

Abstract
Hyperekplexia is a rare human neuromotor disorder caused by mutations that impair the efficacy of glycinergic inhibitory neurotransmission. Loss-of-function mutations in the GLRA1 or GLRB genes, which encode the α1 and β glycine receptor (GlyR) subunits, are the major cause. Paradoxically, gain-of-function GLRA1 mutations also cause hyperekplexia, although the mechanism is unknown. Here we identify two new gain-of-function mutations (I43F and W170S) and characterize these along with known gain-of-function mutations (Q226E, V280M, and R414H) to identify how they cause hyperekplexia. Using artificial synapses, we show that all mutations prolong the decay of inhibitory postsynaptic currents (IPSCs) and induce spontaneous GlyR activation. As these effects may deplete the chloride electrochemical gradient, hyperekplexia could potentially result from reduced glycinergic inhibitory efficacy. However, we consider this unlikely as the depleted chloride gradient should also lead to pain sensitization and to a hyperekplexia phenotype that correlates with mutation severity, neither of which is observed in patients with GLRA1 hyperekplexia mutations. We also rule out small increases in IPSC decay times (as caused by W170S and R414H) as a possible mechanism given that the clinically important drug, tropisetron, significantly increases glycinergic IPSC decay times without causing motor side effects. A recent study on cultured spinal neurons concluded that an elevated intracellular chloride concentration late during development ablates α1β glycinergic synapses but spares GABAergic synapses. As this mechanism satisfies all our considerations, we propose it is primarily responsible for the hyperekplexia phenotype.
Keywords: glycine receptor, inhibition mechanism, patch clamp, site-directed mutagenesis, synapse, glycinergic, startle disease, tropisetron
Introduction
Glycine receptor (GlyR)2 chloride channels mediate fast inhibitory neurotransmission in the spinal cord, brainstem, and retina, and are thus essential for controlling motor and sensory function (1–3). Like other members of the pentameric ligand-gated ion channel family, GlyRs are assembled from five subunits arranged around a central water-filled pore. Each GlyR subunit is composed of an N-terminal extracellular domain, a transmembrane domain containing four α-helical domains (termed M1-M4), and a long intracellular domain linking M3 and M4. Synaptic GlyRs are formed as heteromers of α1–3 and β subunit isoforms with a stoichiometry of 2α:3β or 3α:2β (4, 5). The dominant native synaptic isoform is the α1β heteromer, with the β subunit responsible for anchoring GlyRs at postsynaptic densities by directly binding to the cytoplasmic clustering protein, gephyrin (6–8).
Human hereditary hyperekplexia, or startle disease, is a rare neurological disorder characterized by neonatal hypertonia and an exaggerated startle reflex in response to sudden, unexpected stimuli (9). It is most commonly caused by mutations that reduce the efficacy of glycinergic synaptic signaling. Mutations in the GLRA1 or GLRB genes, which encode the α1 and β GlyR subunits, respectively, are the major cause of hyperekplexia (9–12). The vast majority of these mutations are loss-of-function in that they reduce the ability of GlyRs to flux chloride. Recessive hyperekplexia mutations generally result in the loss of α1 or β GlyR protein expression at the cell surface, whereas dominant mutations usually allow strong surface expression but impair channel function via reduced open probability, single channel conductance, or glycine sensitivity (11, 13). Despite recessive mutations often resulting in the complete loss of α1 subunit expression (14) and dominant-negative mutations often involving only moderate disruption to α1 subunit function, there is no evidence for “major” or “minor” forms of hyperekplexia and no evidence for a correlation between inheritance mode and phenotype (12). Thus, it appears that as long as the deleterious effect of a GLRA1 mutation exceeds a certain threshold, a common hyperekplexia phenotype will result. All genetic forms of hyperekplexia are successfully treated with the benzodiazepine, clonazepam (9, 12). This works by potentiating GABA type-A receptor (GABAAR) chloride channels to compensate for the loss of glycinergic inhibition (15).
Hyperekplexia can also be caused by gain-of-function GLRA1 mutations (16, 17). To date four such mutations have been identified (Y128C, Q226E, V280M, and R414H). All of these result in spontaneous channel activation, whereas Y128C and V280M also significantly enhance glycine sensitivity (16, 17). While characterizing a range of other GLRA1 hyperekplexia mutations, we discovered that spontaneous activity is also induced by the recently characterized W170S mutation (18, 19) and by the as yet uncharacterized I43F mutation (20). From the clinical descriptions, it is evident that the hyperekplexia phenotype induced by gain-of-function mutations is no different to that caused by loss-of-function mutations and clonazepam remains an effective treatment (16, 18, 20).
In this study we sought to provide insight into the mechanism by which gain-of-function mutations cause hyperekplexia. We have recently developed an “artificial” synapse system that allows control over the subunit composition of GlyRs in glycinergic synapses (21). We inserted heteromeric α1β GlyRs incorporating each mutation in turn to evaluate the properties of the inhibitory postsynaptic currents (IPSCs) mediated by each mutant isoform. We then evaluated whether the hyperekplexia phenotype is likely to result from the change in IPSC properties or from the spontaneous channel activity.
Results
Properties of GlyRs Measured by Whole Cell Recording
The locations of the mutations investigated in this study are shown in Fig. 1. The glycine EC50 and Imax values for GlyRs incorporating the Y128C, W170S, Q226E, V280M, and R414H mutations have previously been characterized by whole cell recording (16, 17, 19). The Y128C mutation was found to induce a sufficiently large spontaneous leak current as to degrade HEK293 cell viability (17) and thus we did not study it further. The V280M mutation significantly reduced both the glycine EC50 and Imax values, although the W170S, Q226E, and R414H mutations had little effect of these parameters (16, 19). The Q226E, V280M, and R414H mutations have previously been shown to induce spontaneous single channel activity (16, 22). In this study we filled in the gaps by quantifying the glycine EC50 and Imax values of α1I43Fβ GlyRs by whole cell recording. We also employed outside-out patch clamp recording to characterize the properties of spontaneous single channels mediated by α1I43Fβ and α1W170Sβ GlyRs.
FIGURE 1.
