

Abstract
Cohesin associates with distinct sites on chromosomes to mediate sister chromatid cohesion. Single cohesin complexes are thought to bind by encircling both sister chromatids in a topological embrace. Transcriptionally repressed chromosomal domains in the yeast Saccharomyces cerevisiae represent specialized sites of cohesion where cohesin binds silent chromatin in a Sir2-dependent fashion. In this study, we investigated the molecular basis for Sir2-mediated cohesion. We identified a cluster of charged surface residues of Sir2, collectively termed the EKDK motif, that are required for cohesin function. In addition, we demonstrated that Esc8, a Sir2-interacting factor, is also required for silent chromatin cohesion. Esc8 was previously shown to associate with Isw1, the enzymatic core of ISW1 chromatin remodelers, to form a variant of the ISW1a chromatin remodeling complex. When ESC8 was deleted or the EKDK motif was mutated, cohesin binding at silenced chromatin domains persisted but cohesion of the domains was abolished. The data are not consistent with cohesin embracing both sister chromatids within silent chromatin domains. Transcriptional silencing remains largely intact in strains lacking ESC8 or bearing EKDK mutations, indicating that silencing and cohesion are separable functions of Sir2 and silent chromatin.
INTRODUCTION
Cohesin is best known for its role as the molecular glue that holds sister chromatids together. The ring-shaped complex associates with chromatin in a topological manner with DNA passing through the central void (1, 2). Substantial evidence supports a model in which single cohesin complexes embrace both sister chromatids. Such a double-embrace model, however, cannot account for additional evidence that suggests that the complex mediates cohesion at some locations without encircling both chromatids (3).
Other nuclear processes utilize the ability of cohesin to hold unlinked or distant DNAs together. The complex binds double-stranded DNA breaks to assist in repair using DNA homology from the sister chromatid (4, 5). The complex also binds in and around genes to influence their transcription and provide insulation from neighboring chromosomal domains (6). In the yeast Saccharomyces cerevisiae, the complex binds chromosome arms at sites between genes that are oriented toward one another (7, 8). Convergent transcription is thought to slide the topologically bound complexes toward one another to accumulate between gene pairs.
In budding yeast, cohesin is guided to additional sites by the chromatin-bound, histone deacetylases Sir2 and Hst1 (9, 10). Both enzymes belong to an evolutionarily conserved family of NAD-dependent, protein deacetylases known as sirtuins (11, 12). There are five sirtuins in yeast (Sir2 and Hst1 to Hst4) and seven in humans (SirT1 to SirT7). Both yeast Sir2 and yeast Hst1 repress the transcription of the genes to which they bind, albeit through the assembly of radically different chromatin structures. In humans, deacetylation of histones and other proteins by sirtuins regulates aging, as well as pathologies related to human health and disease. In this study, we focus primarily on Sir2 to understand how the yeast sirtuins participate in sister chromatid cohesion.
Sir2 represses genes at the HM mating-type loci and telomeres through the formation of an extended heterochromatin-like structure known as silent chromatin (13). The protein associates with additional factors, Sir3 and Sir4, to form a complex that is recruited to nucleation sites on chromatin known as silencers. Sir2 deacetylates neighboring nucleosomes to create additional recruitment sites for Sir3 and Sir4, both of which possess an affinity for deacetylated histone tails. Through cycles of histone deacetylation and histone binding, the Sir complex spreads several kilobases from silencers to yield large domains of silent chromatin. Sir2 is also recruited to the rRNA gene cluster (known as the rDNA), where it represses RNA polymerase II transcription by a mechanism that is less well understood.
Cohesin binds to silenced loci, including the well-characterized HMR locus, in a manner that requires silent chromatin assembly (3). At HMR, binding of cohesin was shown to mediate cohesin-dependent cohesion of the silenced sister chromatids. A tRNA gene adjacent to HMR acts as a genomic loading site for cohesin complexes that ultimately accumulate at HMR (14). Evidence that Sir2 was critical for the cohesin function within silent chromatin domains came from experiments that reconstituted cohesion by tethering Sir2 directly to DNA (9). In this context, a nonenzymatic, 63-amino-acid fragment of the Sir2 C terminus was sufficient for cohesin-dependent cohesion. We refer to this fragment as the cohesion-proficient domain of Sir2. How the domain interacts with cohesin is not known.
In this study, we identified two determinants of Sir2-mediated cohesion of silent chromatin: a cluster of charged residues on the surface of Sir2 and the Sir2-interacting factor Esc8, which is a subunit of a presumed ISW1a chromatin-remodeling complex. By mutating each of these factors, we show that robust silencing and cohesion are separable functions of silenced chromosomal domains. Interestingly, neither Esc8 nor the Sir2 surface is required for cohesin binding. Instead, they are required for cohesin to function in promoting cohesion of silent chromatin domains.
MATERIALS AND METHODS
Strains and plasmids.
All plasmids used in this study are listed in Table S1 in the supplemental material. The oligonucleotides used in plasmid or strain construction are listed in Table S2 in the supplemental material. Plasmid pYFC12 was constructed with two overlapping fragments bearing the 3′ end of SIR2, the AKAA mutation, and the SIR2 3′ untranslated region, which were cloned into EcoRI-digested pRS423 by PCR-mediated plasmid gap repair in yeast. The 2μ region of the plasmid was then removed by digestion with AfeI and religation to yield pYFC13. Plasmids pYFC29, pYFC30, and pYFC31 were constructed similarly. All modifications within open reading frames were confirmed by sequencing.
All strains used in this study are listed in Table S3 in the supplemental material. Complete open reading frame deletions were generated by PCR-mediated gene replacement and confirmed by PCR. Strains with mutations in SIR2 were constructed by transformation with StuI-digested plasmids (pYFC13, pYFC29, pYFC30, and pYFC31), which generated tandem alleles of sir2: the first contained a full-length sir2 mutant, and the second lacked the promoter and 5′ end of the gene. Additional strains were derived from crosses and confirmed by genetic selection: YFC8 is a segregant of MC52 × CSW84, YFC101 is a segregant of YCF8 × CSW116, and YFC109 is a segregant of CRC83 × YFC8.
Cohesion assays.
Assays for cohesion of DNA circles were performed as described previously for the native HMR locus and the locus with lexA binding sites (3, 9). Briefly, cells were grown for 8 h in either yeast extract-peptone-dextrose-adenine (YPDA) or synthetic dextrose complete (SC) dropout medium (to select for plasmids) before inoculation of similar media containing 2% raffinose for overnight growth. On the following morning, the cultures were diluted back to an optical density at 600 nm of roughly 0.1 in yeast extract-peptone-adenine containing raffinose. When the cultures reached mid-log phase, nocodazole was added to arrest the cells in M phase (final concentration [Cf], 10 μg/ml; stock concentration, 1 mg/ml in dimethyl sulfoxide [Sigma]). Three hours later, galactose was added to a final concentration of 2% to induce DNA circle formation. After 2 h, cells were harvested and fixed with paraformaldehyde (Cf, 4%). In each case, data from at least three independent trials were pooled because they satisfied χ2 tests of the homogeneity of proportions. Error bars represent the standard errors of proportions. Each reported value was compared to that for an appropriate control by a χ2 test and judged to be significant using a 95% confidence interval.
ChIP assays.
Chromatin immunoprecipitation (ChIP) assays were performed as described in reference 9, with the following exceptions. Cultures were arrested in M phase and either cross-linked immediately to evaluate chromosomal loci or cross-linked 2 h after addition of galactose to evaluate DNA circles. Chromatin was sheared with a Bioruptor UCD-200 bath sonicator (Diagenode) in ice water using 20 cycles, each consisting of 15 s at high power followed by a 15-s rest. Anti-TAP antibody (Open Biosystems) and protein A Dynabeads (Invitrogen) were used for immunoprecipitation. DNA was eluted from the beads at 65°C for 10 min in 100 μl of buffer containing 1% SDS, 50 mM Tris-HCl, pH 7.5, and 10 mM EDTA. Cross-links were reversed by adding an equal volume of 10% Chelex 100 resin (Bio-Rad) and incubating at 95°C for 10 min. Quantitative PCRs were performed with a Rotor-Gene Q real-time PCR system (Qiagen) using the primers listed in Table S2 in the supplemental material. Pairwise Student's t tests relative to an appropriate control in each figure were performed to assess the statistical significance of at least three independent trials.
