

ABSTRACT
The Gram-positive bacterium Actinomyces oris, a key colonizer in the development of oral biofilms, contains 18 LPXTG motif-containing proteins, including fimbrillins that constitute two fimbrial types critical for adherence, biofilm formation, and polymicrobial interactions. Export of these protein precursors, which harbor a signal peptide, is thought to be mediated by the Sec machine and require cleavage of the signal peptide by type I signal peptidases (SPases). Like many Gram-positive bacteria, A. oris expresses two SPases, named LepB1 and LepB2. The latter has been linked to suppression of lethal “glyco-stress,” caused by membrane accumulation of the LPXTG motif-containing glycoprotein GspA when the housekeeping sortase srtA is genetically disrupted. Consistent with this finding, we show here that a mutant lacking lepB2 and srtA was unable to produce high levels of glycosylated GspA and hence was viable. However, deletion of neither lepB1 nor lepB2 abrogated the signal peptide cleavage and glycosylation of GspA, indicating redundancy of SPases for GspA. In contrast, the lepB2 deletion mutant failed to assemble the wild-type levels of type 1 and 2 fimbriae, which are built by the shaft fimbrillins FimP and FimA, respectively; this phenotype was attributed to aberrant cleavage of the fimbrillin signal peptides. Furthermore, the lepB2 mutants, including the catalytically inactive S101A and K169A variants, exhibited significant defects in polymicrobial interactions and biofilm formation. Conversely, lepB1 was dispensable for the aforementioned processes. These results support the idea that LepB2 is specifically utilized for processing of fimbrial proteins, thus providing an experimental model with which to study the basis of type I SPase specificity.
IMPORTANCE Sec-mediated translocation of bacterial protein precursors across the cytoplasmic membrane involves cleavage of their signal peptide by a signal peptidase (SPase). Like many Gram-positive bacteria, A. oris expresses two SPases, LepB1 and LepB2. The latter is a genetic suppressor of lethal “glyco-stress” caused by membrane accumulation of glycosylated GspA when the housekeeping sortase srtA is genetically disrupted. We show here that LepB1 and LepB2 are capable of processing GspA, whereas only LepB2 is required for cleavage of fimbrial signal peptides. This is the first example of a type I SPase dedicated to LPXTG motif-containing fimbrial proteins. Thus, A. oris provides an experimental model with which to investigate the specificity mechanism of type I SPases.
INTRODUCTION
Actinomyces oris is a key colonizer in the development of dental plaque or oral biofilms because of its ability to adhere to many oral colonizers and host surfaces. This multiadhesive property is attributed to the presence of two types of fimbriae or pili called type 1 and 2 fimbriae (1, 2). Type 1 fimbriae—made of the shaft fimbrillin FimP and the tip fimbrillin FimQ—mediate bacterial adherence to proline-rich proteins that coat tooth enamel (2–4). Type 2 fimbriae—composed of the shaft fimbrillin FimA and the tip fimbrillin FimB—promote polymicrobial interactions or bacterial coaggregation, biofilm formation, and adhesion to host cells (2, 5, 6). A. oris also encodes 14 cell wall-anchored proteins (AcaA to AcaN), one of which, CafA (previously named AcaF), has been shown to be the major coaggregation factor that forms a distinct fimbrial tip of type 2 fimbriae (7). It was recently shown that the cell wall-anchored protein AcaC (named GspA) is glycosylated, and its glycosylation appears to be coupled to protein secretion and sortase SrtA-catalyzed cell wall anchoring (8). Shared features found in these cell wall-anchored proteins and pilins (or fimbrillins) are an N-terminal signal peptide and a C-terminal cell wall sorting signal (CWSS). The N-terminal signal peptide of these surface proteins is predicted to contain common characteristics, including an n region with positively charged residues, followed by an h region rich in hydrophobic residues and a c region with residues like the AXA motif that are critical for recognition and processing by type 1 signal peptidases (SPases) (see Table S1 in the supplemental material) (9). The CWSS is composed of the signature LPXTG motif, a hydrophobic domain, and a tail of positively charged residues (10). In all Gram-positive bacteria, a conserved transpeptidase enzyme called the housekeeping sortase carries out the cell wall-anchoring function of surface proteins via the LPXTG motif (11–13).
According to the current models proposed for A. oris (1, 8, 14), membrane translocation of surface protein precursors is mediated by the Sec translocase via their signal peptides, which are removed by a SPase after translocation. Posttranslocational folding of the surface proteins with multiple cysteine residues is catalyzed by the thiol-disulfide oxidoreductase MdbA (14, 15). Folded proteins, embedded into the membrane via the hydrophobic domain of the CWSS, are cleaved at the LPXTG motif between threonine and glycine and then anchored to the cell wall by the housekeeping sortase SrtA. Unlike that in other Gram-positive bacteria studied to date, deletion of srtA in A. oris is lethal to cells (8). The basis for srtA essentiality was identified by transposon mutagenesis screening for srtA genetic suppressors. Molecular and genetic analyses of the suppressors revealed that ΔsrtA lethality was due to the membrane accumulation of glycosylated GspA, which causes membrane stress, altered cell morphology, and eventual cell death. Consistent with these observations, the deletion of srtA in the gspA deletion strain did not generate any detrimental defects, nor did overexpression of GspA lacking the CWSS in the srtA gspA double mutant. Additionally, mutagenesis screening also revealed 12 other suppressors, one of which was mapped to ANA_1190, which encodes a type I SPase (8). Farther upstream of ANA_1190 is another type I SPase-encoding gene (ANA_1188). We renamed ANA_1188 lepB1 and ANA_1190 lepB2. LepB1 and LepB2 appear to be the only two type I SPases, as BLAST searches with LepB1 and LepB2 as queries did not reveal additional SPases (data not shown). The role of LepB1 and LepB2 in the processing of signal peptides and how LepB2 is involved in the suppression of ΔsrtA lethality still remain to be investigated.
We report here the characterization of the two SPases, LepB1 and LepB2, of A. oris. We confirmed that LepB2 is indeed a genetic suppressor of ΔsrtA lethality since srtA can be deleted from the bacterial chromosome lacking lepB2. We also demonstrated that both LepB1 and LepB2 are involved in cleavage of the GspA signal peptide. In contrast, LepB2 is specifically required for processing of FimP and FimA, the two fimbrial shaft proteins of the type 1 and 2 fimbriae, respectively. Activity of LepB2 depends on the two conserved catalytic residues S101 and K169, as alanine substitution of these residues abrogates pilus assembly, biofilm formation, and interspecies interactions. On the other hand, LepB1 is dispensable for the aforementioned processes. Thus, LepB2 appears to be specific for LPXTG motif-containing fimbrial substrates and we propose that A. oris provides an experimental model with which to investigate the specificity of type I SPases.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
Actinomyces strains were grown in heart infusion broth (HIB) and on heart infusion agar (HIA) at 37°C with 5% CO2. Escherichia coli strains were grown in Luria broth or on Luria agar at 37°C. If needed, 50 μg ml−1 kanamycin (Kan) or streptomycin was added to bacterial cultures. Streptococci were grown in HIB supplemented with 1% glucose. E. coli strain DH5α was used in cloning experiments. The restriction enzymes T4 polynucleotide kinase and Phusion DNA polymerase were purchased from New England BioLabs Inc. For the bacterial strains and plasmids used in this study, see Table S1 in the supplemental material.
Plasmid construction. (i) pLepB2.
The promoter regions of the rimM and lepB2 coding sequences were generated by PCR with primers Pro-1192F and Pro-1192R and primers com-lepB2-F and com-lepB2-R, respectively (see Table S2 in the supplemental material). After digestion of the rimM promoter amplicon with EcoRI and KpnI and the lepB2 amplicon with KpnI and HindIII, the digested fragments were ligated into the HindIII and EcoRI sites of E. coli-Actinomyces shuttle vector pJRD215 to generate pLepB2.
(ii) pCWU10.
To generate an RSF1010 derivative that is functional in A. oris, the ampicillin resistance gene of pCVD047, a cyanobacterial broad-host-range vector (16), was replaced with the Kanr cassette from pJRD215 (17). Briefly, with primers pCVD047-noAmp-F and pCVD047-noAmp-R for an inverse PCR, an amplicon containing the pCVD047 sequence without the ampicillin resistance gene was generated. Next, the fragment encompassing the Kanr gene and a multiple cloning site from pJRD215 was amplified with primers 215KanMCS-F and 215KanMCS-R (see Table S2 in the supplemental material). Both amplicons were digested with SacI and HindIII. The isolated products were ligated to generate pCWU10.
