

SUMMARY
Misfolded proteins of the endoplasmic reticulum (ER) are retrotranslocated to the cytosol, where they are polyubiquitinated, extracted from the membrane, and degraded by the proteasome. To investigate how the ER-associated Degradation (ERAD) machinery can accomplish retrotranslocation of a misfolded luminal protein domain across a lipid bilayer, we have reconstituted retrotranslocation with purified S. cerevisiae proteins, using proteoliposomes containing the multi-spanning ubiquitin ligase Hrd1. Retrotranslocation of the luminal domain of a membrane-spanning substrate is triggered by autoubiquitination of Hrd1. Substrate ubiquitination is a subsequent event, and the Cdc48 ATPase that completes substrate extraction from the membrane is not required for retrotranslocation. Ubiquitination of lysines in Hrd1’s RING-finger domain is required for substrate retrotranslocation in vitro and for ERAD in vivo. Our results suggest that Hrd1 forms a ubiquitin-gated protein-conducting channel.
INTRODUCTION
Misfolded proteins in the endoplasmic reticulum (ER) are disposed of by a process called ER-associated protein degradation (ERAD) (for review, see (Christianson and Ye, 2014). In Saccharomyces cerevisiae, and probably in all other eukaryotic cells, substrates are degraded by three different ERAD pathways (ERAD-L, -M, and -C), depending on whether their misfolded domain is localized in the ER lumen, within the membrane, or on the cytosolic side of the ER membrane (Carvalho et al., 2006; Huyer et al., 2004; Vashist and Ng, 2004). Most substrates of these ERAD pathways are polyubiquitinated on the cytosolic side of the ER membrane, but they employ distinct ubiquitin ligase complexes. ERAD-L substrates use the RING-finger ubiquitin ligase Hrd1 in complex with three other membrane proteins (Hrd3, Usa1, and Der1) and a luminal protein (Yos9). ERAD-M substrates also use Hrd1, but only a subset of the other components. ERAD-C substrates require Doa10, another RING-finger ligase. A fourth ERAD pathway has recently been identified that disposes of inner nuclear membrane proteins (Foresti et al., 2014; Khmelinskii et al., 2014). It uses a RING-finger ligase that comprises three membrane proteins (Asi1,2,3). All ERAD pathways converge on the cytosolic side, where they require components of the ubiquitination machinery and an ATPase complex consisting of Cdc48, Ufd1, and Npl4. The Cdc48 complex is thought to move polyubiquitinated substrates into the cytosol, where they are ultimately degraded by the proteasome (Bays et al., 2001; Jarosch et al., 2002; Rabinovich et al., 2002; Ye et al., 2001).
Many aspects of ERAD remain unclear, but arguably the most important unresolved question is how a misfolded protein crosses the ER membrane. This question is particularly relevant for ERAD-L substrates, which have to be inserted into the luminal side of the ER membrane, moved through the membrane, and extracted on the cytosolic side of the ER membrane. It seems unlikely that these ERAD substrates are moved directly through the lipid bilayer. Rather, like in other cases of protein translocation, transport probably occurs through a conduit or channel formed by integral membrane proteins (Collinson et al., 2015; Park and Rapoport, 2012; Wickner and Schekman, 2005). Several multi-spanning membrane proteins have been proposed as channel candidates. These include the Sec61 channel that is normally involved in “forward” translocation of proteins from the cytosol into the ER (Pilon et al., 1997; Schafer and Wolf, 2009; Wiertz et al., 1996; Willer et al., 2008), the mammalian homologs of Der1 (Derlin’s) (Lilley and Ploegh, 2004; Ye et al., 2004), and the Hrd1 ubiquitin ligase (Carvalho et al., 2010).
Hrd1 seems to be a major component of the retrotranslocation channel, as its overexpression in S. cerevisiae bypasses the other membrane components (Hrd3, Usa1, Der1) (Carvalho et al., 2010), whereas overexpression of these proteins, does not bypass the Hrd1 requirement. In overexpressing Hrd1 cells, the downstream cytosolic components, including the Cdc48 complex, are still needed. Hrd1 overexpression makes substrate selection less specific, but the cells remain viable (Denic et al., 2006). Under these conditions, Hrd1 itself is polyubiquitinated and degraded (Gardner et al., 2000). These results suggest that Hrd1 is the only membrane component required for a basic ERAD-L process. However, since these experiments were performed with intact cells they do not exclude the possibility that other, unidentified membrane proteins form the retrotranslocon or cooperate with Hrd1. In vitro reconstitution experiments with purified components are crucial to identify the retrotranslocation channel and elucidate the role of Hrd1.
We have recently made progress towards the goal of reconstituting ERAD with purified components by recapitulating substrate recognition, polyubiquitination, and recruitment of the Cdc48 complex in detergent (Stein et al., 2014). We also demonstrated Cdc48-dependent membrane extraction of a polyubiquitinated protein. However, the proteoliposomes contained a pre-assembled complex of Hrd1 and a soluble ERAD-L substrate in which at least one segment of the substrate was exposed to the outside of the vesicles before addition of Cdc48 complex. Because the misfolded substrate was not exclusively in the lumen of the vesicles, this system was inappropriate to study the retrotranslocation of a substrate across the membrane. Thus, the mechanism of the most important step in ERAD, the retrotranslocation of a polypeptide, has not yet been clarified. The lack of a retrotranslocation assay also makes it impossible to test whether Hrd1 forms a retrotranslocon, and if so, how it is gated.
Here, we have recapitulated Hrd1-dependent retrotranslocation using membrane-spanning substrates. We show that autoubiquitination of Hrd1 on several lysines of its RING-finger domain allows the misfolded luminal domain of a substrate to move across the membrane. The Cdc48 ATPase complex is not required for retrotranslocation. Our results indicate that Hrd1 forms a ubiquitin-gated protein-conducting channel. Other membrane-spanning ubiquitin ligases in the ER may function in an analogous manner.