Silencing assays. (i) RT-PCR assay for expression of HMR a1 gene.
RNA extraction and reverse transcription-PCR (RT-PCR) were performed as described in reference 15 using the oligonucleotides listed in Table S2 in the supplemental material.
(ii) Spotting assays for HML, rDNA, and telomeric silencing.
Strains were grown overnight to saturation in YPDA medium and spotted in 10-fold serial dilutions on appropriate indicator plates.
(iii) Sectoring assay for HMR silencing.
For strains without plasmids, 300 to 500 cells from saturated cultures were spread on yeast extract-peptone-dextrose (YPD) plates. After 2 to 3 days, the plates were transferred to 4°C to enhance the red pigmentation. For strains carrying plasmids, cultures were grown in SC medium lacking Ura supplemented with extra adenine (Cf, 720 mg/liter) overnight at 30°C before they were spread cells on SC medium plates lacking Ura that contained 5 mg/liter of adenine.
Immunoblotting.
Cell pellets were suspended in lysis buffer (100 mM Tris, pH 8.0, 20% glycerol, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride) and agitated with glass beads in a Mini-BeadBeater-16 cell disrupter (BioSpec) at 4°C in a cold room for five cycles, each of which was 1 min long. Protein concentrations were determined by the Bradford method (catalog number 500-0006; Bio-Rad). After SDS-PAGE, protein transfer and immunoblotting were performed as described previously (16). Blots were incubated with anti-PGK1 antibody at a 1:5,000 dilution (catalog number ab-113687; Abcam) and either anti-lexA antibody at a 1:1,000 dilution (catalog number 06-719; EMD Millipore) or anti-Sir2 antibody at a 1:5,000 dilution (catalog number sc-6666; Santa Cruz Biotechnology). Membranes were washed and then incubated with a 1:5,000 dilution of horseradish peroxidase-conjugated (HPC) anti-rabbit immunoglobulin antibodies (catalog number W401B; Promega) and either HPC anti-mouse immunoglobulin antibodies (catalog number W402B, Promega) or HPC anti-goat immunoglobulin antibodies (catalog number sc2033; Santa Cruz Biotechnology). Immunoblots were developed with an enhanced chemiluminescence system (catalog number 32106, Thermo Scientific) according to the manufacturer's specifications.
Yeast two-hybrid screening.
Bait strains L40 (wild type [wt]) and YFC35 (sir2Δ) carrying plasmid pCSW55 (lexA-Sir2243–562-H364Y/TRP1, where Sir2243–562-H364Y indicates Sir2 from amino acids 243 to 562 with the H364Y mutation) were transformed with a LEU2 library of genomic fragments fused to GAL4AD (17). Positive interactors were first selected on SC medium lacking Trp, Leu, and His and containing 3-aminotriazole (0 to 20 mM) and/or nicotinamide (NAM; 0 to 2.5 mM). Positive candidates were then screened with a secondary lacZ, filter lift assay (16). All positive candidates were retransformed into a screening strain and tested again on SC medium lacking Trp, Leu, and His and containing 3-aminotriazole.
RESULTS
Full-length Hst1 mediates cohesion.
To help define regions of Sir2 that mediate cohesion, we sought to make comparisons between the protein sequences of Sir2 and Hst1. Previous work had shown that a small C-terminal fragment of Hst1 was sufficient for cohesion when tethered to DNA (9). To validate that full-length Hst1 also mediates cohesion, we used genetic manipulation to direct the protein to a locus where cohesion can be measured easily. Ordinarily, Hst1 is recruited by Sum1 to target promoters where it represses transcription via localized histone deacetylation (18–20). The well-characterized SUM1-1 mutant gene encodes a protein that gains the ability to bind silencers and spread in an HST1-dependent manner, even in the absence of Sir proteins (21–23). In this context, histone deacetylation and silencing of distal genes require the enzymatic activity of Hst1. We introduced the SUM1-1 mutation into a strain where the cohesion of HMR can be measured with fluorescence microscopy. In this strain, the locus is tagged nearby with a lac operator array that binds green fluorescent protein (GFP)-tagged lacI (GFP-lacI). A pair of target sites for the R site-specific recombinase flanks the construct. Induction of the recombinase in M phase-arrested cells generates a pair of GFP-tagged HMR DNA circles that yield one fluorescent dot if they cohere and two dots if they do not (Fig. 1A) (3).
FIG 1.

Full-length HST1 mediates cohesion at HMR in a SUM1-1 background. (A) Flow chart of assay. Silent chromatin, GFP-lacI, and recombinase sites are depicted with pink bars, green circles, and half-filled boxes, respectively. (B) Cohesion by full-length Hst1. Strains CSW10 (wt), CSW84 (sir2Δ), YFC8 (sir2Δ SUM1-1), and YFC97 (sir2Δ SUM1-1 hst1Δ) were used. NAM, nicotinamide; N, number of cells examined; p, P values obtained by pairwise χ2 tests relative to the results for a benchmark strain (−); ns, not significant. (C) Cohesion by HST1 requires cohesin. Strains YFC8 and YFC109 (sir2Δ SUM1-1 scc1-73) were arrested in M phase, and DNA ring formation was induced at 25°C before the cultures were split and half was shifted to 37°C for 2 h. (D) HST1 is required for cohesin binding at HMR in a SUM1-1 background. ChIP of Mcd1-TAP was performed in strains CSW116 (wt), YFC9 (sir2Δ), YFC101 (sir2Δ SUM1-1), and YFC103 (sir2Δ SUM1-1 hst1Δ). Binding at the both the a2 gene of HMR and the position 549 positive-control site was measured relative to binding at position 534, a negative-control site. P values obtained by Student's t tests relative to the results for a benchmark strain (−) are reported under the other strains.
In the parental strain with wild-type sirtuins and SUM1, cohesion of the HMR circles occurred in 67% of the M-phase cells (Fig. 1B). In the absence of SIR2, the frequency of cohesion dropped to 26%. Importantly, introduction of the SUM1-1 mutation restored cohesion, but this was abolished either by simultaneous omission of HST1 or by addition of nicotinamide (NAM), an inhibitor of sirtuin enzymatic activity. Cohesion in the SUM1-1 strain was also abolished by introduction of a temperature-sensitive allele of MCD1 (SCC1), which encodes a core subunit of cohesin (Fig. 1C). This result indicates that cohesion of HMR by HST1 in the SUM1-1 mutant also requires cohesin. Binding of cohesin at HMR was analyzed by chromatin immunoprecipitation (ChIP) of a TAP-tagged MCD1 allele with an anti-TAP antibody in M-phase-arrested cells. All measurements were made relative to the measurement for a negative-control site and compared to the measurement for a positive-control site (positions 534 kb and 549 kb on chromosome V, respectively). In Fig. 1D, cohesin bound the chromosomal HMR locus in the parental strain, but that binding was lost when SIR2 was deleted (9). Significantly, binding of cohesin was partly restored when the SUM1-1 allele was introduced, but this binding was abolished when HST1 was deleted. At the positive-control site (labeled 549 in Fig. 1D), introduction of SUM1-1 or deletion of HST1 did not influence cohesin binding. Collectively, these results show that full-length Hst1, like Sir2, generates cohesion of silenced sister chromatids by recruiting cohesin.
A Sir2 surface required for heterochromatic cohesion.
Using amino acid conservation between Hst1 and Sir2 homologs in Saccharomyces sensu stricto species as one guiding criterion, we sought mutations within the cohesion-proficient domain of Sir2 that disrupt cohesion (Fig. 2A). Our attention was drawn to a cluster of charged amino acids, each mapping to a compact, solvent-exposed surface of a Sir2 crystal structure, defined by an antiparallel beta strand and adjoining hairpin loop (Fig. 2B). The glutamic and aspartic acids at positions 545 and 547, respectively, are absolutely conserved. The lysine at position 548 is fairly conserved and is replaced by isoleucine only when a second isoleucine is at position 546. Here we describe the consequences of simultaneously mutating the charged residues at E545, D547, and K548 to alanines. Other surface residues meeting our criteria were tested but did not produce effects as strong as those described below. Here, we refer to the charged cluster motif of Sir2 as EKDK and the mutant sir2 allele with the designation AKAA (sir2-AKAA).