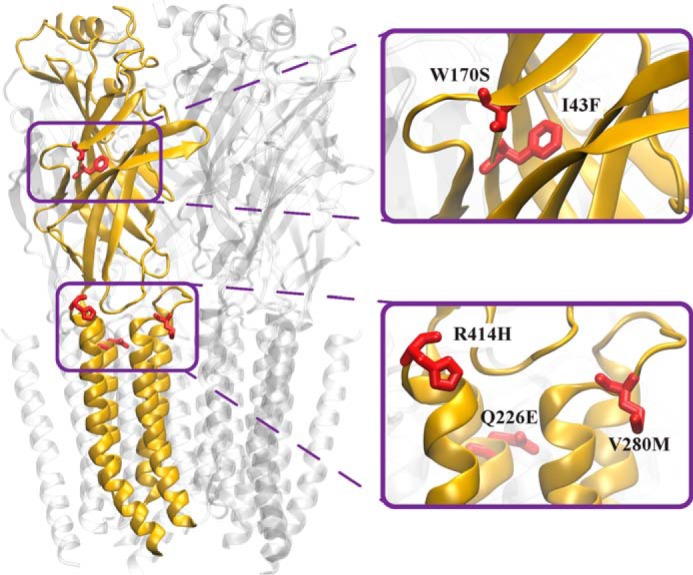
Model of the α1 GlyR viewed from within the membrane. The model is based on the cryoEM structure of zebrafish α1 GlyR in the glycine-bound conformation (PDB access code 3JAE) (39). One subunit is depicted in gold with gain-of-function hyperekplexia mutations (red) shown in stick form.
Examples of whole cell currents activated by increasing glycine concentrations at α1β and α1I43Fβ GlyRs are shown in Fig. 2A. Averaged glycine dose-response relationships for α1, α1I43F, α1β, and α1I43Fβ GlyRs are shown in Fig. 2B, with mean parameters presented in Table 1. In both homomeric and heteromeric GlyRs, the I43F mutation resulted in a drastically increased sensitivity to glycine.
FIGURE 2.

Effect of the α1 subunit I43F mutation examined by whole cell patch clamp recording. Recordings were performed at −60 mV. A, sample whole cell recordings for heteromeric α1β and α1I43Fβ GlyRs in the presence of the indicated glycine concentrations. B, averaged whole cell glycine dose-response curves for α1, α1β, α1I43F, and α1I43Fβ GlyRs. Mean parameters of best fit are provided in Table 1.
TABLE 1.
Effect of the I43F mutation on GlyR functional properties
| α1β | α1 | α1I43Fβ | α1I43F | |
|---|---|---|---|---|
| EC50 (μm) | 29.5 ± 3.7 | 52.8 ± 8.3 | 2.8 ± 1.3a | 1.3 ± 0.3b |
| Hill slope | 1.4 ± 0.3 | 2.0 ± 0.4 | 1.2 ± 0.2 | 1.8 ± 0.7 |
| Imax (nA) | 1.1 ± 0.2 | 1.2 ± 0.1 | 1.0 ± 0.2 | 0.7 ± 0.2 |
| n | 6 | 6 | 9 | 6 |
a p < 0.001 relative to the corresponding homo- or heteromeric wild-type GlyR via unpaired t test.
b p < 0.0001 relative to the corresponding homo- or heteromeric wild-type GlyR via unpaired t test.
Spontaneous Channel Activity in α1W170Sβ and α1I43Fβ GlyRs
To investigate the presence of spontaneous activity, 100 μm picrotoxin alone was applied to whole cells expressing either the α1W170Sβ GlyR (Fig. 3A) or the α1I43Fβ GlyR (Fig. 3B). We observed small but consistent upward current deflections in the presence of picrotoxin, which was particularly prominent for the α1I43Fβ GlyR. To quantify the level of spontaneous activity in both receptors we carried out single channel measurements in excised outside out patches.
FIGURE 3.

Spontaneous activity in α1W170Sβ and α1I43Fβ GlyRs. Whole cell steady-state currents showing the effect of 100 μm picrotoxin (PTX) on α1W170Sβ (A) and α1I43Fβ (B) GlyRs. Paired single channel recordings of the α1W170Sβ GlyR in the absence of applied glycine (C) and in the presence of 100 μm glycine (D). The patch contained an estimate of 6 channels. Paired single channel recordings of the α1I43Fβ GlyR in the absence of applied glycine (E) and in the presence of 100 μm glycine (F). The patch contained an estimate of 4 channels.
In the absence of applied glycine, the α1W170Sβ GlyR exhibited brief spontaneous openings of 1.4-ms mean duration, the majority of which were simple open-shut events (Fig. 3C). We determined the frequency of these openings by dividing their occurrence over time by an estimate of the number of channels contained in each recorded patch (between 3 and 6 channels). This yielded an open frequency of 0.2 Hz per channel. Wild-type α1β GlyRs under the similar recording conditions exhibit negligible spontaneous activity (10, 22–24).
In the presence of 100 μm glycine, single channel currents mediated by α1W170Sβ GlyRs occurred in clusters of tightly spaced open-shut transitions (Fig. 3D). The mean duration of the clusters was 621 ± 69 ms (n = 3), which is only 1.2-fold longer than wild-type α1β GlyRs (∼500 ms at saturating glycine (22)). The mean single channel amplitude measured at −70 mV was 3.31 ± 0.06 pA (n = 4), yielding a conductance of 41.6 ± 0.7 pS. This is similar to that of 45.9 ± 1.4 pS for wild-type receptors (22) and suggests that the α1W170S mutation has no appreciable effect on conductance.
Similar experiments were carried out using the α1I43Fβ GlyR. This receptor also exhibited spontaneous openings, but their mean duration was 11.2 ms, which is 8-fold greater than that of the α1W170Sβ GlyR. Moreover, the activity consisted of brief, single open-shut bursts in addition to a significant number of more complex bursts, where the receptor oscillated between open and shut configurations multiple times before terminating (Fig. 3E). The calculated spontaneous open frequency of this receptor, from patches containing 1–5 channels, was 0.5 Hz. This receptor exhibited a greater level of spontaneous activity than the α1W170Sβ GlyR, with longer, more complex bursts and a 2.5-fold higher frequency of occurrence.
A 100 μm concentration of glycine elicited long clusters of activity that had a mean duration of 1030 ± 73 ms (n = 3), similar to those of the α1Q226Eβ GlyR (22). The single channel mean amplitude the α1I43Fβ GlyR was 3.64 ± 0.05 pA (n = 3), producing a conductance at −70 mV of 45.7 ± 0.7 pS. This value is also close to that of wild-type α1β GlyRs, suggesting that this mutation also has little effect on channel conductance.
