated after chromatography as a yellowish white solid: mp 152–156 °C; Rf = 0.41 (1:1 EtOAc/hexane); [α]D20 = −58.3 (c = 0.125, CHCl3); FTIR (neat) 2962, 2875, 1598, 1467, 1380, 1338, 1163, 1064, 786, 761 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.42 (ddd, J = 8.4, 5.8 Hz, 1H, aromatic), 7.07 (d, J = 6.2 Hz, 1H, NH), 6.89 (ddd, J = 8.5, 1.1, 1.1 Hz, 1H, aromatic), 6.82 (ddd, J = 9.6, 8.4, 1.0 Hz, 1H, aromatic), 4.42 (dd, J = 11.8, 3.2 Hz, 1H, OCHaHbCHN), 3.92 (dd, J = 11.5, 11.2 Hz, 1H, OCHaHbCHN), 3.19–3.04 (m, 2H, NHCHCH2N, NCHCH2O), 3.03–2.93 (m, 1H, NCHaHbCH2CH2), 2.64 (dd, J = 13.8, 5.4 Hz, 1H, NHCHCHaHbN), 2.57 (ddd, J = 9.3, 9.2, 6.1 Hz, 1H, NCHaHbCH2CH2), 2.46 (dd, J = 13.8, 4.9 Hz, 1H, NHCHCHaHbN), 1.99–1.88 (m, 1H, NCH2CHaHbCH2), 1.88–1.61 (m, 4H, NCH2CHaHbCH2, NCH2CHaHbCH2, CH3CHCHaHbCH3), 1.38 (dddd, J = 13.1, 6.8, 3.6, 3.5 Hz, 1H, CH3CHCHaHbCH3), 1.10–1.00 (m, 1H, CH3CHCHaHbCH3), 0.99 (d, J = 6.8 Hz, 3H, CH3CHCH2CH3), 0.89 (t, J = 7.3 Hz, 3H, CH3CHCH2CH3); 13C NMR (126 MHz, CDCl3) δ ppm 161.1 (d, 1JC–F = 259.8 Hz), 155.6, 133.5 (d, 3JC–F = 11.0 Hz), 119.2 (d, 3JC–F = 13.1 Hz), 110.6 (d, 2JC–F = 24.2 Hz), 109.9 (d, 4JC–F = 3.6 Hz), 73.5, 62.1, 58.5, 57.6, 55.6, 36.8, 27.1, 25.6, 24.3, 15.9, 11.5; HRMS calcd for C17H25FN2O3SH (M + H)+ 357.1648, found 357.1635 (TOF MS ES+).
(6S,14aS)-11-Fluoro-6-isopropyl-1,2,3,5,6,7,14,14a-octahydrobenzo[b]pyrrolo[1,2-h][1,4,5,8]oxathiadiazecine 8,8-Dioxide (6l)
According to the reaction protocol described in general procedure B from 4d (47.0 mg), compound 6l (42%, 25.7 mg) was isolated after chromatography as a colorless oil: Rf = 0.1 (1:1 EtOAc/hexane); [α]D20 = +74.4 (c = 0.36, CHCl3); FTIR (thin film) 3286, 2962, 1603, 1587, 1475, 1383, 1329, 1163, 1070, 847, 735, 698 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.94 (dd, J = 8.6, 6.5 Hz, 1H, aromatic), 6.92–6.76 (m, 3H, aromatic, NH), 4.33 (dd, J = 11.9, 3.3 Hz, 1H, OCHaHbCHN), 3.92 (dd, J = 11.8, 11.7 Hz, 1H, OCHaHbCHN), 3.16 (ddd, J = 10.2, 5.5, 4.8 Hz, 1H, NHCHCH2N), 3.06 (ddt, J = 11.9, 8.6, 3.1 Hz, 1H, NCHCH2O), 2.71 (dt, J = 11.9, 5.9 Hz, 1H, NCHaHbCH2CH2), 2.64–2.50 (m, 2H, NCHaHbCH2CH2, NHCHCHaHbN), 2.37 (dd, J = 13.5, 5.0 Hz, 1H, NHCHCHaHbN), 2.02–1.89 (m, 2H, CH3CHCH3, NCH2CH2CHaHb), 1.87–1.78 (m, 2H, NCH2CH2CH2), 1.36 (ddt, J = 12.6, 6.6, 3.4 Hz, 1H, NCH2CH2CHaHb), 0.95 (dd, J = 17.2, 6.8 Hz, 6H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 166.0 (d, 1JC–F = 254.3 Hz), 156.2 (d, 3JC–F = 10.5 Hz), 132.5 (d, 3JC–F = 10.8 Hz), 126.4 (d, 4JC–F = 3.3 Hz), 108.8 (d, 2JC–F = 22.2 Hz), 103.1 (d, 2JC–F = 25.4 Hz), 73.7, 62.0, 59.1, 57.3, 55.2, 30.5, 27.0, 25.6, 19.6, 17.5; HRMS calcd for C16H23FN2O3SH (M + H)+ 343.1492, found 343.1459 (TOF MS ES+).
(R)-10-Chloro-2,3,5,7,14,14a-hexahydro-1H-spiro[benzo[b]pyrrolo[1,2-h][1,4,5,8]oxathiadiazecine-6,1′-cyclohexane] 8,8-Dioxide (6m)
According to the reaction protocol described in general procedure B from 4i (82.0 mg), compound 6m (51%, 53.0 mg) was isolated after chromatography as a white solid: mp 154–159 °C; Rf = 0.37 (1:1 EtOAc/hexane); [α]D20 = −24.0 (c = 0.125, CHCl3); FTIR (neat) 2937, 1585, 1465, 1315, 1228, 1157, 1064, 819, 732 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.92 (d, J = 2.7 Hz, 1H, aromatic), 7.45 (dd, J = 8.8, 2.7 Hz, 1H, aromatic), 7.05 (d, J = 8.8 Hz, 1H, aromatic), 6.30 (s, 1H, NH), 4.46 (dd, J = 11.9, 3.2 Hz, 1H, OCHaHbCHN), 3.93 (dd, J = 11.8, 11.5 Hz, 1H, OCHaHbCHN), 3.24–3.18 (m, 1H, NCHCH2O), 3.13 (dddd, J = 12.2, 9.3, 3.4, 3.3 Hz, 1H, NCHaHbCH2CH2), 2.50–2.37 (m, 2H, NHCCHaHbN, NCHaHbCH2CH2), 2.17 (d, J = 14.1 Hz, 1H, NHCCHaHbN), 2.03–1.86 (m, 2H, NCH2CHaHbCHaHb, NCH2CH2CHaHb), 1.84–1.76 (m, 2H, NCH2CHaHbCHaHb, NCH2CH2CHaHb), 1.75–1.63 (m, 2H, cyclohexyl), 1.58–1.50 (m, 1H, cyclohexyl), 1.50–1.41 (m, 1H, cyclohexyl), 1.41–1.20 (m, 5H, cyclohexyl), 1.14–1.04 (m, 1H, cyclohexyl); 13C NMR (126 MHz, CDCl3) δ ppm 153.0, 133.6, 133.3, 129.6, 126.5, 116.3, 73.6, 63.7, 60.3, 59.6, 38.4, 31.3, 27.3, 26.1, 25.4, 21.6, 21.4 (2C); HRMS calcd for C18H25ClN2O3SH (M + H)+ 385.1353, found 385.1336 (TOF MS ES+).
(7S)-2-Bromo-7-isopropyl-7,8,10,11,12,13,13a,14-octahydro-6H-benzo[b]pyrido[1,2-h][1,4,5,8]oxathiadiazecine 5,5-Dioxide (6n)
According to the reaction protocol described in general procedure C from 4b (95.6 mg), compound 6n (36%, 44.7 mg) was isolated after chromatography as a sticky colorless oil: Rf = 0.52 (1:1 EtOAc/hexane); [α]D20 = +30.1 (c = 0.59, CHCl3); FTIR (thin film) 3259, 2934, 1578, 1464, 1391, 1327, 1159, 1063, 812, 762, 733, 702 cm–1; 1H NMR (400 MHz, CDCl3) δ ppm 7.80 (d, J = 8.3 Hz, 1H, aromatic), 7.29 (d, J = 1.8 Hz, 1H, aromatic), 7.20 (d, J = 1.8 Hz, 1H, aromatic), 4.39 (dd, J = 10.7, 4.6 Hz, 1H, OCHaHbCHN), 3.75–3.60 (m, 1H, OCHaHbCHN), 3.42–3.25 (m, 1H, NCHCH2O), 2.79–2.60 (m, 4H, NCH2CH2CH2CH2, NHCHCH2N, NHCHCHaHbN), 2.49–2.31 (m, 1H, NHCHCHaHbN), 1.99–1.86 (m, 1H, CH3CHCH3), 1.87–1.77 (m, 1H, NCH2CH2CHaHbCH2), 1.59–1.36 (m, 4H, NCH2CH2CHaHbCHaHb), 1.30–1.16 (m, 1H, NCH2CH2CH2CHaHb), 0.94 (d, J = 6.8 Hz, 3H, CH3CHCH3), 0.89 (d, J = 7.0 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 156.5, 131.9, 127.7, 127.7, 125.7, 120.3, 73.7, 59.4, 57.4 (2C), 54.2, 31.2, 23.8, 22.9, 21.6, 18.3, 17.2; HRMS calcd for C17H25BrN2O3SH (M + H)+ 417.0848, found 417.0838 (TOF MS ES+).
(7S)-2-Bromo-7-isobutyl-6,7,8,10,11,12,13,13a,14,15-decahydrobenzo[b]pyrido[1,2-h][1,4,5,8]oxathiadiazacycloundecine 5,5-Dioxide (6o)
According to the reaction protocol described in general procedure C from 4a (53.6 mg), compound 6o (20%, 14.5 mg) was isolated after chromatography as a light yellow oil: Rf = 0.20 (2:1 EtOAc/hexane); [α]D20 = +132.4 (c = 0.69, CHCl3); FTIR (thin film) 3202, 2935, 1580, 1470, 1389, 1325, 1163, 1065, 812, 733 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.82 (d, J = 8.3 Hz, 1H, aromatic), 7.21 (dd, J = 8.3, 1.7 Hz, 1H, aromatic), 7.13 (d, J = 1.7 Hz, 1H, aromatic), 6.68 (s, 1H, NH), 4.56 (ddd, J = 11.9, 9.9, 4.4 Hz, 1H, OCHaHbCH2CHN), 4.43 (ddd, J = 11.9, 4.7, 4.6 Hz, 1H, OCHaHbCH2CHN), 3.20–2.99 (m, 2H, NHCHCHaHbN, NCHaHbCH2CH2CH2), 2.81 (ddd, J = 12.8, 8.9, 4.3 Hz, 1H, NHCHCH2N), 2.50 (dt, J = 14.1, 3.9 Hz, 1H, NCHaHbCH2CH2CH2), 2.35 (dt, J = 12.9, 4.4 Hz, 1H, NCHCH2CH2O), 2.15–2.05 (m, 2H, NHCHCHaHbN, NCHCHaHbCH2O), 2.01 (dt, J = 15.1, 5.0 Hz, 1H, NCHCHaHbCH2O), 1.94 (ddd, J = 13.6, 9.2, 4.1 Hz, 1H, NCH2CH2CHaHbCH2), 1.77–1.68 (m, 1H, NCH2CH2CH2CHaHb), 1.68–1.50 (m, 4H, NCH2CH2CH2CH2, CH3CHCH3, NHCHCHaHb), 1.38–1.28 (m, 2H, NCH2CH2CHaHbCH2, NHCHCHaHb), 1.22–1.13 (m, 1H, NCH2CH2CH2CHaHb), 0.87 (d, J = 6.6 Hz, 3H, CH3CHCH3), 0.79 (d, J = 6.6 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 154.5, 132.3, 128.1, 126.3, 123.7, 116.2, 65.5, 55.5, 50.5 (2C), 49.0, 43.6, 28.1, 24.7, 23.9, 23.5, 22.0, 21.3, 20.6; HRMS calcd for C19H29BrN2O3SH (M + H)+ 445.1161, found 445.1157 (TOF MS ES+).