(iii) pGspAΔCWSS.
With pAcaCΔCWS (8) as a template, primers com-AcaC-F and GspAΔcws-His6-R were used to PCR amplify a fragment encompassing the promoter region of gspA and its open reading frame (ORF) lacking the CWSS while a 6-His tag was appended to the GspA C terminus. The gspA amplicon was digested with NdeI and EcoRI and ligated into pCWU10 precut with the same enzymes to generate pGspAΔCWSS.
(iv) pFimAΔCWSS.
Primers prFimB-BamHI-F and prFimB-R and primers FimA-F and FimAΔcws-His6-EcoRI-R (see Table S2 in the supplemental material) were used in PCRs with MG1 genomic DNA (gDNA) as a template to amplify the promoter region of fimB and the fimA ORF lacking the CWSS with a 6-His tag appended to the FimA C terminus, respectively. The fimB promoter product was digested with BamHI, whereas the fimA coding region was digested with EcoRI and then treated with T4 polynucleotide kinase to phosphorylate the 5′ end. The digested DNA fragments were ligated into pCWU10 precut with BamHI and EcoRI to generate pFimAΔCWSS.
(v) pHTT-lepB2.
The lepB2 coding sequence and the rimM promoter region were amplified from pLepB2 with primers Pro-1192-F and com-lepB2-R. The PCR product was digested with EcoRI and HindIII and ligated into pHTT177 (3) precut with the same enzymes.
Site-directed mutagenesis.
For site-directed mutagenesis of LepB2, pHTT-lepB2 was used as a template for site-directed mutagenesis with mutation sites incorporated into the 5′ end of the synthesized primers (see Table S2 in the supplemental material) for a PCR-based assay as previously described (8). The PCR products were purified by gel extraction and phosphorylated to facilitate circulation of the amplicons. The plasmids generated were then used to transform E. coli DH5α. The lepB2 mutations were verified by DNA sequencing. The fragment encompassing the lepB2 coding sequence with the desired mutations and the rimM promoter region were subcloned into pJRD215 via the EcoRI and HindIII sites.
Generation of nonpolar in-frame deletion mutants of A. oris.
The nonpolar in-frame deletion mutants used in this study were generated in accordance with a previously published protocol (6). Briefly, 1.0-kb DNA fragments upstream and downstream of a target gene were PCR amplified with appropriate primers (see Table S2 in the supplemental material). The two fragments were treated with restriction enzymes and linked together by single-step ligation. Subsequently, the 2.0-kb fragment was cloned into deletion vector pCWU2 (8). The plasmids generated were introduced into the A. oris ΔgalK mutant strain by electroporation. Selection of corresponding in-frame deletion mutants was performed with GalK as a counterselectable marker. The mutants generated were characterized by PCR and/or immunoblotting.
Cell growth assays.
A. oris growth was assessed by a plate assay and optical density at 600 nm (OD600) in accordance with a previous protocol (8). For the plate assay, overnight cultures of various strains were harvested and normalized to the same OD600 in HIB. Equivalent cell suspensions were subjected to 10-fold serial dilution and spotted onto HIA plates with or without supplementation with 100 ng ml−1 anhydrotetracycline (AHT) and 2 mM theophylline. Cell growth at 37°C was recorded after 3 days. For growth in liquid broth, strains were subcultured to an OD600 of ∼0.02. Cell growth was monitored by OD600 over 19 h. The OD values presented are averages of three independent experiments performed in duplicate.
Reverse transcriptase and quantitative real-time PCRs.
Overnight cultures were used to inoculate fresh cultures of various A. oris strains, which were grown to an OD600 of ∼0.25. Cells were harvested by centrifugation, and cell pellets were suspended in TRIzol (Ambion) and lysed by mechanical disruption with 0.1-mm silica spheres (MP Bio). Total RNA was extracted with a Direct-zol RNA MiniPrep kit (Zymo Research). cDNA was synthesized with SuperScript III reverse transcriptase (RT; Invitrogen). For quantitative real-time PCRs, cDNA was mixed with iTAQ SYBR green supermix (Bio-Rad), along with appropriate primer sets (see Table S2 in the supplemental material). Cycle threshold (CT) values were determined, and the relative expression level was calculated by the 2−ΔΔCT method (18), with the 16S rRNA gene serving as a control. Data were obtained from two or three independent experiments performed in triplicate.
Cell fractionation and immunoblotting.
Overnight cultures of A. oris strains were diluted 40-fold in HIB and grown at 37°C to an OD600 of ∼0.4 to 0.5. Normalized aliquots were subject to cell fractionation as described before (19). Protein samples obtained from supernatant (S), cell wall (W), membrane (M), and cytoplasm (C) fractions were analyzed by 15% or 3 to 12% SDS-PAGE and immunoblotted with specific antibodies (a 1:1,333 dilution of the anti-GspA antibody, a 1:10,000 dilution of the anti-FimP antibody, a 1:8,000 dilution of the anti-FimA antibody, and a 1:4,000 dilution of the anti-MdbA antibody), and the proteins were detected by chemiluminescence.
Protein purification and analysis.
Recombinant plasmids pGspAΔCWSS and pFimAΔCWSS, generated as described above, were introduced into various A. oris strains by electroporation. Each strain was inoculated into 200 ml of HIB supplemented with 50 μg ml−1 Kan and grown overnight. Cell-free culture supernatants were obtained by centrifugation and filtration with 0.45-μm-pore-size filters. A 2-ml volume of nickel resin, washed twice with EQ buffer (150 mM NaCl, 50 mM Tris HCl, pH 7.5), was added to the supernatants, and they were gently agitated at 4°C overnight. The entire suspensions were decanted onto a protein purification column, which was then washed with 20 ml of EQ buffer. Bound proteins were eluted in 3-ml fractions with 1 M imidazole. The eluted proteins were desalted with a desalting column (Bio-Rad) and concentrated with an Amicon Ultra 0.5-ml centrifugal unit (Millipore) with a 10,000 (GspA) or 30,000 (FimA) molecular weight cutoff.
Purified proteins were analyzed by SDS-PAGE and visualized by Coomassie blue and periodic acid-Schiff (PAS) staining as previously described (8). For Edman sequencing, the proteins were transferred to a polyvinylidene difluoride membrane. The membrane was rinsed with ultrapure water, stained with 0.02% Coomassie brilliant blue for 30 s, destained in a 40% methanol–5% acetic acid solution for 1 min, and then rinsed with ultrapure water three times. The membrane was completely air dried, and protein bands were excised for Edman sequencing at the TUCF core facility (Tufts University).
Coaggregation and biofilm assays.
Coaggregation of A. oris and Streptococcus oralis was assayed according to a previously published protocol (19). Briefly, stationary-phase cultures of A. oris and S. oralis were collected by centrifugation, washed, and resuspended in coaggregation buffer (19). To quantify coaggregation, the OD600s of individual A. oris and S. oralis cell suspensions in coaggregation buffer were measured. Equal cell volumes (0.5 ml) were mixed and allowed to form aggregates. The resulting aggregates were photographed by an Alpha-Imager or removed by centrifugation at 100 × g, and the OD600s of the remaining supernatants were recorded. Relative coaggregation was determined as previously described (20).
A. oris biofilms were cultivated in vitro as reported before (6). Briefly, cells were inoculated into HIB supplemented with 1% sucrose in a 24-well polystyrene plate and incubated at 37°C with 5% CO2 for 48 h. After incubation, the medium was removed and each well was gently washed with phosphate-buffered saline (PBS) three times and dried overnight at room temperature. Biofilms were stained with 1% crystal violet and washed with water to remove the unbound dye. To quantify biofilm production, the biofilms were destained with 95% ethanol and the released dye was quantified by measuring absorbance at 580 nm with a Tecan Infinite M1000 microplate reader. The assays were performed three times in triplicate.
Whole-cell ELISA.