FIG 2.

The EKDK motif is required for SIR2-mediated cohesion. (A) Alignment of S. cerevisiae Sir2 (residues 499 to 558) and Hst1 (residues 440 to 449) to homologs in Saccharomyces sensu stricto species, with conservation noted according to Clustal Omega criteria (61): asterisks, identical residues; colons, conservative substitutions; periods, semiconservative substitutions. Charged residues that were mutated are highlighted in red. Scer, Saccharomyces cerevisiae; Spar, Saccharomyces paradoxus; Smik, Saccharomyces mikatae; Skud, Saccharomyces kudriavzevii ; Sarb, Saccharomyces arboricola; Sbay, Saccharomyces bayanus. (B) Crystal structure of Sir2 residues 220 to 556. Sir2 crystallizes as a dimer of nearly identical subunits, but only one is shown (PDB accession number 2HJH, deposited by T. Ellenberger). The cohesion-proficient domain is shown in black, and the mutated residues of the EKDK motif are shown in red. (C) Flow chart of tethering assay. (D) The sir2-AKAA mutation abolishes cohesion by tethered Sir2. Strain CSW42 (sir2Δ) expressing lexA-Sir2499–562 (pYFC1), lexA-Sir2499–562-AKAA (pYFC10), and lexA alone (pBTM116H) was evaluated. (E) The sir2-AKAA mutation abolishes the cohesion of HMR. Strains CSW10 (wt), YFC1 (sir2-AKAA), and CSW84 (sir2Δ) were evaluated. (F) Cohesin recruitment by full-length Sir2-AKAA. ChIP of Mcd1-TAP was performed with strains CSW116 (wt), YFC7 (sir2-AKAA), and YFC9 (sir2Δ). P values obtained by Student's t test are reported relative to the results for the wild-type strain.
The role of the EKDK motif in Sir2-mediated cohesion was measured first with a protein-targeting assay (9). In this assay, a protein fragment of interest is tethered via lexA to GFP-tagged DNA circles formed by recombination in M-phase-arrested cells (Fig. 2C). To this end, lexA was fused to the 63-amino-acid cohesion-proficient domain of Sir2 (residues 499 to 562 [Sir2499–562]). The domain is not sufficient to nucleate silencing when tethered directly to DNA (see Fig. S1 in the supplemental material). Cohesion of the DNA circles occurred in 54% of the cells expressing lexA-Sir2499–562, as reported previously (9). When lexA-Sir2499–562-AKAA was expressed, cohesion occurred in only 30% of the cells (Fig. 2D). By comparison, expression of lexA alone yielded cohesion in only 25% of the cells. The negative impact of the AKAA mutation on cohesion was not due to differential protein expression. Immunoblotting with antibodies against lexA showed that both chimeras were expressed equally (see Fig. S2A in the supplemental material). These data show that the EKDK motif mediates cohesion by Sir2.
To test whether the Sir2 EKDK cluster is required for cohesion in a silent chromatin context, HMR DNA circles were examined in a strain bearing the AKAA mutation within full-length SIR2. Immunoblotting with antibodies against Sir2 showed that the mutant was expressed at normal levels, as were all other full-length sir2 alleles used in this study (see Fig. S2B in the supplemental material). As first shown in Fig. 1B, HMR circles cohered in 67% of the parental strain, whereas cohesion dropped to 26% in a sir2Δ negative control (Fig. 2E). The sir2-AKAA allele yielded cohesion in only 37% of the cells. We conclude that the EKDK motif of Sir2 is important for cohesion of native silent chromatin. That cohesion was not entirely eliminated by the AKAA mutation in full-length Sir2 as it was with the tethered cohesion-proficient domain indicates either that the mutation is not fully penetrant or that other features of silent chromatin, perhaps even other domains of Sir2, contribute to cohesion at HMR.
Cohesin recruitment by the Sir2 EKDK motif.
ChIP was used to measure the impact of the AKAA mutation on cohesin binding at the chromosomal HMR locus, as described in the legend to Fig. 1D. Unexpectedly, Mcd1-TAP bound as well at HMR in the sir2-AKAA mutant as it did in the wild-type strain (Fig. 2F). Similar results were obtained when ChIP was performed on the HMR DNA circles used in the cohesion assay (see Fig. S3 in the supplemental material). These data indicate that cohesion loss in the AKAA mutant, as indicated in Fig. 2E, is not due to the loss of cohesin binding. If the EKDK motif is the sole cohesin recruitment surface of Sir2, then mutation to AKAA may result in loading of nonproductive cohesin complexes. Alternatively, additional features of silent chromatin may participate in cohesin recruitment, but those surfaces cannot utilize cohesin without participation of the EKDK motif. Either way, it should be noted that inactivation of Sir2 with an inhibitor during M phase also caused a loss of cohesion without a loss of cohesin binding (3).
Cohesion defects in sir2-AKAA strains are not caused by defects in silencing.
A simple explanation for the loss of cohesion in strains bearing the AKAA mutation in full-length Sir2 is that the alteration interferes with proper silent chromatin assembly. Therefore, RT-PCR was used to determine whether the a1 gene at HMR remained silent when the locus was excised from the chromosome and circularized in the cohesion assay. mRNA from the nonsilent KCC4 locus was used as an internal control. Figure 3A shows that a1 transcripts were not detectable either in a wild-type strain or in the sir2-AKAA mutant. In contrast, a1 transcripts were easily measured in a strain lacking SIR2. These results indicate that the sir2-AKAA mutation does not impair silencing of HMR.
FIG 3.

Persistence of transcriptional silencing in strains with sir2-AKAA. (A) Analysis of HMR a1 by RT-PCR. RNA extracts were prepared from strains YFC150 (wt), YFC152 (sir2-AKAA), YFC153 (esc8Δ), and YFC151 (sir2Δ) that had been grown according to the cohesion assay protocol. (B) Silencing of URA3p-ADE2 reporter at HML. Strains THC74 (wt), YFC24 (sir2-AKAA), and CCC3 (sir2Δ) were spotted in 10-fold serial dilutions on SC medium lacking Ade (SC-ade) to measure silencing and on SC medium as a loading control. (C) Silencing of URA3 at a telomere created at ADH4 by truncation of the chromosome VII left arm (TelVIIL). Strains YDS631 (wt), YFC12 (sir2-AKAA), and CCC1 (sir2Δ) were spotted on SC medium plus 5-FOA (SC+FOA) to measure silencing and on SC medium as a loading control. (D) Silencing of URA3 within a TRP1p-URA3::LEU2 cassette at the IGS1 and IGS2 sites of an rRNA gene repeat. Strains DMY2804 (wt), YFC13 (sir2-AKAA), and DMY2835 (sir2Δ) with the reporter at IGS1 and strains DMY2800 (wt), YFC3 (sir2-AKAA), and DMY2831 (sir2Δ) with the reporter at IGS2 were spotted on SC medium lacking Leu plus 5-FOA (SC-leu+FOA) to measure silencing and SC medium lacking Leu (SC-leu) as a loading control.
Silencing of HMR is particularly robust, which might mask subtle defects in Sir2-mediated transcriptional repression. Therefore, phenotypic assays were used to measure the impact of the AKAA mutation on silencing at other locations where Sir2 acts. At the HML mating-type locus, silencing was measured with an ADE2 reporter gene, which is required for adenine prototrophy (24). Saturated cell cultures were spotted in 10-fold serial dilutions on indicator plates with SC medium lacking Ade. Figure 3B shows that neither the wild-type strain nor the sir2-AKAA strain grew in the absence of adenine, whereas the sir2-null strain grew unhindered. These results show that the sir2-AKAA mutation has no substantial impact on silencing at HML.