(7S,13aS)-2-Bromo-7-isopropyl-6,7,8,10,11,12,13,13a,14,15-decahydrobenzo[b]pyrido[1,2-h][1,4,5,8]oxathiadiazacycloundecine 5,5-Dioxide (6p)
According to the reaction protocol described in general procedure C from 4b (78.1 mg), compound 6p (22%, 23.3 mg) was isolated after chromatography as a white solid: mp 152–157 °C; Rf = 0.21 (2:1 EtOAc/hexane); [α]D20 = +107.4 (c = 0.81, CHCl3); FTIR (thin film) 3205, 2934, 1580, 1470, 1387, 1327, 1163, 1065, 821, 733 cm–1; 1H NMR (400 MHz, CDCl3) δ ppm 7.81 (d, J = 8.3 Hz, 1H, aromatic), 7.20 (dd, J = 8.3, 1.7 Hz, 1H, aromatic), 7.13 (d, J = 1.7 Hz, 1H, aromatic), 6.62 (s, 1H, NH), 4.54 (ddd, J = 11.6, 9.4, 4.4 Hz, 1H, OCHaHbCH2CHN), 4.40 (ddd, J = 11.6, 5.0, 4.9 Hz, 1H, OCHaHbCH2CHN), 3.17–3.03 (m, 1H, NCHaHbCH2CH2CH2), 2.96 (dd, J = 12.9, 4.6 Hz, 1H, NHCHCHaHbN), 2.73 (ddd, J = 10.4, 4.6, 4.4 Hz, 1H, NHCHCH2N), 2.51 (d, J = 14.3 Hz, 1H, NCHaHbCH2CH2CH2), 2.47–2.32 (m, 2H, CH3CHCH3, NCHCH2CH2O), 2.29–2.08 (m, 2H, NHCHCHaHbN, NCHCHaHbCH2O), 1.95 (dddd, J = 14.6, 9.3, 5.1, 4.9 Hz, 1H, NCHCHaHbCH2O), 1.81–1.67 (m, 1H, NCH2CH2CH2CHaHb), 1.67–1.45 (m, 3H, NCH2CHaHbCH2CH2), 1.37–1.20 (m, 1H, NCH2CHaHbCH2CH2), 1.19–1.09 (m, 1H, NCH2CH2CH2CHaHb), 0.98 (d, J = 6.9 Hz, 3H, CH3CHCH3), 0.87 (d, J = 7.2 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 154.8, 132.4, 128.1, 126.3, 123.7, 116.3, 66.0, 55.1, 50.4, 50.2, 49.4, 29.1, 28.4, 23.6, 21.1, 20.5, 18.9, 15.6; HRMS calcd for C18H27BrN2O3SH (M + H)+ 431.1004, found 431.0976 (TOF MS ES+).
(3S,6S,7R)-10-Fluoro-3-isopropyl-5,6-dimethyl-7-phenyl-2,3,4,5,6,7-hexahydrobenzo[b][1,4,5,8]oxathiadiazecine 1,1-Dioxide (8a)
According to the reaction protocol described in general procedure C from 4d (52.0 mg), compound 8a (46%, 37.2 mg) was isolated after chromatography as a sticky colorless oil: Rf = 0.67 (1:1 EtOAc/hexane); [α]D20 = +43.1 (c = 1.145, CHCl3); FTIR (thin film) 3267, 2962, 1603, 1585, 1475, 1454, 1371, 1323, 1163, 1068, 812, 764, 737, 704 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 8.02 (dd, J = 8.8, 6.5 Hz, 1H, aromatic), 7.60–7.54 (m, 2H, aromatic), 7.48 (s, 1H, NH), 7.45–7.35 (m, 3H, aromatic), 6.94 (dd, J = 10.3, 2.4 Hz, 1H, aromatic), 6.87 (ddd, J = 8.7, 7.7, 2.3 Hz, 1H, aromatic), 5.25 (d, J = 2.5 Hz, 1H, OCHPh), 2.93 (qd, J = 7.1, 2.6 Hz, 1H, NCHCH3), 2.63 (ddd, J = 11.3, 4.6, 4.5 Hz, 1H, NHCHCH2N), 2.55 (dd, J = 12.9, 5.2 Hz, 1H, NHCHCHaHbN), 2.32 (m, 1H, CH3CHCH3), 2.11 (dd, J = 12.1, 12.0 Hz, 1H, NHCHCHaHbN), 1.51 (s, 3H, NCH3), 1.09 (d, J = 7.1 Hz, 3H, NCHCH3), 0.98 (d, J = 6.9 Hz, 3H, CH3CHCH3), 0.78 (d, J = 7.2 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 166.3 (d, 1JC–F = 254.6 Hz), 155.6 (d, 3JC–F = 10.7 Hz), 135.0, 134.0 (d, 3JC–F = 10.8 Hz), 128.4, 128.3 (2C), 127.1 (2C), 125.1 (d, 4JC–F = 3.4 Hz), 109.5 (d, 2JC–F = 22.0 Hz), 104.3 (d, 2JC–F = 25.2 Hz), 85.3, 56.7, 55.1, 52.9, 38.9, 28.6, 18.3, 15.5, 10.6; HRMS calcd for C21H27FN2O3SH (M + H)+ 407.1805, found 407.1790 (TOF MS ES+).
(3S,6R,7R)-10-Fluoro-3-isopropyl-5,6-dimethyl-7-phenyl-2,3,4,5,6,7-hexahydrobenzo[b][1,4,5,8]oxathiadiazecine 1,1-Dioxide (8b)
According to the reaction protocol described in general procedure B from 4d (53.0 mg), compound 8b (47%, 39.0 mg) was isolated after chromatography as a white solid: mp 87–93 °C; Rf = 0.72 (1:1 EtOAc/hexane); [α]D20 = +12.8 (c = 0.69, CHCl3); FTIR (thin film) 3300, 2962, 1603, 1583, 1479, 1456, 1369, 1325, 1157, 1068, 843, 770, 735, 700 cm–1; 1H NMR (400 MHz, CDCl3) δ ppm 7.97 (dd, J = 8.8, 6.6 Hz, 1H, aromatic), 7.56–7.45 (m, 2H, aromatic), 7.45–7.32 (m, 3H, aromatic), 6.76 (ddd, J = 8.7, 7.7, 2.4 Hz, 1H, aromatic), 6.66 (dd, J = 10.6, 2.4 Hz, 1H, aromatic), 6.47 (s, 1H, NH), 4.78 (d, J = 9.9 Hz, 1H, OCHPh), 3.10 (dq, J = 9.8, 6.5 Hz, 1H, NCHCH3), 2.82–2.59 (m, 2H, NHCHCH2N, NHCHCHaHbN), 2.40 (dd, J = 13.9, 7.2 Hz, 1H, NHCHCHaHbN), 2.15 (s, 3H, NCH3), 1.98–1.79 (m, 1H, CH3CHCH3), 0.97 (d, J = 6.9 Hz, 6H, CH3CHCH3), 0.86 (d, J = 6.6 Hz, 3H, NCHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 165.9 (d, 1JC–F = 253.7 Hz), 158.7 (d, 3JC–F = 10.8 Hz), 138.5, 133.0 (d, 3JC–F = 10.7 Hz), 129.0 (2C), 128.8, 127.1 (2C), 124.2 (d, 4JC–F = 3.2 Hz), 108.5 (d, 2JC–F = 22.0 Hz), 104.7 (d, 2JC–F = 25.3 Hz), 88.3, 67.8, 58.4, 52.5, 37.7, 32.1, 19.0, 17.7, 10.6; HRMS calcd for C21H27FN2O3SH (M + H)+ 407.1805, found 407.1765 (TOF MS ES+).
(3S,6S,7S)-10-Bromo-3-isopropyl-5,6-dimethyl-7-phenyl-2,3,4,5,6,7-hexahydrobenzo[b][1,4,5,8]oxathiadiazecine 1,1-Dioxide (8c)
According to the reaction protocol described in general procedure C from 4b (52.3 mg), compound 8c (13%, 9.7 mg) was isolated after chromatography as a colorless oil: Rf = 0.55 (1:1 EtOAc/hexane); [α]D20 = +73.2 (c = 0.335, CHCl3); FTIR (thin film) 3285, 2964, 1578, 1468, 1452, 1319, 1155, 1064, 804, 756, 727, 702 cm–1; 1H NMR (400 MHz, CDCl3) δ ppm 7.78 (d, J = 8.4 Hz, 1H, aromatic), 7.52–7.47 (m, 2H, aromatic), 7.46–7.40 (m, 2H, aromatic), 7.40–7.33 (m, 1H, aromatic), 7.07 (dd, J = 8.4, 1.8 Hz, 1H, aromatic), 6.95 (d, J = 1.8 Hz, 1H, aromatic), 4.71 (d, J = 9.3 Hz, 1H, OCHPh), 4.41 (s, 1H, NH), 4.01 (bs, 1H, NHCHCH2N), 3.19–3.08 (m, 1H, NCHCH3), 2.78 (dd, J = 13.3, 2.9 Hz, 1H, NHCHCHaHbN), 2.23 (m, 4H, NHCHCHaHbN, NCH3), 1.96–1.79 (m, 1H, CH3CHCH3), 1.09 (d, J = 6.8 Hz, 3H, CH3CHCH3), 1.00 (d, J = 6.9 Hz, 3H, CH3CHCH3), 0.75 (d, J = 7.0 Hz, 3H, NCHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 157.8, 138.5, 131.0, 130.8, 128.9 (2C), 128.6, 127.7, 126.7 (2C), 123.4, 118.8, 87.0, 60.4, 58.8, 58.0, 37.3, 32.3, 19.0, 18.0, 10.4; HRMS calcd for C21H27BrN2O3SH (M + H)+ 467.1004, found 467.1004 (TOF MS ES+).
(3S,6S,7S)-10-Fluoro-3-isobutyl-5,6-dimethyl-7-phenyl-2,3,4,5,6,7-hexahydrobenzo[b][1,4,5,8]oxathiadiazecine 1,1-Dioxide (8d)
According to the reaction protocol described in general procedure C from 4c (71.9 mg), compound 8d (30%, 33.2 mg) was isolated after chromatography as a white solid: mp 165–169 °C; Rf = 0.52 (1:1 EtOAc/hexane); [α]D20 = +20.0 (c = 0.145, CHCl3); FTIR (thin film) 3265, 2960, 1602, 1586, 1473, 1451, 1373, 1323, 1163, 1066, 815, 762, 734, 706 cm–1; 1H NMR (400 MHz, CDCl3) δ ppm 7.94 (dd, J = 8.8, 6.6 Hz, 1H, aromatic), 7.51–7.31 (m, 5H, aromatic), 6.65 (ddd, J = 8.5, 8.3, 2.4 Hz, 1H, aromatic), 6.52 (dd, J = 10.4, 2.4 Hz, 1H, aromatic), 4.68 (d, J = 9.3 Hz, 1H, OCHPh), 4.40 (d, J = 7.3 Hz, 1H, NH), 4.18 (bs, 1H, NHCHCH2N), 3.17 (dd, J = 8.8, 7.1 Hz, 1H, NCHCH3), 2.79 (dd, J = 13.6, 3.4 Hz, 1H, NHCHCHaHbN), 2.26 (s, 3H, NCH3), 2.22–2.14 (m, 1H, NHCHCHaHbN), 1.94 (dt, J = 13.3, 6.7 Hz, 1H, CH3CHCH3), 1.50 (dd, J = 14.0, 7.0 Hz, 1H, NHCHCHaHb), 1.32 (ddd, J = 13.9, 7.7, 6.0 Hz, 1H, NHCHCHaHb), 1.04 (d, J = 6.6 Hz, 3H, CH3CHCH3), 1.02 (d, J = 6.8 Hz, 3H, CH3CHCH3), 0.77 (d, J = 7.0 Hz, 3H, NCHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 165.9 (d, 1JC–F = 253.3 Hz), 158.7 (d, 3JC–F = 11.0 Hz), 138.6, 131.6 (d, 3JC–F = 10.9 Hz), 129.0 (2C), 128.6, 127.9 (d, 4JC–F = 3.5 Hz), 126.7 (2C), 107.3 (d, 2JC–F = 22.3 Hz), 103.2 (d, 2JC–F = 25.5 Hz), 86.9, 60.9, 60.7, 52.5, 45.4, 37.2, 24.7, 23.1, 22.8, 10.9; HRMS calcd for C22H29FN2O3SH (M + H)+ 421.1956, found 421.1956 (TOF MS ES+).