Hyperekplexia Mutations Prolong the Duration of IPSCs
Next we characterized the mean amplitudes, 10–90% rise times, and decay time constants of spontaneous IPSCs mediated by recombinant α1β, α1I43Fβ, α1W170Sβ, α1Q226Eβ, α1V280Mβ, and α1R414Hβ GlyRs (Fig. 4). Sample recordings from artificial synapses incorporating each isoform are shown at three progressively increasing temporal resolutions in Fig. 4A with a globally averaged, normalized IPSC shown for each isoform in Fig. 4B. The averaged IPSC amplitudes, 10–90% rise times, and decay time constants for each isoform are presented in Fig. 4C and Table 2. Relative to α1β GlyRs, IPSCs mediated by α1I43Fβ and α1V280Mβ GlyRs exhibited significantly larger amplitudes, whereas those mediated by α1W170Sβ, α1Q226Eβ, and α1R414Hβ GlyRs exhibited no significant change (Fig. 4C). All five mutations resulted in either modest (W170S and R414H) or dramatic (I43F, Q226E, and V280M) increases in the mean IPSC decay time constant (Fig. 4C). The α1I43Fβ and α1V280Mβ GlyRs also exhibited significantly slower rise times relative to the α1β GlyR (Fig. 4C). As illustrated in Fig. 5, the IPSC amplitudes and decay time constants were monotonically distributed for all isoforms, suggesting a single postsynaptic GlyR population in each case. It is likely that the prolonged IPSC rise and decay times in the mutant GlyRs is due either to altered intrinsic channel gating or to altered geometric factors within the synapse (e.g. synaptic cleft width, GlyR clustering propensity) that may slow access of neurotransmitter to the receptors. Our next experiments sought to distinguish between these possibilities by comparing the IPSC rise and decay rates with the intrinsic channel opening and closing rates for each mutant GlyR.
FIGURE 4.

Properties of spontaneous IPSCs mediated by wild-type and mutant GlyRs in artificial synapses. Recordings were performed at −60 mV. A, representative recordings of glycinergic IPSCs in HEK293 cells expressing the indicated homomeric GlyRs at three different temporal resolutions. B, averaged, normalized IPSCs each averaged from >50 events from the corresponding cell in A. C, comparison of mean IPSC amplitude, decay time constant, and 10–90% rise time for the indicated GlyRs. Statistical significance was determined via one-way analysis of variance followed by Bonferroni's post hoc correction with significance represented by *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001 relative to α1β GlyRs.
TABLE 2.
Comparison of 10–90% rise times and decay time constants of IPSCs and macropatch currents mediated by the indicated wild-type and mutant GlyRs
For IPSCs, the n values refer to the total number of cells from which data were collected. For each cell, parameters were analyzed from a single IPSC waveform that was digitally averaged from >50 individual events.
| α1β | α1R414Hβ | α1W170Sβ | α1Q226Eβ | α1I43Fβ | α1V280Mβ | ||
|---|---|---|---|---|---|---|---|
| 10–90% rise time (ms) | IPSCs | 1.6 ± 0.1 (n = 17) | 1.2 ± 0.1 (7) | 2.6 ± 0.3 (12) | 2.7 ± 0.4 (5) | 5.1 ± 0.6a (13) | 7.0 ± 0.9a (10) |
| Macropatch currents | 0.8 ± 0.1 (11) | 0.9 ± 0.2 (8) | 1.2 ± 0.2 (6) | 0.8 ± 0.1 (11) | 0.7 ± 0.1 (7) | 0.5 ± 0.1 (9) | |
| Deactivation time constant (ms) | IPSCs | 7.2 ± 0.5 | 10.9 ± 0.8 | 18.7 ± 1.5 | 102 ± 5b | 96 ± 12a | 181 ± 31a |
| Macropatch currents | 26.2 ± 3.4 | 32.4 ± 4.1. | 30.4 ± 3.1. | 221 ± 23a | 311 ± 46a | 139 ± 21b | |
a p < 0.0001 relative to the wild-type α1β GlyR via one-way ANOVA followed by Bonferroni's post hoc correction.
b p < 0.001 relative to the wild-type α1β GlyR via one-way ANOVA followed by Bonferroni's post hoc correction.
FIGURE 5.
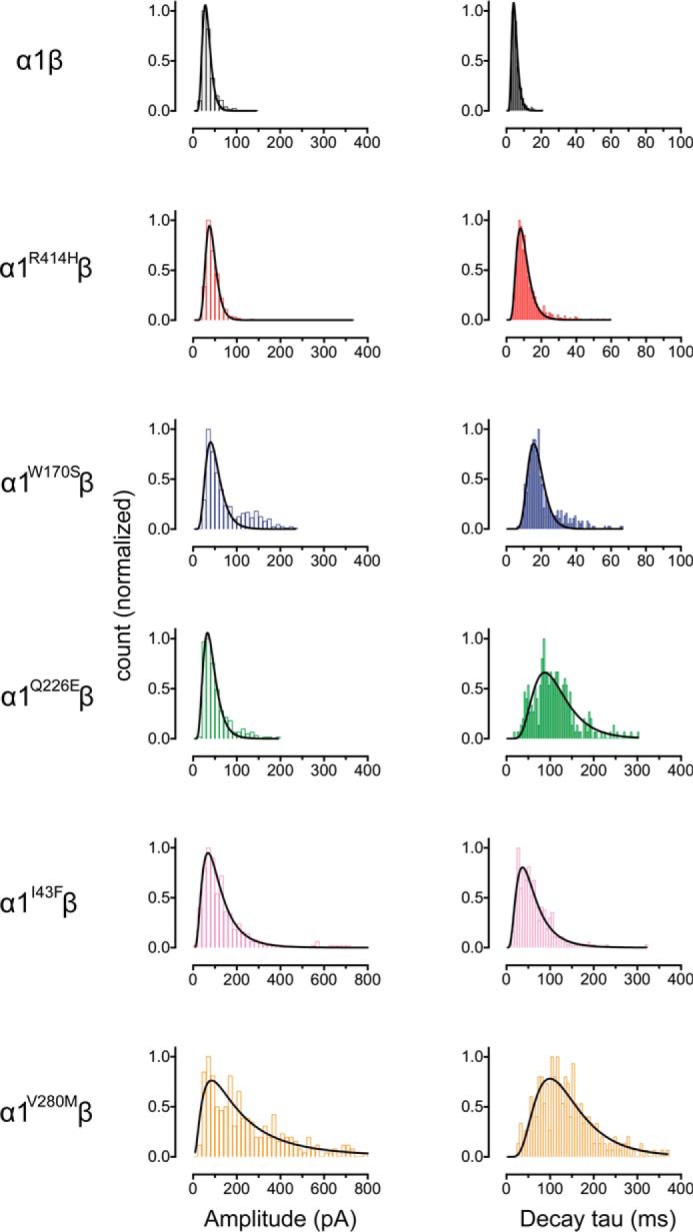
Distribution histograms for the peak amplitude and decay time constants of IPSCs recorded from wild-type and mutant GlyRs (n = 6 cells for each subtype). Curves represent single Log(Gaussian) functions of best fit to the data.