Silencing at telomeres and the rDNA was measured with URA3 reporter genes inserted either at the terminus of a chromosome VII truncation or within the intergenic regions of the rRNA gene repeat unit (IGS1 and IGS2). Tenfold serial dilutions of saturated cultures were spotted on SC medium plates containing 5-fluoro-orotic acid (5-FOA), a toxic metabolite of the URA3 gene product (25). In each case, the wild-type strain and the sir2-AKAA mutant grew equally well, whereas the sir2-null strain did not grow at all (Fig. 3C and D). These results indicate that silencing at the rDNA and telomeres is not affected by the sir2-AKAA mutation. If it is presumed that the AKAA mutation causes a loss of cohesion at all of these loci as it does at HMR, the results indicate that robust transcriptional silencing does not require cohesion of silent chromatin.
Silencing and cohesion are separable functions of Sir2.
Collectively, the silencing assays described in the preceding section indicate that mutation of the Sir2 EKDK motif does not cause a gross alteration of silent chromatin structure. To evaluate silencing at HMR with greater sensitivity, we turned to an assay that measures persistence of the silent state during colony formation (26). The assay utilizes an attenuated HMR locus with a partially disabled silencer (hmrΔa) and an ADE2 reporter gene that suppresses accumulation of a red pigment on medium with low levels of adenine. Lineages of cells that have switched from the silent to the nonsilent state produce white sectors in an otherwise red background, whereas lineages of cells that restore silencing yield red sectors in a white background. We binned colonies into four categories based on visual inspection: (i) fully red, (ii) mostly red, (iii) mostly white, and (iv) fully white. Figure 4A shows that the hmrΔa::ADE2 reporter was fully silenced (completely red) in 72% of the colonies of a wild-type strain, whereas no red pigment was observed in a sir2Δ strain. Significantly, only 1% of the colonies were fully silenced in the AKAA mutant. Thus, sir2-AKAA shifts the silencing equilibrium toward the derepressed state in this sensitized assay.
FIG 4.

Separation of silencing and cohesion functions of Sir2 with the AKAA mutation. (A) Variegated silencing of an attenuated HMR locus by the sir2-AKAA mutation. Strains GCY317 (wt), YFC44 (sir2-AKAA), and YFC116 (sir2Δ) bearing the hmrΔa::ADE2 allele were used. (B) Suppression of silencing defects in the strains described for panel A by a centromeric SIR4 expression plasmid (pJR368). Empty vector (pRS416) was used as a control. At least 300 cells for each strain-plasmid combination were scored in at least three independent trials. (C) Extra SIR4 does not suppress the sir2-AKAA cohesion defect at HMR. HMR DNA circles were evaluated in strains CSW10 (wt), YFC1 (sir2-AKAA), and CSW84 (sir2Δ) bearing either a centromeric SIR4 expression plasmid (pJR368) or empty vector (pRS416). (D) Point mutations within the Sir2 EKDK motif disrupt cohesion of HMR DNA circles. Strains CSW10 (wt), YFC1 (sir2-AKAA), YFC137 (sir2-AKAK), YFC138 (sir2-AKDK), and YFC139 (sir2-EKAK) were evaluated. (E) Point mutations of the Sir2 EKDK motif impact silencing minimally. Silencing of the hmrΔa::ADE2 reporter was evaluated in strains GCY317 (wt), YFC44 (sir2-AKAA), YFC140 (sir2-AKAK), YFC141 (sir2-AKDK), and YFC142 (sir2-EKAK).
The silencing defects highlighted by the sectoring assay raised the possibility that the loss of cohesion was due to subtle differences in silencing rather than a cohesin-specific defect introduced by the AKAA mutation. In a recent cocrystal of Sir2 and Sir4 fragments, the last residue of the EKDK motif (K548) was found to be precariously close to a Sir2 cleft occupied by Sir4 (27). Binding of Sir4 displaces the K548 residue from the cleft (see Fig. S4 in the supplemental material). Thus, it was conceivable that the AKAA mutation destabilizes the silent chromatin structure by perturbing the Sir2-Sir4 interaction. To disentangle the roles of the EKDK motif in silencing and silent chromatin cohesion, we attempted to suppress the silencing defect of the AKAA mutant by providing additional Sir4. The protein is limiting for silencing, and even subtle increases in the SIR4 gene dosage can improve transcriptional repression (26, 28–30). In the case of the sectoring assay, extra SIR4 provided by a low-copy-number centromere plasmid in a wild-type strain increased the fraction of completely red colonies from 65% to nearly 100% (Fig. 4B). Significantly, the additional SIR4 also restored silencing in a sir2-AKAA mutant, increasing the number of completely red colonies from 1% to 86%. These results show that roughly doubling the level of SIR4 suppresses the silencing defect caused by sir2-AKAA.
We next asked whether suppression of the AKAA silencing defect also suppresses the cohesion defect. To address this question, we evaluated cohesion of HMR DNA circles in strains bearing extra SIR4 on a low-copy-number centromere plasmid. Irrespective of the SIR2 allele tested (SIR2, sir2Δ, or sir2-AKAA), the additional SIR4 did not improve cohesion (Fig. 4C). These results demonstrate that cohesion defects imposed by sir2-AKAA persist even when silencing is restored to full capacity. We conclude that silencing and cohesion are separable functions of SIR2 and that the sir2-AKAA mutation affects the cohesion of silent chromatin directly.
To analyze the EKDK motif in finer detail, individual residues of Sir2 were mutated. Replacement of E545 with alanine yielded the sir2-AKDK mutant, replacement of D547 with alanine yielded the sir2-EKAK mutant, and replacement of both amino acids with alanine yielded the sir2-AKAK mutant. Importantly, the sir2-AKDK mutant altered the EKDK residue that lies furthest from Sir4 in the cocrystal (see Fig. S4 in the supplemental material). Figure 4D shows that each of the three new mutants disrupted the cohesion of silent chromatin to the same extent as the original AKAA mutation. Additionally, each of the new mutants yielded a derepression phenotype that was intermediate between that of the wild type and that of the original sir2-AKAA mutant in the sensitive hmrΔa::ADE2 sectoring assay (Fig. 4E). Collectively, the results obtained with the new mutants show that cohesion defects do not scale linearly with silencing defects. These results reinforce the notion that mutations of the EKDK domain do not disrupt silent chromatin cohesion by interfering with Sir4.
Esc8: a Sir2-interacting partner required for silent chromatin cohesion.
Traditional two-hybrid screening was performed to identify Sir2-interacting factors responsible for Sir2-mediated cohesion. A bait protein containing the Sir2 enzymatic core and the adjacent cohesion-proficient domain (collectively spanning amino acids 243 to 562) recovered a number of previously identified Sir2-interacting factors from a genomic library. Our studies focused on one factor that was isolated, Esc8, which was first identified in a screen for proteins that restore silencing when tethered to a mutated silencer (31). Esc8 has been shown to associate directly with Sir2 in glutathione S-transferase pulldown experiments. Our two-hybrid studies showed that the C-terminal domain of Esc8 (amino acids 640 to 714) contains a Sir2 interaction domain. Additionally, Esc8 and Sir2 interacted when the AKAA mutation was present in Sir2, suggesting that the EKDK motif is not required for the association of the two proteins (see Fig. S5 in the supplemental material).
Esc8 is a paralog of Ioc3, a protein that associates with the Isw1 ATPase to form ISW1a, a chromatin-remodeling complex (32). Homology modeling with a crystal structure of ISW1a suggests that Esc8 retains the residues necessary for Isw1 binding (33). Indeed, Esc8 and Isw1 interact in proteome-scale studies (34). Thus, we infer that Esc8 associates with Isw1 in vivo to form a variant of the ISW1a chromatin-remodeling complex.
Cohesion of HMR DNA circles was measured in strains lacking ESC8, IOC3, or ISW1. Figure 5A shows that cohesion of the circles was disrupted in the esc8 and isw1 mutants and to a less severe extent in the ioc3 mutant. Isw1 associates with Ioc2 and Ioc4 to form a distinct chromatin-remodeling complex named ISW1b. Deletion of IOC4 affected HMR cohesion to a similar extent as deletion of IOC3 (Fig. 5A). These results suggest that the ISW1 chromatin-remodeling complexes facilitate the cohesion of HMR but that ISW1a complexes containing Esc8 play the most prominent role.