(3S,6S)-10-Fluoro-3,6-diisobutyl-5-methyl-2,3,4,5,6,7-hexahydrobenzo[b][1,4,5,8]oxathiadiazecine-1,1-Dioxide (8e)
According to the reaction protocol described in general procedure B from 4c (49.2 mg), compound 8e (39%, 26.6 mg) was isolated after chromatography as a white solid: mp 133–137 °C; Rf = 0.66 (1:1 EtOAc/hexane); [α]D20 = +106.8 (c = 0.825, CHCl3); FTIR (thin film) 2957, 1601, 1585, 1475, 1387, 1325, 1165, 1068, 849, 731 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 8.04–7.96 (m, 1H, aromatic), 7.22 (s, 1H, NH), 6.89–6.83 (m, 2H, aromatic), 4.22–4.08 (m, 2H, OCH2CHN), 2.86 (dd, J = 13.0, 4.8 Hz, 1H, NHCHCHaHbN), 2.74–2.63 (m, 1H, NHCHCH2N), 2.51 (tdd, J = 9.0, 5.3, 3.5 Hz, 1H, NCHCH2), 2.35 (s, 3H, NCH3), 2.13 (dd, J = 13.0, 11.0 Hz, 1H, NHCHCHaHbN), 1.87 (ddd, J = 13.7, 9.8, 3.7 Hz, 1H, NHCHCHaHb), 1.64–1.44 (m, 2H, NHCHCH2CH, NCHCH2CH), 1.39–1.20 (m, 2H, NHCHCHaHb, NCHCHaHb), 1.10 (ddd, J = 14.2, 8.7, 5.8 Hz, 1H, NCHCHaHb), 0.91 (d, J = 6.6 Hz, 3H, CH3CHCH3), 0.85 (d, J = 6.6 Hz, 3H, CH3CHCH3), 0.77 (d, J = 6.6 Hz, 3H, CH3CHCH3), 0.71 (d, J = 6.5 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 166.2 (d, 1JC–F = 254.8 Hz), 156.6 (d, 3JC–F = 10.5 Hz), 133.6 (d, 3JC–F = 10.6 Hz), 126.1 (d, 4JC–F = 3.4 Hz), 109.6 (d, 2JC–F = 22.1 Hz), 104.6 (d, 2JC–F = 25.0 Hz), 71.4, 57.7, 55.0, 49.4, 42.8, 36.1, 34.8, 25.3, 24.4, 23.7, 23.1, 22.0, 21.5; HRMS calcd for C19H31FN2O3SH (M + H)+ 387.2118, found 387.2093 (TOF MS ES+).
(S)-10-Fluoro-3-isobutyl-5-methyl-3,4,5,7-tetrahydro-2H-spiro[benzo[b][1,4,5,8]oxathiadiazecine-6,1′-cyclohexane] 1,1-Dioxide (8f)
According to the reaction protocol described in general procedure B from 4c (46.2 mg), compound 8f (46%, 30.9 mg) was isolated after chromatography as a sticky colorless oil: Rf = 0.58 (1:1 EtOAc/hexane); [α]D20 = +8.7 (c = 0.695, CHCl3); FTIR (thin film) 2953, 1605, 1589, 1468, 1425, 1391, 1317, 1159, 1070, 847, 733 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.97 (dd, J = 8.7, 6.5 Hz, 1H, aromatic), 6.84 (ddd, J = 8.8, 7.9, 2.4 Hz, 1H, aromatic), 6.79 (dd, J = 9.9, 2.4 Hz, 1H, aromatic), 4.69 (d, J = 10.2 Hz, 1H, OCHaHbC), 3.70 (d, J = 9.8 Hz, 1H, OCHaHbC), 2.99–2.84 (m, 2H, NHCH, NCHaHb), 2.43 (s, 3H, NCH3), 2.25–2.12 (m, 1H, NCHaHb), 1.94–1.66 (m, 8H, NHCHCH2CH, NHCHCHaHb, cyclohexyl), 1.64–1.47 (m, 1H, cyclohexyl), 1.43 (d, J = 12.8 Hz, 1H, cyclohexyl), 1.36–1.11 (m, 3H, NHCHCHaHb, cyclohexyl), 0.84 (dd, J = 6.6, 6.5 Hz, 6H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 165.9 (d, 1JC–F = 254.3 Hz), 157.7 (d, 3JC–F = 10.6 Hz), 132.5 (d, 3JC–F = 10.7 Hz), 124.8, 109.3 (d, 2JC–F = 22.0 Hz), 104.2 (d, 2JC–F = 24.9 Hz), 73.2, 59.8, 52.5, 49.3, 47.3, 36.8, 30.6, 28.1, 25.5, 24.3, 23.1, 22.8, 22.7, 22.4; HRMS calcd for C20H31FN2O3SH (M + H)+ 399.2118, found 399.2126 (TOF MS ES+).
(S)-3-Isopropyl-7-methyl-5-propyl-2,3,4,5-tetrahydrobenzo[f][1,2,5]thiadiazepine 1,1-Dioxide (10a)
According to the reaction protocol described in general procedure B from 4j (65.3 mg), compound 10a (44%, 33.1 mg) was isolated after chromatography as a yellowish oil: Rf = 0.40 (1:1 EtOAc/hexane); [α]D20= −140.3 (c = 0.125, CHCl3); FTIR (neat) 3267, 2927, 1595, 1461, 1325, 1161, 790, 732 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.77 (d, J = 8.1 Hz, 1H, aromatic), 6.84 (s, 1H), 6.81 (ddd, J = 8.0, 1.5, 0.7 Hz, 1H), 4.18 (d, J = 9.2 Hz, 1H, NHCHCH2N), 3.46 (dd, J = 14.8, 2.5 Hz, 1H, NHCHCH2N), 3.41–3.24 (m, 2H, NHCHCHaHbN, NCHaHbCH2CH3), 3.18 (ddd, J = 13.1, 7.1, 6.9 Hz, 1H, NCHaHbCH2CH3), 3.01 (dd, J = 14.9, 9.4 Hz, 1H, NHCHCHaHbN), 2.35 (s, 3H, PhCH3), 2.02–1.85 (m, 1H, CH3CHCH3), 1.66 (ddddd, J = 7.3, 7.3, 7.3, 7.3, 7.3 Hz, 2H, NCH2CH2CH3), 1.04 (dd, J = 6.8, 5.0 Hz, 6H, CH3CHCH3), 0.99 (t, J = 7.3 Hz, 3H, NCH2CH2CH3); 13C NMR (126 MHz, CDCl3) δ ppm 148.6, 143.5, 131.1, 128.4, 121.7, 119.8, 61.4, 56.9, 56.0, 30.3, 21.7, 21.5, 19.6, 19.0, 11.5; HRMS calcd for C15H24N2O2SH (M + H)+ 297.1637, found 297.1615 (TOF MS ES+).
(S)-7-Bromo-5-butyl-3-isobutyl-2,3,4,5-tetrahydrobenzo[f][1,2,5]thiadiazepine 1,1-Dioxide (10b)
According to the reaction protocol described in general procedure B from 4a (50.4 mg), compound 10b (50%, 29.0 mg) was isolated after chromatography as a colorless oil: Rf = 0.48 (1:4 EtOAc/hexane); [α]D20 = −90.2 (c = 3.3, CHCl3); FTIR (neat) 3258, 2957, 1578, 1468, 1369, 1319, 1151, 802, 733 cm–1; 1H NMR (400 MHz, CDCl3) δ ppm 7.68 (d, J = 8.5 Hz, 1H, aromatic), 7.12 (d, J = 1.8 Hz, 1H, aromatic), 7.07 (dd, J = 8.5, 1.7 Hz, 1H, aromatic), 4.33 (d, J = 8.1 Hz, 1H, NH), 3.68–3.55 (m, 1H, NHCHCH2N), 3.51 (dd, J = 15.2, 2.9 Hz, 1H, NHCHCHaHbN), 3.45–3.34 (m, 1H, NCHaHbCH2CH2CH3), 3.28–3.17 (m, 1H, NCHaHbCH2CH2CH3), 3.05 (dd, J = 15.2, 8.3 Hz, 1H, NHCHCHaHbN), 1.86 (ddq, J = 12.9, 8.3, 6.5 Hz, 1H, CH3CHCH3), 1.68–1.57 (m, 2H, NCH2CH2CH2CH3), 1.57–1.49 (m, 1H, NHCHCHaHbCH), 1.48–1.36 (m, 2H, NCH2CH2CH2CH3), 1.29 (ddd, J = 13.9, 8.5, 5.5 Hz, 1H, NHCHCHaHbCH), 1.01–0.94 (m, 9H); 13C NMR (126 MHz, CDCl3) δ ppm 149.3, 131.9, 129.7, 127.0, 123.1, 121.4, 58.5, 54.3, 53.9, 40.8, 29.8, 24.6, 23.0, 21.9, 20.1, 13.9; HRMS calcd for C16H25BrN2O2SH (M + 2+H)+ 391.0873, found 391.0872 (TOF MS ES+).
(S)-5-Benzyl-7-bromo-3-isobutyl-2,3,4,5-tetrahydrobenzo[f][1,2,5]thiadiazepine 1,1-Dioxide (10c)
According to the reaction protocol described in general procedure C from 4a (65.4 mg), compound 10c (50%, 41.1 mg) was isolated after chromatography as a white solid: mp 140–144 °C; Rf = 0.46 (1:3 EtOAc/hexane); [α]D20 = −75.5 (c = 0.14, CHCl3); FTIR (thin film) 3275, 2957, 1578, 1458, 1325, 1155, 800, 783, 698 cm–1; 1H NMR (400 MHz, CDCl3) δ ppm 7.76 (d, J = 8.5 Hz, 1H, aromatic), 7.42–7.29 (m, 5H, aromatic), 7.23 (d, J = 1.8 Hz, 1H, aromatic), 7.16 (dd, J = 8.4, 1.8 Hz, 1H, aromatic), 4.65 (d, J = 14.3 Hz, 1H, NCHaHbPh), 4.38 (d, J = 14.3 Hz, 1H, NCHaHbPh), 4.30–4.17 (m, 1H, NH), 3.51–3.28 (m, 2H, NHCHCH2N, NHCHCHaHbN), 3.01–2.79 (m, 1H, NHCHCHaHbN), 1.59–1.48 (m, 1H, CH3CHCH3), 1.35 (ddd, J = 14.2, 7.4, 7.0 Hz, 1H, NHCHCHaHb), 1.09–0.96 (m, 1H, NHCHCHaHb), 0.79 (d, J = 6.5 Hz, 3H, CH3CHCH3), 0.70 (d, J = 6.6 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 149.8, 136.7, 129.7, 128.9 (2C), 128.3 (2C), 127.9, 127.4, 127.35 124.2, 122.4, 58.3, 57.8, 53.7, 41.0, 24.5, 22.4, 22.0; HRMS calcd for C19H23BrN2O2SH (M + H)+ 423.0742, found 423.0742 (TOF MS ES+).
(S)-7-Bromo-3-isobutyl-5-(4-isopropylbenzyl)-2,3,4,5-tetrahydrobenzo[f][1,2,5]thiadiazepine 1,1-Dioxide (10d)
According to the reaction protocol described in general procedure C from 4a (58.0 mg), compound 10d (42%, 23.5 mg) was isolated after chromatography as a light yellow solid: mp 155–161 °C; Rf = 0.54 (1:3 EtOAc/hexane); [α]D20 = −102.9 (c = 0.485, CHCl3); FTIR (thin film) 3258, 2959, 1578, 1468, 1375, 1325, 1155, 843, 798, 700 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.74 (dd, J = 8.4, 2.0 Hz, 1H, aromatic), 7.32 (d, J = 7.9 Hz, 2H, aromatic), 7.26 (d, J = 5.0 Hz, 2H, aromatic), 7.27–7.21 (m, 1H, aromatic), 7.19–7.11 (m, 1H, aromatic), 4.60 (d, J = 13.9 Hz, 1H, NCHaHbPh), 4.32 (d, J = 13.9 Hz, 1H, NCHaHbPh), 4.23 (s, 1H, NH), 3.41 (dd, J = 14.9, 2.4 Hz, 1H, NHCHCH2N), 3.38–3.30 (m, 1H, NHCHCHaHbN), 3.03–2.80 (m, 1H, NHCHCHaHbN), 1.60–1.46 (m, 1H, CH3CHCH3), 1.40–1.26 (m, 1H CCHCH3), 1.25 (d, J = 6.9 Hz, 6H, CH3CHCH3), 1.06–0.93 (m, 1H, NHCHCHaHb), 0.95–0.83 (m, 1H, NHCHCHaHb), 0.76 (d, J = 6.6 Hz, 3H, CH3CHCH3), 0.64 (d, J = 6.6 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 150.0 (2C), 148.7, 134.0, 129.6, 128.5 (2C), 127.4, 126.9 (2C), 124.0, 122.3, 57.9, 57.5, 53.8, 41.0, 33.9, 24.6, 24.0, 23.9, 22.3, 22.1; HRMS calcd for C22H29BrN2O2SH (M + H)+ 465.1211, found 465.1184 (TOF MS ES+).