Characterization of Intrinsic Channel Closing Rates
The peak synaptic glycine concentration has been estimated to reach 1–3 mm in embryonic zebrafish neurons (25) and 2.2–3.5 mm in adult rat spinal neurons (23). Glycine is removed from the cleft with a time constant of 0.6–0.9 ms (23). Considering these parameters, we simulated synaptic activation conditions by rapidly applying 1 mm glycine for a period of ∼1 ms to outside-out macropatches expressing each receptor. We regularly calibrated the speed of the solution exchange system by rapidly switching the solution perfusing an open patch pipette between standard extracellular solution and an extracellular solution that had been diluted by 50% with distilled water. By monitoring the resulting pipette current, we were able to ensure that the solution perfusing the macropatch was completely exchanged within 200 μs (26). Under these conditions, the α1β GlyR activated robustly with a mean 10–90% rise time of 0.8 ± 0.1 ms and mean decay time constant of 26.2 ± 3.4 ms (Fig. 6, Table 2). This rise time was conserved for all five mutant GlyRs, which is surprising given that the mean IPSC rise times for the mutant GlyRs were all significantly slower than the wild-type α1β GlyR value (Table 2). We also observed a systematic discrepancy between the IPSC decay time constants and the macropatch current deactivation time constants for the wild-type and mutant GlyRs (Fig. 6, Table 2). With the exception of the α1V280Mβ GlyR, the intrinsic channel deactivation time constants were a factor of 1.7–3.6 slower than the IPSC decay time constants. The discrepancies between the macropatch and synaptic rise and decay times are presumably caused by factors specific to the synapse. One possibility is that synaptic GlyRs are clustered by a binding interaction with a protein that alters their gating properties. It is also possible that we have made an incorrect assumption about the temporal profile and/or peak glycine concentration in the synaptic cleft. However, given the strong correlation between IPSC decay time constants and macropatch deactivation time constants in the α1I43Fβ, α1Q226Eβ, and α1V280Mβ GlyRs, we infer that the dramatic slowing in their IPSC decay rates is dominated by changes in their intrinsic channel gating properties. Indeed, we have characterized the molecular basis of these gating changes in the α1Q226Eβ and α1V280Mβ GlyRs (10, 22).
FIGURE 6.
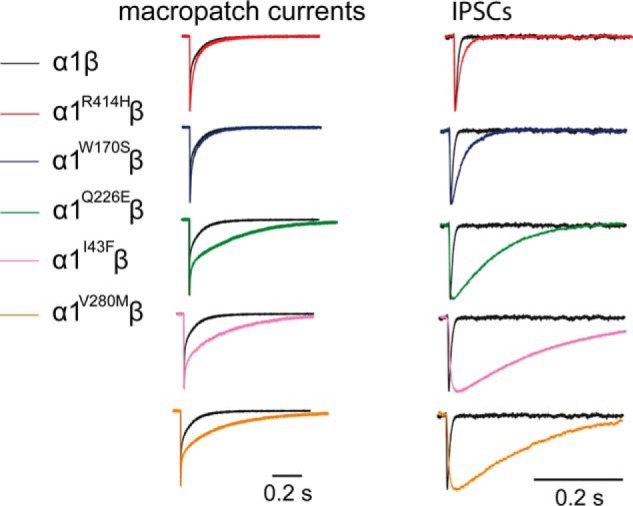
Comparison of the IPSC decay rate with the corresponding intrinsic receptor deactivation rate. Averaged macropatch currents recorded from outside-out patches containing the indicated GlyR. The currents were activated by brief (∼1 ms) exposure to saturating (1 mm) glycine. Recordings were performed at −60 mV. Corresponding averaged IPSCs (right panel, black traces), reproduced from Fig. 3B, are included for comparison.
Tropisetron Significantly Increased α1β GlyR-mediated IPSC Decay Time Constant
The R414H mutation results in a full hyperekplexia phenotype (16) despite causing a relatively small increase in IPSC decay time constant (from 7.2 to 11.4 ms). Can a small increase in the IPSC decay time constant alone result in a motor impairment? We addressed this question by investigating the effects 1 nm tropisetron on IPSCs mediated by α1β GlyRs.
Tropisetron, a 5-hydroxytryptamine type-3 receptor antagonist, is clinically important for the treatment of postoperative and chemotherapy-induced emesis and, importantly, it causes no motor side effects (27). As tropisetron inhibits the 5-hydroxytryptamine type-3 receptor with an IC50 near 1 nm (28, 29), we assume this concentration is clinically relevant. This concentration is also known to potentiate recombinant α1β GlyRs activated by a low (e.g. EC20) glycine concentration (4, 30), although its effect on glycinergic IPSCs has never been reported. Sample recordings of α1β GlyR-mediated IPSCs in the absence and presence of 1 nm tropisetron are shown in Fig. 7A. The globally averaged and normalized IPSCs suggest that 1 nm tropisetron exerted a prolonging effect on IPSC decay times (Fig. 7B). The averaged data presented in Fig. 7C reveal that 1 nm tropisetron had no significant effect on IPSC amplitude or 10–90% rise times, although it significantly increased the decay time constant from 5.8 ± 0.3 to 7.6 ± 0.4 ms (p < 0.05, paired t test; n = 6 cells). The percentage increase (32%) in the IPSC decay time constant in 1 nm tropisetron was modestly lower than that caused by either the R414H or W170S mutations (59 and 56%, respectively).
FIGURE 7.

A clinically relevant concentration of tropisetron prolongs the IPSC duration. A, representative IPSC recordings from an α1β GlyR-expressing cell before, during, and after the application of 1 nm tropisetron. Recordings were performed at −60 mV. B, superimposed averaged traces of IPSCs (n > 200 events) were recorded in control (black trace), 1 nm tropisetron exposure (red trace), and 5 min after the tropisetron washout (gray trace). Currents have been normalized. C, the decay time constant of IPSCs mediated by α1β GlyRs was significantly prolonged by 1 nm tropisetron, whereas the amplitude and 10–90% rise times were unchanged. *, p < 0.05 relative to drug-free control.
Discussion
Validation of the Artificial Synapse Technique
We have previously validated our artificial synapse system by showing that the decay rates of IPSCs mediated by α1β, α2β, and α3β GlyRs in artificial synapses are similar to those mediated by the same isoforms in native neuronal synapses (21). We have also compared the effects of α1 GlyR subunit mutations in artificial and native synapses. The α1 GlyR subunit loss-of-function D80A and A52S mutations have previously been shown to cause hyperekplexia-like phenotypes in mice (31, 32). Artificial synapses incorporating recombinant α1D80Aβ and α1A52Sβ mutant GlyRs exhibited accelerated IPSC decay rates that were quantitatively similar to those recorded in native synapses in mutant mice homozygous for the same mutations (21). The main trends in our artificial synapse data are also supported by the fast agonist application experiments (Fig. 6). Thus, the possible existence of ultrastructural or protein expression differences between native and artificial synapses are unlikely to seriously affect the IPSC decay rates as reported here.
Summary of Results
We have identified two new GLRA1 gain-of-function hyperekplexia mutations, I43F and W170S. The I43F mutation resulted in prolonged bursts of spontaneous channel activity (Fig. 3E) similar to that previously demonstrated for the Q226E mutation (22). These bursts most likely underlie the observed slow IPSC decay rates. Indeed, synaptic currents mediated by other ligand-gated ion channels have also been shown to deactivate as a function of the durations of single channel burst activity (26, 33–36). Conversely, the short-lived spontaneous bursts induced by α1W170Sβ and α1R414Hβ GlyRs most likely underlie their faster IPSC decay rates, compared with those mediated by α1I43Fβ and α1Q226Eβ GlyRs.