Surface expression of A. oris tip proteins was quantified by whole-cell enzyme-linked immunosorbent assay (ELISA) as previously described, with some modifications (21, 22). Briefly, mid-log-phase cells of A. oris MG1 and isogenic mutant strains were harvested by centrifugation and washed twice with PBS. The cells were suspended in carbonate-bicarbonate buffer (15 mM sodium carbonate, 35 mM sodium bicarbonate, pH 9.6) and normalized to an OD600 of 1.0. Cell suspension aliquots of 100 μl were dispensed into 96-well high-binding ELISA plates (Corning Costar EIA/RIA plates) to allow cell binding to wells at 4°C overnight. Unbound cells were removed by washing with PBS containing 0.05% Tween 20 (PBST). Adhered cells were blocked with 2% bovine serum albumin in PBST prior to incubation with antibodies specific to CafA and FimB (1:10,000) for 2 h at 25°C, followed by washing with PBST and staining with horseradish peroxidase (HRP)-linked anti-rabbit IgG antibodies (1:20,000; Cell Signaling) for 1 h at 25°C. Washed cells were treated with 100 μl of the chromogenic 3,3′,5,5′-tetramethylbenzidine substrate (Affymetrix; eBioscience) prior to the measurement of absorbance at 450 nm with a Tecan M1000 plate reader. The absorbance of strain MG1 and the ΔlepB1 and ΔlepB2 mutants, compared with that of the ΔcafA and ΔfimB mutants used as the background, was determined in at least two independent experiments performed in triplicate.
EM.
Fimbriae were visualized by electron microscopy (EM) as previously described (14). Briefly, A. oris cells grown in HIB were pelleted, washed once, and suspended in 0.1 M NaCl. For negative staining, a drop of a bacterial suspension was placed onto a carbon-coated nickel grid and stained with 1% uranyl acetate before being viewed with a JEM1400 electron microscope. For immuno-EM (IEM), after cells were immobilized on a nickel grid, samples were stained with specific antibodies (anti-FimP antibody, 1:100 dilution; anti-FimA antibody, 1:100 dilution) and then incubated with 12-nm colloidal gold–goat anti-rabbit IgG complexes (Jackson ImmunoResearch). Finally, the samples were washed five times with water before being stained with 1% uranyl acetate.
RESULTS
The type I SPase LepB2 is a genetic suppressor of srtA essentiality.
We have previously reported that A. oris srtA is an essential gene and identified the Tn5::lepB2 mutation as one of the genetic suppressors of srtA essentiality (8). lepB2 is genetically linked to trmD and rimM, which encode a tRNA [guanine(37)-N(1)]-methyltransferase and a 16S rRNA processing protein, respectively (Fig. 1A). According to the BioCyc databases (http://biocyc.org/), the three genes are predicted to be expressed as one transcription unit in Actinomyces naeslundii Howell 279 (23). Farther upstream of lepB2 is another SPase-encoding gene annotated as lepB1 (Fig. 1A). To exclude the possibility that the Tn5::lepB2 mutation has a polar effect on adjacent genes, we created in-frame nonpolar deletion mutants by allelic exchange (6), including the lepB2 srtA double deletion mutant (ΔlepB2 ΔsrtA). First, we generated a lepB2 mutant and used it to delete srtA from the bacterial chromosome without ectopic expression of SrtA. Of note, we were unable to delete srtA in the ΔlepB1 background. The ΔlepB2 ΔsrtA double mutant strain and others were assessed for the ability to grow under laboratory conditions with both plate and liquid broth assays (8). In the plate assay, normalized cell cultures were spotted in serial dilution onto HIA plates and grown at 37°C. Compared to the conditional srtA deletion mutant, which failed to grow in the absence of two inducers, AHT and theophylline (8), the ΔlepB2 ΔsrtA mutant did not display any visible defects when grown under any conditions (Fig. 1B). The growth of the ΔgspA, ΔlepB2, ΔlepB1, and ΔlepB1 ΔlepB2 mutants was comparable to that of parental strain MG1 (Fig. 1B). Nonetheless, when grown in liquid broth, the ΔlepB2 and ΔlepB1 ΔlepB2 mutants displayed a slight growth defect compared to MG1 and the ΔlepB1 mutant strain (Fig. 1C).
FIG 1.
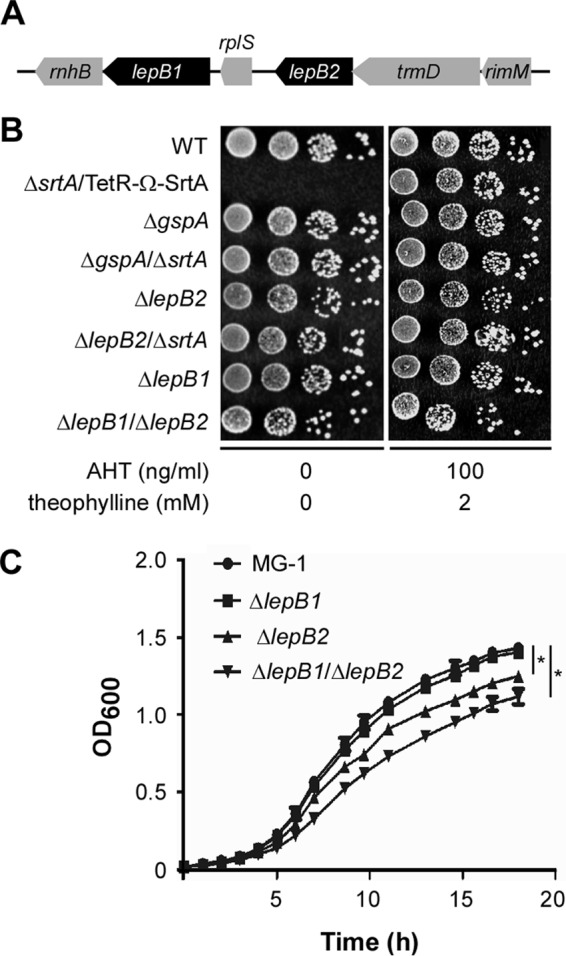
SPase LepB2 is a genetic suppressor of SrtA essentiality. (A) Graphic representation of the lepB2 gene locus of A. oris MG1. This locus encodes RNase H (rnhB), SPase I (lepB1), ribosomal protein L19 (rplS), SPase I (lepB2), a tRNA (guanine-N1)-methyltransferase (trmD), and a 16S rRNA processing protein (rimM). (B) Growth of WT MG1 and conditional srtA mutant and nonpolar deletion mutant strains on solid medium in the presence or absence of the inducers AHT and theophylline. (C) Growth of A. oris MG1 and the ΔlepB1, ΔlepB2, and ΔlepB1 ΔlepB2 mutant strains in liquid medium. The OD600s presented are averages of three independent experiments done in duplicate. *, P < 0.05 (determined by the paired, two-tailed t test with GraphPad Prism).
We next examined if lepB1 and lepB2 are part of a transcription unit by reverse transcription-PCR as previously described (24). Total RNA isolated from MG1 was used in reverse transcription reactions to produce cDNA. As shown in Fig. 2A and B, reverse transcription-PCR detected the presence of lepB1 or lepB2 transcripts with probes specific for lepB1 (primers P1 and P2) or lepB2 (primers P3 and P4), respectively. Neither transcript was observed in the absence of RT, indicative of no gDNA contamination. No transcripts were detected with probes specific for the region encompassing lepB1 and lepB2 (primers P5 and P6), whereas the same probes enabled amplification of this region from gDNA (Fig. 2B, g lanes). To ascertain that deletion of lepB2 does not affect lepB1 expression, we examined the expression levels of both genes by quantitative real-time PCR (see Materials and Methods). Compared to that in the wild-type (WT) strain, the relative expression of lepB1 or lepB2 was not altered when lepB2 or lepB1 was deleted, respectively (Fig. 2C), supporting the idea that lepB1 and lepB2 are independently expressed. Altogether, the results in Fig. 1 and 2 confirm that LepB2 is a genetic suppressor of srtA deficiency and suggest that LepB2 is required for processing of substrates that are involved in the SrtA-mediated glycosylation pathway.
FIG 2.