(S)-9-Fluoro-3-isopropyl-5-(4-(trifluoromethyl)benzyl)-2,3,4,5-tetrahydrobenzo[f][1,2,5]thiadiazepine 1,1-Dioxide (10e)
According to the reaction protocol described in general procedure B from 4k (70.5 mg), compound 10e (53%, 59.6 mg) was isolated after chromatography as a brown oil: Rf = 0.42 (1:1 EtOAc/hexane); [α]D20 = −149.9 (c = 0.125, CHCl3); FTIR (neat) 3267, 2968, 1604, 1573, 1477, 1433, 1325, 1161, 854, 742 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.93 (dd, J = 8.8, 6.4 Hz, 1H, aromatic), 7.65 (d, J = 8.0 Hz, 2H, aromatic), 7.55 (d, J = 8.0 Hz, 2H, aromatic), 6.78–6.69 (m, 2H, aromatic), 4.68 (d, J = 14.7 Hz, 1H, NCHaHbPh), 4.44 (d, J = 14.7 Hz, 1H, NCHaHbPh), 4.32 (d, J = 9.1 Hz, 1H, NH), 3.43 (dd, J = 14.8, 2.2 Hz, 1H, NHCHCH2N), 3.29–3.14 (m, 1H, NHCHCHaHbN), 3.13–2.96 (m, 1H, NHCHCHaHbN), 1.85–1.66 (m, 1H, CH3CHCH3), 0.86 (d, J = 6.8 Hz, 3H, CH3CHCH3), 0.79 (d, J = 6.7 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 165.3 (d, 1JC–F = 253.5 Hz), 150.6 (d, 3JC–F = 10.1 Hz), 140.9 (d, 4JC–F = 0.9 Hz), 130.8 (d, 3JC–F = 10.9 Hz), 130.2 (q, 2JC–CF3 = 32.6 Hz), 130.1, 128.5 (2C), 125.8 (q, 3JC–CF3 = 3.7 Hz, 2C), 124.0 (q, 1JC–CF3 = 273.1 Hz), 108.7 (d, 2JC–F = 22.6 Hz), 106.5 (d, 2JC–F = 24.4 Hz), 60.7, 58.0, 56.4, 30.0, 19.0, 18.7; HRMS calcd for C19H20F4N2O2SH (M + H)+ 417.1260, found 417.1271 (TOF MS ES+).
(S)-3-Isopropyl-7-methyl-5-(oxetan-3-yl)-2,3,4,5-tetrahydrobenzo[f][1,2,5]thiadiazepine 1,1-Dioxide (10f)
According to the reaction protocol described in general procedure B from 4j (69.5 mg), compound 10f (55%, 46.1 mg) was isolated after chromatography as a white solid: mp 145–148 °C; Rf = 0.38 (1:1 EtOAc/hexane); [α]D20 = −27.0 (c = 0.125, CHCl3); FTIR (neat) 3267, 2960, 1602, 1471, 1369, 1326, 1218, 1145, 1068, 815, 729 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.79 (d, J = 8.0 Hz, 1H, aromatic), 7.27–7.24 (m, 1H, aromatic), 7.13 (bs, 1H, aromatic), 3.80 (ddd, J = 14.6, 7.7, 1.6 Hz, 1H, NCHCHaHbOCH2), 3.70–3.62 (m, 1H, NCHCH2OCHaHb), 3.59 (dd, J = 10.9, 9.2 Hz, 1H, NCHCHaHbOCH2), 3.54–3.42 (m, 1H, NCHCH2OCHaHb), 3.31 (dddd, J = 9.7, 9.7, 7.7, 1.6 Hz, 1H, NHCHCH2N), 3.20–3.12 (m, 1H, NCHCH2OCH2), 2.94 (dd, J = 14.5, 9.6 Hz, 1H, NHCHCHaHbN), 2.70 (dd, J = 14.5, 10.0 Hz, 1H, NHCHCHaHbN), 2.56 (s, 1H, NH), 2.40 (s, 3H, PhCH3), 1.92–1.80 (m, 1H CH3CHCH3), 1.09 (d, J = 6.5 Hz, 3H, CH3CHCH3), 0.84 (d, J = 6.6 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 144.5, 141.1, 140.1, 130.4, 129.1, 128.7, 61.9, 60.1, 58.7, 47.6, 41.1, 29.3, 21.3, 20.6, 18.5; HRMS calcd for C15H22N2O3SH (M + H)+ 311.1429, found 311.1415 (TOF MS ES+).
(S)-9-Fluoro-3-isopropyl-5-(3-methoxypropyl)-2,3,4,5-tetrahydrobenzo[f][1,2,5]thiadiazepine 1,1-Dioxide (10g)
According to the reaction protocol described in general procedure B from 4k (70.6 mg), compound 10g (56%, 50.0 mg) was isolated after chromatography as a yellow oil: Rf = 0.37 (1:1 EtOAc/hexane); [α]D20 = −140.7 (c = 0.125, CHCl3); FTIR (neat) 3263, 2962, 1608, 1569, 1456, 1386, 1319, 1201, 1149, 1068, 723 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.85 (dd, J = 8.8, 6.5 Hz, 1H, aromatic), 6.74 (dd, J = 11.5, 2.4 Hz, 1H, aromatic), 6.66 (ddd, J = 8.8, 7.5, 2.4 Hz, 1H, aromatic), 4.54 (d, J = 7.2 Hz, 1H, NH), 3.58–3.42 (m, 4H, NHCHCHaHbN, NHCHCH2N, NCH2CH2CH2OCH3), 3.34 (s, 3H, NCH2CH2CH2OCH3), 3.33–3.30 (m, 1H, NCHaHbCH2CH2OCH3), 3.28–3.19 (m, 2H, NHCHCHaHbN, NCHaHbCH2CH2OCH3), 2.01–1.93 (m, 1H, CH3CHCH3), 1.92–1.83 (m, 2H, NCH2CH2CH2OCH3), 1.06 (d, J = 6.8 Hz, 3H, CH3CHCH3), 1.03 (d, J = 6.7 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 165.2 (d, 1JC–F = 252.1 Hz), 150.3 (d, 3JC–F = 10.5 Hz), 130.8 (d, 3JC–F = 10.9 Hz), 129.2, 107.7 (d, 2JC–F = 22.7 Hz), 105.6 (d, 2JC–F = 24.7 Hz), 69.7, 61.6, 58.7, 56.8, 51.0, 30.2, 28.0, 19.6, 19.0; HRMS calcd for C15H23FN2O3SH (M + H)+ 331.1492, found 331.1481 (TOF MS ES+).
(S)-3-((S)-sec-Butyl)-8-chloro-5-(3-methoxypropyl)-2,3,4,5-tetrahydrobenzo[f][1,2,5]thiadiazepine 1,1-Dioxide (10h)
According to the reaction protocol described in general procedure B from 4f (78.7 mg), compound 10h (46%, 44.8 mg) was isolated after chromatography as a colorless oil: Rf = 0.42 (1:1 EtOAc/hexane); [α]D20 = +132.5 (c = 0.125, CHCl3); FTIR (neat) 3267, 2931, 1506, 1488, 1458, 1386, 1326, 1220, 1157, 1058, 821 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.83 (d, J = 2.5 Hz, 1H, aromatic), 7.34 (dd, J = 8.8, 2.6 Hz, 1H, aromatic), 7.05 (d, J = 8.8 Hz, 1H, aromatic), 4.49 (d, J = 9.4 Hz, 1H, NH), 3.60–3.48 (m, 2H, NHCHCH2N, NCH2CH2CHaHbOCH3), 3.48–3.42 (m, 2H, NCH2CH2CHaHbOCH3, NCHaHbCH2CH2OCH3), 3.38 (dd, J = 14.8, 2.5 Hz, 1H, NHCHCHaHbN), 3.33 (s, 3H, NCH2CH2CH2OCH3), 3.29 (dd, J = 13.5, 6.8 Hz, 1H, NCHaHbCH2CH2OCH3), 3.04 (dd, J = 14.8, 9.6 Hz, 1H, NHCHCHaHbN), 1.90–1.78 (m, 2H, NCH2CH2CH2OCH3), 1.74–1.64 (m, 1H, CH3CHCH2CH3), 1.59–1.49 (m, 1H, CH3CHCHaHbCH3), 1.36–1.25 (m, 1H, CH3CHCHaHbCH3), 1.01–0.93 (m, 6H, CH3CHCH2CH3); 13C NMR (126 MHz, CDCl3) δ ppm 146.7, 134.8, 132.6, 128.1, 125.9, 120.7, 77.2, 69.8, 61.5, 58.7, 57.1, 51.2, 30.2, 28.2, 19.6, 18.9; HRMS calcd for C16H25ClN2O3SH (M + H)+ 361.1353, found 361.1338 (TOF MS ES+).
9-Fluoro-5-(3-methoxypropyl)-4,5-dihydro-2H-spiro[benzo[f][1,2,5]thiadiazepine-3,1′-cyclohexane] 1,1-Dioxide (10i)
According to the reaction protocol described in general procedure B from 4g (77.7 mg), compound 10i (56%, 54.0 mg) was isolated after chromatography as a brownish oil: Rf = 0.38 (1:1 EtOAc/hexane); FTIR (neat) 3326, 2928, 1612, 1573, 1469, 1338, 1147, 1041, 773 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.27–7.22 (m, 1H, aromatic), 6.97 (s, 1H, NH), 6.48 (d, J = 8.8, Hz, 1H, aromatic), 6.35 (ddd, J = 11.2, 8.1, 1.0 Hz, 1H, aromatic), 5.61–5.54 (m, 1H, NCH2CH2CHaHbOCH3,), 4.95 (dd, J = 6.6, 6.5 Hz, 1H, NCH2CH2CHaHbOCH3), 3.54–3.44 (m, 4H, NHCCH2N, NCH2CH2CH2OCH3), 3.36 (s, 3H, OCH3), 3.25 (ddd, J = 6.7, 6.6, 5.0 Hz, 2H, NCH2CH2CH2OCH3), 2.00–1.84 (m, 5H, cyclohexyl), 1.65–1.55 (m, 1H, cyclohexyl), 1.54–1.39 (m, 4H, cyclohexyl); 13C NMR (126 MHz, CDCl3) δ ppm 161.1 (d, 1JC–F = 248.9 Hz), 148.6 (d, 4JC–F = 3.4 Hz), 134.2 (d, 3JC–F = 12.7 Hz), 109.4 (d, 3JC–F = 14.0 Hz), 107.8, 101.1 (d, 2JC–F = 23.5 Hz), 70.2, 58.8, 50.0, 40.8, 29.0, 26.2 (2C), 25.0, 22.3, 21.9 (2C); HRMS calcd for C17H25FN2O3SH (M + H)+ 357.1648, found 357.1627 (TOF MS ES+).
7-Bromo-5-(2-hydroxyethyl)-4,5-dihydro-2H-spiro[benzo[f][1,2,5]thiadiazepine-3,1′-cyclohexane] 1,1-Dioxide (10j)
According to the reaction protocol described in general procedure C from 4l (47.6 mg), compound 10j (46%, 24.8 mg) was isolated after chromatography as a white solid: mp 178–182 °C; Rf = 0.44 (1:1 EtOAc/hexane); FTIR (thin film) 3454, 3263, 2934, 1580, 1487, 1369, 1312, 1150, 1057, 795, 733, 694 cm–1; 1H NMR (400 MHz, CDCl3) δ ppm 7.68 (dd, J = 8.4, 1.7 Hz, 1H, aromatic), 7.25 (s, 1H, aromatic), 7.19 (dd, J = 8.4, 1.7 Hz, 1H, aromatic), 4.44 (s, 1H, NH), 3.81–3.66 (m, 2H, NCH2CH2OH), 3.57 (bs, 2H, NCH2CH2OH), 3.26 (bs, 2H, NCH2CNH), 2.85 (s, 1H, OH), 1.75–1.53 (m, 6H, cyclohexyl), 1.50–1.27 (m, 4H, cyclohexyl); 13C NMR (126 MHz, CDCl3) δ ppm 148.1, 129.5, 127.1, 125.8, 125.3, 122.6, 65.3, 59.5, 59.1, 56.5, 25.6 (2C), 21.0 (3C); HRMS calcd for C15H21BrN2O3SH (M – H)+ 387.0383, found 387.0372 (TOF MS ES–).