RESULTS
Polyubiquitination of integral membrane ERAD substrates by Hrd1
Our previous experiments employed a well-established soluble ERAD-L substrate, misfolded carboxypeptidase Y (CPY*), which differs from the native protein by a single point mutation (Finger et al., 1993). However, this substrate is difficult to use in retrotranslocation assays, as it needs to be exclusively located inside proteoliposomes at the beginning of the reaction. This is technically demanding, none the least because the reconstituted vesicles have a small internal volume. To circumvent this problem, we used a membrane-spanning substrate. CPY* was fused to a C-terminal transmembrane (TM) segment derived from the multi-spanning protein Pdr5, followed by a short tail (CPY*-TM; Figure 1A)(Taxis et al., 2003). In contrast to CPY*, CPY*-TM can be reconstituted with high efficiency into proteoliposomes because the TM segment preferentially partitions into the lipid bilayer. The protein is expected to sit in the lipid bilayer in both orientations (Figure 1B). These proteoliposomes can be used to establish a retrotranslocation assay in which the movement of the CPY* domain from the lumen to the outside of the vesicles can be observed (see scheme in Figure 1B).
Figure 1. Membrane-bound ERAD substrates of the Hrd1 ubiquitin ligase.
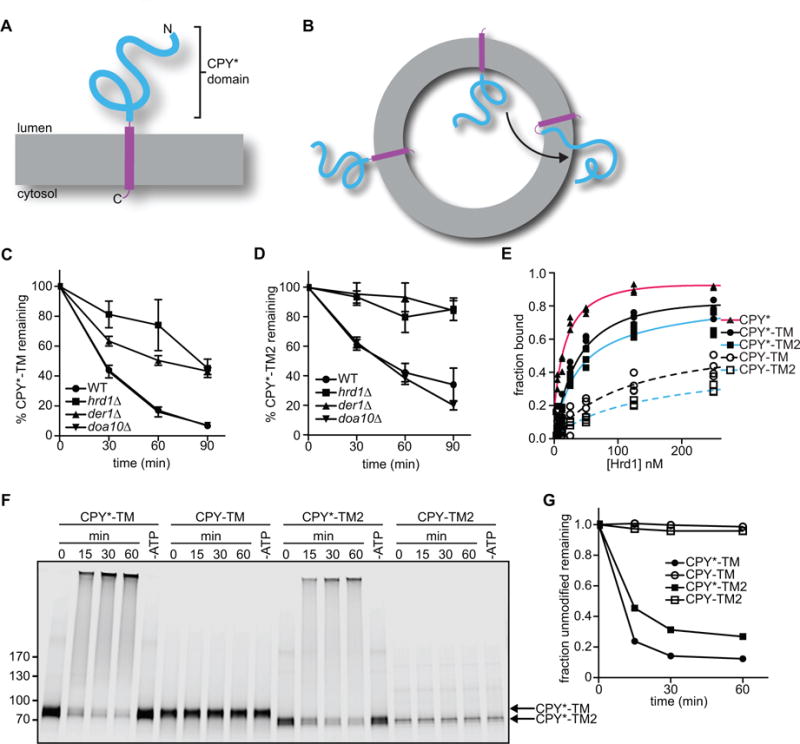
(A) Scheme of CPY*-TM, a fusion of the misfolded ER luminal substrate CPY* (blue) with a transmembrane (TM) segment derived from the multi-spanning membrane protein Pdr5 (purple).
(B) CPY*-TM is reconstituted into proteoliposomes in both orientations. Retrotranslocation is defined as the movement of a segment of the luminal domain from the inside to the outside of the vesicles (arrow).
(C) The degradation of CPY*-TM with a C-terminal HA tag was tested after addition of cycloheximide to wild-type S. cerevisiae cells or strains lacking the indicated ERAD components. At each time point, proteins were analyzed by SDS-PAGE and immunoblotting with HA antibodies. Full-length protein and a degradation product were observed (Figure S1A). The quantification shows the sum of the intensity of both bands. Data are represented as mean +/− SEM from at least three experiments.
(D) As in (C), but with CPY*-TM2, which contains the TM segment from the single-spanning membrane protein Spt23 (see also Figure S1B).
(E) Purified, DyLight800-labeled CPY*-TM, CPY*-TM2, or CPY* (each 100 nM) was incubated with different concentrations of purified Hrd1-SBP immobilized on streptavidin beads. The bound material was analyzed by SDS-PAGE and fluorescence scanning. For comparison, the binding of fusions of wild-type CPY with TM or TM2 (CPY-TM and CPY-TM2, respectively) was tested (dotted lines). The apparent affinities for CPY*, CPY*-TM, CPY*-TM2, CPY-TM, and CPY-TM2 were estimated to be 20 nM, 40 nM, 30 nM, 120 nM, and 110 nM, respectively.
(F) The indicated DyLight800-labeled proteins were incubated with Hrd1 and ubiquitination machinery in detergent. Controls were performed in the absence of ATP. The samples were analyzed by SDS-PAGE and fluorescence scanning.
(G) Quantification of the experiments in (F).
See also Figure S1.