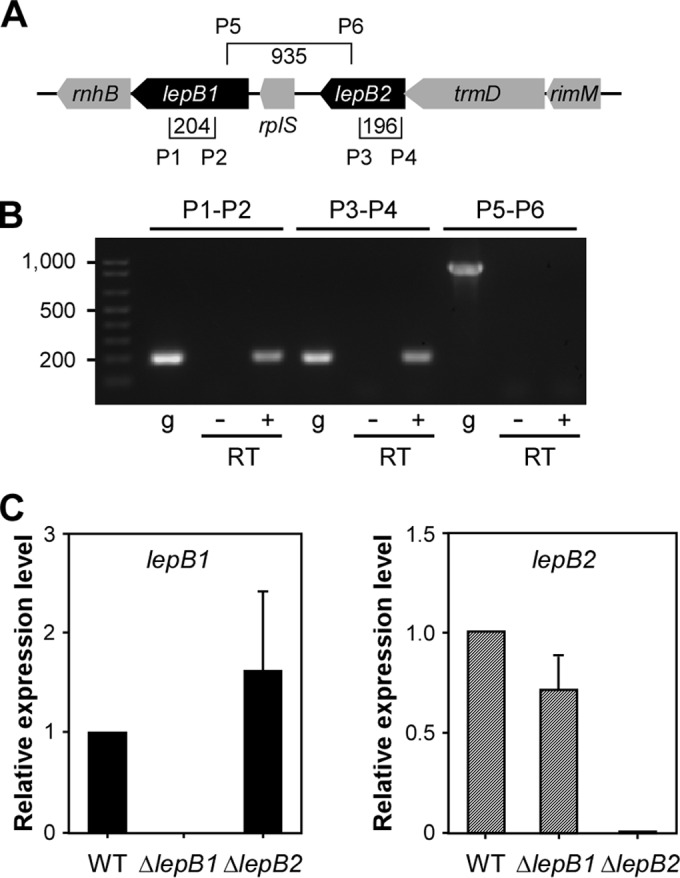
lepB1 and lepB2 are independently expressed. (A) Specific primers (P) were designed to detect lepB1, lepB2, and a region encompassing both. Brackets show primer positions, and values specify the sizes of amplicons in base pairs. (B) With the specific primers shown in panel A, reverse transcription-PCR was performed to determine if lepB1 and lepB2 are independently expressed. A plus or minus sign indicates that reverse transcription-PCRs were performed in the presence or absence of RT. PCRs with gDNA (g) were used as a control. (C) Quantitative real-time PCR used to determine the expression levels of lepB1 and lepB2 relative to those in parental strain MG1 by the 2−ΔΔCT method, where 16S rRNA served as the internal control. The data presented are averages of three independent experiments performed in triplicate; error bars represent standard deviations.
Involvement of SPases LepB1 and LepB2 in GspA glycosylation.
We previously showed that glycosylated GspA is a substrate of SrtA since depletion of srtA leads to the membrane accumulation of glycosylated GspA, causing lethal phenotypes (8). Subsequent Tn5 mutagenesis identified viable mutants that lack both srtA and gspA or both srtA and lepB2 (8). In this work, we have confirmed that LepB2 is a genetic suppressor of srtA essentiality by generating a nonpolar deletion mutant devoid of srtA and lepB2 (Fig. 1). Therefore, we hypothesized that LepB2 works on the same GspA glycosylation pathway by processing GspA before it is glycosylated and then anchored to the cell wall by sortase SrtA. To examine this possibility, we first examined the status of GspA glycosylation in the absence of SPases by analyzing GspA proteins isolated from the culture medium (S), cell wall (W), membrane (M), and cytoplasmic (C) fractions of the WT and SPase mutant strains. Protein samples were immunoblotted with antibodies to GspA and MdbA, a membrane-bound protein used as a control (14). In the WT MG1 strain, glycosylated GspA polymers (GspAP) as smeared bands were detected in the cell wall fraction and no GspA signal was observed in the ΔgspA mutant (Fig. 3A, first eight lanes), as previously reported (8). While the lepB1, lepB2, and ΔlepB1 ΔlepB2 mutants did not exhibit significant defects in GspA glycosylation, the ΔlepB2 ΔsrtA double mutant failed to produce WT levels of GspA polymers. Instead, the accumulation of three low-molecular-weight (LMW) species was detected in the cell wall fraction, in addition to that of a 50-kDa species observed in the culture medium and membrane fractions (Fig. 3A, lanes ΔlepB2 ΔsrtA). To ascertain that the observed defects were not due to low gspA expression levels, we determined gene expression by quantitative real-time PCR (see Materials and Methods). Overall, no significant difference in gene expression levels was observed between the WT and mutant strains (Fig. 3B).
FIG 3.

Involvement of SPases in sortase-associated GspA glycosylation. (A) Cells were grown to mid-log phase, and normalized by OD. Culture supernatant (S), cell wall (W), membrane (M), and cytoplasm (C) fractions were separated by SDS-PAGE and immunoblotted with antibodies to GspA and membrane protein MdbA, which served as loading and fractionation controls. Brackets designate glycosylated HMW species of GspA polymers (GspAP). (B) The relative expression levels of gspA in the strains indicated were determined by quantitative real-time PCR as described in the legend to Fig. 2. Error bars show the standard deviations of two independent experiments performed in triplicate.
Altogether, the results suggest that either LepB1 or LepB2 is sufficient for cleavage of the cell wall-anchored glycoprotein GspA and that LepB2 may process factors involving GspA glycosylation.
Specificity of A. oris SPases for processing of LPXTG motif-containing proteins.
GspA is 1 of 18 cell wall-anchored proteins with a CWSS and a putative signal peptide. A typical signal peptide is composed of a net positively charged (n) region, followed by a hydrophobic (h) region and a cleavage (c) region (25) (Fig. 4A). The c region harbors a typical cleavage site, i.e., an AXA motif, that is recognized and cleaved by a type I SPase after the second Ala residue (25). To determine the cleavage site of GspA and the roles of LepB1 and LepB2 in this process, we purified GspA by affinity chromatography from A. oris strains lacking either gene or both and analyzed the N termini of the purified proteins by Edman degradation. Because GspA is heavily glycosylated and its glycosylation sites are not known (8), we expressed in these strains a GspA variant lacking its membrane-bound CWSS and containing a His tag at the C terminus (Fig. 4A, pGspAΔCWSS). Using this approach, we could avoid the formation of high-molecular-weight (HMW) glycosylated GspA by forcing the protein to be released as a monomer to facilitate the detection of small changes in preprotein processing. After harvesting of the cell-free culture supernatants of A. oris strains expressing this construct by centrifugation and filtration, His-tagged proteins were captured by Ni-nitrilotriacetic acid agarose and purified according to a previously published protocol (7). Purified proteins were analyzed by SDS-PAGE and then Coomassie blue and periodic acid-Schiff (PAS) staining (see Materials and Methods). In the WT strain, the predominant GspA band and a weaker band migrated below and above the 37-kDa marker, respectively, in addition to the last one smeared between the 50- and 75-kDa markers by Coomassie blue staining (Fig. 4B, left side). As predicted, this predominant GspA species was negative for PAS staining, while some trace of PAS staining was detected with the smeary band (Fig. 4B, bracket 1). The same phenotypes were observed in the ΔlepB1 mutant; intriguingly, in the ΔlepB2 mutant, a smeary band migrating around 37 kDa was positive for PAS staining (Fig. 4B, bracket 2), while the HMW band positive for PAS staining remained weakly visible (Fig. 4B, bracket 1). Finally, in the lepB1 lepB2 double mutant, a further upshifted band with positive PAS staining was detected; in addition, it appeared that the HMW species of GspA showed a slight increase in PAS staining (Fig. 4B, bracket 1, last lane). These results indicate that glycosylation of GspA still occurs, albeit weakly, in the absence of the CWSS. From these findings, together with the data presented in Fig. 3A, we conclude that the deletion of neither lepB1 nor lepB2 affects GspA glycosylation.
FIG 4.