FIG 5.

ESC8 in silent chromatin cohesion and transcriptional silencing. (A) Cohesion of HMR requires ESC8. HMR DNA circles were produced in strains CSW10 (wt), YFC18 (esc8Δ), YFC133 (isw1Δ), YFC130 (ioc3Δ), YFC144 (ioc4Δ), and CSW84 (sir2Δ). (B) Cohesion by tethered Sir2 does not require ESC8 or the IOC genes. DNA circles bearing the lexA binding sites were produced in strains CSW42 (sir2Δ), YFC135 (sir2Δ isw1Δ), YFC21 (sir2Δ esc8Δ), YFC128 (sir2Δ ioc3Δ esc8Δ), and YFC145 (sir2Δ ioc4Δ) expressing lexA-Sir2499–562 (pYFC1). Strain CSW42 expressing lexA alone (pBTM116H) was included as a negative control. (C) Silencing defects in the absence of ESC8 are suppressed by extra SIR4. Assays were performed with strains GCY317 (wt) and GCY310 (esc8Δ) bearing either a centromeric SIR4 expression vector (pJR368) or empty vector (pRS416). (D) Extra SIR4 does not suppress cohesion defects caused by the loss of ESC8. HMR DNA circles were produced in strains CSW10 (wt), CSW84 (sir2Δ), and YFC18 (esc8Δ) bearing either an empty vector (pRS416) or a centromeric SIR4 expression vector (pJR368).
Cohesion by tethered lexA-Sir2499–562 was also measured in strains lacking subunits of the Isw1 remodeling complexes. Whereas cohesion by the chimeric protein was abolished in a strain lacking ISW1, deletion of ESC8 alone or in combination with IOC3 bore no effect (Fig. 5B). Deletion of IOC4 also had no impact. Similar results were obtained with lexA-Sir278–562, which nucleates silent chromatin domains when tethered to DNA (see Fig. S1 and S6 in the supplemental material). Why do the two assays for cohesion, one using native chromatin and the other using tethered proteins, display different dependencies on ESC8? We suspect that the distinction lies in the cis-acting silencers of the loci under study. The native silent chromatin assay includes the E and I silencers that normally flank HMR, whereas the tethering assay replaces both silencers with a single highly engineered construct that contains lexA sites (35). Apparently, the replacement of bona fide silencers with sites for artificial Sir2 recruitment interferes with the specification for the ISW1a chromatin remodeling complex. The results suggest that Isw1 acts without Ioc proteins on the synthetic construct, as was found for Isw1 in transcriptional silencing at the rDNA (36).
Disentangling of the roles of ESC8 in silencing and sister chromatid cohesion.
Previous characterization of ISW1 complexes in transcriptional silencing demonstrated that deletion of ESC8 yielded phenotypes less severe than those yielded by deletion of ISW1 (31, 37). In support, the RT-PCR assay in Fig. 3A shows that the gene was not required for silencing of HMR DNA circles. Deletion of ESC8, however, did produce silencing defects when the sensitive hmrΔa::ADE2 reporter was used, as confirmed in Fig. 5C (compare the result to that for the wild type in Fig. 4B). Given the minimal but measurable role of Esc8 in silencing, we focused on esc8 mutants for the remainder of the study.
If an esc8 mutant compromises silencing even in subtle ways, then the diminution of silencing could be responsible for the loss of cohesion. To investigate this possibility, we tested whether extra SIR4 could suppress the silencing defect of an esc8 mutant. Figure 5C shows that a centromeric plasmid bearing SIR4 restored silencing to normal levels. Importantly, restoration of silencing by extra SIR4 did not suppress the cohesion defect in the absence of ESC8 (Fig. 5D). This result suggests that ISW1a complexes containing Esc8 affect the cohesion of silent chromatin, in addition to the role that the complexes play in transcriptional silencing.
ChIP was used to test whether ESC8 is required for cohesin binding at silenced loci. Figure 6A shows that the association of the Mcd1 cohesin subunit with HMR was not diminished by the loss of ESC8. Similar results were obtained when ChIP was performed on the HMR DNA circles used in the cohesion assay (see Fig. S3 in the supplemental material). As with the AKAA mutation, these data indicate that ESC8 is required for the formation of productive cohesin complexes at silent chromatin but not recruitment of the complex to the locus.
FIG 6.
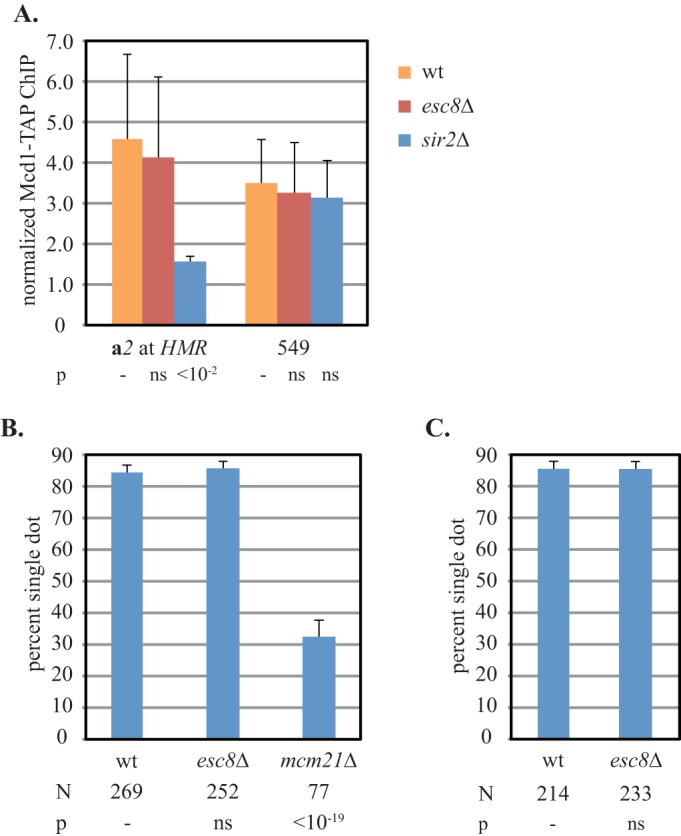
Loss of ESC8 disrupts the cohesion of silent chromatin selectively without causing a loss of cohesin. (A) Binding of cohesin at HMR does not require ESC8. ChIP of Mcd1-TAP was measured in strains CSW116 (wt), YFC9 (sir2Δ), and YFC95 (esc8Δ). (B) ESC8 is not required for pericentric cohesion. Cohesion of plasmid pPCM14tet0 was measured in nocodazole-arrested strains pMY127 (wt), YFC96 (esc8Δ), and YFC108 (mcm21Δ). (C) ESC8 is not required for cohesion of the right arm of chromosome III. The cohesion of the HMR locus in strains CSW10 (wt) and YFC18 (esc8Δ) was evaluated immediately after arrest in the M phase without forming DNA circles.
To test whether ESC8 is a global cohesion regulator, cohesion at other loci was tested. Centromeric cohesion was evaluated with a centromeric plasmid bearing tet operators in a strain expressing tetR-GFP (38). Figure 6B shows that the plasmid exhibited a high level of cohesion (84%) in wild-type cells arrested in M phase. Cohesion was not diminished by deletion of ESC8. As a positive control, MCM21, a gene required for pericentromeric cohesion, was deleted (39). Accordingly, cohesion of the centromeric plasmid was abolished. These results indicate that ESC8 is not required for pericentromeric cohesion. Cohesion of euchromatic sites distal to pericentromeric domains was tested in two ways. First, we evaluated chromosome arm cohesion using the strain described above with a GFP-tagged HMR locus. In the absence of the galactose-induced recombinase, the GFP tag reports on cohesion of the right arm of chromosome III. In cells arrested in M phase, chromosome III arm cohesion persists in the absence of ESC8, even if the local cohesion of HMR is compromised (Fig. 6C). As a second measure of the role of ESC8 in cohesion, we examined cohesion of the euchromatic gene URA3. Concurrent studies in our lab have shown that URA3 is sufficient for cohesion when the gene is present in DNA circles (J. S. Campor, M. S. Borrie, and M. R. Gartenberg, unpublished data). Deletion of ESC8 did not disrupt URA3-mediated cohesion either (data not shown). Together, these results suggest that the ISW1a complex containing Esc8 is not a general cohesion factor but, rather, one that acts specifically on a subset of chromosomal domains that include Sir2-mediated silent chromatin.