We first characterized CPY*-TM in vivo. A variant of CPY*-TM with lysines in the cytosolic tail has been reported to be an ERAD-M substrate, as its degradation in S. cerevisiae was found to be dependent on Hrd1, but not Der1 (Taxis et al., 2003). Degradation of our CPY*-TM substrate was dependent on Hrd1 and partially on Der1, as shown by cycloheximide-chase experiments (Figure 1C; representative blot in Figure S1A). Deletion of DOA10 had no effect. Soluble CPY* and membrane-bound KWW were tested in parallel and, as reported (Knop et al., 1996; Vashist and Ng, 2004), behaved as pure ERAD-L substrates, since they were completely dependent on both Hrd1 and Der1 (data not shown). Thus, it seems that CPY*-TM can use both ERAD-L and –M pathways. We reasoned that CPY*-TM might be converted into a pure ERAD-L substrate by replacing the TM segment of the multi-spanning integral membrane protein Pdr5 with the TM segment of the single-spanning Spt23 protein. Indeed, this substrate (CPY*-TM2) became completely dependent on Hrd1 and Der1 (Figure 1D; representative blot in Figure S1B). These results support the idea that ERAD-L substrates are misfolded in luminal domains, whereas ERAD-M substrates have unfavorable sequence features inside the membrane (Carvalho et al., 2006). It should be noted that the TMs and C-terminal tails of CPY*-TM and CPY*-TM2 do not contain lysines to which ubiquitin chains could be attached (Figure S1C). CPY*-TM, but not CPY*-TM2, has a single serine in the tail, which could serve as an alternative modification site. Thus, most or all ubiquitination occurs in the CPY* domain of the substrates.
We tested whether purified CPY*-TM and CPY*-TM2 interact with purified Hrd1, as observed previously for CPY* (Stein et al., 2014). The proteins were labeled with a fluorescent dye at the C-terminus using sortase (Popp et al., 2007; Stein et al., 2014) and incubated with increasing concentrations of Hrd1 immobilized on beads. CPY*-TM and CPY*-TM2 bound to Hrd1 with an apparent affinity in the nanomolar range, although not as strongly as CPY* (Figure 1E). In contrast, CPY-TM and CPY-TM2 (a fusion of wild-type CPY to the same C-terminal domains as in CPY*-TM and CPY*-TM2) did not bind as efficiently (Figure 1E). These results confirm that Hrd1 has a higher affinity for misfolded proteins (Stein et al., 2014), and indicate that Hrd1 primarily recognizes the luminal CPY* domain. Next, Hrd1 was incubated with the substrates in the presence of purified ubiquitination machinery, i.e. ubiquitin activating enzyme Uba1 (E1), ubiquitin conjugating enzyme Ubc7 (E2), its activator Cue1, ubiquitin, and ATP. Polyubiquitination was observed with CPY*-TM and CPY*-TM2, but not with CPY-TM or CPY-TM2 (Figure 1F, G), indicating that the discrimination between unfolded and folded proteins is more pronounced in the ubiquitination reaction than in the binding assay.
To examine the Hrd1-substrate interaction in a membrane context, we reconstituted fluorescently labeled CPY*-TM (red, DyLight680) and Hrd1 (blue, DyLight800) into proteoliposomes (Figure 2A). CPY*-TM was added to a mixture of phosphatidylcholine and Triton X-100, the detergent was removed with Bio-Beads, and the vesicles floated in a glycerol gradient. They were then incubated with Hrd1 at a low concentration of the detergent decyl maltose neopentyl glycol (DMNG), such that the liposomes were only partially solubilized and Hrd1 reconstituted into them in a unidirectional manner (see scheme in Figure 2A). The detergent was removed and the vesicles were floated again in a glycerol gradient. Both the labeled substrate and Hrd1 floated to the same position in the gradient (Figure 2B). Essentially all Hrd1 molecules were oriented with their C-terminus on the outside of the vesicles, as demonstrated by the accessibility of a unique TEV protease recognition motif in the cytosolic domain; cleavage was observed regardless of whether the vesicles were intact or solubilized with detergent (Figure 2C). The orientation of Hrd1 is equivalent to that in ER membranes, with the outside of vesicles corresponding to the cytosol.
Figure 2. Hrd1 polyubiquitinates CPY*-TM in reconstituted proteoliposomes.
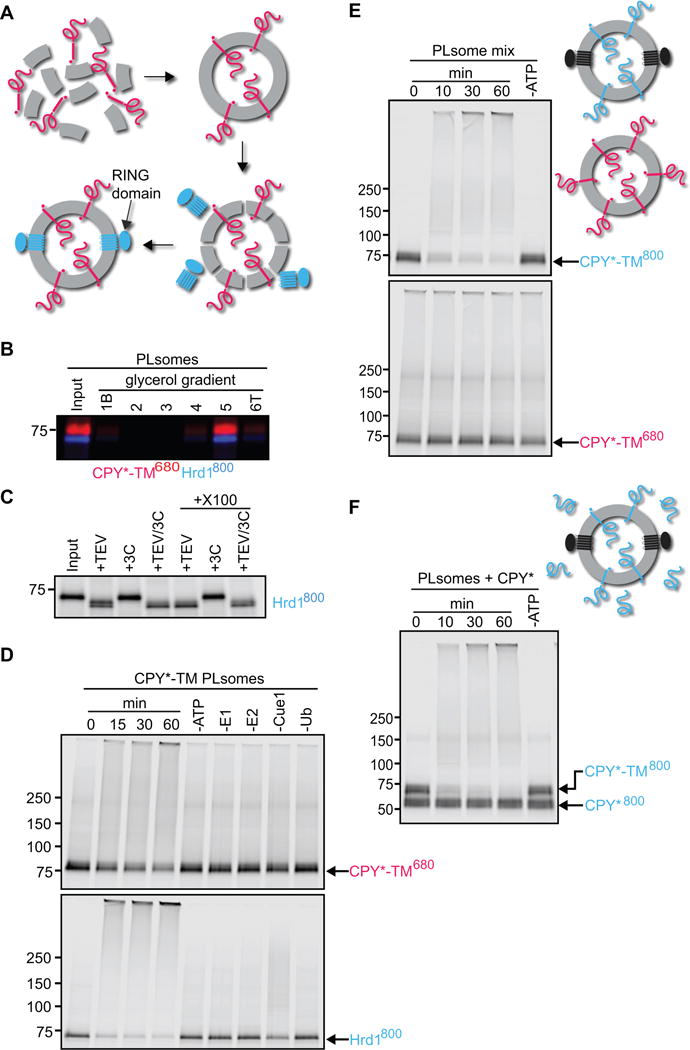
(A) Scheme of the reconstitution protocol. CPY*-TM (red) was mixed with phospholipids totally solubilized in Triton X-100 and the detergent was removed. Then, the vesicles were partially solubilized in the detergent DMNG, Hrd1 (blue) was added, and the detergent was removed.