(S)-7-Bromo-5-(2-hydroxyethyl)-3-isopropyl-2,3,4,5-tetrahydrobenzo[f][1,2,5]thiadiazepine 1,1-Dioxide (10k)
According to the reaction protocol described in general procedure C from 4b (97.6 mg), compound 10k (46%, 50.4 mg) was isolated after chromatography as a colorless oil: Rf = 0.35 (1:1 EtOAc/hexane); [α]D20 = −175.5 (c = 0.125, CHCl3); FTIR (thin film) 3466, 3252, 2964, 1578, 1470, 1371, 1319, 1157, 1059, 795, 731, 698 cm–1; 1H NMR (400 MHz, CDCl3) δ ppm 7.74 (dd, J = 8.6, 2.1 Hz, 1H, aromatic), 7.37 (d, J = 2.0 Hz, 1H, aromatic), 7.29–7.22 (m, 1H, aromatic), 4.17 (d, J = 9.4 Hz, 1H, NH), 3.88–3.76 (m, 1H, HOCHaHb), 3.68–3.57 (m, 2H, NCH2CH2), 3.51–3.36 (m, 3H, NCHaHBCHNH, NHCHCH2, OH), 3.33–3.22 (m, 1H, HOCHaHb), 2.85 (dd, J = 15.0, 10.4 Hz, 1H, NCHaHBCHNH), 1.92–1.79 (m, 1H, CH3CHCH3), 1.06 (d, J = 6.8 Hz, 3H, CH3CHCH3), 1.02 (d, J = 6.9 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 148.9, 135.6, 129.6, 127.7, 126.1, 125.5, 60.9, 60.0, 58.9, 57.9, 30.0, 19.5, 18.3; HRMS calcd for C13H19BrN2O3SH (M + H)+ 363.0378, found 363.0375 (TOF MS ES+).
(S)-7-Bromo-5-(2-hydroxyethyl)-3-isobutyl-2,3,4,5-tetrahydrobenzo[f][1,2,5]thiadiazepine 1,1-Dioxide (10l)
According to the reaction protocol described in general procedure C from 4a (63.5 mg), compound 10l (43%, 31.0 mg) was isolated after chromatography as a colorless oil: Rf = 0.45 (1:1 EtOAc/hexane); [α]D20 = −120.8 (c = 0.085, CHCl3); FTIR (thin film) 3454, 3250, 2957, 1576, 1470, 1367, 1319, 1155, 1061, 808, 789, 745, 700 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.69 (d, J = 8.4 Hz, 1H, aromatic), 7.36 (d, J = 1.8 Hz, 1H, aromatic), 7.23 (dd, J = 8.5, 1.8 Hz, 1H, aromatic), 4.27 (d, J = 9.0 Hz, 1H, NH), 3.82 (ddd, J = 13.6, 5.6, 3.9 Hz, 1H, HOCHaHb), 3.73–3.57 (m, 3H, NHCHCH2, NCH2CH2OH), 3.42 (s, 1H, OH), 3.36 (dd, J = 15.0, 2.1 Hz, 1H, NHCHCHaHbN), 3.32–3.21 (m, 1H, HOCHaHb), 2.85–2.71 (m, 1H, NHCHCHaHbN), 1.93–1.79 (m, 1H, CH3CHCH3), 1.36 (ddd, J = 14.3, 9.1, 5.7 Hz, 1H, NHCHCHaHb), 1.30–1.18 (m, 1H, NHCHCHaHb), 0.98 (dd, J = 6.5, 6.4 Hz, 6H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 148.9, 135.6, 129.6, 127.7, 126.1, 125.5, 61.3, 60.1, 58.0, 54.1, 40.7, 24.6, 23.0, 21.9; HRMS calcd for C14H21BrN2O3SH (M + H)+ 377.0535, found 377.0515 (TOF MS ES+).
(S)-7-Bromo-5-((R)-1-hydroxypropan-2-yl)-3-isobutyl-2,3,4,5-tetrahydrobenzo[f][1,2,5]thiadiazepine 1,1-Dioxide (10m)
According to the reaction protocol described in general procedure C from 4a (146.3 mg), compound 10m (12%, 20.2 mg) was isolated after chromatography as a white solid: mp 150–154 °C; Rf = 0.55 (1:1 EtOAc/hexane); [α]D20 = −143.5 (c = 0.365, CHCl3); FTIR (thin film) 3475, 3253, 2957, 1576, 1470, 1381, 1321, 1161, 1055, 808, 777, 727, 694 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.76 (d, J = 8.4 Hz, 1H, aromatic), 7.35 (d, J = 1.8 Hz, 1H, aromatic), 7.27–7.24 (m, 1H, aromatic), 3.98 (s, 1H, NH), 3.88–3.74 (m, 1H, NCHCH3), 3.62 (s, 1H, OH), 3.58–3.47 (m, 3H, NHCHCH2N, NHCHCHaHbN, HOCHaHb), 3.40 (dd, J = 11.0, 10.4 Hz, 1H, HOCHaHb), 2.47–2.26 (m, 1H, NHCHCHaHbN), 1.95–1.78 (m, 1H, CH3CHCH3), 1.40–1.19 (m, 5H, CH3CHN, NHCHCH2), 1.00 (dd, J = 6.3 Hz, 6H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 150.7, 134.6, 129.5, 127.8, 125.7, 125.5, 65.6, 60.9, 54.0, 51.7, 41.1, 24.7, 23.0, 21.9, 13.9; HRMS calcd for C15H23BrN2O3SH (M + H)+ 391.0691, found 391.0656 (TOF MS ES+).
(S)-7-Bromo-5-(1-(hydroxymethyl)cyclohexyl)-3-isopropyl-2,3,4,5-tetrahydrobenzo[f][1,2,5]thiadiazepine 1,1-Dioxide (10n)
According to the reaction protocol described in general procedure C from 4b (96.4 mg), compound 10n (26%, 34.0 mg) was isolated after chromatography as a white solid: mp 164–168 °C; Rf = 0.66 (1:1 EtOAc/hexane); [α]D20 = −187.1 (c = 1.29, CHCl3); FTIR (thin film) 3445, 3261, 2959, 1574, 1462, 1398, 1321, 1163, 1063, 808, 733 cm–1; 1H NMR (400 MHz, CDCl3) δ ppm 7.76 (d, J = 8.5 Hz, 1H, aromatic), 7.68 (d, J = 1.8 Hz, 1H, aromatic), 7.35 (dd, J = 8.4, 1.8 Hz, 1H, aromatic), 4.06 (d, J = 9.2 Hz, 1H, NH), 3.92–3.77 (m, 2H, NCHaHbCHNH, CHaHbOH), 3.68 (dd, J = 12.6, 4.8 Hz, 1H, CHaHbOH), 3.45 (ddd, J = 8.9, 8.8, 8.6 Hz, 1H, NHCHCH2N), 3.36–3.21 (m, 1H, OH), 2.35 (dd, J = 15.6, 10.4 Hz, 1H, NCHaHbCHNH), 2.09 (d, J = 12.9 Hz, 1H, cyclohexyl), 1.88–1.68 (m, 6H, cyclohexyl, CH3CHCH3), 1.57 (ddd, J = 13.1, 12.8, 3.7 Hz, 1H, cyclohexyl), 1.48–1.34 (m, 2H, cyclohexyl), 1.31–1.17 (m, 1H, cyclohexyl), 1.06 (d, J = 6.8 Hz, 3H, CH3CHCH3), 1.00 (d, J = 6.9 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 147.9, 139.0, 131.8, 129.3, 127.6, 126.2, 63.6, 61.5, 61.2, 50.3, 31.6, 31.2, 30.1, 25.5, 23.0, 22.8, 19.3, 18.2; HRMS calcd for C18H27BrN2O3SH (M + H)+ 431.1004, found 431.1019(TOF MS ES+).
(S)-7-Bromo-5-((S)-1-hydroxy-4-methylpentan-2-yl)-3-isobutyl-2,3,4,5-tetrahydrobenzo[f]-[1,2,5]thiadiazepine 1,1-Dioxide (10o)
According to the reaction protocol described in general procedure C from 4a (258.0 mg), compound 10o (21%, 70.0 mg) was isolated after chromatography as a white solid: mp 86–90 °C; Rf = 0.47 (1:1 EtOAc/hexane); [α]D20 = +117.3 (c = 0.92, CHCl3); FTIR (thin film) 3470, 3250, 2955, 1578, 1470, 1402, 1323, 1163, 1068, 876, 812 cm–1; 1H NMR (400 MHz, CDCl3) δ ppm 7.83 (d, J = 8.1 Hz, 1H, aromatic), 7.25–7.14 (m, 2H, aromatic), 6.70 (d, J = 6.4 Hz, 1H, NH), 4.41 (dd, J = 11.7, 2.9 Hz, 1H, NCHCHaHbOH), 3.87 (dd, J = 11.8, 11.0 Hz, 1H, NCHCHaHbOH), 3.16–2.99 (m, 1H, NHCHCH2N), 2.77–2.57 (m, 2H, NCHCH2OH, NCHaHbCHNH), 2.40 (dd, J = 13.6, 4.8 Hz, 1H, NCHaHbCHNH), 1.80–1.68 (m, 2H, CH3CHCH3, CH3CHCH3), 1.63 (ddd, J = 14.1, 7.2, 7.1 Hz, 1H, NHCHCHaHb), 1.24 (dd, J = 7.8, 6.3 Hz, 2H, NCHCH2), 1.13 (ddd, J = 13.7, 7.0, 6.9 Hz, 1H, NHCHCHaHb), 0.98–0.88 (m, 9H, CH3CHCH3, CH3CHCH3), 0.84 (d, J = 6.6 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 155.2, 131.8, 129.7, 128.0, 124.9, 118.5, 75.2, 54.0, 51.4, 51.3, 43.4, 42.0, 24.9, 24.3, 23.0, 22.7, 22.5, 22.4; HRMS calcd for C18H29BrN2O3SH (M + H)+ 435.1136, found 435.1147 (TOF MS ES+).
(S)-7-Bromo-5-((R)-4-hydroxy-3-methylbutyl)-3-isobutyl-2,3,4,5-tetrahydrobenzo[f][1,2,5]thiadiazepine 1,1-Dioxide (10p)
According to the reaction protocol described in general procedure C from 4a (81.1 mg), compound 10p (43%, 43.1 mg) was isolated after chromatography as a colorless oil: Rf = 0.45 (1:1 EtOAc/hexane); [α]D20 = −103.1 (c = 0.66, CHCl3); FTIR (thin film) 3512, 3252, 2957, 1578, 1543, 1470, 1371, 1315, 1150, 1040, 800, 731, 700 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.67 (d, J = 8.5 Hz, 1H, aromatic), 7.20 (d, J = 1.8 Hz, 1H, aromatic), 7.10 (dd, J = 8.5, 1.8 Hz, 1H, aromatic), 4.41 (d, J = 8.2 Hz, 1H, NH), 3.68–3.56 (m, 2H, NHCHCH2N, NCHaHbCH2CHMe), 3.55–3.49 (m, 1H, HOCHaHbCHMe), 3.46 (dd, J = 15.1, 2.7 Hz, 1H, NHCHCHaHbN), 3.43–3.39 (m, 1H, HOCHaHbCHMe), 3.21 (ddd, J = 13.7, 8.3, 5.9 Hz, 1H, NCHaHbCH2CHMe), 2.95 (dd, J = 15.1, 9.0 Hz, 1H, NHCHCHaHbN), 1.91–1.79 (m, 2H, HOCH2CHMe, CH3CHCH3), 1.79–1.70 (m, 2H, NCH2CHaHbCHMe, OH), 1.53–1.43 (m, 2H, NHCHCHaHbCH, NCH2CHaHbCHMe), 1.27 (ddd, J = 14.0, 8.4, 5.6 Hz, 1H, NHCHCHaHbCH), 1.03–0.93 (m, 9H, CH3CHCH3, HOCH2CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 149.3, 132.4, 129.7, 127.3, 123.8, 122.1, 67.7, 59.2, 54.0, 52.3, 40.9, 33.4, 31.4, 24.6, 22.9, 21.9, 17.0; HRMS calcd for C17H27BrN2O3SH (M + H)+ 421.0979, found 421.0980 (TOF MS ES+).
