

Abstract
In humans approximately 60 mg of ascorbic acid (AA) breaks down in the body each day and has to be replaced by a dietary intake of 70 mg in females and 90 mg in males to maintain optimal health and AA homeostasis. The breakdown of AA is non-enzymatic and results in oxalate formation. The exact amount of oxalate formed has been difficult to ascertain primarily due to the limited availability of healthy human tissue for such research and the difficulty in measuring AA and its breakdown products. The breakdown of 60 mg of AA to oxalate could potentially result in the formation of up to 30 mg oxalate per day. This exceeds our estimates of the endogenous production of 10 – 25 mg oxalate per day, indicating that degradative pathways that do not form oxalate exist. In this review we examine what is known about the pathways of AA metabolism and how oxalate forms. We further identify how gaps in our knowledge may be filled to more precisely determine the contribution of AA breakdown to oxalate production in humans. The use of stable isotopes of AA to directly assess the conversion of vitamin to oxalate should help fill this void.
INTRODUCTION
Despite over half a century of research on the relationship between ascorbic acid (AA; also known as vitamin C) intake and calcium oxalate stone disease the nature of this association is still blurred. The first report that AA could be converted to oxalate in humans was in 1958 by Hellman and Burns1. They reported that oxalate was the major product of AA excreted in urine. This report clearly triggered interest that AA consumption and the oxalate it could produce was involved in calcium oxalate stone formation. In the mid-1960’s Atkins et al and Baker et al quantified the breakdown of ingested AA to oxalate using isotopes2, 3. They estimated AA breakdown contributed > 40% of the oxalate excreted in urine. Their analytical tools were limited by today’s standards and it is unclear whether they took adequate steps to limit the in vitro conversion of AA to oxalate during analysis. Subsequent research confirmed that breakdown of AA to oxalate could occur in urine and increased with time, temperature and pH, but also revealed that it could be partially prevented by EDTA and urine acidification4, 5. More recent research has indicated that with even low levels of dietary AA consumption, small increases in intake (> 281 mg/day vs < 105 mg/day) in male health professionals increased stone risk by 31%6, and total AA intake increased stone risk 2 fold in a large Swedish population7. At the other extreme, individuals constantly consuming large oral doses of AA or receiving intravenous infusions, have been reported to develop oxalate nephropathy in several case reports8–12. In contrast, a short term intravenous infusion of up to 100 g AA over a 6 hr period has been used as a pilot cancer treatment without any reported short term adverse events13. A large increase in urinary oxalate excretion did accompany such infusions. In between these extremes, a significant number of individuals consume daily AA supplements between 1 and 10 g for extended periods of time as either a protective or therapeutic agent and have an increased urinary oxalate excretion14. We will review this data, identifying its limitations and weaknesses, and will describe the current view on the breakdown of AA to oxalate, the only mechanism that has been seriously considered as a cause for the oxalate nephropathy and oxalate stone disease associated with AA ingestion.
FUNCTIONAL ROLE OF AA
Humans similar to other primates, guinea pigs, some fish and some bats require AA because in these species the activity of a key enzyme involved in AA synthesis, gulonolactone oxidase, is absent15, 16. The gene encoding this enzyme still exists in the human genome but contains several mutations that make it non-functional16. It is believed this loss of function occurred over 45 million years ago and that it was probably the result of some evolutionary pressure. Halliwell speculatively proposed that the loss of this enzyme and urate oxidase in humans were steps to lower hydrogen peroxide production15. Grano and De Tullio further suggested that the inability to synthesize AA could make it an oxidative stress sensor as it is required for the hydroxylation of the hypoxia inducible factor (HIF) to induce an hypoxic stress response17.
The major functional role usually ascribed to AA is its role as a water soluble antioxidant. It has the ability to scavenge free radicals forming a more stable ascorbyl radical. Two of these radicals can react to form AA and dehydroascorbic acid (DHA), the oxidized form of AA18, 19. Due to the multiplicity of antioxidant defense mechanisms in the body, however, it is not clear that this important activity is compromised by AA deficiency alone. The hallmark symptoms of AA deficiency (scurvy) result from an abnormal collagen synthesis due to impaired proline and lysine hydroxylation. AA is required for a variety of biosynthetic pathways, particularly those involving hydroxylation and amidation reactions. These reactions, besides those involved in collagen biosynthesis, include the hydroxylation of dopamine to norepinephrine in chromaffin granules of adrenal medulla, the α-amidation of glycine in pituitary gland peptides, prolyl hydroxylation in HIF, and histone demethylation18.
DIETARY SOURCES OF AA
The daily intake of AA currently recommended by the Institute of Medicine in the U.S. is 75 mg/day for adult women and 90 mg/day for adult men. According to the NHANES survey in 2009 – 2010 the average AA intake for adult males was 96 mg/day and 87 mg/day for adult females. If the assumption is made that ~20 mg/day of AA is excreted undegraded and the remainder is broken down in the body, the yield of oxalate from breakdown of AA could potentially be as high as 39 mg/day in males and 34 mg/day in females. This is clearly an overestimate as the average urinary oxalate excretion is ~30 mg/day20. Dietary oxalate and the metabolism of hydroxyproline, glycine and glyoxal also contribute to urinary oxalate excretion20–22. Dietary oxalate absorption on average may contribute 12 mg/day (the absorption of 8% of an average daily intake of 150 mg)23, 24. Our analyses of the contributions of hydroxyproline, glycine and phenylalanine degradation to urinary oxalate excretion21 (unpublished observations) indicate that they contribute at most 25% (7.5 mg) to the oxalate excreted by normal individuals. These estimates indicate that AA must be metabolized by pathways other than those that result in oxalate formation and that its breakdown could lead to the formation of up to 10.5 mg oxalate/day.