(B) Reconstituted proteoliposomes (PLsomes) containing fluorescently labeled CPY*-TM (red; DyLight680) and Hrd1 (blue; DyLight800) were subjected to flotation in a glycerol gradient. Fractions were analyzed by SDS-PAGE and fluorescence scanning (bottom (B) is fraction 1 and top (T) is fraction 6).
(C) PLsomes containing amine-labeled Hrd1 (DyLight800) with a unique TEV-cleavage site near its C-terminus were incubated with TEV protease in the presence or absence of Triton X-100 (X100). Controls were performed with 3C protease, a related protease that should not cleave Hrd1. The samples were analyzed by SDS-PAGE and fluorescence scanning. Note that all Hrd1 molecules have their C-terminus on the outside of the vesicles.
(D) PLsomes as in (B) were incubated with ubiquitination machinery for different time periods. Controls were performed in the absence of the indicated components. The samples were analyzed by SDS-PAGE and fluorescence scanning at two different wavelengths.
(E) PLsomes containing fluorescently labeled CPY*-TM (blue; DyLight800) and unlabeled Hrd1 were mixed with PLsomes containing labeled CPY*-TM (red; DyLight680), but lacking Hrd1. Ubiquitination machinery was added for different time periods, with a control lacking ATP. Note that substrate is modified only if co-reconstituted with Hrd1.
(F) PLsomes containing Hrd1 and DyLight800-labeled CPY*-TM were incubated with CPY* and ubiquitination machinery for different time periods. Note that CPY* remains unmodified.
See also Figure S2.
Incubation of the proteoliposomes with the ubiquitination machinery resulted in the polyubiquitination of both CPY*-TM and Hrd1 (Figure 2D). As observed previously in detergent, autoubiquitination was faster and more complete than substrate ubiquitination (see also Stein et al., 2014). In the absence of any one of the components of the ubiquitination machinery, the modification reaction was abolished or, in the case of Cue1, reduced. The rate of Hrd1p autoubiquitination was about the same in the presence or absence of substrate (Figure S2A). Similar results were obtained with proteoliposomes containing labeled CPY*-TM2 and Hrd1 (Figure S2B). The efficiency of polyubiquitination was slightly lower (50% modified for CPY*-TM2 versus 60% for CPY*-TM, respectively), but the ratio of the two membrane topologies, determined by protease protection, was about the same for both constructs (Figure S2C). CPY*-TM was used in the initial characterization of the in vitro system described below, but most experiments were also performed with CPY*-TM2.
Substrate and Hrd1 have to be located in the same vesicle for substrate ubiquitination to occur, as no modification was observed when CPY*-TM and Hrd1 were reconstituted into separate vesicles (Figure 2E). Moreover, when proteoliposomes containing CPY*-TM were incubated with soluble CPY*, only the membrane-bound substrate was modified (Figure 2F). These results show that substrate is only polyubiquitinated when it interacts with Hrd1 in the same membrane.
Hrd1 allows retrotranslocation of a substrate domain
We used reconstituted proteoliposomes containing membrane-bound substrate and Hrd1 to test whether the CPY* domain moves from the inside to the outside of the vesicles, i.e. retrotranslocates through the membrane (see scheme in Figure 1B). We first prepared vesicles with C-terminally sortase-labeled CPY*-TM in both orientations (blue, DyLight800; see scheme in Figure 3A). After reconstitution, the vesicles were incubated again with sortase and a peptide coupled to DyLight680, so that the color was switched in all CPY*-TM molecules that had their C-terminus on the outside (red; Figure 3A). Finally, Hrd1 was incorporated into the liposomes as before. This procedure generates Hrd1-containing proteoliposomes in which the two differently oriented substrate populations carry distinct labels. The labeled substrate molecules were in the same vesicles as SBP-tagged Hrd1, as both the blue and red CPY*-TM molecules were quantitatively captured by streptavidin beads (Figure S3A). Co-reconstitution was also tested in the converse experiment, in which the vesicles were captured through interaction with substrate (Figure S3B). To this end, CPY*-TM molecules with their C-terminus on the outside were color-switched from DyLight800 to Alexa488 (green) using sortase. After incorporation of DyLight680-labeled Hrd1, the vesicles were incubated with antibodies to Alexa488 and protein G beads. Labeled substrate and Hrd1 were quantitatively bound to the beads (Figure S3B), confirming that they were in the same vesicles.
Figure 3. Hrd1 retrotranslocates the misfolded CPY* domain of CPY*-TM.