(3S)-7-Bromo-3-isobutyl-3,4-dihydro-2,5-ethanobenzo[f][1,2,5]thiadiazepine 1,1-Dioxide (11a)
According to the reaction protocol described in general procedure D from 10l (17.6 mg), compound 11a (87%, 14.6 mg) was isolated after chromatography as a colorless oil: Rf = 0.53 (1:2 EtOAc/hexane); [α]D20 = −29.1 (c = 0.945, CHCl3); FTIR (thin film) 2957, 1574, 1448, 1391, 1333, 1165, 839, 797, 694 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.74 (d, J = 8.4 Hz, 1H, aromatic), 7.56 (dd, J = 8.4, 2.0 Hz, 1H, aromatic), 7.48 (d, J = 2.0 Hz, 1H, aromatic), 3.97–3.82 (m, 1H, NCHCH2N), 3.72 (dddd, J = 14.7, 8.6, 2.4, 1.7 Hz, 1H, SNCHaHb), 3.42 (ddd, J = 14.1, 7.7, 2.5 Hz, 1H, CNCHaHbCH2NS), 3.39–3.32 (m, 1H, CNCHaHbCH2NS), 3.26–3.21 (m, 1H, NCHaHbCHN), 3.21–3.16 (m, 1H, SNCHaHb), 2.73 (dd, J = 14.4, 9.1 Hz, 1H, NCHaHbCHN), 1.89–1.75 (m, 1H, CH3CHCH3), 1.68 (ddd, J = 14.3, 9.8, 4.7 Hz, 1H, NCHCHaHbCH), 1.14 (ddd, J = 13.8, 9.0, 4.6 Hz, 1H, NCHCHaHbCH), 0.95 (d, J = 6.8 Hz, 3H, CH3CHCH3), 0.93 (d, J = 6.5 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 148.1, 143.2, 133.9, 131.3, 129.8, 126.8, 53.3, 51.1, 51.07, 39.1, 38.3, 24.6, 23.2, 21.6; HRMS calcd for C14H19BrN2O2SH (M + H)+ 359.0429, found 359.0430 (TOF MS ES+).
(4R,11S)-7-Bromo-11-isobutyl-4-methyl-3,4-dihydro-2,5-ethanobenzo[f][1,2,5]thiadiazepine 1,1-dioxide (11b)
According to the reaction protocol described in general procedure D from 10m (28.5 mg), compound 11b (46%, 12.5 mg) was isolated after chromatography as a colorless oil: Rf = 0.66 (1:2 EtOAc/hexane); [α]D20 = −2.1 (c = 0.25, CHCl3); FTIR (thin film) 2957, 1574, 1456, 1381, 1331, 1161, 816, 789, 756, 698 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.72 (d, J = 8.4 Hz, 1H, aromatic), 7.59 (dd, J = 8.4, 2.0 Hz, 1H, aromatic), 7.45 (d, J = 1.9 Hz, 1H, aromatic), 4.09–3.90 (m, 1H, NCHCH2N), 3.51–3.29 (m, 4H, SNCH2CHCH3, NCHCHaHbN), 2.81 (dd, J = 14.3, 6.6 Hz, 1H, NCHCHaHbN), 1.84–1.77 (m, 1H, CH3CHCH3), 1.73 (ddd, J = 13.9, 10.2, 4.7 Hz, 1H, NCHCHaHbCH), 1.21 (ddd, J = 14.1, 9.0, 5.0 Hz, 1H, NCHCHaHbCH), 0.96 (dd, J = 6.5, 3.8 Hz, 9H, NCHCH3, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 144.3, 143.6, 136.0, 131.6, 129.1, 126.6, 56.2, 54.5, 49.2, 44.8, 40.3, 25.1, 23.2, 21.5, 20.2; HRMS calcd for C15H21BrN2O2SH (M + H)+ 373.0585, found 373. 0565 (TOF MS ES+).
7-Bromo-4H-spiro[2,5-ethanobenzo[f][1,2,5]thiadiazepine-3,1′-cyclohexane] 1,1-Dioxide (11c)
According to the reaction protocol described in general procedure D from 10j (21.2 mg), compound 11c (40%, 8.1 mg) was isolated after chromatography as a white solid: Rf = 0.51 (1:2 EtOAc/hexane); mp 182–185 °C; FTIR (thin film) 2935, 1574, 1452, 1393, 1327, 1167, 804, 694 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.67 (d, J = 8.4 Hz, 1H, aromatic), 7.53 (dd, J = 8.4, 2.0 Hz, 1H, aromatic), 7.47 (d, J = 2.0 Hz, 1H, aromatic), 3.88 (ddd, J = 14.8, 6.9, 5.7 Hz, 1H, SNCHaHb), 3.44 (ddd, J = 14.7, 8.5, 7.1 Hz, 1H, SNCHaHb), 3.38–3.30 (m, 2H, CNCH2CH2NS), 3.15 (ddd, J = 14.3, 1.3, 1.2 Hz, 1H, NCHaHbCNS), 2.94 (dd, J = 14.5, 0.9 Hz, 1H, NCHaHbCNS), 2.20–2.09 (m, 1H, cyclohexyl), 1.89–1.77 (m, 3H, cyclohexyl), 1.75–1.65 (m, 1H, cyclohexyl), 1.49–1.35 (m, 3H, cyclohexyl), 1.34–1.21 (m, 2H, cyclohexyl); 13C NMR (126 MHz, CDCl3) δ ppm 148.9, 144.8, 133.7, 131.2, 128.7, 126.8, 60.5, 58.8, 49.8, 41.3, 37.1, 36.9, 25.1, 22.8, 22.4; HRMS calcd for C15H19BrN2O2SH (M + 2 + H)+ 373.0403, found 373. 0399 (TOF MS ES+).
(3S,7R)-7-((Benzyloxy)methyl)-10-fluoro-5-((R)-1-hydroxypropan-2-yl)-3-isobutyl-2,3,4,5,6,7-hexahydrobenzo[b][1,4,5,8]oxathiadiazecine 1,1-Dioxide (18)
According to the reaction protocol described in general procedure B from 4c (44.2 mg), compound 18 (42%, 33.4 mg) was isolated after chromatography as a white solid: mp 50–55 °C; Rf = 0.50 (1:1 EtOAc/hexane); [α]D20 = +22.5 (c = 1.315, CHCl3); FTIR (thin film) 3514, 2953, 1601, 1587, 1477, 1454, 1389, 1321, 1155, 1070, 808, 741, 700 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.98 (dd, J = 8.8, 6.5 Hz, 1H, aromatic), 7.41–7.25 (m, 5H, aromatic), 6.79 (ddd, J = 8.7, 7.8, 2.4 Hz, 1H, aromatic), 6.72 (dd, J = 9.9, 2.4 Hz, 1H, aromatic), 5.75 (s, 1H, NH), 4.46 (d, J = 11.8 Hz, 1H, OCHaHbC), 4.40 (d, J = 11.9 Hz, 1H, OCHaHbC), 4.37 (dd, J = 12.3, 5.1 Hz, 1H, OCHCHaHbO), 3.90 (dd, J = 11.9 Hz, 1H, OCHCHaHbO), 3.64 (bs, 1H, NHCHCH2N), 3.37–3.23 (m, 2H, OCHCH2O, NCHCH3), 3.14 (dd, J = 9.4, 4.1 Hz, 1H, HOCHaHbCH), 3.09–2.99 (m, 2H, NHCHCHaHbN, HOCHaHbCH), 2.53 (d, J = 12.7 Hz, 1H, NCHaHbCHO), 2.36–2.28 (m, 2H, NCHaHbCHO, OH), 2.14 (dd, J = 15.1, 10.8 Hz, 1H, NHCHCHaHbN), 1.95–1.81 (m, 1H, CH3CHCH3), 1.42 (ddd, J = 13.7, 7.7, 6.0 Hz, 1H, NHCHCHaHb), 1.14 (ddd, J = 13.6, 7.9, 5.6 Hz, 1H, NHCHCHaHb), 0.98 (d, J = 6.5 Hz, 3H, NCHCH3), 0.93 (d, J = 6.5 Hz, 3H, CH3CHCH3), 0.89 (d, J = 6.6 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 165.7 (d, 1JC–F = 254.0 Hz), 157.2, 137.7, 132.2 (d, 3JC–F = 10.8 Hz), 128.5 (2C), 127.9 (2C), 127.7 (2C), 108.6 (d, 2JC–F = 22.2 Hz), 104.1 (d, 2JC–F = 24.5 Hz), 73.4, 72.2, 71.9, 69.4 (2C), 57.4, 56.3, 53.7, 46.1, 24.2, 23.0, 22.5, 9.5; HRMS calcd for C25H35FN2O5SH (M + H)+ 495.2329, found 495.2348 (TOF MS ES+).
(3S,7S)-7-((Benzyloxy)methyl)-10-fluoro-5-((R)-1-hydroxypropan-2-yl)-3-isobutyl-2,3,4,5,6,7-hexahydrobenzo[b][1,4,5,8]oxathiadiazecine 1,1-Dioxide (19)
According to the reaction protocol described in general procedure B from 4c (40.0 mg), compound 19 (42%, 30.5 mg) was isolated after chromatography as a colorless sticky oil: Rf = 0.35 (1:1 EtOAc/hexane); [α]D20 = +45.9 (c = 0.535, CHCl3); FTIR (thin film) 3450, 3259, 2957, 1603, 1587, 1477, 1454, 1387, 1323, 1161, 1070, 808, 733, 698 cm–1; 1H NMR (500 MHz, CDCl3) δ ppm 7.96 (ddd, J = 8.6, 6.4, 1.6 Hz, 1H, aromatic), 7.53–7.44 (m, 2H, aromatic), 7.44–7.37 (m, 2H, aromatic), 7.38–7.29 (m, 1H, aromatic), 6.87–6.79 (m, 1H, aromatic), 6.79 (ddd, J = 10.0, 1.8 Hz, 1H, aromatic), 6.42 (s, 1H, NH), 4.66 (s, 2H, OCH2C), 4.56 (dd, J = 5.4, 3.1 Hz, 1H, OCHCH2O), 4.01 (dd, J = 10.5, 5.1 Hz, 1H, OCHCHaHbO), 3.98–3.89 (m, 1H, OCHCHaHbO), 3.44–3.26 (m, 2H, HOCH2CH), 3.06–2.96 (m, 1H, NHCHCH2N), 2.95–2.87 (m, 3H, NCH2CHO, OH), 2.77–2.64 (m, 1H, NCHCH3), 2.33 (dd, J = 14.8, 4.9 Hz, 1H, NHCHCHaHbN), 2.26 (dd, J = 14.9, 3.8 Hz, 1H, NHCHCHaHbN), 1.65–1.53 (m, 1H, CH3CHCH3), 1.54–1.43 (m, 1H, NHCHCHaHb), 0.96 (ddd, J = 13.5, 7.9, 5.6 Hz, 1H, NHCHCHaHb), 0.84 (dd, J = 6.5, 1.5 Hz, 3H, CH3CHCH3), 0.81 (dd, J = 6.7, 1.6 Hz, 3H, NCHCH3), 0.72 (dd, J = 6.6, 1.5 Hz, 3H, CH3CHCH3); 13C NMR (126 MHz, CDCl3) δ ppm 165.9 (d, 1JC–F = 254.8 Hz), 155.5, 136.2, 132.3 (d, 3JC–F = 10.2 Hz), 129.0 (2C), 128.6 (2C), 128.4, 127.3, 109.2 (d, 2JC–F = 22.0 Hz), 104.1 (d, 2JC–F = 25.2 Hz), 78.6, 75.0, 71.0, 64.8, 61.4, 55.8, 53.5, 51.6, 43.4, 24.1, 23.1, 21.8, 10.0; HRMS calcd for C25H35FN2O5SH (M + H)+ 495.2329, found 495.2298 (TOF MS ES+).