Some of the major dietary sources of AA are shown in Table 1. Fruits and vegetables are significant sources. Some meats such as liver can also contribute substantially. The content may vary depending on the cultivar, growth and storage conditions, as well as the method of food preparation. For example, one third of the AA was lost on storage of spinach at 4°C for 2 weeks25. As the AA in food degrades, oxalate may very well be a product. Oxalate was a major breakdown product of AA added to drinking water that had become contaminated with copper after flowing through copper pipes26. AA could also potentially breakdown to oxalate in any food or beverage fortified with this vitamin. Perusal of the shelves at grocery stores reveals an ever increasing supply of juices, baby foods and snack bars that contain supplemental AA.
TABLE 1.
Vitamin C Content of Selected Raw Fruits and Vegetables
| Food | Total Vitamin C (mg/100g) |
|---|---|
| Broccoli | 89.2 |
| Chili Pepper Green | 242.5 |
| Cauliflower | 48.2 |
| Guava | 228.3 |
| Kiwi Fruit | 92.7 |
| Lemon Peeled | 53.0 |
| Orange Peeled | 53.2 |
| Pepper Green | 80.4 |
| Pepper Red | 127.7 |
| Parsley | 133.0 |
| Strawberry | 58.8 |
Values taken from the USDA Database (http://ndb.nal.usda.gov/ndb/search/list)
ABSORPTION, CIRCULATION, TISSUE UPTAKE AND URINARY EXCRETION
The absorption of AA in the human gut has not been fully characterized. A sodium dependent AA transporter (SVTC1) encoded by the gene SLCA23A1 has been identified in the small intestine of mammals and appears to play a significant role in AA absorption27. However, knockout mice lacking SVTC1 have a normal plasma AA. This led Corpe et al to identify the glucose transporters, GLUT2 and GLUT8, as also contributing to AA absorption28. Interestingly, plasma AA levels in these mice responded much better to the gavage of the oxidized form of AA, dehydroascorbic acid (DHA), than they did to gavage with AA. One interpretation of these results is that normally DHA is produced from ingested AA in the gut and then absorbed. It is then converted back into AA after internalization within epithelial cells. Oral doses of AA up to 200 mg/day are well absorbed (70 – 90%) but above this dose absorption declines and at 1250 mg/day it is ~33%29, 30. Ingested AA begins to be observed in urine at doses above 50 mg/day30. Factors that may influence the oxidation of AA in the gut and its absorption are largely unknown. Whether non-absorbed AA can breakdown in the gut to oxalate is also unknown.
Assessments of the AA distribution in tissues have been limited. Studies using single oral doses of 13C- and 14C-isotopes have suggested that there are 2 major pools of AA in the body31, 32. Our estimates of the size of these pools and average 24 hr urinary excretions are shown in Fig. 1. The first pool consisting of plasma and the extracellular fluid compartment contains 150 mg of AA. The second compartment presumably represents the AA within cells and is ~ 1 – 1.5 g. Muscle is the largest tissue store containing ~ two thirds of the tissue pool despite containing one of the lowest AA concentrations of all tissues33. The concentration in each tissue compartment may differ due to its complement of transporters and the unique AA metabolism that might occur. Given the long interval taken for scurvy to surface after consumption of an AA-deficient diet it is clear that tissue cells have a remarkable capacity to retain and recycle AA. This slow flux appears to be bi-directional with 3 – 5 weeks taken for individuals to reach a stable concentration when adjusting from one level of dietary AA to another30, 34. For comparative purposes, estimates of oxalate compartmentation are also provided in Fig. 1. These estimates are based on values obtained for human liver tissue35, mouse kidney36 and food table values for beef muscle (0.1 mg/100 g).
Figure 1.
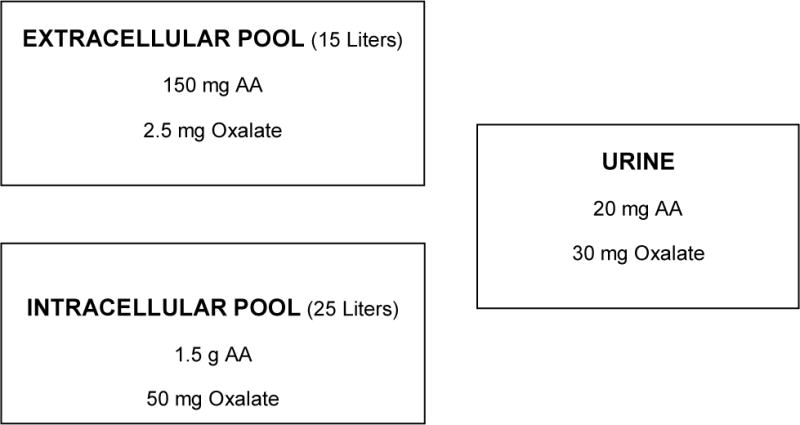
Estimated average compartment sizes of the AA and oxalate pools in humans.
As discussed above in the gut, AA uptake in cells appears to result from the activities of GLUT transporters that have a higher affinity for DHA than AA, and specific AA transporters (SVTC). With the recommended daily intake of AA these transporters result in a plasma AA concentration of 50 – 60 μM and a DHA concentration of ~1μM. Efflux of AA from cells can occur by several mechanisms with the main contributors believed to be volume-sensitive channels and Ca2+ -dependent channels37.