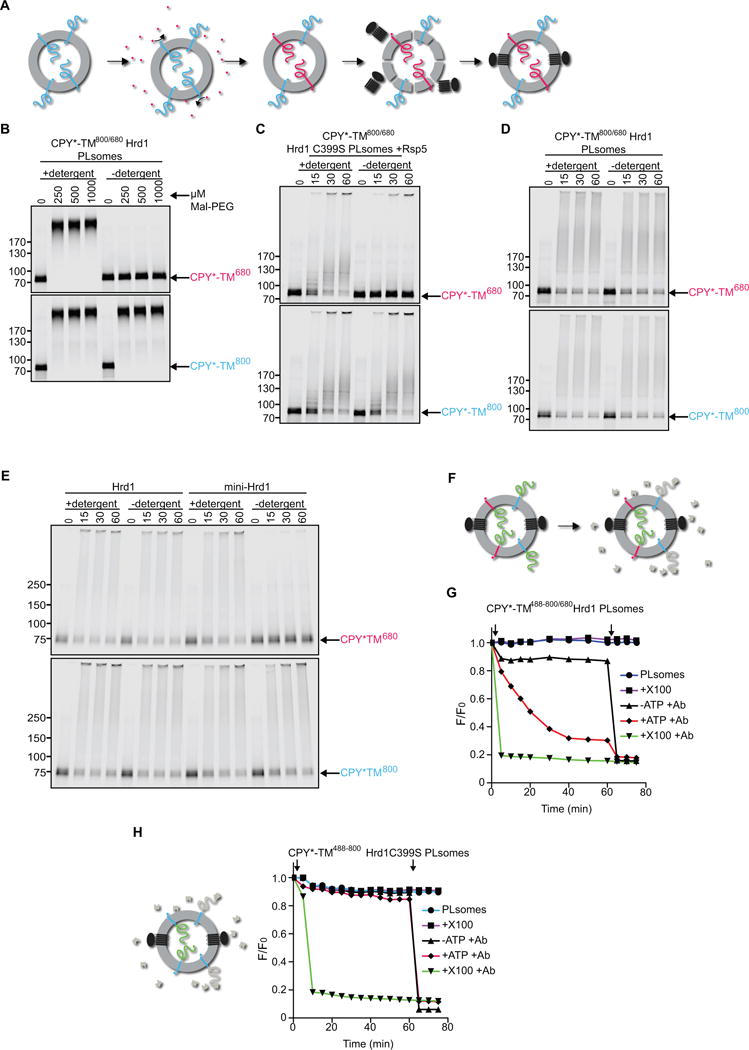
(A) Specific labeling of differently oriented substrate molecules. CPY*-TM was labeled by sortase with a fluorescent probe (blue; DyLight800) at the C-terminus, and incorporated into proteoliposomes (PLsomes) in both orientations. The vesicles were then incubated with sortase and peptide coupled to another fluorescent dye (red; DyLight680), switching the color of substrate molecules that have their C-terminus accessible. Finally, Hrd1 was incorporated into the vesicles.
(B) The orientation of substrate molecules in reconstituted PLsomes was tested by addition of different concentrations of the cysteine-modifying reagent Mal-PEG. Controls were performed in the presence of detergent. The samples were analyzed by SDS-PAGE and fluorescence scanning. Note that the CPY* domain of the red molecules can be modified only after addition of detergent.
(C) The orientation of substrate molecules was tested in PLsomes containing inactive Hrd1 ligase (Hrd1 C399S). Substrate lysine ubiquitination by the ligase Rsp5 was followed over time. Controls were performed in the presence of detergent. Note that the CPY* domain of the red molecules is modified by Rsp5 only after addition of detergent.
(D) PLsomes containing wild-type Hrd1 and CPY*-TM in both orientations were incubated with ubiquitination machinery for different time periods in the presence or absence of detergent. Note that the CPY* domain of the red molecules is modified even in the absence of detergent.
(E) As in (D), but with PLsomes containing either wild-type Hrd1 or a mutant that lacks TMs 3-6 (mini-Hrd1). Note that mini-Hrd1 is active in polyubiqitination, but does not support modification of the CPY* domain of the red molecules unless detergent is added.
(F) Scheme of the fluorescence quenching retrotranslocation assay. PLsomes were generated as in (D), but CPY*-TM also contained an additional lysine-attached fluorescent dye on the CPY* domain (Alexa488; indicated by green domain) to which a quenching antibody (shown in gray) is available. The antibody quenched the fluorescence of substrate molecules that had their CPY* domain on the outside of the vesicles (gray domains). Substrate molecules of the opposite orientation can only be quenched after retrotranslocation of their CPY* domain.
(G) PLsomes as in (F) were incubated with antibody and ubiquitination machinery in the absence of ATP. The sample was placed in a fluorescence plate reader, ATP was added (open arrow), and the decrease in fluorescence was followed over time. A control was performed by addition of Triton X-100 (X100) before the reaction (open arrow). Each sample received Triton X-100 after the reaction (closed arrow).
(H) PLsomes were generated with catalytically inactive Hrd1 (C399S) and CPY*-TM that was labeled at the C-terminus with Dylight 800 (blue) and at lysines in the CPY* domain with Alexa488 (green). Retrotranslocation was assayed as in (G).
See also Figure S3.
Next, we verified that the two labeled substrate populations had the expected opposite orientations. We first used the accessibility of cysteines to a membrane-impermeable modification reagent (Maleimide-PEG5000, Mal-PEG). All cysteines are located in the CPY* domain and their modification results in a size increase of the substrate. As expected, the CPY* domain of the red molecules remained inaccessible to Mal-PEG, unless detergent was added (Figure 3B). In contrast, the blue molecules were modified regardless of whether the vesicles were intact or solubilized in detergent.
A second test for the orientation of the labeled substrate populations employed the ubiquitin ligase Rsp5 (Wang et al., 1999). CPY*-TM contains several Rsp5 recognition motifs in the misfolded domain. Hrd1 lacks Rsp5-recognition motifs. To prevent substrate modification by Hrd1, these experiments were performed with proteoliposomes containing a purified inactive Hrd1 mutant (C399S) (Bordallo and Wolf, 1999). The results showed that the red CPY*-TM molecules were labeled by Rsp5 only after addition of detergent, whereas the blue molecules were labeled even in intact vesicles (Figure 3C). Taken together, these experiments show that the CPY* domain of red molecules is located inside the vesicles, separated from the surrounding environment by the lipid bilayer. In contrast, the blue molecules have their CPY* domain on the outside of the reconstituted vesicles.