The original electrophysiological characterization of the α1W170Sβ GlyR showed that zinc potentiation was abolished (19). Physiological levels of zinc potentiate GlyRs (37, 38), and the loss of zinc potentiation via the α1D80A mutation accelerated the IPSC decay rate and thus caused hyperekplexia in a knock-in mouse model (31). Thus, it was reasonable to hypothesize that the W170S-mediated hyperekplexia phenotype was due to a diminished capacity of IPSCs to transfer chloride ions (19). However, in the present study we found that the decay time constant of IPSCs mediated by α1W170Sβ GlyRs was actually longer than that of α1β GlyRs (Fig. 4C). Thus, the loss of zinc potentiation does not explain the hyperekplexia phenotype.
It seems surprising that the R414H mutation also resulted in a full hyperekplexia phenotype, especially given its autosomal dominant inheritance mode (16). This mutation resulted in a modest level of spontaneous channel activity (16) and a modestly prolonged IPSC duration (Fig. 4C), similar to that induced by W170S. Due to its inheritance mode, the magnitudes of these effects in heterozygote patients may be further reduced by the equivalent levels of expression of mutant and wild-type α1 subunits.
Molecular Mechanisms of the Mutations
Gln-226 lies at the top of the M1 domain (Fig. 1). As the channel opens, Gln-226 moves closer to Arg-271 at the top of M2 (39). We have previously demonstrated that channel activation is facilitated by an energetic interaction between these residues (10, 22). This interaction is strengthened by the Q226E mutation, resulting in the longer active periods that underlie the gain-of-function startle phenotype (10, 22). Val-280 is located in the M2-M3 loop (Fig. 1) where it is closely apposed to Ile-225 at the top of M1. We previously concluded that the bulky V280M substitution exerts a steric repulsion against Ile-225, thereby tilting the M2 outwards to facilitate spontaneous channel opening (10, 22). Ile-49 and Trp-170 lie in close proximity in the inner and outer β-sheets, respectively, of the extracellular domain (Fig. 1). It is possible that mutations to either of these residues disrupt the relative orientations of the β-sheets and this could lead to allosteric defects including aberrant receptor activation. The molecular mechanism of R414H, which lies at the extracellular C terminus of M4 (Fig. 1), remains elusive.
Proposed Mechanism of Hyperekplexia by Gain-of-function GLRA1 Mutants
Synaptic inhibition in adult spinal motor neurons is mediated exclusively by glycine (40, 41) or by a combination of glycine and GABA (42, 43). The chloride concentration in adult neurons is normally very low, and as a result small increases in the chloride influx rate can have a significant effect on the strength, or even polarity, of glycinergic signaling (44). If a tonic increase in intracellular chloride concentration in otherwise healthy adult spinal motor neurons causes hyperekplexia, then the I43F, Q226E, or V280M mutations should cause a more severe phenotype than the W170S or R414H mutations, due to the large disparity in their rates of spontaneous and glycine-mediated chloride influx (44). Furthermore, if spontaneous GlyR activity results in increased chloride accumulation in spinal motor neurons then it should do likewise in spinal dorsal horn nociceptive neurons that also receive glycinergic inputs (45), and this should result in chronic pain (44). As neither chronic pain nor a correlation between GLRA1 mutation severity and startle phenotype have ever been reported in hyperekplexia patients (12), we consider it unlikely that hyperekplexia is predominantly due to an elevated chloride concentration in adult motor neurons.
Another possibility is that the mutation-induced increase in IPSC decay time constant directly elicits hyperekplexia, perhaps by a presynaptic effect on an excitatory or inhibitory input onto motor neurons. We demonstrate that a clinically relevant (1 nm) concentration of tropisetron significantly prolongs the decay time constant of α1β-mediated IPSCs (Fig. 7). This is notable because tropisetron has never been reported to cause hyperekplexia or other motor side effects (27, 46). As the tropisetron-induced increase in IPSC decay time constant almost reaches the levels induced by the W170S and R414H mutations, it is unlikely that the modest slowing in the IPSC decay rate induced by these mutations could cause hyperekplexia.
Prenatally in the rat, the α2 subunit is the predominant GlyR isoform. However, a developmental switch occurs during the first three postnatal weeks, whereby most of the α2 subunits are replaced by α1 (3). Simultaneously, the intracellular chloride concentration is reduced due to chloride extrusion by the KCC2 and KCC3 chloride-potassium co-transporters to the point where it supports efficient synaptic inhibition (47). A recent study that inhibited KCC2 function in cultured spinal neurons during this period found that the elevated chloride concentration reduced the number and size of α1 GlyR dendritic clusters without affecting neighboring α2 GlyR clusters (48). Concomitantly, glycinergic miniature IPSCs were reduced in amplitude and frequency although GABAergic IPSCs were not affected. This mechanism, which is thought to be active during the final maturation stage of spinal motor neurons (48), provides a mechanism for explaining how gain-of-function hyperekplexia mutations can cause a single common hyperekplexia phenotype without necessarily requiring an elevated chloride concentration in adult neurons.
Relevance of Findings to Pain Drug Development
Glycinergic synapses on pain sensory neurons in the spinal cord superficial laminae are unique in that they incorporate both α1 and α3 GlyR subunits (49). Inflammatory mediators (notably prostaglandin E2) induce chronic inflammatory pain by phosphorylating, and inhibiting, α3-containing GlyRs (49, 50). This reduces the magnitude of IPSCs in spinal pain sensory neurons, which in turn “disinhibits” them and thereby increases the rate of transmission of pain impulses to the brain. This mechanism, which provides a paradigm for chronic inflammatory pain sensitization, implies that drugs that selectively potentiate (i.e. restore) α3β GlyR-mediated IPSCs should be analgesic. It is generally assumed that such drugs should avoid potentiating α1β GlyRs due to their broader distribution and the consequent risk of motor and other side effects (45, 51). However, developing drugs specific for α3-containing GlyRs has proved difficult (52), due largely to the high amino acid sequence identity of α1 and α3 subunits, particularly in the known ligand binding sites. Our demonstration that a clinically relevant tropisetron concentration potentiates α1β GlyR-mediated IPSCs without causing motor or other side effects suggests the policy of avoiding α1 potentiation during analgesic drug development may be overemphasized.