Elimination of AA from the body is believed to be almost entirely through the kidney38. Proximal tubules contain SVTC1 which appears to play a role in the reabsorption of filtered AA in conjunction with GLUT transporters27. With oral doses of AA of < 60 mg/day, < 0.4 mg/day is excreted30 although 1500 mg/day is filtered. When the oral dose reaches 100 mg/day, ~25 mg/day is excreted30. The maximal plasma AA level reached with oral doses is ~0.25 mM39. The combined data on absorption and excretion indicate with oral doses over 1 g/day, a significant portion is not absorbed and the bulk of that which is absorbed is excreted unchanged in urine30, 38. AA may have clinical utility in treating cancer and infections but to be effective it must be given intravenously to raise the plasma AA to therapeutic levels39. Levels of 25 – 50 mM have been obtained with this approach13, 40. Recent research suggests it may act by inhibiting glycolysis through its effect on glyceraldehyde 3-phosphate dehydrogenase41.
METABOLISM OF AA
Circulating AA can be taken up and concentrated in cells and tissues where it can act as an antioxidant resulting in the formation of the ascorbyl radical18. The loss of a further electron results in the formation of DHA. Two ascorbyl radicals can readily dismute to AA and DHA. DHA can also be converted back to AA through an interaction with reduced glutathione. Glutaredoxin and possibly other enzymes have been reported to play a role in this reduction19, 42. The formation and removal of DHA is potentially important for oxalate formation from AA as a fraction of the DHA irreversibly forms the open chain diketogulonic acid (DKG), an unstable molecule that can break down to oxalate43. The conditions that promote or limit the breakdown of both DHA and DKG have not been well characterized. This is a major limitation in determining the contribution of AA metabolism to oxalate synthesis. The only work published is that from over half a century ago using outdated methodology coupled with a lack of awareness of the susceptibility of AA to breakdown at alkaline pH1–3. Studies on the breakdown of DHA and DKG in phosphate buffer by Simpson et al have indicated that DKG rapidly splits into L-erythrulose and oxalate43. In lens homogenates it was reported that erythrulose can be converted to L-threitol by sorbitol dehydrogenase44. Possible pathways that may be involved in AA breakdown are shown in Fig. 2.
Figure 2.

Major products in AA degradation pathway.
The analysis of AA and its degradation products has been an analytical challenge for decades. Poor stability in solution and the lack of suitable detection techniques have hampered achieving high selectivity and sensitivity in analyses. Classical analytical techniques such as titrimetric45, spectrophotometric46 and enzymatic methods47 employing the AA/DHA redox chemistry have been used in the past but they lack specificity and are prone to the interferences inherently present in biological samples.
High performance liquid chromatography (HPLC) has become the preferred method of AA analysis in the past two decades. UV-Vis Absorption detection has been extensively used but coulometric electrochemical detection (ECD) has proven to have much higher specificity and sensitivity and is now widely used for the determination of AA in biological samples48. An electrochemical detector only visualizes analytes that are active at the electrode potential applied during the detection process and therefore is less susceptible to interferences present in the sample matrix. Even though ECD is believed to be less prone to interferences, during our recent studies on plasma AA content, we have observed that in plasma uric acid is also visualized at the same potential and elutes close to AA. It is essential that chromatographic conditions be optimized to obtain the separation required.
Considering the challenges in detecting and accurately measuring AA and its degradation products in biological specimens, mass spectrometry (MS) appears to offer superior detection and specification49. For this reason, presently an MS detector coupled to an HPLC system (LC-MS) is the most commonly applied approach for the analysis of AA derived from biological specimens, where analytes are separated on a HPLC column and then identified initially by their mass:charge (m/z) ratio. The main requirement for MS detection is the ability for the analyte(s) of interest to form positive or negative ions in the gaseous state, in which case AA can form both, and the parent mass can be measured with high accuracy depending on the MS instrument of choice. In the case of AA specifically, this analyte is generally quantified by LCMS using electro-spray ionization (ESI) in negative ion mode, as this provides optimal sensitivity49–51. With the current availability of more sophisticated MS instruments, high mass accuracy combined with tandem mass spectrometry MS(MS2) has become very common. The latest MS(MS2) instruments offer very high specificity and quantification for analyte(s) in complex matrices due to the ability to selectively monitor the parent analyte fragment ions (otherwise known as daughter ions). In this regard, Fenoll et al. reported a method to simultaneously determine AA and DHA in fruit extracts using LCMS(MS2) operated in negative ion mode while monitoring the resultant daughter ions in what is referred to as selected reaction monitoring (SRM)52. This is most likely the method of choice to measure DHA in plasma and tissues as current methods rely on DHA being converted to AA in samples and measuring the subsequent change in total AA48. This approach is inaccurate due to the large differential between AA and DHA concentrations. GCMS or GCMS(MS2) is another mass spectrometric technique capable of analyzing AA and degradation products. Derivatization will be required to make these molecules volatile. Bluck et al highlighted the promise of this approach with a constant intravenous infusion of 13C1-AA for 24 hrs in human subjects31. The slow decline in isotope enrichment in the extracellular pool was consistent with the remarkable ability of the body to retain AA.