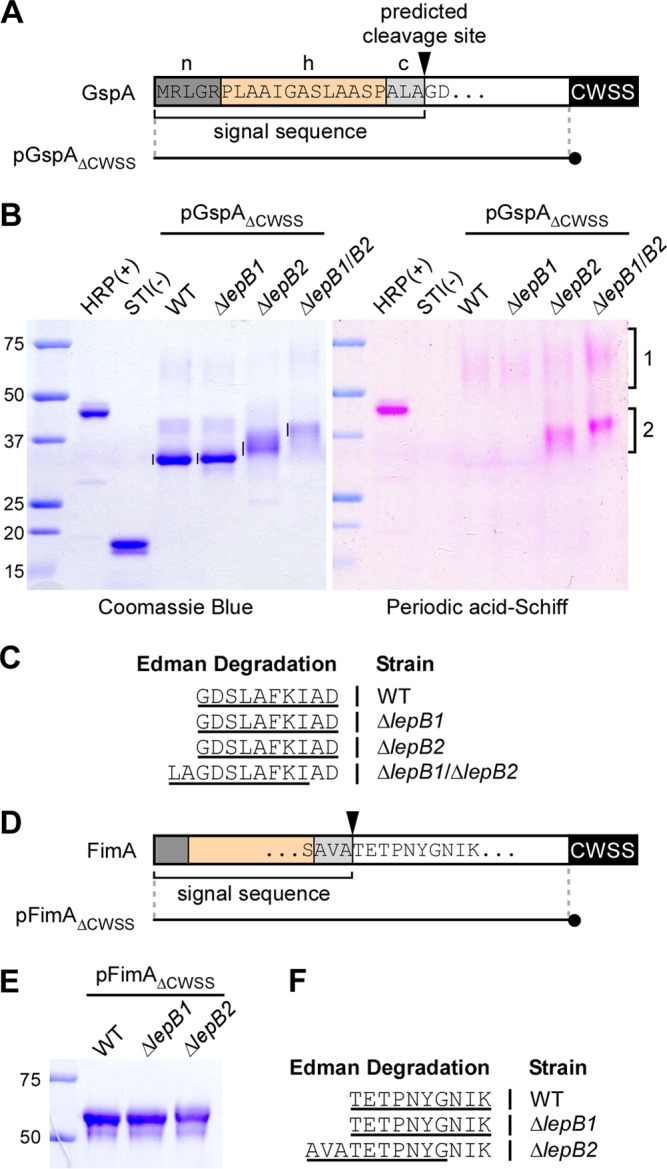
Mapping of the signal peptide cleavage site by Edman degradation. (A) Shown is a graphic representation of GspA. The N-terminal sequence of GspA contains a typical signal peptide composed of a positively charged polar region (n), a hydrophobic central region (h), and a region with the SPase recognition motif AXA (c). The arrowhead marks the predicted SPase I cleavage site. pGpsAΔCWSS is a plasmid expressing a GspA variant in which the CWSS is replaced with a 6-His tag (black circle). (B) His-tagged GspA proteins from the culture medium of the strains indicated were purified by affinity chromatography, separated by SDS-PAGE, and visualized by Coomassie blue (∼1.25 μg of proteins in all lanes) and PAS (2.5 μg of proteins) staining. Glycosylated HRP and nonglycosylated soybean trypsin inhibitor (STI) were used as positive and negative controls. Numbered brackets indicate glycosylated forms of GspA. (C) The bands indicated by vertical lines in panel B were subjected to Edman degradation. The N-terminal sequence of GspA was deduced from the first 10 sequencing cycles (underlined). Samples from the ΔlepB1 ΔlepB2 double mutant produced ragged N-terminal sequencing with the major sequence shown. (D) Like GspA, FimA harbors a signal peptide with the conserved cleavage site motif AXA. pFimAΔCWSS is a plasmid expressing a FimA molecule in which the CWSS is replaced with a 6-His tag (black circle). (E) His-tagged FimA proteins from the culture medium of the strains indicated were purified by affinity chromatography, separated by SDS-PAGE, and stained with Coomassie blue. (F) The purified proteins were subjected to N-terminal sequencing. The sequences that resulted from first 10 sequencing cycles are underlined. The lepB2 mutant also produced ragged N-terminal sequencing with the major sequence shown. The values to the left of panels B and E are molecular sizes in kilodaltons.
If either SPase is sufficient to process GspA, the cleaved products in each deletion mutant should contain the same N-terminal sequence. To determine if this is the case, we excised the aforementioned major GspA bands indicated in Fig. 4B for Edman sequencing. As shown in Table S4 in the supplemental material, the first 10 cycles of Edman degradation for GspA samples purified from the WT and the ΔlepB1 and ΔlepB2 mutant strains revealed the matching sequence GDSLAFKIAD (Fig. 4C), consistent with the predicted cleavage site between Ala (−1 position) and Gly (+1 position) (Fig. 4A). In contrast, the same analysis of GspA samples isolated from the ΔlepB1 ΔlepB2 double mutant produced a mixture of residues in the majority of the first 10 Edman sequencing cycles (see Table S4), indicative of multiple polypeptides present in these samples; with the major polypeptide with the sequence LAGDSLAFKI (Fig. 4C). These results indicated that LepB1 and LepB2 are capable of cleaving the GspA signal peptide. In the absence of both SPases, the GspA signal peptide might be proteolytically degraded by an unidentified protease(s), causing ragged N-terminal sequencing.
LepB2 is required for pilus assembly.
As previously mentioned, in addition to GspA and 13 other cell wall-anchored proteins (7), the A. oris MG1 strain expresses four pilus proteins, FimP/Q and FimA/B, which constitute type 1 and type 2 fimbriae, respectively (3, 4, 6). We asked if the function of the two SPases is extended to these LPXTG motif-containing pilus proteins. Using the same approach as for GspA, whereby the CWSS of FimA was replaced with a His tag (Fig. 4D), we purified secreted FimA proteins from the culture medium and analyzed them by SDS-PAGE and Edman degradation. While the FimA proteins isolated from the WT and ΔlepB1 mutant strains migrated with the same mobility by SDS-PAGE, the FimA proteins isolated from the ΔlepB2 mutant migrated slightly more slowly than the first two (Fig. 4E). Consistently, Edman degradation revealed the same sequence, TETPNYGNIK, in the first two samples, supportive of the cleavage site between Ala (−1) and Thr (+1) (Fig. 4D), whereas the last one had ragged sequences, with the major species AVATETPNYG (Fig. 4F; see Table S5 in the supplemental material). The results indicate that LepB1 is not required for proper cleavage of the FimA signal peptide and that LepB2 is essential for FimA preprotein processing.
We next investigated the impact of LepB2-mediated cleavage of FimA precursors by examining pilus assembly on the bacterial cell surface by IEM, whereby A. oris cells were immobilized on nickel grids; washed cells were stained with an antibody to FimA, followed by a secondary IgG antibody conjugated to gold particles. Samples were then stained with 1% uranyl acetate and viewed with an electron microscope. As expected, FimA-labeled pili were abundant on the surface of WT cells but absent from a mutant lacking fimA (Fig. 5A and B); note that visible unstained pili in the ΔfimA mutant belonged to FimP pili (6). While deletion of lepB1 did not affect FimA assembly, deletion of lepB2 severely affected FimA pilus assembly (compare Fig. 5C and D). Ectopic expression of LepB2 in the lepB2 mutant rescued this assembly defect to the WT level (Fig. 5E). To examine if LepB2 is required for the assembly process of tip fimbrial proteins, we quantified the protein level of FimB and CafA on the bacterial surface by whole-cell ELISA. Consistent with the results described above, the ΔlepB2 mutant displayed a drastic FimB and CafA signal reduction, whereas no significant defect was observed in the ΔlepB1 mutant compared to the WT (see Fig. S1 in the supplemental material).
FIG 5.

The SPase LepB2 is required for pilus assembly. A. oris cells were immobilized on nickel grids and reacted with antiserum specific to the type 2 major pilin subunit FimA (A to E) or the type 1 major pilin subunit FimP (F to J), followed by goat anti-rabbit IgG conjugated to 12-nm gold particles, and stained with 1.0% uranyl acetate. Samples were viewed by transmission EM. Scale bars, 0.2 μm.
To determine if LepB2 also acts on type 1 fimbriae, we analyzed pilus assembly by IEM with an antibody to the fimbrial shaft FimP. As shown in Fig. 5F to J, the FimP phenotypes mirrored those of FimA presented above. Altogether, these results indicate that LepB2 is specifically utilized for the processing of pilus precursors in A. oris.
Requirement of the conserved catalytic dyad for LepB2 activity.