Conclusion
Given that hyperekplexia is normally associated with a loss of glycinergic inhibitory tone, it seems paradoxical that an increase in glycinergic tone elicits an identical phenotype. We demonstrate here that all known GLRA1 gain-of-function hyperekplexia mutations act similarly to induce spontaneous GlyR activity and slow the decay rate of glycinergic IPSCs. We argue that mutations with modest IPSC prolonging effects (W170S, R414H) are unlikely to cause hyperekplexia by simply slowing the IPSC decay rate. We also argue that intracellular chloride accumulation in otherwise healthy adult neurons is unlikely to be solely responsible, although it may contribute to the phenotype. It has been shown that an elevated intracellular chloride concentration late in development selectively ablates α1β GlyR-containing synapses on spinal neurons. We consider this provides a plausible mechanism for gain-of-function GLRA1 hyperekplexia mutations because the loss of synapses would result in a single common hyperekplexia phenotype without requiring significant chloride accumulation in adult neurons.
Experimental Procedures
Cell Culture and Molecular Biology
The human GlyR α1 (pCIS) and β (pCIS) plasmid DNAs were combined in a ratio of 1α:50β (artificial synapse and macropatch recordings) or 1α:100β (single channel recordings) and transfected into HEK293 cells via calcium phosphate-DNA co-precipitation. This resulted in the expression of α1β heteromeric GlyRs (21). For artificial synapse experiments, mouse neuroligin-2A (pNice) and rat gephyrin (pCIS) were co-transfected along with GlyR plasmids to facilitate the formation of artificial synapses. Empty pEGFP plasmid was also transfected as an expression marker. Mutagenesis was performed using the QuikChange mutagenesis kit, and the successful incorporation of mutations was confirmed by DNA sequencing.
Artificial Synapse Formation
E15 timed-pregnant rats were euthanized via CO2 inhalation in accordance with procedures approved by the University of Queensland Animal Ethics Committee. Primary cultures of embryonic spinal cord neurons were prepared as previously described (21, 53). Cells were plated at a density of ∼80,000 cells per 18-mm poly-d-lysine-coated coverslip in DMEM with 10% fetal bovine serum. After 24 h, the plating medium was changed to Neurobasal medium supplemented with 2% B27 and 1% Glutamax, and a second feed after 1 week replaced half of this medium. Neurons were grown for 1–4 weeks in vitro and the heterosynaptic co-cultures were prepared by directly introducing transfected HEK293 cells onto the primary neuronal cultures 1–3 days prior to recordings.
Electrophysiology
Whole cell recordings were performed in voltage-clamp mode using a HEKA EPC10 amplifier (HEKA Electronics, Lambrecht, Germany) and Patchmaster software (HEKA), at room temperature. Cells were continuously perfused with extracellular solution comprising (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 d-glucose, adjusted to pH 7.4 with NaOH. Patch pipettes (1–3 MΩ resistance), made from borosilicate glass (GC150F-7.5, Harvard apparatus), were filled with an intracellular solution containing the following (in mm): 145 CsCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 10 EGTA, and 2 MgATP, adjusted to pH 7.4 with NaOH. Glycine-gated currents were recorded at a holding potential of −40 mV, digitized at 2.9 kHz, and filtered at 10 kHz. For IPSC recordings, the patch pipette resistance was adjusted to 4–6 MΩ and filled with the same internal solution. Series resistance was routinely compensated to 60% of maximum and was monitored throughout the recording. Spontaneous glycinergic IPSCs in HEK293 cells were recorded at a holding potential at −60 mV and signals were filtered at 4 kHz and sampled at 10 kHz. As these IPSCs were completely abolished by 1 μm tetrodotoxin (not shown), we infer they were induced by spontaneous action potentials.
Single-channel currents were recorded from outside-out excised patches at a clamped potential of −70 mV. Glass electrodes were pulled from borosilicate glass (G150F-3; Warner Instruments), coated with a silicone elastomer (Sylgard-184; Dow Corning), and heat-polished to a final tip resistance of 8–15 MΩ when filled with an intracellular solution containing (in mm) 145 CsCl, 2 MgCl2, 2 CaCl2, 10 HEPES, and 5 EGTA, pH 7.4. Excised patches were directly perfused with extracellular solution by placing them in front of one barrel of a double-barreled glass tube. Single channel currents were either recorded while the patch was exposed to extracellular solution (without added glycine) or elicited by exposing the patch continuously to glycine (100 μm) containing solution. Experiments were recorded using an Axopatch 200B amplifier (Molecular Devices), filtered at 5 kHz and digitized at 20 kHz using Clampex (pClamp 10, Molecular Devices) via a Digidata 1440A digitizer. The currents were filtered off-line at 3 kHz for making figures.
In the pharmacological studies, tropisetron (Sigma) was applied by bath perfusion. After waiting 2 min for drug effects to stabilize, we compared at least 3 min of spontaneous activity before, during, and after drug application.
Macropatch recordings were performed in the excised outside-out patch clamp configuration. Patch pipettes were fired-polished to a resistance of ∼10 MΩ and filled with the same internal solution. Macroscopic currents in outside-out patches pulled from transfected HEK293 cells were activated by brief (<1 ms) exposure to agonists using a piezo-electric translator (Siskiyou). We regularly calibrated the speed of the solution exchange system by rapidly switching the solution perfusing an open patch pipette between standard extracellular solution and an extracellular solution that had been diluted by 50% with distilled water. By monitoring the resulting pipette current, we were able to ensure that the solution perfusing the macropatch was completely exchanged within 200 μs (26). Recordings were performed using a Multiclamp 700B amplifier and pClamp9 software (Molecular Devices), filtered at 4 kHz and sampled at 10 kHz.
Analysis
Analyses of IPSC amplitudes, 10–90% rise times, and weighted decay time constants were performed using Axograph (Axograph Scientific). Only cells with a stable series resistance of <25 MΩ throughout the recording period were included in the analysis. Single peak IPSCs with amplitudes of at least three times above the background noise were detected using a semi-automated sliding template. Each detected event was visually inspected and only well separated IPSCs with no inflections in the rising or decay phases were included. To calculate macroscopic current decay time constants, averaged macroscopic traces were fitted with double-exponential functions in Axograph X, and a weighted time constant was calculated from individual time constants (τ1, τ2) and their relative amplitude (A1, A2) as followings: τweighted = (τ1 × A1 + τ2 × A2)/(A1 + A2). The averaged data from individual cells were then pooled to obtain group data. Statistical analysis and plotting were performed with Prism 5 (GraphPad Software). The fitting of single Log(Gaussian) functions to IPSC amplitude and decay time constant distributions was also performed using Prism 5. All data are presented as mean ± S.E. Student's t tests or one-way analysis of variance, as appropriate, were employed for comparisons. For all tests, the number of asterisks corresponds to level of significance: *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001.
Single channel recordings were analyzed with pClamp 10 (Clampfit) or QuB software. Segments of single channel activity separated by long periods of baseline were idealized into noise-free open and shut events using a temporal resolution of 70 μs. Single channel activations were separated using a between-burst closure period ranging between 6 and 30 ms.
Author Contributions
J. L. conceived the project. Y. Z., A. B., B. N., and A. K. all performed experiments and analyzed data. Y. Z. and J. L. wrote the manuscript. All authors reviewed the content of the manuscript.