With the use of LCMS(MS2) technologies, Nemet et al. recently identified five degradation products of AA, including DHA, DKG, 3-deoxythreosone, xylosone and threosone, in the human lens by derivatization with o-phenylenediamine and analysis in positive ion mode44. This group was able to identify 3-deoxythreosone, which originates from the decomposition of erythrulose as a major non-oxidative in vivo degradation product of AA, and this observation is consistent with those of Simpson et al43. Nemet et al also concluded that under oxidative conditions erythrulose can be converted to threosone. Wang et al. reported an LCMS(MS)2 method for the determination of L-threonate present in human plasma and urine using the ion transition from the parent ion of 134.5 m/z to the fragmented daughter ion of 74.7 m/z (134.5 → 74.7) as measured in negative ion mode53. They also observed that threonate is stable for 24 hr at room temperature and up to 3 months at −30°C. This observation is consistent with threonate being one of the major end-products of AA degradation. In a similar fashion, Szultka et al. more recently reported the identification of DHA, DKG, threonate and oxalic acid in aqueous AA solutions at different pH values and H2O2 concentrations, as measured by LCMS(MS2) in negative ion mode51. They proposed an AA degradation pathway similar to previous reports and observed products associated with hydration of DHA. Deutsch et al. also reported a mechanism for the hydrated hemiketal form of DHA54. These reports coincide with our recent observations using NMR (nuclear magnetic resonance) spectroscopy of aqueous DHA solutions (Fig. 3), which revealed the presence of two distinct forms of DHA (also depicted in the NMR databases: http://www.hmdb.ca/spectra/nmr_one_d/1674). A better understanding of the chemistry of DHA is vital, as this molecule is believed to give rise to the very unstable DKG, which spontaneously breaks down to oxalate. A clearer picture of the other degradation products formed from DKG is also required to characterize what could be several different degradative processes. We propose that by infusing healthy subjects with a stable 13C6 isotope of AA, as we have previously performed with 13C2-glycine infusion21, coupled with an optimized LCMS(MS2) technique, we will be able to more precisely determine the contribution of AA degradation to endogenous oxalate production.
Figure 3.
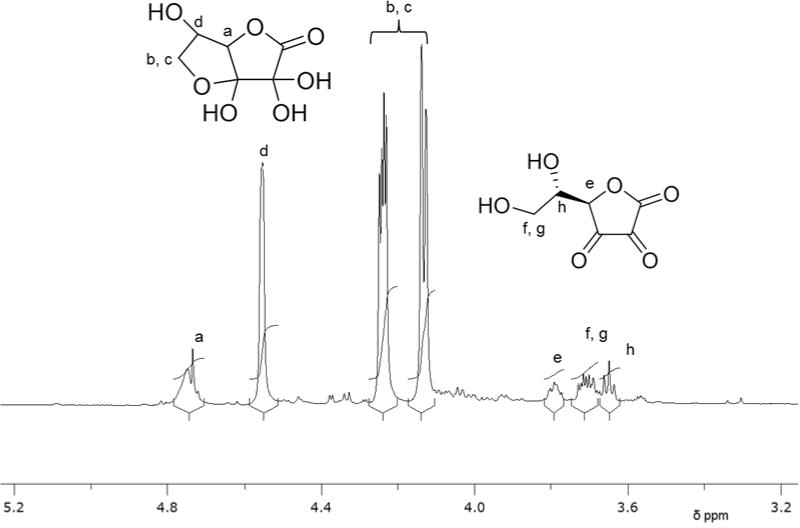
1HNMR spectrum (Bruker 850 Mhz NMR spectrometer) of DHA (3 mM) in D2O, showing proton assignments for hydrated bicyclic (a–d) and open chain (e–h) forms.
EPIDEMIOLOGICAL EVIDENCE THAT AA CONTRIBUTES TO KIDNEY STONE DISEASE
The association between AA intake and kidney stone formation was first reported by Taylor et al in a prospective study of ~ 50,000 male health professionals6. Men with an intake >218 mg/day had a 31% higher risk of forming stones than those consuming < 105 mg/day (P = 0.01). Supplemental AA also increased risk with an intake > 1000 mg/day associated with a 16% increase over those taking no supplemental AA. In a similarly sized study in Sweden, supplemental AA intake was associated with a near 2-fold increase in stone risk7. This risk appeared to be associated with the number of supplemental tablets taken each day, but the dose relationship was not accurately quantified. These findings suggest that AA intake could be a significant risk factor for the development of kidney stones and indicate that studies to determine the effects of both dietary and supplemental AA on stone risk factors, particularly urinary oxalate excretion, in normal subjects and stone formers consuming controlled diets are clearly warranted.
ASCORBIC ACID INGESTION AND URINARY OXALATE EXCRETION
Because oxalate is formed from the breakdown of AA, an increased formation of AA resulting in an increased urinary excretion of oxalate is the only mechanism that has been considered for the stone risk associated with AA ingestion. It remains possible that some other less obvious mechanism is involved. Numerous studies have examined the association between supplemental AA ingestion and stone risk parameters and have reported an increased urinary oxalate excretion. The validity of many of these studies can be questioned, however, due to the possible formation of oxalate in urine samples either during storage or during analysis. Levine et al showed that when 400 mg of supplemental AA was given to normal subjects on a controlled, AA-deplete diet, urinary oxalate excretion began to increase30. As dietary oxalate was not controlled in these studies, it is possible that this limited their ability to detect the effects of levels of AA below 400 mg/day on urinary oxalate excretion. Traxer et al observed a 21.8% increase in urinary oxalate excretion in normal subjects receiving 2 g of AA on a controlled diet and a 34.4% increase in stone formers on the same diet55. Baxmann et al reported a 56% increase in urinary oxalate excretion in normal subjects consuming 1 g/day, a 61% increase in stone formers consuming 1 g/day and a 75% increase in stone formers consuming 2 g/day56. It is possible that more AA breaks down to oxalate in stone formers due to the increased oxidative stress associated with the disease57.