When the reconstituted proteoliposomes containing CPY*-TM and wild-type Hrd1 were incubated with the ubiquitination machinery, substrate molecules of both orientations were efficiently polyubiquitinated, regardless of whether the vesicles were intact or solubilized in detergent (Figure 3D). While the CPY* domain of the blue molecules was located on the outside before the start of the ubiquitination reaction and could therefore be modified by Hrd1 directly, the CPY* domain of the red molecules was previously inside the vesicles and some segment must have moved across the membrane to become accessible to Hrd1. Thus, Hrd1 can mediate retrotranslocation of a misfolded protein.
Next, we tested whether the entire membrane-spanning domain of Hrd1 is required. To this end, we used a Hrd1 variant in which TMs 3–6 were deleted (mini-Hrd1); mini-Hrd1 interacts with all other components of the Hrd1 complex (Carvalho et al., 2010). When purified mini-Hrd1 was reconstituted into proteoliposomes, it was still capable of polyubiquitinating substrate molecules that had their CPY* domain on the outside (Figure 3E). However, mini-Hrd1 did not modify molecules of the opposite orientation, unless detergent was added. These experiments suggest that the membrane-spanning domain of Hrd1 forms a conduit for substrate movement through the membrane.
To validate retrotranslocation of the CPY* domain, we generated Hrd1-containing proteoliposomes containing blue and red CPY*-TM molecules in opposite orientations. In addition, lysines in the CPY* domains of both populations were labeled with Alexa488 before reconstitution (to an average of 1.5 dye molecules per substrate molecule; indicated in green, Figure 3F). The vesicles were floated in the presence of an antibody to Alexa488, which quenches the Alexa488 fluorescence of all substrate molecules that have their CPY* domain accessible (Figure 3F, indicated in grey). The remaining green fluorescence should originate from molecules that have their CPY* domain in the lumen, inaccessible to the antibodies. Indeed, when Alexa488 antibodies were added to the isolated vesicles (together with ubiquitination machinery, but without ATP), the fluorescence showed an immediate small decrease, but remained constant thereafter (Figure 3G; black trace). The immediate drop was variable between different experiments and may have been caused by some proteoliposome rupture. When antibodies and detergent were added together, the fluorescence immediately dropped to a low level (Figure 3G; green trace). Importantly, when ubiquitination machinery and ATP were added together with Alexa488 antibodies, the fluorescence gradually decreased over a period of ~40 min. Parallel analysis by SDS-PAGE showed that both the blue and red CPY*-TM molecules were polyubiquitinated during this period (Figure S3C). These experiments indicate that polyubiquitination allowed the movement of the CPY* domain across the membrane, where it became accessible to the quenching antibody. Consistent with this conclusion, an enzymatically inactive Hrd1 mutant (Hrd1 C399S) did not allow retrotranslocation (Figure 3H), ruling out the possibility that the presence of active ubiquitin machinery alone was sufficient.
To exclude that the antibody moved across the membrane or that the vesicles were disrupted, we generated proteoliposomes containing CPY*-TM molecules labeled with Alexa488 only at the C-terminus (Figure S3D; green). CPY*-TM molecules that had their C-terminus outside were color-switched as before (Figure S3D; red). Importantly, the green CPY*-TM molecules that had their C-terminus inside the vesicles remained inaccessible to the quenching Alexa488-antibodies, unless detergent was added (Figure S3D). Again, parallel SDS-PAGE analysis showed that the red CPY*-TM molecules were polyubiquitinated (Figure S3E). Thus, the vesicles remain intact during retrotranslocation of the CPY* domain. Similar results were obtained using reconstituted CPY*-TM2 (Figures S3F and S3G). Taken together, these results suggest that polyubiquitination by Hrd1 is sufficient to mediate the retrotranslocation of a misfolded substrate segment. Because of the low labeling density of the substrate with fluorescent dyes, it seemed possible that only a small part of the substrate moved through the membrane. However, we obtained similarly high quenching efficiencies with CPY*-TM2 that contained an average of 8 fluorophores (Figure S3H), indicating that a substantial portion of the polypeptide chain must have crossed the membrane.
Substrate polyubiquitination is not essential for retrotranslocation
To test whether substrate- or Hrd1- ubiquitination is essential for retrotranslocation, we generated CPY* and CPY*-TM variants that lack all lysines (CPY*-noK and CPY*-TM-noK). Both substrates were degraded in S. cerevisiae cells with similar kinetics as the original CPY* and CPY*-TM proteins, as shown by cycloheximide-chase experiments (Figure 4A; and Figures 4B versus 1C, representative blots in Figures S4A and S4B). Lysine-free substrates showed the same dependence on ERAD components as their lysine-containing counterparts. These results suggest that ERAD does not require the modification of substrate, at least not on lysine residues, consistent with previous observations (Bernardi et al.; Hassink et al., 2006; Wang et al., 2013; Yu and Kopito, 1999).
Figure 4. Hrd1 retrotranslocates a lysine-free substrate in vitro and in vivo.