This work was supported by Australian Research Council Research Grant DP150102428 and National Health and Medical Research Council Grants 1058542 and 1080976. The authors declare that they have no conflict of interest with the contents of this article.
- GlyR
- glycine receptor
- IPSC
- inhibitory postsynaptic current
- MΩ
- megaohms.
References
- 1. Dutertre S., Becker C. M., and Betz H. (2012) Inhibitory glycine receptors: an update. J. Biol. Chem. 287, 40216–40223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Legendre P. (2001) The glycinergic inhibitory synapse. Cell Mol. Life Sci. 58, 760–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lynch J. W. (2004) Molecular structure and function of the glycine receptor chloride channel. Physiol. Rev. 84, 1051–1095 [DOI] [PubMed] [Google Scholar]
- 4. Yang Z., Ney A., Cromer B. A., Ng H. L., Parker M. W., and Lynch J. W. (2007) Tropisetron modulation of the glycine receptor: femtomolar potentiation and a molecular determinant of inhibition. J. Neurochem. 100, 758–769 [DOI] [PubMed] [Google Scholar]
- 5. Durisic N., Godin A. G., Wever C. M., Heyes C. D., Lakadamyali M., and Dent J. A. (2012) Stoichiometry of the human glycine receptor revealed by direct subunit counting. J. Neurosci. 32, 12915–12920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Choii G., and Ko J. (2015) Gephyrin: a central GABAergic synapse organizer. Exp. Mol. Med. 47, e158. [DOI] [PubMed] [Google Scholar]
- 7. Meyer G., Kirsch J., Betz H., and Langosch D. (1995) Identification of a gephyrin binding motif on the glycine receptor beta subunit. Neuron 15, 563–572 [DOI] [PubMed] [Google Scholar]
- 8. Tyagarajan S. K., and Fritschy J. M. (2014) Gephyrin: a master regulator of neuronal function? Nat. Rev. Neurosci. 15, 141–156 [DOI] [PubMed] [Google Scholar]
- 9. Bakker M. J., van Dijk J. G., van den Maagdenberg A. M., and Tijssen M. A. (2006) Startle syndromes. Lancet Neurol. 5, 513–524 [DOI] [PubMed] [Google Scholar]
- 10. Bode A., and Lynch J. W. (2013) Analysis of hyperekplexia mutations identifies transmembrane domain rearrangements that mediate glycine receptor activation. J. Biol. Chem. 288, 33760–33771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harvey R. J., Topf M., Harvey K., and Rees M. I. (2008) The genetics of hyperekplexia: more than startle!. Trends Genet. 24, 439–447 [DOI] [PubMed] [Google Scholar]
- 12. Thomas R. H., Chung S. K., Wood S. E., Cushion T. D., Drew C. J., Hammond C. L., Vanbellinghen J. F., Mullins J. G., and Rees M. I. (2013) Genotype-phenotype correlations in hyperekplexia: apnoeas, learning difficulties and speech delay. Brain 136, 3085–3095 [DOI] [PubMed] [Google Scholar]
- 13. Bode A., and Lynch J. W. (2014) The impact of human hyperekplexia mutations on glycine receptor structure and function. Mol. Brain 7, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brune W., Weber R. G., Saul B., von Knebel Doeberitz M., Grond-Ginsbach C., Kellerman K., Meinck H. M., and Becker C. M. (1996) A GLRA1 null mutation in recessive hyperekplexia challenges the functional role of glycine receptors. Am. J. Hum. Genet. 58, 989–997 [PMC free article] [PubMed] [Google Scholar]
- 15. Schaefer N., Vogel N., and Villmann C. (2012) Glycine receptor mutants of the mouse: what are possible routes of inhibitory compensation? Front Mol. Neurosci. 5, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bode A., Wood S. E., Mullins J. G., Keramidas A., Cushion T. D., Thomas R. H., Pickrell W. O., Drew C. J., Masri A., Jones E. A., Vassallo G., Born A. P., Alehan F., Aharoni S., Bannasch G., et al. (2013) New hyperekplexia mutations provide insight into glycine receptor assembly, trafficking, and activation mechanisms. J. Biol. Chem. 288, 33745–33759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chung S. K., Vanbellinghen J. F., Mullins J. G., Robinson A., Hantke J., Hammond C. L., Gilbert D. F., Freilinger M., Ryan M., Kruer M. C., Masri A., Gurses C., Ferrie C., Harvey K., Shiang R., Christodoulou J., Andermann F., Andermann E., Thomas R. H., Harvey R. J., Lynch J. W., and Rees M. I. (2010) Pathophysiological mechanisms of dominant and recessive GLRA1 mutations in hyperekplexia. J. Neurosci. 30, 9612–9620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Al-Futaisi A. M., Al-Kindi M. N., Al-Mawali A. M., Koul R. L., Al-Adawi S., and Al-Yahyaee S. A. (2012) Novel mutation of GLRA1 in Omani families with hyperekplexia and mild mental retardation. Pediatr. Neurol. 46, 89–93 [DOI] [PubMed] [Google Scholar]
- 19. Zhou N., Wang C. H., Zhang S., and Wu D. C. (2013) The GLRA1 missense mutation W170S associates lack of Zn2+ potentiation with human hyperekplexia. J. Neurosci. 33, 17675–17681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Horváth E., Farkas K., Herczegfalvi A., Nagy N., and Széll M. (2014) Identification of a novel missense GLRA1 gene mutation in hyperekplexia: a case report. J. Med. Case Rep. 8, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y., Dixon C. L., Keramidas A., and Lynch J. W. (2015) Functional reconstitution of glycinergic synapses incorporating defined glycine receptor subunit combinations. Neuropharmacology 89, 391–397 [DOI] [PubMed] [Google Scholar]
- 22. Scott S., Lynch J. W., and Keramidas A. (2015) Correlating structural and energetic changes in glycine receptor activation. J. Biol. Chem. 290, 5621–5634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beato M. (2008) The time course of transmitter at glycinergic synapses onto motoneurons. J. Neurosci. 28, 7412–7425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lewis T. M., Sivilotti L. G., Colquhoun D., Gardiner R. M., Schoepfer R., and Rees M. (1998) Properties of human glycine receptors containing the hyperekplexia mutation alpha1(K276E), expressed in Xenopus oocytes. J. Physiol. 507, 25–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Legendre P. (1998) A reluctant gating mode of glycine receptor channels determines the time course of inhibitory miniature synaptic events in zebrafish hindbrain neurons. J. Neurosci. 18, 2856–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dixon C., Sah P., Lynch J. W., and Keramidas A. (2014) GABAA receptor α- and γ-subunits shape synaptic currents via different mechanisms. J. Biol. Chem. 289, 5399–5411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haus U., Späth M., and Färber L. (2004) Spectrum of use and tolerability of 5-HT3 receptor antagonists. Scand. J. Rheumatol. Suppl. 119, 12–18 [PubMed] [Google Scholar]