In 2008 Taylor and Curhan reported on factors that influenced urinary oxalate excretion in over 3,000 participants who submitted 24 hr urine samples as part of their epidemiological studies of stone formation in male health professionals and female nurses14. Approximately two thirds had a history of kidney stone formation. Of the dietary factors analyzed, which included calcium, oxalate, animal protein, vitamin B6 and AA, and urinary excretions of potassium, sodium, and phosphorus which reflect the intake of these minerals, AA had the greatest effect on urinary oxalate excretion. Compared with participants consuming < 90 mg/day, participants with an intake > 1000 mg/day had a urinary oxalate excretion that was 6.8 mg/day higher when adjusted for confounding variables. This was much greater than the effect of dietary calcium which reduced urinary oxalate by only 2.3 mg/day in an analysis of quartile groups of intake with similar adjustments. These results alone suggest that calcium oxalate stone formers should cease taking supplemental AA should decrease their intake of AA-rich foods. Clearly the effects of AA intake warrant much closer scrutiny for its influence on urinary oxalate excretion and calcium oxalate stone formation.
PARENTERAL AND ENTERAL SOURCES OF ASCORBIC ACID AND URINARY OXALATE EXCRETION
Individuals receiving parenteral and enteral fluids to meet their nutritional needs require a source of AA. Due to the lability of AA, care has to be taken that it does not breakdown to oxalate during preparation or storage. Ribeiro et al have reported that 15% of the AA in a pediatric parenteral formulation was lost after storage for 3 days at 25 °C58. This loss depended on the storage temperature, storage time and photoprotection. We have previously suggested that the oxalate we detected in the defined formula diet, Ensure®, is derived from AA during its preparation59. The Food and Drug Administration requires that 200 mg AA be added to multivitamin preparations used in parenteral solutions to meet daily nutritional requirements. Pena de la Vega et al found that the increase from 100 – 200 mg/d was associated with a 30% increase in urinary oxalate excretion60. Despite this increase in oxalate excretion, studies have not yet associated it with an increased stone incidence in individuals receiving parenteral nutrition.
ASCORBIC ACID INGESTION AND OXALATE NEPHROPATHY
There have been a number of reports of oxalate nephropathy that suggest the oral ingestion or intravenous administration of moderate to large amounts of AA was a contributing factor (Table 2). The majority were treated with dialysis and in all but two, dialysis was temporary. Two of the reported cases resulted in mortality. Intratubular calcium oxalate deposits were demonstrated in all of the renal biopsies and other findings included mesangial hypercellularity, tubular atrophy, interstitial fibrosis, tubulointerstitial nephritis and edema. A number of these subjects took mega-doses of AA in hopes that it would improve their general health or provide relief or cure of ongoing diseases. This approach was popularized a number of years ago by Noble Prize recipient, Dr. Linus Pauling61. The benefits of consuming large amounts of AA have not been substantiated and the risk associated with an increased oxalate production is substantial. AA has been demonstrated to have anti-proliferative and anti-tumor effects in cell culture and animal models. To achieve cytotoxic concentrations of AA some investigators have administered large doses of intravenous AA to patients afflicted with advanced cancer13, 40. While oncologic benefits have not yet been proven, patients have experienced limited toxicity in trials of a short duration40. However, such infusions could potentially result in oxalate nephropathy. The public needs to be informed of this risk as there is an increasing pool of candidates receiving large doses of intravenous AA under the guidance of complementary and alternative medicine practitioners for purported health benefits62. The patient in Table 2 administered the unspecified dose of AA IV received it in such a clinic63. Over 30,000 individuals may have received such infusions with an average of 22 treatments per patient for the treatment of infection, fatigue, cancer and other conditions with unknown consequences. An assessment of whether there is any decline in kidney function associated with these treatments is warranted.
Table 2.
Vitamin C ingestion and oxalate nephropathy
| Ref. | Sex | Age | Reason for adminstration | Amount | Highest serum Cr mg/dL | Dialysis | Final serum Cr mg/dL |
|---|---|---|---|---|---|---|---|
| 64 | M | 73 | Patient choice | 680 mg/dy High Ox diet | 8.4 | Y Temp | 1.8 |
| 65 | M | 51 | Metastatic prostate cancer | 60 g IV | 13.3 | N | 2.9 |
| 63 | F | 51 | Lupus | 2 IV doses (Amount ?) | 7.2 | Y Temp | 1.1 |
| 8 | F | 58 | Nephrotic Syndrome | 45 g IV | 3.5 | Y | Death |
| 12 | M | 58 | Burn injury followed by parenteral nutrition | 1.6 g/dy for 50 dys | Y Temp | 1.4 | |
| 11 | M | 31 | Patient choice | 2 – 5 g/dy several dys | 10.1 | Y Temp | 2.2 |
| 10 | F | 49 | Patient choice | 4 g/dy several mths | 4.5 | N | 1.1 |
| 9 | M | 72 | Patient choice | 480 – 960 mg/dy several mths | 10.6 | Y Temp | 1.9 |
| 66 | M | 72 | Patient choice | Several grams daily (Period ?) | 15 | Y | Death |
| 67 | F | 58 | Patient choice | 3 – 6.5 g/dy for 1 mth | 9.1 | Y | Awaiting transplant |
CONCLUSION
Epidemiological evidence indicates that the amount of AA ingested is a risk factor for calcium oxalate stone disease and that this risk is associated with the amount of oxalate excreted in urine. Short term experiments with human subjects have confirmed that supplemental AA increases urinary oxalate excretion. The available evidence presented above indicates that the oxidized form of AA, DHA, can form an open chain isomer, DKA, which is unstable and breaks down to oxalate and 4-carbon sugars. What is not clear is how much AA and DHA is metabolized in cells and tissues, and how much is converted to DKA and ultimately oxalate. Each of these pathways may be influenced by the diet, physiological effectors or by genetic variation. How the amount converted to oxalate is modulated is important knowledge to accrue to decrease endogenous oxalate synthesis and to identify optimal intake levels for individuals with calcium oxalate stone disease. While our investigations on the sources of endogenous oxalate synthesis indicate that hydroxyproline metabolism may account for 30 – 40%, it remains possible that AA is also a major source. Finally, it is clear that compromised renal function coupled with excessive AA ingestion can result in oxalate nephropathy in susceptible individuals.