Bacterial type 1 SPases utilize a conserved Ser-Lys catalytic dyad present within conserved box B and box D, respectively, for their proteolytic activities (26). Our sequence alignment of LepB1 and LepB2 with many other Gram-positive SPases revealed the conservation of this catalytic dyad (Fig. 6A; see Fig. S2 in the supplemental material). In LepB2, Ser is located at position 101 and Lys is at 169. To determine if these residues are indeed required for LepB2 activity, we generated recombinant plasmids expressing LepB2 with an alanine substitution at S101 or K169. The resulting plasmids were introduced into the ΔlepB2 mutant, and the effects of these mutations on pilus assembly were analyzed by immunoblotting and EM.
FIG 6.
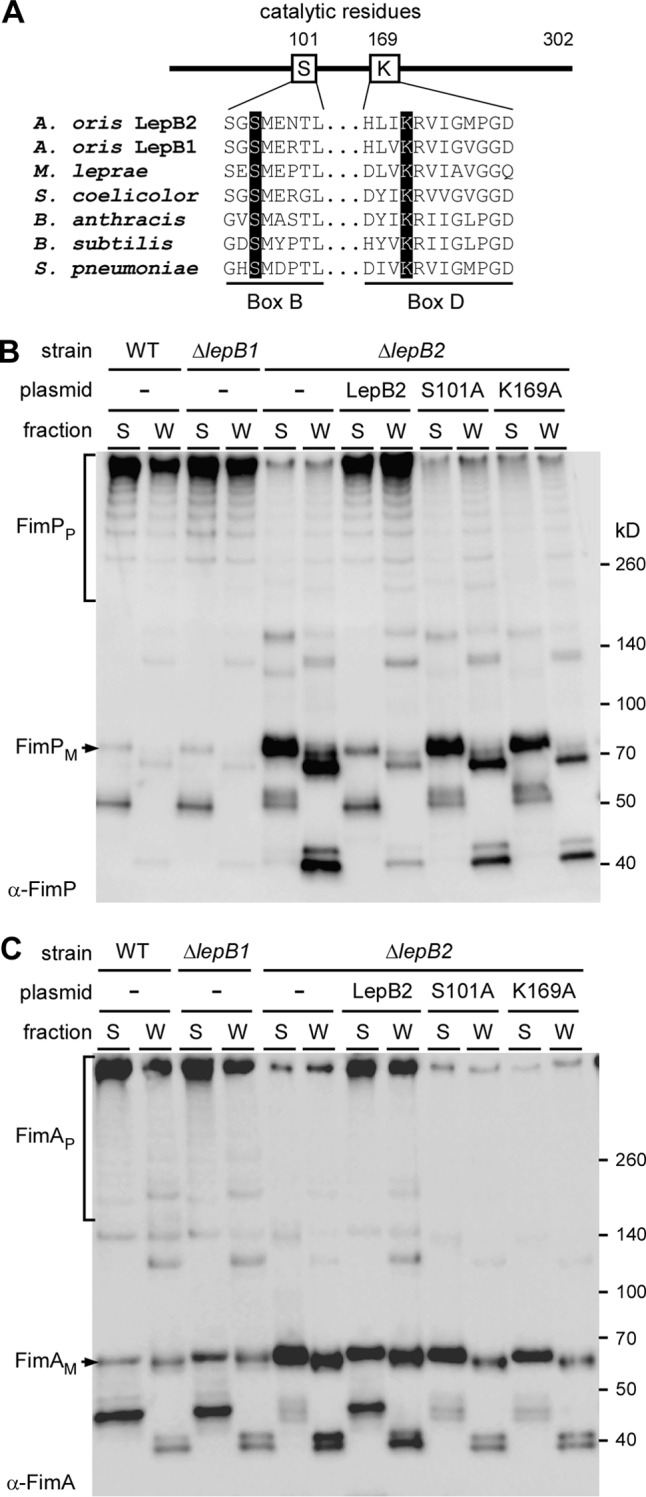
The Ser-Lys catalytic dyad of LepB2 is necessary for pilus assembly. (A) Multiple-sequence alignment of type 1 SPases from A. oris (LepB1 and LepB2), Mycobacterium leprae (LepB), Streptomyces coelicolor (Sip1), Bacillus anthracis (SipS), Bacillus subtilis (SipS), and Streptococcus pneumoniae (Spi) performed with CLUSTAL W (33). Conserved box B and box D, which contain the catalytic Ser and Lys residues (highlighted in black), respectively, are shown. Numbers indicate Ser and Lys positions in A. oris LepB2. (B) Supernatant (S) and cell wall (W) fractions were collected from MG1 and its isogenic derivatives. Equivalent protein samples were subjected to immunoblotting with anti-FimP antibody. (C) The same samples as in panel B were immunoblotted with anti-FimA antibody. The positions of fimbrial monomer (FimPM), HMW polymers (FimPP), and molecular mass markers are indicated.
By Western blotting, we observed HMW species of FimP and FimA, indicative of pilus polymers (designated P), in the culture medium (S) and cell wall (W) fractions of WT cells (Fig. 6B and C, first two lanes), as previously reported (3). While deletion of lepB1 did not affect pilus assembly (Fig. 6B and C, next two lanes), deletion of lepB2 greatly reduced pilus polymers and increased the accumulation of LMW products, presumably degradation products, in the membrane, as well as their secretion into the culture medium (Fig. 6B and C, lanes ΔlepB2). The defects of this mutant were rescued by ectopic expression of lepB2 (Fig. 6B and C, lanes ΔlepB2/pLepB2). As expected, alanine substitutions at S101 and K169 resulted in the same defects as deletion of lepB2 (Fig. 6B and C, last four lanes). The effects of the catalytic dyad mutations on pilus assembly were also confirmed by negative-staining EM (data not shown). Thus, LepB2 contains a canonical catalytic dyad present in type I SPase enzymes, and this dyad is critical for LepB2 activity.
Because LepB2 is required for pilus assembly and type 2 fimbriae are involved in polymicrobial interactions (or coaggregation) and biofilm formation, we then examined if LepB2 is also important for these processes. To test for polymicrobial interactions, Actinomyces cells were mixed with S. oralis in equal numbers and coaggregation was determined both visually (5) and quantitatively as previously reported (19). As shown in Fig. 7A and B, Actinomyces coaggregation with S. oralis was dependent on CafA, as deletion of cafA abrogated this interaction, consistent with our previous report (7). Compared to the parental MG1 strain, the lepB1 mutant did not display any noticeable defect in coaggregation. In contrast, deletion of lepB2 significantly reduced bacterial coaggregation; this defect was rescued by the overexpression from a plasmid of WT LepB2 but not by that of the catalytically inactive LepB2 variants, i.e., S101A and K169A. Finally, the ability of the LepB2 mutants to form biofilms was evaluated by using an established protocol (19) whereby Actinomyces biofilms were cultivated in the presence of 1% sucrose and quantified by staining with crystal violet (see Materials and Methods). Unlike the lepB1 mutant, which produced biofilms at the WT level, the lepB2 mutant was unable to form biofilms. The biofilm defect of the lepB2 mutant was restored when WT LepB2, but not the S101A or K169A mutant, was expressed ectopically (Fig. 7C and D). Altogether, these results support the notion that LepB2 is the SPase specific for LPXTG motif-containing pilus proteins in A. oris.
FIG 7.