- 28. Lankiewicz S., Lobitz N., Wetzel C. H., Rupprecht R., Gisselmann G., and Hatt H. (1998) Molecular cloning, functional expression, and pharmacological characterization of 5-hydroxytryptamine3 receptor cDNA and its splice variants from guinea pig. Mol. Pharmacol. 53, 202–212 [DOI] [PubMed] [Google Scholar]
- 29. Miyake A., Mochizuki S., Takemoto Y., and Akuzawa S. (1995) Molecular cloning of human 5-hydroxytryptamine3 receptor: heterogeneity in distribution and function among species. Mol. Pharmacol. 48, 407–416 [PubMed] [Google Scholar]
- 30. Supplisson S., and Chesnoy-Marchais D. (2000) Glycine receptor beta subunits play a critical role in potentiation of glycine responses by ICS-205,930. Mol. Pharmacol 58, 763–770 [DOI] [PubMed] [Google Scholar]
- 31. Hirzel K., Müller U., Latal A. T., Hülsmann S., Grudzinska J., Seeliger M. W., Betz H., and Laube B. (2006) Hyperekplexia phenotype of glycine receptor α1 subunit mutant mice identifies Zn2+ as an essential endogenous modulator of glycinergic neurotransmission. Neuron 52, 679–690 [DOI] [PubMed] [Google Scholar]
- 32. Graham B. A., Schofield P. R., Sah P., Margrie T. W., and Callister R. J. (2006) Distinct physiological mechanisms underlie altered glycinergic synaptic transmission in the murine mutants spastic, spasmodic, and oscillator. J. Neurosci. 26, 4880–4890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dixon C. L., Harrison N. L., Lynch J. W., and Keramidas A. (2015) Zolpidem and eszopiclone prime α1β2γ2 GABAA receptors for longer duration of activity. Br. J. Pharmacol. 172, 3522–3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Elenes S., Ni Y., Cymes G. D., and Grosman C. (2006) Desensitization contributes to the synaptic response of gain-of-function mutants of the muscle nicotinic receptor. J. Gen. Physiol. 128, 615–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sine S. M., and Engel A. G. (2006) Recent advances in Cys-loop receptor structure and function. Nature 440, 448–455 [DOI] [PubMed] [Google Scholar]
- 36. Wyllie D. J., Béhé P., and Colquhoun D. (1998) Single-channel activations and concentration jumps: comparison of recombinant NR1a/NR2A and NR1a/NR2D NMDA receptors. J. Physiol. 510, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Laube B., Kuhse J., Rundström N., Kirsch J., Schmieden V., and Betz H. (1995) Modulation by zinc ions of native rat and recombinant human inhibitory glycine receptors. J. Physiol. 483, 613–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miller P. S., Da Silva H. M., and Smart T. G. (2005) Molecular basis for zinc potentiation at strychnine-sensitive glycine receptors. J. Biol. Chem. 280, 37877–37884 [DOI] [PubMed] [Google Scholar]
- 39. Du J., Lü W., Wu S., Cheng Y., and Gouaux E. (2015) Glycine receptor mechanism elucidated by electron cryo-microscopy. Nature 526, 224–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bhumbra G. S., Moore N. J., Moroni M., and Beato M. (2012) Co-release of GABA does not occur at glycinergic synapses onto lumbar motoneurons in juvenile mice. Front. Cell Neurosci. 6, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiang J., and Alstermark B. (2015) Not GABA but glycine mediates segmental, propriospinal, and bulbospinal postsynaptic inhibition in adult mouse spinal forelimb motor neurons. J. Neurosci. 35, 1991–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gao B. X., Stricker C., and Ziskind-Conhaim L. (2001) Transition from GABAergic to glycinergic synaptic transmission in newly formed spinal networks. J. Neurophysiol. 86, 492–502 [DOI] [PubMed] [Google Scholar]
- 43. Martin L. J., and Chang Q. (2012) Inhibitory synaptic regulation of motoneurons: a new target of disease mechanisms in amyotrophic lateral sclerosis. Mol. Neurobiol. 45, 30–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. De Koninck Y. (2007) Altered chloride homeostasis in neurological disorders: a new target. Curr. Opin. Pharmacol. 7, 93–99 [DOI] [PubMed] [Google Scholar]
- 45. Zeilhofer H. U. (2005) The glycinergic control of spinal pain processing. Cell Mol. Life Sci. 62, 2027–2035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carlisle J. B., and Stevenson C. A. (2006) Drugs for preventing postoperative nausea and vomiting. Cochrane Database Syst Rev. CD004125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Blaesse P., Airaksinen M. S., Rivera C., and Kaila K. (2009) Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838 [DOI] [PubMed] [Google Scholar]
- 48. Schwale C., Schumacher S., Bruehl C., Titz S., Schlicksupp A., Kokocinska M., Kirsch J., Draguhn A., and Kuhse J. (2016) KCC2 knockdown impairs glycinergic synapse maturation in cultured spinal cord neurons. Histochem. Cell Biol. 145, 637–646 [DOI] [PubMed] [Google Scholar]
- 49. Harvey R. J., Depner U. B., Wässle H., Ahmadi S., Heindl C., Reinold H., Smart T. G., Harvey K., Schütz B., Abo-Salem O. M., Zimmer A., Poisbeau P., Welzl H., Wolfer D. P., Betz H., Zeilhofer H. U., and Müller U. (2004) GlyR α3: an essential target for spinal PGE2-mediated inflammatory pain sensitization. Science 304, 884–887 [DOI] [PubMed] [Google Scholar]
- 50. Ahmadi S., Lippross S., Neuhuber W. L., and Zeilhofer H. U. (2002) PGE2 selectively blocks inhibitory glycinergic neurotransmission onto rat superficial dorsal horn neurons. Nat. Neurosci. 5, 34–40 [DOI] [PubMed] [Google Scholar]
- 51. Lynch J. W., and Callister R. J. (2006) Glycine receptors: a new therapeutic target in pain pathways. Curr. Opin. Investig. Drugs 7, 48–53 [PubMed] [Google Scholar]
- 52. Balansa W., Islam R., Fontaine F., Piggott A. M., Zhang H., Xiao X., Webb T. I., Gilbert D. F., Lynch J. W., and Capon R. J. (2013) Sesterterpene glycinyl-lactams: a new class of glycine receptor modulator from Australian marine sponges of the genus Psammocinia. Org. Biomol. Chem. 11, 4695–4701 [DOI] [PubMed] [Google Scholar]
- 53. Dixon C. L., Zhang Y., and Lynch J. W. (2015) Generation of functional inhibitory synapses incorporating defined combinations of GABA(A) or glycine receptor subunits. Front. Mol. Neurosci. 8, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]