Acknowledgments
Funding: This work was supported in part by NIH grant and DK73732.
Footnotes
Conflict of Interest: Authors declare no conflict of interest.
Ethical Approval: This article does not contain any studies with human participants or animals performed by the investigators.
References
- 1.Hellman L, Burns JJ. Metabolism of L-ascorbic acid-1-C14 in man. J Biol Chem. 1958;230:923–930. [PubMed] [Google Scholar]
- 2.Atkins GL, Dean BM, Griffin WJ, Watts RWE. Quantitative aspects of ascorbic acid metabolism in man. J Biol Chem. 1964;239:2975–2980. [PubMed] [Google Scholar]
- 3.Baker EM, Saari JC, Tolbert BM. Ascorbic acid metabolism in man. Amer J Clin Nutr. 1966;19:371–378. doi: 10.1093/ajcn/19.5.371. [DOI] [PubMed] [Google Scholar]
- 4.Chalmers AH, Cowley DM. Stability of ascorbate in urine: relevance to analyses for ascorbate and oxalate. Clinical chemistry. 1985;31:1703–1705. [PubMed] [Google Scholar]
- 5.Mazzachi BC, Teubner JK, Ryall RL. Factors affecting measurement of urinary oxalate. Clinical chemistry. 1984;30:1339–1343. [PubMed] [Google Scholar]
- 6.Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. Journal of the American Society of Nephrology: JASN. 2004;15:3225–3232. doi: 10.1097/01.ASN.0000146012.44570.20. [DOI] [PubMed] [Google Scholar]
- 7.Thomas LD, Elinder CG, Tiselius HG, Wolk A, Akesson A. Ascorbic acid supplements and kidney stone incidence among men: a prospective study. JAMA internal medicine. 2013;173:386–388. doi: 10.1001/jamainternmed.2013.2296. [DOI] [PubMed] [Google Scholar]
- 8.Lawton JM, Conway LT, Crosson JT, Smith CL, Abraham PA. Acute oxalate nephropathy after massive ascorbic acid administration. Arch Intern Med. 1985;145:950–951. [PubMed] [Google Scholar]
- 9.Lamarche J, Nair R, Peguero A, Courville C. Vitamin C-induced oxalate nephropathy. International journal of nephrology. 2011;2011:146927. doi: 10.4061/2011/146927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nasr SH, Kashtanova Y, Levchuk V, Markowitz GS. Secondary oxalosis due to excess vitamin C intake. Kidney international. 2006;70:1672. doi: 10.1038/sj.ki.5001724. [DOI] [PubMed] [Google Scholar]
- 11.Mashour S, Turner JF, Jr, Merrell R. Acute renal failure, oxalosis, and vitamin C supplementation: a case report and review of the literature. Chest. 2000;118:561–563. doi: 10.1378/chest.118.2.561. [DOI] [PubMed] [Google Scholar]
- 12.Alkhunaizi AM, Chan L. Secondary oxalosis: a cause of delayed recovery of renal function in the setting of acute renal failure. Journal of the American Society of Nephrology: JASN. 1996;7:2320–2326. doi: 10.1681/ASN.V7112320. [DOI] [PubMed] [Google Scholar]
- 13.Robitaille L, Mamer OA, Miller WH, Jr, Levine M, Assouline S, Melnychuk D, Rousseau C, Hoffer LJ. Oxalic acid excretion after intravenous ascorbic acid administration. Metabolism: clinical and experimental. 2009;58:263–269. doi: 10.1016/j.metabol.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor EN, Curhan GC. Determinants of 24-hour urinary oxalate excretion. Clinical journal of the American Society of Nephrology: CJASN. 2008;3:1453–1460. doi: 10.2215/CJN.01410308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Halliwell B. Vitamin C and genomic stability. Mutation research. 2001;475:29–35. doi: 10.1016/s0027-5107(01)00072-0. [DOI] [PubMed] [Google Scholar]
- 16.Nishikimi M, Yagi K. Molecular basis for the deficiency in humans of gulonolactone oxidase, a key enzyme for ascorbic acid biosynthesis. Am J Clin Nutr. 1991;54:1203S–1208S. doi: 10.1093/ajcn/54.6.1203s. [DOI] [PubMed] [Google Scholar]
- 17.Grano A, De Tullio MC. Ascorbic acid as a sensor of oxidative stress and a regulator of gene expression: the Yin and Yang of vitamin C. Medical hypotheses. 2007;69:953–954. doi: 10.1016/j.mehy.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 18.Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta. 2012;1826:443–457. doi: 10.1016/j.bbcan.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Linster CL, Van Schaftingen E. Vitamin C. Biosynthesis, recycling and degradation in mammals. The FEBS journal. 2007;274:1–22. doi: 10.1111/j.1742-4658.2006.05607.x. [DOI] [PubMed] [Google Scholar]
- 20.Holmes RP, Goodman HO, Assimos DG. Contribution of dietary oxalate to urinary oxalate excretion. Kidney international. 2001;59:270–276. doi: 10.1046/j.1523-1755.2001.00488.x. [DOI] [PubMed] [Google Scholar]
- 21.Knight J, Assimos DG, Callahan MF, Holmes RP. Metabolism of primed, constant infusions of [1,2-(13)C(2)] glycine and [1-(13)C(1)] phenylalanine to urinary oxalate. Metabolism: clinical and experimental. 2011;60:950–956. doi: 10.1016/j.metabol.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knight J, Jiang J, Assimos DG, Holmes RP. Hydroxyproline ingestion and urinary oxalate and glycolate excretion. Kidney international. 2006;70:1929–1934. doi: 10.1038/sj.ki.5001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmes RP, Kennedy M. Estimation of the oxalate content of foods and daily oxalate intake. Kidney international. 2000;57:1662–1667. doi: 10.1046/j.1523-1755.2000.00010.x. [DOI] [PubMed] [Google Scholar]