Acknowledgments
This investigation was supported by funds provided by the NIH Center for Chemical Methodologies and Library Development at the University of Kansas (P50 GM069663) and NIGMS Pilot-Scale Libraries Program (NIH P41 GM076302). We thank Justin Douglas and Sarah Neuenswander in the University of Kansas NMR Laboratory (NSF Grant Nos. 9512331, 9977422, and 0320648 and NIH Center Grant Nos. P20 GM103418, S10RR024664, and S10OD016360) and Victor Day in the X-ray Crystallography Laboratory (NSF-MRI Grant No. CHE-0923449). We also acknowledge Patrick Porubsky and Benjamin Neuenswander for providing assistance with mass spectrometry as well as Gerald Lushington for computational analysis and data. We also thank The University of Kansas and the State of Kansas for partial student support (J.K.L., N.A., and T.B.S.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.5b01429.
The authors declare no competing financial interest.
Supplementary Material
References
- For reviews, see:; a Nubbemeyer U. Top. Curr. Chem. 2001, 216, 125–196. 10.1007/3-540-44726-1_4. [DOI] [Google Scholar]; and refs cited therein; b Sharma A.; Appukkuttan P.; Van der Eycken E. Chem. Commun. 2012, 48, 1623–1637. 10.1039/C1CC15238F. [DOI] [PubMed] [Google Scholar]; For some recent methods, see:; c Bauer R. A.; Wenderski T. A.; Tan D. S. Nat. Chem. Biol. 2013, 9, 21–29. 10.1038/nchembio.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Bogdan A. R.; Jerome S. V.; Houk K. N.; James K. J. Am. Chem. Soc. 2012, 134, 2127–2138. 10.1021/ja208503y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review, see:; a Cossy J. C. R. Chim. 2008, 11, 1303–1305. 10.1016/j.crci.2008.10.003. [DOI] [Google Scholar]; b For selected examples of bioactive lactams, see:; c Lou L.; Qian G.; Xie Y.; Hang J.; Chen H.; Zaleta-Rivera K.; Li Y.; Shen Y.; Dussault P. H.; Liu F.; Du L. J. Am. Chem. Soc. 2011, 133, 643–645. 10.1021/ja105732c. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Yang S.; Xi Y.; Zhu R.; Wang L.; Chen J.; Yang Z. Org. Lett. 2013, 15, 812–815. 10.1021/ol400038h. [DOI] [PubMed] [Google Scholar]; e Floss H. G.; Yu T.-W. Chem. Rev. 2005, 105, 621–632. 10.1021/cr030112j. [DOI] [PubMed] [Google Scholar]; f Ksander G. M.; de Jesus R.; Yuan A.; Ghai R. D.; McMartin C.; Bohacek R. J. Med. Chem. 1997, 40, 506–514. 10.1021/jm960583g. [DOI] [PubMed] [Google Scholar]; g Bach T.; Lemarchand A. Synlett 2002, 1302–1304. 10.1055/s-2002-32958. [DOI] [Google Scholar]; h Kawamura T.; Tashiro E.; Shindo K.; Imoto M. J. Antibiot. 2008, 61, 312–317. 10.1038/ja.2008.44. [DOI] [PubMed] [Google Scholar]; i Laumen K.; Machauer R.; Tintelnot-Blomley M.; Veenstra S. J.. WO2008009750 A2 20080124, 2008.; j Stamford A. W.; Huang Y.; Li G.; Strickland C. O.; Voigt J. H.. WO2006014944 A1 20060209, 2006.
- a Schreiber S. L. Science 2000, 287, 1964–1969. 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]; b Nielsen T. E.; Schreiber S. L. Angew. Chem., Int. Ed. 2008, 47, 48–56. 10.1002/anie.200703073. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Burke M. D.; Schreiber S. L. Angew. Chem., Int. Ed. 2004, 43, 46–58. 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]
- a Udugamasooriya D. G.; Spaller M. R. Biopolymers 2008, 89, 653–667. 10.1002/bip.20983. [DOI] [PubMed] [Google Scholar]; b Gilon C.; Halle D.; Chorev M.; Selincer Z.; Byk G. Biopolymers 1991, 31, 745–750. 10.1002/bip.360310619. [DOI] [PubMed] [Google Scholar]; c Veber D. F.; Johnson S. R.; Cheng H. Y.; Smith B. R.; Ward K. W.; Kopple K. D. J. Med. Chem. 2002, 45, 2615–2623. 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]; d Adessi C.; Soto C. Curr. Med. Chem. 2002, 9, 963–978. 10.2174/0929867024606731. [DOI] [PubMed] [Google Scholar]
- a McGeary R. P.; Fairlie D. P. Curr. Opin. Drug Discovery Dev. 1998, 1, 208–217. [PubMed] [Google Scholar]; b Fairlie D. P.; Abbenante G.; March D. R. Macrocyclic peptidomimetics - forcing peptides into bioactive conformations. Curr. Med. Chem. 1995, 2, 654–686. [Google Scholar]; c Driggers E. M.; Hale S. P.; Lee J.; Terrett N. K. Nat. Rev. Drug Discovery 2008, 7, 608–624. 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]; d Marsault E.; Peterson M. L. J. Med. Chem. 2011, 54, 1961–2004. 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]
- a Lee D.; Sello J. K.; Schreiber S. L. J. Am. Chem. Soc. 1999, 121, 10648–10649. 10.1021/ja992658m. [DOI] [Google Scholar]; b Wessjohann L. A. Curr. Opin. Chem. Biol. 2000, 4, 303–309. 10.1016/S1367-5931(00)00093-4. [DOI] [PubMed] [Google Scholar]; c Su Q.; Beeler A. B.; Lobkovsky E.; Porco J. A. Jr.; Panek J. S. Org. Lett. 2003, 5, 2149–2152. 10.1021/ol034608z. [DOI] [PubMed] [Google Scholar]; d Clardy J.; Walsh C. Nature 2004, 432, 829–837. 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]; e Wessjohann L. A.; Ruijter E. Mol. Diversity 2005, 9, 159–169. 10.1007/s11030-005-1313-y. [DOI] [PubMed] [Google Scholar]; f de Greef M.; Abeln S.; Belkasmi K.; Dömling A.; Orru R. V. A.; Wessjohann L. A. Synthesis 2006, 23, 3997–4004. 10.1055/s-2006-950335. [DOI] [Google Scholar]
- Lovering F. J.; Bikker J.; Humblet C. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- Vendeville S.; Cummings M. D. Annu. Rep. Med. Chem. 2013, 48, 371–386. 10.1016/B978-0-12-417150-3.00023-5. [DOI] [Google Scholar]
- a Villar E. A.; Beglov D.; Chennamadhavuni S.; Porco J. A. Jr.; Kozakov D.; Vajda S.; Whitty A. Nat. Chem. Biol. 2014, 10, 723–731. 10.1038/nchembio.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nero T. L.; Morton C. J.; Holien J. K.; Wielens J.; Parker M. W. Nat. Rev. Cancer 2014, 14, 248–262. 10.1038/nrc3690. [DOI] [PubMed] [Google Scholar]
- a Mai A. ChemMedChem 2014, 9, 415–417. 10.1002/cmdc.201400084. [DOI] [PubMed] [Google Scholar]; b Knapp S.; Weinmann H. ChemMedChem 2013, 8, 1885–1891. 10.1002/cmdc.201300344. [DOI] [PubMed] [Google Scholar]
- a Evans P. A.; Holmes A. B. Tetrahedron 1991, 47, 9131–9166. 10.1016/S0040-4020(01)96203-9. [DOI] [Google Scholar]; b See refs (1) and (2).
- Hegde V. R.; Patel M. G.; Gullo V. P.; Ganguly A. K.; Sarre O.; Puar M. S. J. Am. Chem. Soc. 1990, 112, 6403–6405. 10.1021/ja00173a042. [DOI] [Google Scholar]
- a Ayers S.; Zink D. L.; Mohn K.; Powell J. S.; Brown C. M.; Murphy T.; Grund A.; Genilloud O.; Salazar O.; Thompson D.; Singh S. B. J. Nat. Prod. 2007, 70, 1371–1373. 10.1021/np070131e. [DOI] [PubMed] [Google Scholar]; b Ayers S.; Zink D. L.; Powell J. S.; Brown C. M.; Grund A.; Genilloud O.; Salazar O.; Thompson D.; Singh S. B. J. Antibiot. 2008, 61, 59–62. 10.1038/ja.2008.110. [DOI] [PubMed] [Google Scholar]
- Johnson E. P.; Chen G.-P.; Fales K. R.; Lenk B. E.; Szendroi R. J.; Wang X.-J.; Carlson J. A. J. Org. Chem. 1996, 60, 6595–6598. 10.1021/jo00125a056. [DOI] [Google Scholar]
- Lin T.-I.; Lenz O.; Fanning G.; Verbinnen T.; Delouvroy F.; Scholliers A.; Vermeiren K.; Rosenquist A.; Edlund M.; Samuelsson B.; Vrang L.; de Kock H.; Wigerinck P.; Raboisson P.; Simmen K. Antimicrob. Agents Chemother. 2009, 53, 1377–1385. 10.1128/AAC.01058-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kaul R.; Surprenant S.; Lubell W. D. J. Org. Chem. 2005, 70, 3838–3844. 10.1021/jo0477648. [DOI] [PubMed] [Google Scholar]; b Lesma G.; Colombo A.; Silvani A.; Sacchetti A. Tetrahedron Lett. 2008, 49, 7423–7425. 10.1016/j.tetlet.2008.10.072. [DOI] [Google Scholar]; c Yu X.; Sun D. Molecules 2013, 18, 6230–6268. 10.3390/molecules18066230. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ksander G. M.; de Jesus R.; Yuan A.; Ghai R. D.; McMartin C.; Bohacek R. J. Med. Chem. 1997, 40, 506–514. 10.1021/jm960583g. [DOI] [PubMed] [Google Scholar]; e Ding G.; Liu F.; Yang T.; Fu H.; Zhao Y.; Jiang Y. Bioorg. Med. Chem. 2006, 14, 3766–3774. 10.1016/j.bmc.2006.01.050. [DOI] [PubMed] [Google Scholar]
- Velten R.; Erdelen C.; Gehling M.; Gohrt A.; Gondol D.; Lenz J.; Lockhoff O.; Wachendorff U.; Wendisch D. Tetrahedron Lett. 1998, 39, 1737–1740. 10.1016/S0040-4039(98)00057-4. [DOI] [Google Scholar]
- Baldauf C.; Günther R.; Hofmann H.-J. J. Mol. Struct.: THEOCHEM 2004, 675, 19–28. 10.1016/j.theochem.2003.12.029. [DOI] [Google Scholar]
- Gerard B.; Duvall J. R.; Lowe J. T.; Murillo T.; Wei J.; Akella L. B.; Marcaurelle L. A. ACS Comb. Sci. 2011, 13, 365–374. 10.1021/co2000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lücking U.; Siemeister G.; Schäfer M.; Briem H.; Krüger M.; Lienau P.; Jautelat R. ChemMedChem 2007, 2, 63–77. 10.1002/cmdc.200600199. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Kulkarni S.; Anderson D. D.; Hong L.; Baldridge A.; Wang Y.-F.; Chumanevich A. A.; Kovalevsky A. Y.; Tojo Y.; Amano M.; Koh Y.; Tang J.; Weber I. T.; Mitsuya H. J. Med. Chem. 2009, 52, 7689–7705. 10.1021/jm900695w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanessian S.; Larsson A.; Fex T.; Knecht W.; Blomberg N. Bioorg. Med. Chem. Lett. 2010, 20, 6925–6928. 10.1016/j.bmcl.2010.09.141. [DOI] [PubMed] [Google Scholar]