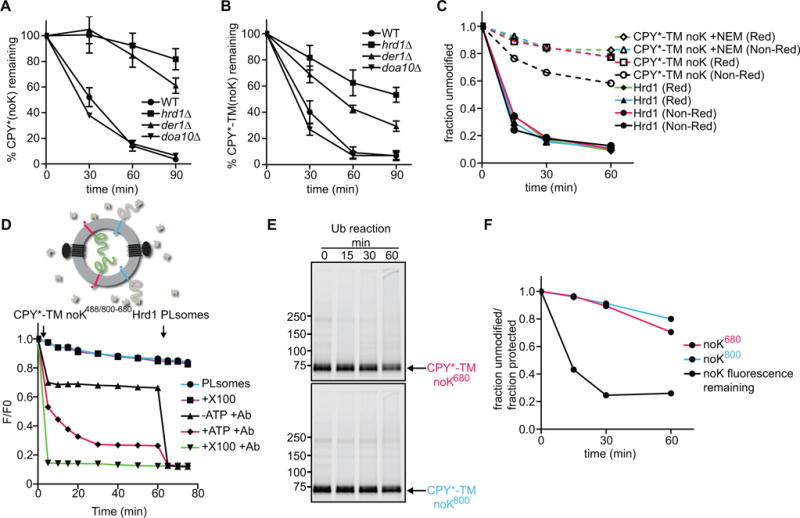
(A) The degradation of lysine-free CPY* was tested after addition of cycloheximide in wild-type S. cerevisiae cells and strains lacking the indicated ERAD components. At each time point, proteins were analyzed by SDS-PAGE and immunoblotting with HA antibodies (see also Figure S4A). Data are represented as mean +/− SEM from at least three experiments.
(B) As in (A), but with lysine-free CPY*-TM. Full-length protein and a C-terminal degradation product were observed (Figure S4B). The quantification shows the sum of both.
(C) Lysine-free CPY*-TM (noK) was incubated with Hrd1 and ubiquitination machinery for different time periods. Where indicated, the substrate was pre-modified with the cysteine-modifying reagent N-ethylmaleimide (NEM). The samples were analyzed by non-reducing (Non-Red) or reducing (Red) SDS-PAGE. Shown is the quantification of non-ubiquitinated Hrd1 and substrate (see also Figure S4C).
(D) The retrotranslocation of CPY*-TM (no K) was tested with the fluorescence quenching assay as in Figure 3F. Note that the lysine-free CPY* domain gradually becomes accessible to the quenching antibodies.
(E) The samples in (D) were analyzed for substrate polyubiquitination by SDS-PAGE and fluorescence scanning.
(F) The fraction of retrotranslocated CPY* domain (black line), as assessed by fluorescence quenching in (D), is compared with the fraction that remains non-ubiquitinated (red line) in the same reaction (E). The blue line shows the fraction of unmodified substrate molecules that have their CPY* domain on the outside of the vesicles.
See also Figure S4.
We purified the lysine-free substrate CPY*-TM-noK and tested its polyubiquitination in vitro with purified components. Substrate modification was drastically reduced, although not completely abolished (Figure 4C; Figure S4C). Most of the residual modification was reversed when the samples were treated with a reducing reagent before SDS-PAGE, and polyubiquitination was further diminished after blocking cysteines by pre-treatment of the substrate with N-ethylmaleimide (NEM). These results suggest that the lysine-free substrate is modified by Hrd1 on cysteines, as observed in other cases (Cadwell and Coscoy, 2005; Shimizu et al., 2010). In addition, there may be some even lower degree of modification of the N-terminal amino group or of hydroxyl groups in serines or threonines (Wang et al., 2007). Modification on residues other than lysines might explain why the substrates can still be degraded in vivo.
Next, we determined whether the drastically reduced level of substrate ubiquitination of CPY*-TM-noK affects retrotranslocation in vitro. We generated Hrd1-containing proteoliposomes containing blue and red CPY*-TM-noK molecules in opposite orientations. In addition, the N-termini of substrate molecules were labeled with Alexa488 before reconstitution. The vesicles were isolated in the presence of Alexa488-antibodies to quench the fluorescence of all substrate molecules that have their N-termini on the outside of the vesicles. Further addition of Alexa488-antibodies in the presence of ubiquitination machinery, but absence of ATP, led to only a small decrease in fluorescence (Figure 4D; black trace). In contrast, in the presence of ATP, the fluorescence decreased gradually over time (Figure 4D; red trace). The fluorescence reached a final level of ~30% after subtraction of the background seen in the presence of detergent, implying that ~70% of all substrate molecules were retrotranslocated (green trace; Figure S3I explains the quantification). Importantly, SDS-PAGE showed that the majority of substrate molecules of both orientations remained unmodified (>75%; Figures 4E and 4F). Thus, many unmodified substrate molecules were translocated across the membrane. These results indicate that Hrd1-, but not substrate-, modification is essential for retrotranslocation.
Autoubiquitination of Hrd1 allows retrotranslocation
Consistent with the assumption that autoubiquitination activates Hrd1 for retrotranslocation, we found that polyubiquitinated Hrd1 had a higher affinity for substrate than unmodified Hrd1 (Figure 5A). When polyubiquitinated Hrd1 was treated with a de-ubiquitinating enzyme prior to the binding assay, its affinity for substrate was reduced to the original level. The affinity of Hrd1 for CPY*-TM gradually increased with the length of ubiquitin chains attached to it (Figure 5B), although even the attachment of tetra-ubiquitin did not fully convert Hrd1 into the high-affinity state. The oligomeric state of Hrd1 modified with mono-, di-, or tetra-ubiquitin did not appear to change, as assessed by gel filtration experiments (Figure S5).
Figure 5. Autoubiquitination of Hrd1 is required for retrotranslocation.