- 24.von Unruh GE, Voss S, Sauerbruch T, Hesse A. Reference range for gastrointestinal oxalate absorption measured with a standardized [13C2] oxalate absorption test. The Journal of urology. 2003;169:687–690. doi: 10.1097/01.ju.0000051637.63068.92. [DOI] [PubMed] [Google Scholar]
- 25.Fan X, Sokorai KJ. Changes in quality, liking, and purchase intent of irradiated fresh-cut spinach during storage. Journal of food science. 2011;76:S363–368. doi: 10.1111/j.1750-3841.2011.02207.x. [DOI] [PubMed] [Google Scholar]
- 26.Jansson PJ, Jung HR, Lindqvist C, Nordstrom T. Oxidative decomposition of vitamin C in drinking water. Free radical research. 2004;38:855–860. doi: 10.1080/10715760410001700497. [DOI] [PubMed] [Google Scholar]
- 27.Burzle M, Suzuki Y, Ackermann D, Miyazaki H, Maeda N, Clemencon B, Burrier R, Hediger MA. The sodium-dependent ascorbic acid transporter family SLC23. Molecular aspects of medicine. 2013;34:436–454. doi: 10.1016/j.mam.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 28.Corpe CP, Eck P, Wang J, Al-Hasani H, Levine M. Intestinal dehydroascorbic acid (DHA) transport mediated by the facilitative sugar transporters, GLUT2 and GLUT8. J Biol Chem. 2013;288:9092–9101. doi: 10.1074/jbc.M112.436790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kallner A, Hartmann D, Hornig D. On the absorption of ascorbic acid in man. International journal for vitamin and nutrition research. Internationale Zeitschrift fur Vitamin-und Ernahrungsforschung. Journal international de vitaminologie et de nutrition. 1977;47:383–388. [PubMed] [Google Scholar]
- 30.Levine M, Conry-Cantilena C, Wang Y, Welch RW, Washko PW, Dhariwal KR, Park JB, Lazerev A, Graumlich JF, King J, Cantilena LR. Vitamin C pharmacokinetics in healthy volunteers: Evidence for a recommended dietary allowance. Proc Natl Acad Sci. 1996;93:3704–3709. doi: 10.1073/pnas.93.8.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bluck LJ, Izzard AP, Bates CJ. Measurement of ascorbic acid kinetics in man using stable isotopes and gas chromatography/mass spectrometry. Journal of mass spectrometry: JMS. 1996;31:741–748. doi: 10.1002/(SICI)1096-9888(199607)31:7<741::AID-JMS352>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 32.Kallner A, Hartmann D, Hornig D. Steady-state turnover and body pool of ascorbic acid in man. Am J Clin Nutr. 1979;32:530–539. doi: 10.1093/ajcn/32.3.530. [DOI] [PubMed] [Google Scholar]
- 33.Omaye ST, Schaus EE, Kutnink MA, Hawkes WC. Measurement of vitamin C in blood components by high-performance liquid chromatography. Implication in assessing vitamin C status. Annals of the New York Academy of Sciences. 1987;498:389–401. doi: 10.1111/j.1749-6632.1987.tb23776.x. [DOI] [PubMed] [Google Scholar]
- 34.Levine M, Wang Y, Padayatty SJ, Morrow J. A new recommended dietary allowance of vitamin C for healthy young women. Proc Natl Acad Sci U S A. 2001;98:9842–9846. doi: 10.1073/pnas.171318198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baker PR, Cramer SD, Kennedy M, Assimos DG, Holmes RP. Glycolate and glyoxylate metabolism in HepG2 cells. Amer J Physiol. 2004;287:C1359–C1365. doi: 10.1152/ajpcell.00238.2004. [DOI] [PubMed] [Google Scholar]
- 36.Knight J, Holmes RP, Cramer SD, Takayama T, Salido E. Hydroxyproline metabolism in mouse models of primary hyperoxaluria. American journal of physiology. Renal physiology. 2012;302:F688–693. doi: 10.1152/ajprenal.00473.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Corti A, Casini AF, Pompella A. Cellular pathways for transport and efflux of ascorbate and dehydroascorbate. Archives of biochemistry and biophysics. 2010;500:107–115. doi: 10.1016/j.abb.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 38.Rumsy SC, Levine M. Absorption, transport, and disposition of ascorbic acid in humans. Nutr Biochem. 1998;9:116–130. [Google Scholar]
- 39.Levine M, Padayatty SJ, Espey MG. Vitamin C: a concentration-function approach yields pharmacology and therapeutic discoveries. Advances in nutrition. 2011;2:78–88. doi: 10.3945/an.110.000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stephenson CM, Levin RD, Spector T, Lis CG. Phase I clinical trial to evaluate the safety, tolerability, and pharmacokinetics of high-dose intravenous ascorbic acid in patients with advanced cancer. Cancer chemotherapy and pharmacology. 2013;72:139–146. doi: 10.1007/s00280-013-2179-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, Roper J, Chio II, Giannopoulou EG, Rago C, Muley A, Asara JM, Paik J, Elemento O, Chen Z, Pappin DJ, Dow LE, Papadopoulos N, Gross SS, Cantley LC. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015 doi: 10.1126/science.aaa5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park JB, Levine M. Purification, cloning and expression of dehydroascorbic acid-reducing activity from human neutrophils: identification as glutaredoxin. The Biochemical journal. 1996;315(Pt 3):931–938. doi: 10.1042/bj3150931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simpson GL, Ortwerth BJ. The non-oxidative degradation of ascorbic acid at physiological conditions. Biochim Biophys Acta. 2000;1501:12–24. doi: 10.1016/s0925-4439(00)00009-0. [DOI] [PubMed] [Google Scholar]