DISCUSSION
Sirtuin-mediated sister chromatid cohesion.
This study extends our understanding of the specialized form of sister chromatid cohesion that occurs at Sir2-related, silent chromatin domains. To help identify features of Sir2 that are important for cohesion, we first evaluated the cohesive properties of another sirtuin, Hst1. We showed that full-length Hst1, like Sir2, promoted cohesion when the protein was redirected to form a repressive chromatin structure at HMR. By extrapolation, we propose that both Sir2 and Hst1 similarly promote cohesion at other locations in the genome where the two proteins bind (10). Since the sirtuins are not required for all sister chromatid cohesion, the sirtuin-bound locations constitute a distinct class of chromosome arm cohesion sites.
A surface motif of Sir2 that supports sister chromatid cohesion.
By mapping the cohesion-proficient domains of the sirtuins to a Sir2 crystal structure, we identified a surface motif necessary for Sir2-mediated cohesion. Mutating conserved residues in the EKDK motif abolished HMR cohesion with virtually no impact on silencing at any of the loci where Sir2 acts. Silencing defects at HMR were detected only with a highly sensitive colony formation assay, and the defects were easily suppressed, as described below. These results indicate that silencing and cohesion are separable functions of Sir2 and that cohesion of silent chromatin is not an obligatory prerequisite for transcriptional silencing.
The subtle silencing defects associated with mutations in EKDK were reversed by mild increases in Sir4, a factor whose quantity is limiting for silencing (30). Thus, if cohesion promotes silencing in any way, it might be to increase the local concentration of Sir4. According to this argument, the emergent silent chromatin of each sister chromatid recruits dynamically equilibrating Sir4 proteins, which are then enriched locally for both sister chromatids that are held together by cohesion. This so-called Circe effect has also been invoked to explain the benefits to silencing of clustered silent chromatin domains at the nuclear periphery (40).
Previous reports documented a role for cohesin as an inhibitor of silent chromatin in assays for the establishment and expansion of silent chromatin domains (41–43). In those cases, the chromatin association of cohesin was modulated by experimental means. Here, cohesin function is disrupted by mutations that do not disrupt cohesin binding. Thus, the negative regulation of silent chromatin by cohesin in the previous studies may arise from the presence of the cohesin complex and not from the cohesion of sister chromatids that the complex imparts.
We propose that Sir2 associates directly with cohesin or an intermediary adaptor protein and that the EKDK motif of Sir2 controls the function of that factor. It is unlikely that the EKDK motif is the sole cohesin recruitment surface because cohesin still accumulates at HMR when the EKDK domain is mutated. Thus, we propose that a second cohesin recruitment site exists within Sir2 or some other component of silent chromatin.
Esc8 and the ISW1a chromatin-remodeling complex in silent chromatin cohesion.
Our attempts to find Sir2-interacting factors that mediate cohesion did not yield cohesin or other intuitive adaptor proteins. Direct tests for interactions between Sir2 and the Smc1 and Scc3 cohesin subunits by coimmunoprecipitation yielded negative results (data not shown). We did, however, find that Esc8, an understudied Sir2-interacting factor, facilitates silent chromatin cohesion. Esc8 associates with the Isw1 ATPase and is thought to form an alternative ISW1a chromatin-remodeling complex. In general, members of the ISWI family create regularly spaced nucleosome arrays, maintain chromatin organization across transcribed regions, and negatively affect transcription (32, 44, 45). ISW1a, in particular, has been shown to oppose chromatin opening at promoters by repositioning nucleosomes near the nucleosome-depleted regions at the ends of genes (46–48). Certain transcription factors, like Rap1 and Abf1, create nucleosome-depleted regions that recruit ISW1 complexes for their subsequent roles in adjacent chromatin domains (49). Both Rap1 and Abf1 bind the nucleosome-free silencers that flank the HM loci (50, 51). If HM silencers recruit ISW1 complexes, then it seems likely that Sir2 at silencers favors recruitment of ISW1a complexes bearing Esc8. Recruitment of Esc8 by Sir2 may explain why ISW1a complexes bearing Esc8 are not required for cohesion at the other genomic locations tested.
We propose that ISW1a complexes bearing Esc8 promote the cohesion of silenced sister chromatids by remodeling nucleosomes within silent chromatin domains. While the specific changes in chromatin structure are unknown, they must be subtle. Silencing remained essentially intact in the absence of ESC8, and the micrococcal nuclease digestion patterns of silent chromatin were essentially unchanged in the absence of ISW1 (37). Nevertheless, the stability of silent chromatin, as measured by a DNA supercoiling assay, was diminished. It is possible that ISW1a complexes bearing Esc8 provide a remodeling activity that counteracts the disruption of silent chromatin by other remodelers, such as RSC (48).
Previous work has identified roles for chromatin remodeling in sister chromatid cohesion, primarily at the step of cohesin loading. In humans, cohesin loading requires ISW1, a homolog of the yeast ISW1 remodeling complexes (52). In yeast, cohesin loading requires the evolutionarily conserved Scc2/Scc4 complex and RSC chromatin remodeler, which together create nucleosome-free regions (53). Indeed, much of the cohesin at HMR arrives via an adjacent, nucleosome-depleted tRNA gene, tT(AGU)C, that binds Scc2/Scc4 and RSC (14, 54, 55). At HMR, Esc8 is required for the function of cohesin but not loading of the complex. Thus, our data imply that there are roles for chromatin remodeling in cohesion beyond cohesin loading, at least within silent chromatin domains.
The cohesin embrace at silent chromatin domains.
Our initial analysis of silent chromatin cohesion identified a topological component of cohesin binding by using a small-molecule inhibitor of Sir2: cohesin complexes were lost from HMR if the inhibitor was added before DNA circularization, but the complexes were trapped at the locus if the inhibitor was added after circularization (3). Importantly, the trapped cohesin complexes on DNA circles could no longer mediate cohesion. In the double-embrace model, a loss of cohesion arises from opening of the cohesin ring, which should release cohesin from DNA. Thus, an alternative model was proposed in which single cohesin complexes bind one sister primarily through topological engagement but the second sister through silent chromatin-specific interactions (Fig. 7). In this study, mutation of the EKDK motif or Esc8 eliminated silent chromatin cohesion without a loss of cohesin binding. In the model shown in Fig. 7, we propose that these perturbations disrupt one of the two linkages that underpin cohesion of silent chromatin domains. At present, it is not known whether the residual binding of cohesin in either of these mutants is topologically based or not. A recent study proposed that cohesin complexes bind single chromatids stably and then mediate cohesion through cohesin-cohesin interactions (56). Whether such interactions also operate at silent chromatin domains has not yet been tested.
FIG 7.
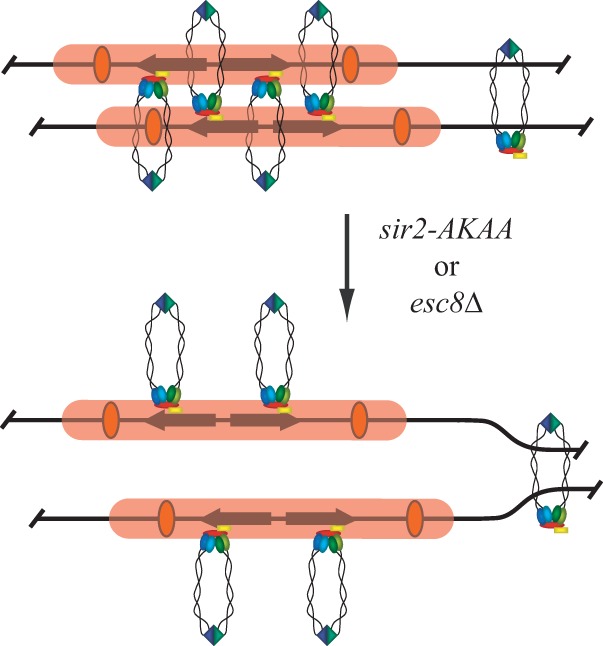
A model for cohesin binding at HMR and the impact of mutation of the Sir2 EKDK domain or loss of Esc8. Silent chromatin, silencers, and cohesin complexes are represented by pink bars, orange ovals, and multicolored rings, respectively. On the basis of evidence from earlier work (3), cohesin embraces one chromatid topologically and associates with silent chromatin of the second chromatid directly or through an adaptor protein. The sir2-AKAA mutation or deletion of ESC8 breaks one of the two linkages between cohesin and the chromatids, leaving cohesin bound but unable to perform cohesion. It is unknown whether the residual cohesin binds chromatin topologically.