- Aldrich L. N.; Kuo S.-Y.; Castoreno A. B.; Goel G.; Petric Kuballa P.; Rees M. G.; Seashore-Ludlow B. A.; Cheah J. H.; Latorre I. J.; Stuart L.; Schreiber S. L.; Shamji A. F.; Xavier R. J. J. Am. Chem. Soc. 2015, 137, 5563–5568. 10.1021/jacs.5b02150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a See refs (1) and (6e) and references cited therein; b Hassan H. M. A. Chem. Commun. 2010, 46, 9100–9106. 10.1039/c0cc03122d. [DOI] [PubMed] [Google Scholar]; c White C. J.; Yudin A. K. Nat. Chem. 2011, 3, 509–524. 10.1038/nchem.1062. [DOI] [PubMed] [Google Scholar]; d Parenty A.; Moreau X.; Niel G.; Campagne J.-M. Chem. Rev. 2013, 113, PR1–PR40. and references cited therein 10.1021/cr300129n. [DOI] [PubMed] [Google Scholar]; e Marcaurelle L. A.; Comer E.; Dandapani S.; Duvall J. R.; Gerard B.; Kesavan S.; Lee M. D.; Liu H.; Lowe J. T.; Marie J.-C.; Mulrooney C. A.; Pandya B. A.; Rowley A.; Ryba T. D.; Suh B.-C.; Wei J.; Young D. W.; Akella L. B.; Ross N. T.; Zhang Y.-L.; Fass D. M.; Reis S. A.; Zhao W.-N.; Haggarty S. J.; Palmer M.; Foley M. A. J. Am. Chem. Soc. 2010, 132, 16962–16967. 10.1021/ja105119r. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Wessjohann L. A.; Ruijter E. Top. Curr. Chem. 2005, 243, 137–184. [Google Scholar]; For selected examples of heterocycles derived from target-oriented samples, see:; g Lou L.; Qian G.; Xie Y.; Hang J.; Chen H.; Zaleta-Rivera K.; Li Y.; Shen Y.; Dussault P. H.; Liu F.; Du L. J. Am. Chem. Soc. 2011, 133, 643–645. 10.1021/ja105732c. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Yang S.; Xi Y.; Zhu R.; Wang L.; Chen J.; Yang Z. Org. Lett. 2013, 15, 812–815. 10.1021/ol400038h. [DOI] [PubMed] [Google Scholar]; i Floss H. G.; Yu T.-W. Chem. Rev. 2005, 105, 621–632. 10.1021/cr030112j. [DOI] [PubMed] [Google Scholar]; For selected examples of heterocycles derived from diversity-oriented samples, see:; j Tan ref (1c).; k Hussain A.; Yousuf S. K.; Sharma D. K.; Mallikharjuna Rao L.; Singh B.; Mukherjee D. Tetrahedron 2013, 69, 5517–5524. 10.1016/j.tet.2013.04.090. [DOI] [Google Scholar]
- Wessjohann L. A.; Ruijter E. Top. Curr. Chem. 2005, 243, 137–184. [Google Scholar]
- Complementary ambiphile pairing (CAP):; a Samarakoon T. B.; Hur M. Y.; Kurtz R. D.; Hanson P. R. Org. Lett. 2010, 12, 2182–2185. 10.1021/ol100495w. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rolfe A.; Samarakoon T. B.; Hanson P. R. Org. Lett. 2010, 12, 1216–1219. 10.1021/ol100035e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For work related to the build–couple–pair paradigm, see:; a Nielsen T. E.; Schreiber S. L. Angew. Chem., Int. Ed. 2008, 47, 48–56. 10.1002/anie.200703073. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Uchida T.; Rodriquez M.; Schreiber S. L. Org. Lett. 2009, 11, 1559–1562. 10.1021/ol900173t. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Luo T.; Schreiber S. L. J. Am. Chem. Soc. 2009, 131, 5667–5674. 10.1021/ja900414s. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Marcaurelle L. A.; Comer E.; Dandapani S.; Duvall J. R.; Gerard B.; Kesavan S.; Lee M. D. IV; Liu H.; Lowe J. T.; Marie J.-C.; Mulrooney C. A.; Pandya B. A.; Rowley A.; Ryba T. D.; Suh B.-C.; Wei J.; Young D. W.; Akella L. B.; Ross N. T.; Zhang Y.-L.; Fass D. M.; Reis S. A.; Zhao W.-N.; Haggarty S. J.; Palmer M.; Foley M. A. J. Am. Chem. Soc. 2010, 132, 16962–16976. 10.1021/ja105119r. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Gerard B.; Duvall J. R.; Lowe J. T.; Murillo T.; Wei J.; Akella L. B.; Marcaurelle L. A. ACS Comb. Sci. 2011, 13, 365–374. 10.1021/co2000218. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Beckmann H. S. G.; Nie F.; Hagerman C. E.; Johansson H.; Tan Y. S.; Wilcke D.; Spring D. R. Nat. Chem. 2013, 5, 861–867. 10.1038/nchem.1729. [DOI] [PubMed] [Google Scholar]; g Comer E.; Beaudoin J. A.; Kato N.; Fitzgerald M. E.; Heidebrecht R. W.; Lee M. d.; Masi D.; Mercier M.; Mulrooney C.; Muncipinto G.; Rowley A.; Crespo-Llado K.; Serrano A. E.; Lukens A. K.; Wiegand R. C.; Wirth D. F.; Palmer M. A.; Foley M. A.; Munoz B.; Scherer C. A.; Duvall J. R.; Schreiber S. L. J. Med. Chem. 2014, 57, 8496–8502. 10.1021/jm500994n. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Mamidala R.; Babu Damerla V. S.; Gundla R.; Chary M. T.; Murthy Y. L. N.; Sen S. RSC Adv. 2014, 4, 10619–10626. 10.1039/c3ra47714b. [DOI] [Google Scholar]; i Flagstad T.; Hansen M. R.; Le Quement S. T.; Givskov M.; Nielsen T. E. ACS Comb. Sci. 2015, 17, 19–23. 10.1021/co500091f. [DOI] [PubMed] [Google Scholar]; j For a review of different pairing approaches to scaffold synthesis, see:Dow M.; Fisher M.; James T.; Marchetti F.; Nelson A. Org. Biomol. Chem. 2012, 10, 17–28. and references cited therein 10.1039/C1OB06098H. [DOI] [PubMed] [Google Scholar]
- For an account of the collective work in this area, see:He Z.; Zajdlik A.; Yudin A. K. Acc. Chem. Res. 2014, 47, 1029–1040. and references cited therein 10.1021/ar400210c. [DOI] [PubMed] [Google Scholar]; Note: The nucleophilic aziridine functionality is retained in their synthesis.
- a Rolfe A.; Young K.; Hanson P. R. Eur. J. Org. Chem. 2008, 2008, 5254–5262. 10.1002/ejoc.200800651. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhou A.; Hanson P. R. Org. Lett. 2008, 10, 2951–2954. 10.1021/ol8009072. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rolfe A.; Young K.; Volp K.; Schoenen F.; Neuenswander B.; Lushington G. H.; Hanson P. R. J. Comb. Chem. 2009, 11, 732–738. 10.1021/cc900025e. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zhou A.; Rayabarapu D. K.; Hanson P. R. Org. Lett. 2009, 11, 531–534. 10.1021/ol802467f. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Rayabarapu D. K.; Zhou A.; Jeon K.-O.; Samarakoon T. B.; Rolfe A.; Siddiqui H.; Hanson P. R. Tetrahedron 2009, 65, 3180–3188. 10.1016/j.tet.2008.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Ullah F.; Samarakoon T.; Rolfe A.; Kurtz R. D.; Hanson P. R.; Organ M. G. Chem. - Eur. J. 2010, 16, 10959–10962. 10.1002/chem.201001651. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Rolfe A.; Lushington G. H.; Hanson P. R. Org. Biomol. Chem. 2010, 8, 2198–2203. 10.1039/b927161a. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Samarakoon T. B.; Loh J. K.; Yoon S. Y.; Rolfe A.; Le L. S.; Hanson P. R. Org. Lett. 2011, 13, 5148–5151. 10.1021/ol201962n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring opening of sulfonyl aziridines and epoxides with amines are among the original “Click” reactions detailed by Sharpless in their seminal paper.Kolb H. C.; Finn M. G.; Sharpless K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Chen C.-Y.; Reamer R. A. Tetrahedron Lett. 2009, 50, 1529–1532. 10.1016/j.tetlet.2009.01.049. [DOI] [Google Scholar]
- Wang Z.; Bois-Choussy M.; Jia Y.; Zhu J. Angew. Chem., Int. Ed. 2010, 49, 2018–2022. 10.1002/anie.200906797. [DOI] [PubMed] [Google Scholar]
- a Beugelmans R.; Singh G. P.; Bois-Choussy M.; Chastanet J.; Zhu J. J. Org. Chem. 1994, 59, 5535–5542. 10.1021/jo00098a010. [DOI] [Google Scholar]; b Ma N.; Jia Y.; Liu Z.; Gonzalez-Zamora E.; Bois-Choussy M.; Malabarba A.; Brunati C.; Zhu J. Bioorg. Med. Chem. Lett. 2005, 15, 743–746. 10.1016/j.bmcl.2004.11.014. [DOI] [PubMed] [Google Scholar]; c Boger D. L.; Borzilleri R. M.; Nukui S.; Beresis R. T. J. Org. Chem. 1997, 62, 4721–4736. 10.1021/jo970560p. [DOI] [PubMed] [Google Scholar]
- a Boisnard S.; Zhu J. Tetrahedron Lett. 2002, 43, 2577–2580. 10.1016/S0040-4039(02)00307-6. [DOI] [Google Scholar]; b Temal-Laïb T.; Chastanet J.; Zhu J. J. Am. Chem. Soc. 2002, 124, 583–590. 10.1021/ja0170807. [DOI] [PubMed] [Google Scholar]
- a Giannotti D.; Viti G.; Sbraci P.; Pestellini V.; Volterra G.; Borsini F.; Lecci A.; Meli A.; Dapporto P.; Paoli P. J. Med. Chem. 1991, 34, 1356–1362. and references cited therein 10.1021/jm00108a018. [DOI] [PubMed] [Google Scholar]; b Viti G.; Nannicini R.; Pestellini V.; Bellarosa D.; Giannotti D. Bioorg. Med. Chem. Lett. 1995, 5, 1461–1466. 10.1016/0960-894X(95)00257-T. [DOI] [Google Scholar]
- a Baxter C. A.; O’Hagan M.; O’Riordan T. J. C.; Sheen F. J.; Stewart G. W.; Cleator E. Tetrahedron Lett. 2010, 51, 1079–1082. 10.1016/j.tetlet.2009.12.099. [DOI] [Google Scholar]; b Pizzirani D.; Kaya T.; Clemons P. A.; Schreiber S. L. Org. Lett. 2010, 12, 2822–2825. 10.1021/ol100914b. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gerard B.; Duvall J. R.; Lowe J. T.; Murillo T.; Wei J.; Akella L. B.; Marcaurelle L. A. ACS Comb. Sci. 2011, 13, 365–374. 10.1021/co2000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meutermans W. D. F.; Gregory T.; Bourne G. T.; Golding S. T.; Horton D. A.; Campitelli M. R.; Craik D.; Scanlon M.; Smythe M. L. Org. Lett. 2003, 5, 2711–2714. 10.1021/ol034907o. [DOI] [PubMed] [Google Scholar]
- Li X.; Chen N.; Xu J. Synthesis 2010, 20, 3423–3428. 10.1055/s-0030-1257913. [DOI] [Google Scholar]
- a Beddoes R. L.; Dalton L.; Joule J. A.; Mills O. S.; Street J. D.; Watt C. I. F. J. Chem. Soc., Perkin Trans. 2 1986, 787–797. 10.1039/p29860000787. [DOI] [Google Scholar]; b Klug H. P. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1968, 24, 792–802. 10.1107/S0567740868003201. [DOI] [PubMed] [Google Scholar]; c Oppolzer W.; Rodriguez I.; Starkemann C.; Walther E. Tetrahedron Lett. 1990, 31, 5019–5022. 10.1016/S0040-4039(00)97794-3. [DOI] [Google Scholar]
- a Barange D. K.; Tu Y.-C.; Kavala V.; Kuo C.-W.; Yao C.-F. Adv. Synth. Catal. 2011, 353, 41–48. 10.1002/adsc.201000465. [DOI] [Google Scholar]; b Chambers C. S.; Patel N.; Hemming K. Tetrahedron Lett. 2010, 51, 4859–4861. 10.1016/j.tetlet.2010.07.050. [DOI] [Google Scholar]
- As noted previously, this particular bis-nucleophile (2-piperidinemethanol) is racemic, and only one of two possible diastereomeric products was isolated resulting in a slightly lower yield (36% over two reactions), suggesting only one diastereomer intermediate underwent cyclization reaction (or potentially the aziridine ring opening was diastereoselective).
- Pangborn A. B.; Giardello M. A.; Grubbs R. H.; Rosen P. K.; Timmers F. J. Organometallics 1996, 15, 1518–1520. 10.1021/om9503712. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.