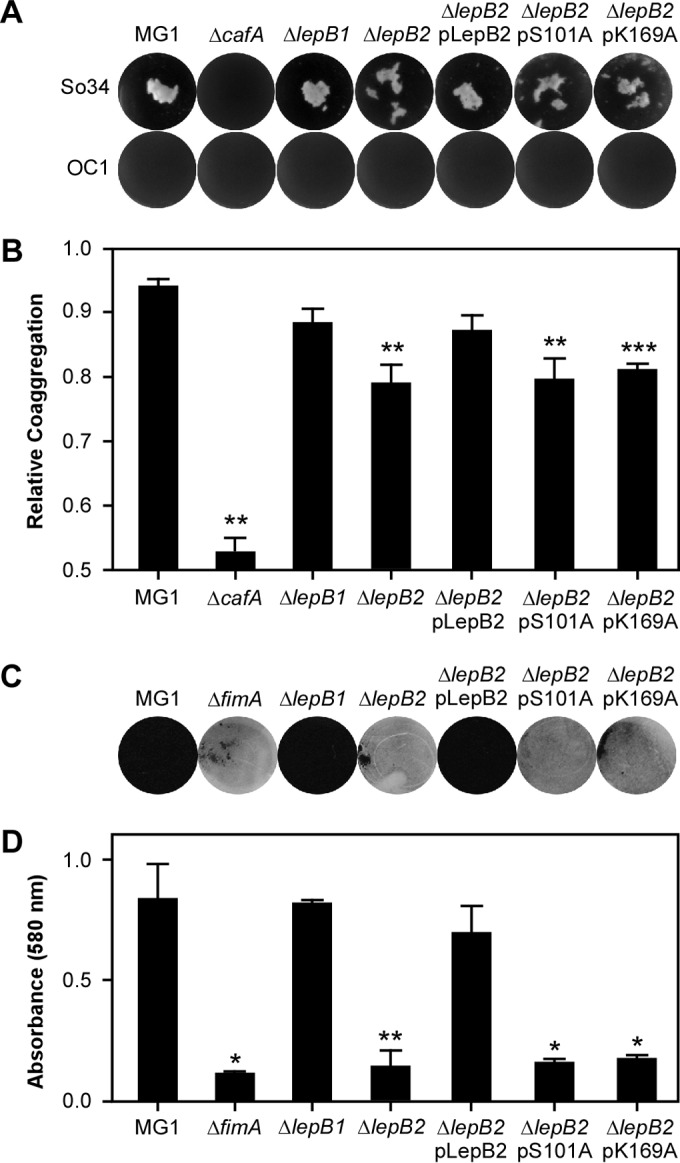
Requirement of LepB2 for polymicrobial interactions and biofilm formation. (A and B) The parental A. oris MG1 strain and variants were examined for the ability to interact with S. oralis (So34, positive for receptor polysaccharide [RPS]; OC1, RPS negative). Coaggregation was scored visually (A) or quantitatively by OD (B). (C and D) To cultivate biofilms, Actinomyces cells were grown in microtiter plates in the presence of 1% sucrose at 37°C with 5% CO2. Generated biofilms were stained with crystal violet (C) and subsequently quantified by measuring absorbance at 580 nm (D). The values shown are averages of three independent experiments performed in triplicate. *, P < 0.05; **, P < 0.01; *** P < 0.005 (determined by paired, two-tailed t test with GraphPad Prism).
DISCUSSION
We recently reported that the SPase LepB2 is a genetic suppressor of srtA essentiality in A. oris, a lethal phenotype associated with “glyco-stress” caused by accumulation of the glycosylated protein GspA in the cytoplasmic membrane when SrtA is disabled (8). The suppressor was found when we recovered a viable srtA deletion mutant with the Tn5 transposon inserted into the lepB2 gene (8). In this study, we confirmed the suppression phenotype of lepB2 by deleting chromosomal srtA in a strain already devoid of lepB2 (Fig. 1). It is important to note that deletion of srtA in the absence of gspA is also nonlethal (8), suggesting that LepB2 acts on the GspA glycosylation pathway. Indeed, in the ΔlepB2 ΔsrtA double mutant, GspA glycosylation was severely defective (Fig. 3), unlike the phenotype caused by srtA depletion, which does not affect GspA glycosylation (8). lepB2 is part of a gene locus that contains another SPase gene, i.e., lepB1 (Fig. 1A). This raises the question of redundancy, as multiple copies of SPases are typically present in a single Gram-positive bacterial species (26).
To address this question, we mapped the cleavage site of the GspA signal peptide by Edman degradation. Because GspA is heavily glycosylated, leading to a smeary migration pattern on SDS-PAGE (Fig. 3A), detecting the cleavage of the GspA signal peptide was challenging. To circumvent this problem, we constructed a GspA molecule lacking its CWSS (designated GspAΔCWSS), causing it to be secreted into the extracellular milieu with less chance of glycosylation. To our surprise, the GspAΔCWSS precursor was properly processed when expressed in the absence of LepB1 or LepB2 (Fig. 4B and C), considering that LepB2 is linked to GspA glycosylation. These results support the idea that both SPases are capable of cleaving the GspA signal peptide. This is consistent with the observation that, in the absence of both lepB1 and lepB2, the GspA signal peptide was not properly processed (Fig. 4C). It is intriguing, however, that the glycosylation level of GspA detected in the cell wall fraction of this double mutant was comparable to that of the parental and single deletion strains (Fig. 3A). This seems contradictory to a general belief that processing of the signal peptide is a prerequisite for protein maturation (27). We speculate that GspA glycosylation occurs immediately as the protein precursors emerge from the Sec translocon. This is in line with the “pilusosome hypothesis” that sortase and its substrates are in close proximity to a protein secretion machine for efficient assembly (28). It is also possible that in the absence of the two SPases, aberrant cleavage of the protein precursors, probably by a membrane-bound protease(s), is sufficient to release the polypeptides from the secretion machine for sortase and glycosyltransferases to be able to perform their functions.
While both LepB1 and LepB2 are capable of processing GspA, we demonstrated that only LepB2 is specific for cleavage of fimbrillin signal peptides. Using the major fimbrillin shaft protein FimA as an experimental model, we showed by N-terminal sequencing that cleavage of the FimA signal peptide depends on LepB2 (Fig. 4D to F). In the absence of the cognate SPase LepB2, the FimA signal peptide was proteolytically cleaved, producing ragged polypeptides (see Table S5 in the supplemental material). Unlike GspA, this failure of signal peptide cleavage severely affected fimbrial assembly (Fig. 5). Consequently, the abilities of this mutant to interact with oral streptococci and to form biofilm are significantly hindered (Fig. 7). LepB2 function is not limited to type 2 fimbriae, as the lepB2 mutant also fails to assemble type 1 fimbriae with FimP as the fimbrial shaft protein; in contrast, LepB1 is dispensable for these processes (Fig. 5 and 6).
The notion that a SPase is involved in pilus formation has been previously reported for the SPase-like protein SipA in the Gram-positive pathogen Streptococcus pyogenes (29). SipA is expressed by the gene locus that encodes the T3 pilin components in many strains of S. pyogenes (29). Although it has the same core fold as the type I SPase of E. coli (30), it lacks the catalytic Ser and Lys residues typical of type I SPases (31) (Fig. 6A). In place of Ser and Lys are Asp and Gly, but they do not play any role in SPase activity or cleavage of T3 prepilins (31). Unlike SipA, LepB2 contains a canonical catalytic dyad with conserved S101 and K169 (Fig. 6A). Alanine substitution of these residues abrogates pilus assembly, as well as biofilm formation (Fig. 6 and 7). Altogether, it is clear that LepB2 is a type I SPase that may serve as the prototype of type I SPases specific for pilus assembly in Gram-positive bacteria.
Intriguingly, it appears that LepB2 is specialized for fimbrial proteins, but it can process the GspA signal peptide. This raises the question of how specificity for a SPase is determined. It is worth pointing out that no distinct motifs or features in the signal peptide sequence of GspA and fimbrillins are apparent (see Table S3 in the supplemental material). In addition, all of the LPXTG motif-containing surface proteins in A. oris do not harbor a YSIRK-G/S motif, which has been shown to be involved in signal peptide processing in the low-GC-content Gram-positive bacterium Staphylococcus aureus (32). Although sequence alignment analysis of A. oris LepB1 and LepB2 reveals a conserved region encompassing the catalytic domains (Fig. 6), LepB1 has extended N- and C-terminal domains that are absent from LepB2. Whether these domains are involved in SPase specificity remains to be investigated. Nonetheless, in light of the pilusosome hypothesis, it is possible that the close proximity of LepB2 to sortase substrates within the assembly center confers SPase specificity. Thus, A. oris potentially provides an experimental model with which to examine this specificity mechanism.
Supplementary Material
ACKNOWLEDGMENTS
We thank Melissa Reardon-Robinson and our lab members for discussions and critical review of the manuscript and Michael Berne (Tufts Medical School) for Edman degradation.
We have no conflict of interest to declare.
Research reported in this publication was supported by the National Institute of Dental and Craniofacial Research of the National Institutes of Health under award DE017382 to H.T.-T.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00353-16.