While our studies have been limited to silent chromatin and silencing-related factors, it should be noted that numerous studies have also created situations where cohesin bound without generating cohesion (see reference 57 and references therein). For example, conditional depletion of cohesin accessory protein Pds5 caused the dissolution of cohesion genome-wide without a loss of cohesin from the chromatids (58–60). That cohesin can release one but not both sisters under these conditions, like the case with Sir2 inactivation described above, may suggest that the mode of cohesin binding at silent chromatin domains is at least thematically related to the mode of cohesin binding elsewhere in the genome.
Supplementary Material
ACKNOWLEDGMENTS
We thank Rolf Sternglanz, Jasper Rine, and David Shore for sharing yeast strains and plasmids, Mike Hampsey and Steven Zheng for sharing supplies and equipment, and Tom Ellenberger for sharing unpublished data. We thank John Everett for help with the figures.
M.R.G. is member of The Rutgers Cancer Institute of New Jersey, New Brunswick, NJ.
Funding Statement
This work was funded by HHS | NIH | National Institute of General Medical Sciences (NIGMS) (51402).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00057-16.
REFERENCES
- 1.Onn I, Heidinger-Pauli JM, Guacci V, Ünal E, Koshland DE. 2008. Sister chromatid cohesion: a simple concept with a complex reality. Annu Rev Cell Dev Biol 24:105–129. doi: 10.1146/annurev.cellbio.24.110707.175350. [DOI] [PubMed] [Google Scholar]
- 2.Nasmyth K, Haering CH. 2009. Cohesin: its roles and mechanisms. Annu Rev Genet 43:525–558. doi: 10.1146/annurev-genet-102108-134233. [DOI] [PubMed] [Google Scholar]
- 3.Chang CR, Wu CS, Hom Y, Gartenberg MR. 2005. Targeting of cohesin by transcriptionally silent chromatin. Genes Dev 19:3031–3042. doi: 10.1101/gad.1356305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ünal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, Koshland D. 2004. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell 16:991–1002. doi: 10.1016/j.molcel.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 5.Ström L, Lindroos HB, Shirahige K, Sjögren C. 2004. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell 16:1003–1015. doi: 10.1016/j.molcel.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 6.Dorsett D, Merkenschlager M. 2013. Cohesin at active genes: a unifying theme for cohesin and gene expression from model organisms to humans. Curr Opin Cell Biol 25:327–333. doi: 10.1016/j.ceb.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lengronne A, Katou Y, Mori S, Yokobayashi S, Kelly GP, Itoh T, Watanabe Y, Shirahige K, Uhlmann F. 2004. Cohesin relocation from sites of chromosomal loading to places of convergent transcription. Nature 430:573–578. doi: 10.1038/nature02742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glynn EF, Megee PC, Yu HG, Mistrot C, Ünal E, Koshland DE, DeRisi JL, Gerton JL. 2004. Genome-wide mapping of the cohesin complex in the yeast Saccharomyces cerevisiae. PLoS Biol 2:E259. doi: 10.1371/journal.pbio.0020259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu CS, Chen YF, Gartenberg MR. 2011. Targeted sister chromatid cohesion by Sir2. PLoS Genet 7:e1002000. doi: 10.1371/journal.pgen.1002000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li M, Valsakumar V, Poorey K, Bekiranov S, Smith JS. 2013. Genome-wide analysis of functional sirtuin chromatin targets in yeast. Genome Biol 14:R48. doi: 10.1186/gb-2013-14-5-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sebastian C, Satterstrom FK, Haigis MC, Mostoslavsky R. 2012. From sirtuin biology to human diseases: an update. J Biol Chem 287:42444–42452. doi: 10.1074/jbc.R112.402768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vaquero A. 2009. The conserved role of sirtuins in chromatin regulation. Int J Dev Biol 53:303–322. doi: 10.1387/ijdb.082675av. [DOI] [PubMed] [Google Scholar]
- 13.Rusche LN, Kirchmaier AL, Rine J. 2003. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem 72:481–516. doi: 10.1146/annurev.biochem.72.121801.161547. [DOI] [PubMed] [Google Scholar]
- 14.Dubey RN, Gartenberg MR. 2007. A tDNA establishes cohesion of a neighboring silent chromatin domain. Genes Dev 21:2150–2160. doi: 10.1101/gad.1583807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chou CC, Li YC, Gartenberg MR. 2008. Bypassing Sir2 and O-acetyl-ADP-ribose in transcriptional silencing. Mol Cell 31:650–659. doi: 10.1016/j.molcel.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (ed). 2010. Current protocols in molecular biology. John Wiley & Sons, Inc, New York, NY. [Google Scholar]
- 17.James P, Halladay J, Craig EA. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144:1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie J, Pierce M, Gailus-Durner V, Wagner M, Winter E, Vershon AK. 1999. Sum1 and Hst1 repress middle sporulation-specific gene expression during mitosis in Saccharomyces cerevisiae. EMBO J 18:6448–6454. doi: 10.1093/emboj/18.22.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bedalov A, Hirao M, Posakony J, Nelson M, Simon JA. 2003. NAD+-dependent deacetylase Hst1p controls biosynthesis and cellular NAD+ levels in Saccharomyces cerevisiae. Mol Cell Biol 23:7044–7054. doi: 10.1128/MCB.23.19.7044-7054.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li M, Petteys BJ, McClure JM, Valsakumar V, Bekiranov S, Frank EL, Smith JS. 2010. Thiamine biosynthesis in Saccharomyces cerevisiae is regulated by the NAD+-dependent histone deacetylase Hst1. Mol Cell Biol 30:3329–3341. doi: 10.1128/MCB.01590-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rusche LN, Rine J. 2001. Conversion of a gene-specific repressor to a regional silencer. Genes Dev 15:955–967. doi: 10.1101/gad.873601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sutton A, Heller RC, Landry J, Choy JS, Sirko A, Sternglanz R. 2001. A novel form of transcriptional silencing by Sum1-1 requires Hst1 and the origin recognition complex. Mol Cell Biol 21:3514–3522. doi: 10.1128/MCB.21.10.3514-3522.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lynch PJ, Fraser HB, Sevastopoulos E, Rine J, Rusche LN. 2005. Sum1p, the origin recognition complex, and the spreading of a promoter-specific repressor in Saccharomyces cerevisiae. Mol Cell Biol 25:5920–5932. doi: 10.1128/MCB.25.14.5920-5932.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng T-H, Gartenberg MR. 2000. Yeast heterochromatin is a dynamic structure that requires silencers continuously. Genes Dev 14:452–463. [PMC free article] [PubMed] [Google Scholar]
- 25.Boeke JD, LaCroute F, Fink GR. 1984. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet 197:345–346. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]
- 26.Sussel L, Vannier D, Shore D. 1993. Epigenetic switching of transcriptional states: cis- and trans-acting factors affecting establishment of silencing at the HMR locus in Saccharomyces cerevisiae. Mol Cell Biol 13:3919–3928. doi: 10.1128/MCB.13.7.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsu HC, Wang CL, Wang M, Yang N, Chen Z, Sternglanz R, Xu RM. 2013. Structural basis for allosteric stimulation of Sir2 activity by Sir4 binding. Genes Dev 27:64–73. doi: 10.1101/gad.208140.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maillet L, Boscheron C, Gotta M, Marcand S, Gilson E, Gasser SM. 1996. Evidence of silencing compartments within the yeast nucleus: a role for telomere proximity and Sir protein concentration in silencer-mediated repression. Genes Dev 10:1796–1811. doi: 10.1101/gad.10.14.1796. [DOI] [PubMed] [Google Scholar]