- 44.Nemet I, Monnier VM. Vitamin C degradation products and pathways in the human lens. J Biol Chem. 2011;286:37128–37136. doi: 10.1074/jbc.M111.245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gál I. Estimation of Ascorbic Acid (Vitamin C) by Titration. Nature. 1936;138:799. [Google Scholar]
- 46.Moeslinger T, Brunner M, Volf I, Spieckermann PG. Spectrophotometric determination of ascorbic acid and dehydroascorbic acid. Clinical chemistry. 1995;41:1177–1181. [PubMed] [Google Scholar]
- 47.Lee W, Roberts SM, Labbe RF. Ascorbic acid determination with an automated enzymatic procedure. Clinical chemistry. 1997;43:154–157. [PubMed] [Google Scholar]
- 48.Lykkesfeldt J. Ascorbate and dehydroascorbic acid as reliable biomarkers of oxidative stress: analytical reproducibility and long-term stability of plasma samples subjected to acidic deproteinization. Cancer epidemiology, biomarkers & prevention. 2007;16:2513–2516. doi: 10.1158/1055-9965.EPI-07-0639. [DOI] [PubMed] [Google Scholar]
- 49.Chen Z, Chen B, Yao S. High-performance liquid chromatography/electrospray ionization-mass spectrometry for simultaneous determination of taurine and 10 water-soluble vitamins in multivitamin tablets. Analytica Chimica Acta. 2006;569:169–175. [Google Scholar]
- 50.Shui G, Leong LP. Analysis of polyphenolic antioxidants in star fruit using liquid chromatography and mass spectrometry. Journal of chromatography. A. 2004;1022:67–75. doi: 10.1016/j.chroma.2003.09.055. [DOI] [PubMed] [Google Scholar]
- 51.Szultka M, Buszewska-Forajta M, Kaliszan R, Buszewski B. Determination of ascorbic acid and its degradation products by high-performance liquid chromatography-triple quadrupole mass spectrometry. Electrophoresis. 2014;35:585–592. doi: 10.1002/elps.201300439. [DOI] [PubMed] [Google Scholar]
- 52.Fenoll J, Martinez A, Hellin P, Flores P. Simultaneous determination of ascorbic and dehydroascorbic acids in vegetables and fruits by liquid chromatography with tandem mass spectrometry. Food chemistry. 2011;127:340–344. [Google Scholar]
- 53.Wang H, Jiang J, Hu P. Determination of L-threonate in human plasma and urine by high performance liquid chromatography-tandem mass spectrometry. Journal of chromatography. B, Analytical technologies in the biomedical and life sciences. 2006;834:155–162. doi: 10.1016/j.jchromb.2006.02.057. [DOI] [PubMed] [Google Scholar]
- 54.Deutsch JC. Spontaneous hydrolysis and dehydration of dehydroascorbic acid in aqueous solution. Analytical biochemistry. 1998;260:223–229. doi: 10.1006/abio.1998.2700. [DOI] [PubMed] [Google Scholar]
- 55.Traxer O, Huet B, Poindexter J, Pak CY, Pearle MS. Effect of ascorbic acid consumption on urinary stone risk factors. The Journal of urology. 2003;170:397–401. doi: 10.1097/01.ju.0000076001.21606.53. [DOI] [PubMed] [Google Scholar]
- 56.Baxmann AC, De OGMC, Heilberg IP. Effect of vitamin C supplements on urinary oxalate and pH in calcium stone-forming patients. Kidney international. 2003;63:1066–1071. doi: 10.1046/j.1523-1755.2003.00815.x. [DOI] [PubMed] [Google Scholar]
- 57.Khan SR. Is oxidative stress, a link between nephrolithiasis and obesity, hypertension, diabetes, chronic kidney disease, metabolic syndrome? Urological research. 2012;40:95–112. doi: 10.1007/s00240-011-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ribeiro DO, Pinto DC, Lima LM, Volpato NM, Cabral LM, de Sousa VP. Chemical stability study of vitamins thiamine, riboflavin, pyridoxine and ascorbic acid in parenteral nutrition for neonatal use. Nutrition journal. 2011;10:47. doi: 10.1186/1475-2891-10-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Holmes RP, Goodman HO, Assimos DG. Dietary oxalate and its intestinal absorption. Scan Microsc. 1995;9:1109–1120. [PubMed] [Google Scholar]
- 60.Pena de la Vega L, Lieske JC, Milliner D, Gonyea J, Kelly DG. Urinary oxalate excretion increases in home parenteral nutrition patients on a higher intravenous ascorbic acid dose. JPEN. Journal of parenteral and enteral nutrition. 2004;28:435–438. doi: 10.1177/0148607104028006435. [DOI] [PubMed] [Google Scholar]
- 61.Pauling L. Evolution and the need for ascorbic acid. Proc Natl Acad Sci U S A. 1970;67:1643–1648. doi: 10.1073/pnas.67.4.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Padayatty SJ, Sun AY, Chen Q, Espey MG, Drisko J, Levine M. Vitamin C: intravenous use by complementary and alternative medicine practitioners and adverse effects. PloS one. 2010;5:e11414. doi: 10.1371/journal.pone.0011414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cossey LN, Rahim F, Larsen CP. Oxalate Nephropathy and Intravenous Vitamin C. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2013 doi: 10.1053/j.ajkd.2013.01.025. [DOI] [PubMed] [Google Scholar]