REFERENCES
- 1.Ton-That H, Das A, Mishra A. 2011. Actinomyces oris fimbriae: an adhesive principle in bacterial biofilms and tissue tropism, p 63–77. In Kolenbrander PE. (ed), Genomic inquiries into oral bacterial communities. ASM Press, Washington, DC. [Google Scholar]
- 2.Yeung MK. 1999. Molecular and genetic analyses of Actinomyces spp. Crit Rev Oral Biol Med 10:120–138. doi: 10.1177/10454411990100020101. [DOI] [PubMed] [Google Scholar]
- 3.Mishra A, Das A, Cisar JO, Ton-That H. 2007. Sortase-catalyzed assembly of distinct heteromeric fimbriae in Actinomyces naeslundii. J Bacteriol 189:3156–3165. doi: 10.1128/JB.01952-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu C, Mishra A, Yang J, Cisar JO, Das A, Ton-That H. 2011. Dual function of a tip fimbrillin of Actinomyces in fimbrial assembly and receptor binding. J Bacteriol 193:3197–3206. doi: 10.1128/JB.00173-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mishra A, Devarajan B, Reardon ME, Dwivedi P, Krishnan V, Cisar JO, Das A, Narayana SV, Ton-That H. 2011. Two autonomous structural modules in the fimbrial shaft adhesin FimA mediate Actinomyces interactions with streptococci and host cells during oral biofilm development. Mol Microbiol 81:1205–1220. doi: 10.1111/j.1365-2958.2011.07745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mishra A, Wu C, Yang J, Cisar JO, Das A, Ton-That H. 2010. The Actinomyces oris type 2 fimbrial shaft FimA mediates co-aggregation with oral streptococci, adherence to red blood cells and biofilm development. Mol Microbiol 77:841–854. doi: 10.1111/j.1365-2958.2010.07252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reardon-Robinson ME, Wu C, Mishra A, Chang C, Bier N, Das A, Ton-That H. 2014. Pilus hijacking by a bacterial coaggregation factor critical for oral biofilm development. Proc Natl Acad Sci U S A 111:3835–3840. doi: 10.1073/pnas.1321417111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu C, Huang IH, Chang C, Reardon-Robinson ME, Das A, Ton-That H. 2014. Lethality of sortase depletion in Actinomyces oris caused by excessive membrane accumulation of a surface glycoprotein. Mol Microbiol 94:1227–1241. doi: 10.1111/mmi.12780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paetzel M. 2014. Structure and mechanism of Escherichia coli type I signal peptidase. Biochim Biophys Acta 1843:1497–1508. doi: 10.1016/j.bbamcr.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Ton-That H, Marraffini LA, Schneewind O. 2004. Protein sorting to the cell wall envelope of Gram-positive bacteria. Biochim Biophys Acta 1694:269–278. doi: 10.1016/j.bbamcr.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 11.Marraffini LA, Dedent AC, Schneewind O. 2006. Sortases and the art of anchoring proteins to the envelopes of Gram-positive bacteria. Microbiol Mol Biol Rev 70:192–221. doi: 10.1128/MMBR.70.1.192-221.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spirig T, Weiner EM, Clubb RT. 2011. Sortase enzymes in Gram-positive bacteria. Mol Microbiol 82:1044–1059. doi: 10.1111/j.1365-2958.2011.07887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dramsi S, Trieu-Cuot P, Bierne H. 2005. Sorting sortases: a nomenclature proposal for the various sortases of Gram-positive bacteria. Res Microbiol 156:289–297. doi: 10.1016/j.resmic.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 14.Reardon-Robinson ME, Osipiuk J, Chang C, Wu C, Jooya N, Joachimiak A, Das A, Ton-That H. 2015. A disulfide bond-forming machine is linked to the sortase-mediated pilus assembly pathway in the Gram-positive bacterium Actinomyces oris. J Biol Chem 290:21393–21405. doi: 10.1074/jbc.M115.672253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reardon-Robinson ME, Ton-That H. 2015. Disulfide-bond-forming pathways in Gram-positive bacteria. J Bacteriol 198:746–754. doi: 10.1128/JB.00769-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taton A, Unglaub F, Wright NE, Zeng WY, Paz-Yepes J, Brahamsha B, Palenik B, Peterson TC, Haerizadeh F, Golden SS, Golden JW. 2014. Broad-host-range vector system for synthetic biology and biotechnology in cyanobacteria. Nucleic Acids Res 42:e136. doi: 10.1093/nar/gku673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu C, Ton-That H. 2010. Allelic exchange in Actinomyces oris with mCherry fluorescence counterselection. Appl Environ Microbiol 76:5987–5989. doi: 10.1128/AEM.00811-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 19.Wu C, Mishra A, Reardon ME, Huang IH, Counts SC, Das A, Ton-That H. 2012. Structural determinants of Actinomyces sortase SrtC2 required for membrane localization and assembly of type 2 fimbriae for interbacterial coaggregation and oral biofilm formation. J Bacteriol 194:2531–2539. doi: 10.1128/JB.00093-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaplan A, Kaplan CW, He X, McHardy I, Shi W, Lux R. 2014. Characterization of aid1, a novel gene involved in Fusobacterium nucleatum interspecies interactions. Microb Ecol 68:379–387. doi: 10.1007/s00248-014-0400-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Broadway MM, Rogers EA, Chang C, Huang IH, Dwivedi P, Yildirim S, Schmitt MP, Das A, Ton-That H. 2013. Pilus gene pool variation and the virulence of Corynebacterium diphtheriae clinical isolates during infection of a nematode. J Bacteriol 195:3774–3783. doi: 10.1128/JB.00500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montealegre MC, La Rosa SL, Roh JH, Harvey BR, Murray BE. 2015. The Enterococcus faecalis EbpA pilus protein: attenuation of expression, biofilm formation, and adherence to fibrinogen start with the rare initiation codon ATT. mBio 6:e00467-15. doi: 10.1128/mBio.00467-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caspi R, Altman T, Billington R, Dreher K, Foerster H, Fulcher CA, Holland TA, Keseler IM, Kothari A, Kubo A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Subhraveti P, Weaver DS, Weerasinghe D, Zhang P, Karp PD. 2014. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res 42:D459-471. doi: 10.1093/nar/gkt1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jakubovics NS, Robinson JC, Samarian DS, Kolderman E, Yassin SA, Bettampadi D, Bashton M, Rickard AH. 2015. Critical roles of arginine in growth and biofilm development by Streptococcus gordonii. Mol Microbiol 97:281–300. doi: 10.1111/mmi.13023. [DOI] [PubMed] [Google Scholar]
- 25.Auclair SM, Bhanu MK, Kendall DA. 2012. Signal peptidase I: cleaving the way to mature proteins. Protein Sci 21:13–25. doi: 10.1002/pro.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Roosmalen ML, Geukens N, Jongbloed JD, Tjalsma H, Dubois JY, Bron S, van Dijl JM, Anne J. 2004. Type I signal peptidases of Gram-positive bacteria. Biochim Biophys Acta 1694:279–297. doi: 10.1016/j.bbamcr.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 27.Schneewind O, Missiakas D. 2014. Sec-secretion and sortase-mediated anchoring of proteins in Gram-positive bacteria. Biochim Biophys Acta 1843:1687–1697. doi: 10.1016/j.bbamcr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guttilla IK, Gaspar AH, Swierczynski A, Swaminathan A, Dwivedi P, Das A, Ton-That H. 2009. Acyl enzyme intermediates in sortase-catalyzed pilus morphogenesis in Gram-positive bacteria. J Bacteriol 191:5603–5612. doi: 10.1128/JB.00627-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zähner D, Scott JR. 2008. SipA is required for pilus formation in Streptococcus pyogenes serotype M3. J Bacteriol 190:527–535. doi: 10.1128/JB.01520-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young PG, Kang HJ, Baker EN. 2013. An arm-swapped dimer of the Streptococcus pyogenes pilin specific assembly factor SipA. J Struct Biol 183:99–104. doi: 10.1016/j.jsb.2013.05.021. [DOI] [PubMed] [Google Scholar]
- 31.Young PG, Proft T, Harris PW, Brimble MA, Baker EN. 2014. Structure and activity of Streptococcus pyogenes SipA: a signal peptidase-like protein essential for pilus polymerisation. PLoS One 9:e99135. doi: 10.1371/journal.pone.0099135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bae T, Schneewind O. 2003. The YSIRK-G/S motif of staphylococcal protein A and its role in efficiency of signal peptide processing. J Bacteriol 185:2910–2919. doi: 10.1128/JB.185.9.2910-2919.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.