- 29.Dodson AE, Rine J. 2015. Heritable capture of heterochromatin dynamics in Saccharomyces cerevisiae. eLife 4:e05007. doi: 10.7554/eLife.05007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larin ML, Harding K, Williams EC, Lianga N, Dore C, Pilon S, Langis E, Yanofsky C, Rudner AD. 2015. Competition between heterochromatic loci allows the abundance of the silencing protein, Sir4, to regulate de novo assembly of heterochromatin. PLoS Genet 11:e1005425. doi: 10.1371/journal.pgen.1005425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cuperus G, Shore D. 2002. Restoration of silencing in Saccharomyces cerevisiae by tethering of a novel Sir2-interacting protein, Esc8. Genetics 162:633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vary JC Jr, Gangaraju VK, Qin J, Landel CC, Kooperberg C, Bartholomew B, Tsukiyama T. 2003. Yeast Isw1p forms two separable complexes in vivo. Mol Cell Biol 23:80–91. doi: 10.1128/MCB.23.1.80-91.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamada K, Frouws TD, Angst B, Fitzgerald DJ, DeLuca C, Schimmele K, Sargent DF, Richmond TJ. 2011. Structure and mechanism of the chromatin remodelling factor ISW1a. Nature 472:448–453. doi: 10.1038/nature09947. [DOI] [PubMed] [Google Scholar]
- 34.Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Kuster B, Neubauer G, Superti-Furga G. 2002. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 35.Li Y-C, Cheng T-H, Gartenberg MR. 2001. Establishment of transcriptional silencing in the absence of DNA replication. Science 291:650–653. doi: 10.1126/science.291.5504.650. [DOI] [PubMed] [Google Scholar]
- 36.Mueller JE, Bryk M. 2007. Isw1 acts independently of the Isw1a and Isw1b complexes in regulating transcriptional silencing at the ribosomal DNA locus in Saccharomyces cerevisiae. J Mol Biol 371:1–10. doi: 10.1016/j.jmb.2007.04.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu Q, Zhang X, Bi X. 2011. Roles of chromatin remodeling factors in the formation and maintenance of heterochromatin structure. J Biol Chem 286:14659–14669. doi: 10.1074/jbc.M110.183269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Megee PC, Koshland D. 1999. A functional assay for centromere-associated sister chromatid cohesion. Science 285:254–257. doi: 10.1126/science.285.5425.254. [DOI] [PubMed] [Google Scholar]
- 39.Ng TM, Waples WG, Lavoie BD, Biggins S. 2009. Pericentromeric sister chromatid cohesion promotes kinetochore biorientation. Mol Biol Cell 20:3818–3827. doi: 10.1091/mbc.E09-04-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gasser SM, Hediger F, Taddei A, Neumann FR, Gartenberg MR. 2004. The function of telomere clustering in yeast: the Circe effect. Cold Spring Harbor Symp Quant Biol 69:327–337. doi: 10.1101/sqb.2004.69.327. [DOI] [PubMed] [Google Scholar]
- 41.Donze D, Adams CR, Rine J, Kamakaka RT. 1999. The boundaries of the silenced HMR domain in Saccharomyces cerevisiae. Genes Dev 13:698–708. doi: 10.1101/gad.13.6.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lau A, Blitzblau H, Bell SP. 2002. Cell-cycle control of the establishment of mating-type silencing in S. cerevisiae. Genes Dev 16:2935–2945. doi: 10.1101/gad.764102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martins-Taylor K, Sharma U, Rozario T, Holmes SG. 2011. H2A.Z (Htz1) controls the cell-cycle-dependent establishment of transcriptional silencing at Saccharomyces cerevisiae telomeres. Genetics 187:89–104. doi: 10.1534/genetics.110.123844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gkikopoulos T, Schofield P, Singh V, Pinskaya M, Mellor J, Smolle M, Workman JL, Barton GJ, Owen-Hughes T. 2011. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science 333:1758–1760. doi: 10.1126/science.1206097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tirosh I, Sigal N, Barkai N. 2010. Widespread remodeling of mid-coding sequence nucleosomes by Isw1. Genome Biol 11:R49. doi: 10.1186/gb-2010-11-5-r49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morillon A, Karabetsou N, O'Sullivan J, Kent N, Proudfoot N, Mellor J. 2003. Isw1 chromatin remodeling ATPase coordinates transcription elongation and termination by RNA polymerase II. Cell 115:425–435. doi: 10.1016/S0092-8674(03)00880-8. [DOI] [PubMed] [Google Scholar]
- 47.Yen K, Vinayachandran V, Batta K, Koerber RT, Pugh BF. 2012. Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell 149:1461–1473. doi: 10.1016/j.cell.2012.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parnell TJ, Schlichter A, Wilson BG, Cairns BR. 2015. The chromatin remodelers RSC and ISW1 display functional and chromatin-based promoter antagonism. eLife 4:e06073. doi: 10.7554/eLife.06073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zentner GE, Tsukiyama T, Henikoff S. 2013. ISWI and CHD chromatin remodelers bind promoters but act in gene bodies. PLoS Genet 9:e1003317. doi: 10.1371/journal.pgen.1003317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weiss K, Simpson RT. 1998. High-resolution structural analysis of chromatin at specific loci: Saccharomyces cerevisiae silent mating-type locus HMLα. Mol Cell Biol 18:5392–5403. doi: 10.1128/MCB.18.9.5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ravindra A, Weiss K, Simpson RT. 1999. High-resolution structural analysis of chromatin at specific loci: Saccharomyces cerevisiae silent mating-type locus HMRa. Mol Cell Biol 19:7944–7950. doi: 10.1128/MCB.19.12.7944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hakimi MA, Bochar DA, Schmiesing JA, Dong Y, Barak OG, Speicher DW, Yokomori K, Shiekhattar R. 2002. A chromatin remodelling complex that loads cohesin onto human chromosomes. Nature 418:994–998. doi: 10.1038/nature01024. [DOI] [PubMed] [Google Scholar]
- 53.Lopez-Serra L, Kelly G, Patel H, Stewart A, Uhlmann F. 2014. The Scc2-Scc4 complex acts in sister chromatid cohesion and transcriptional regulation by maintaining nucleosome-free regions. Nat Genet 46:1147–1151. doi: 10.1038/ng.3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ng HH, Robert F, Young RA, Struhl K. 2002. Genome-wide location and regulated recruitment of the RSC nucleosome-remodeling complex. Genes Dev 16:806–819. doi: 10.1101/gad.978902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.D'Ambrosio C, Schmidt CK, Katou Y, Kelly G, Itoh T, Shirahige K, Uhlmann F. 2008. Identification of cis-acting sites for condensin loading onto budding yeast chromosomes. Genes Dev 22:2215–2227. doi: 10.1101/gad.1675708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eng T, Guacci V, Koshland D. 2015. Interallelic complementation provides functional evidence for cohesin-cohesin interactions on DNA. Mol Biol Cell 26:4224–4235. doi: 10.1091/mbc.E15-06-0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharma U, Stefanova D, Holmes SG. 2013. Histone variant H2A. Z functions in sister chromatid cohesion in Saccharomyces cerevisiae. Mol Cell Biol 33:3473–3481. doi: 10.1128/MCB.00162-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chan KL, Gligoris T, Upcher W, Kato Y, Shirahige K, Nasmyth K, Beckouet F. 2013. Pds5 promotes and protects cohesin acetylation. Proc Natl Acad Sci U S A 110:13020–13025. doi: 10.1073/pnas.1306900110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tong K, Skibbens RV. 2014. Cohesin without cohesion: a novel role for Pds5 in Saccharomyces cerevisiae. PLoS One 9:e100470. doi: 10.1371/journal.pone.0100470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eng T, Guacci V, Koshland D. 2014. ROCC, a conserved region in cohesin's Mcd1 subunit, is essential for the proper regulation of the maintenance of cohesion and establishment of condensation. Mol Biol Cell 25:2351–2364. doi: 10.1091/mbc.E14-04-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. 2010. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res 38:W695–W699. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.