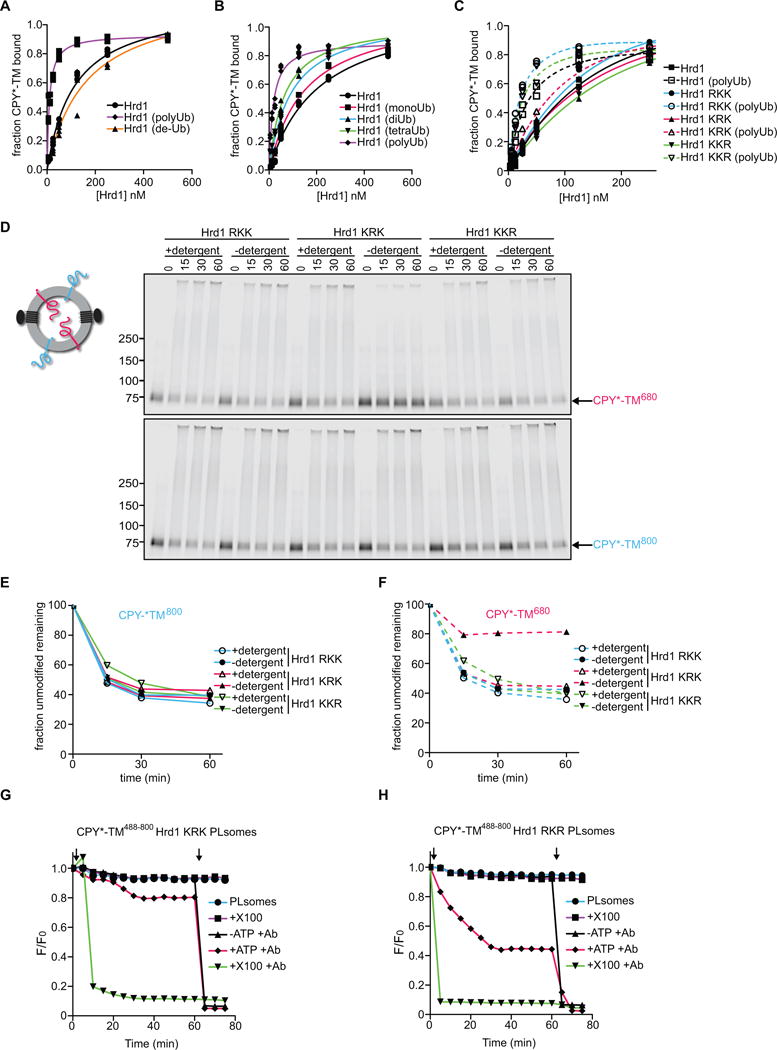
(A) DyLight800-labeled CPY*-TM was incubated with different concentrations of unmodified Hrd1, pre-polyubiquitinated Hrd1 (poly-Ub), or polyubiquitinated Hrd1 treated with the de-ubiquitinating enzyme Usp2 (de-Ub). After incubation with streptavidin beads, the amount of bound material was analyzed by SDS-PAGE and fluorescence scanning. Note that modified Hrd1 has a higher affinity for substrate.
(B) As in (A), but Hrd1 was also pre-modified with mono-, di- or tetra-ubiquitin.
(C) As in (A), but Hrd1 variants were used, which contained lysine-to-arginine substitutions in either the transmembrane domain (RKK), the RING-finger domain (KRK), or the C-terminal tail (KKR). In each case, Hrd1 was tested with and without polyubiquitination. Note that the ubiquitination-mediated increase of substrate affinity is blunted for the KRK mutant.
(D) Proteoliposomes containing substrate in both orientations and one of the Hrd1 mutants described in (C) were incubated with ubiquitination machinery for different time periods in the presence or absence of detergent. The samples were analyzed by SDS-PAGE and fluorescence scanning. Note that the CPY* domain of the red molecules is not efficiently retrotranslocated by the KRK mutant.
(E) Quantification of the fluorescence at 800 nm in (D).
(F) Quantification of the fluorescence at 680 nm in (D).
(G) Retrotranslocation of CPY*-TM was tested with the fluorescence quenching assay as in Figure 3H, but with Hrd1 carrying lysine to arginine substitutions in the RING domain (KRK mutant).
(H) As in (G), but with Hrd1 containing lysines only in the RING domain (Hrd1 RKR).
See also Figure S5.
Based on the affinity change of Hrd1, we tested if a specific Hrd1 domain must be polyubiquitinated to cause the observed change in substrate affinity. We used purified Hrd1 variants with lysine to arginine substitutions in either the N-terminal transmembrane region (RKK), the RING-finger domain (KRK), or the C-terminal tail (KKR) (Stein et al., 2014). Prior to autoubiquitination, all Hrd1 variants had a similar affinity for CPY*-TM (Figure 5C). However, after the polyubiquitination reaction, only the KRK variant failed to adopt a high-affinity state. It should be noted that all variants are active in autoubiquitination and substrate ubiquitination in detergent (Stein et al., 2014). Thus, modification of the RING-finger domain appears to be important for Hrd1’s increased substrate affinity.
To test the role of the RING-finger modification in retrotranslocation, we reconstituted the Hrd1 variants into proteoliposomes containing CPY*-TM in both orientations. All three Hrd1 variants were capable of ubiquitinating substrate molecules that had their CPY* domain on the outside (Figure 5D; 5E). The RKK and KKR variants also allowed retrotranslocation of the CPY* domain of the oppositely oriented substrate molecules. In contrast, the RING domain variant (KRK) was defective in retrotranslocation; it modified the CPY* domain efficiently only after solubilization of the membrane in detergent (Figure 5F). The KRK variant was also defective in the fluorescence-quenching retrotranslocation assay (Figure 5G). In contrast, a variant that contains lysines only in the RING domain (RKR) allowed retrotranslocation of a fluorescent substrate, albeit with somewhat lower efficiency than wild-type Hrd1 (Figure 5H).
The KRK variant was also defective in CPY* degradation in vivo (Figure 6A) although the purified protein has only a slightly reduced ubiquitination activity in vitro (Stein et al., 2014). In contrast, the RKK, KKR, and RKR variants showed undiminished CPY* degradation activity in vivo (Figure 6A, S6A). Although most of the Hrd1 lysine to arginine variants were functional, the steady-state levels were somewhat reduced and the RKR mutant was less stable (Figure S6B).
Figure 6. Hrd1 autoubiquitination affects ERAD.
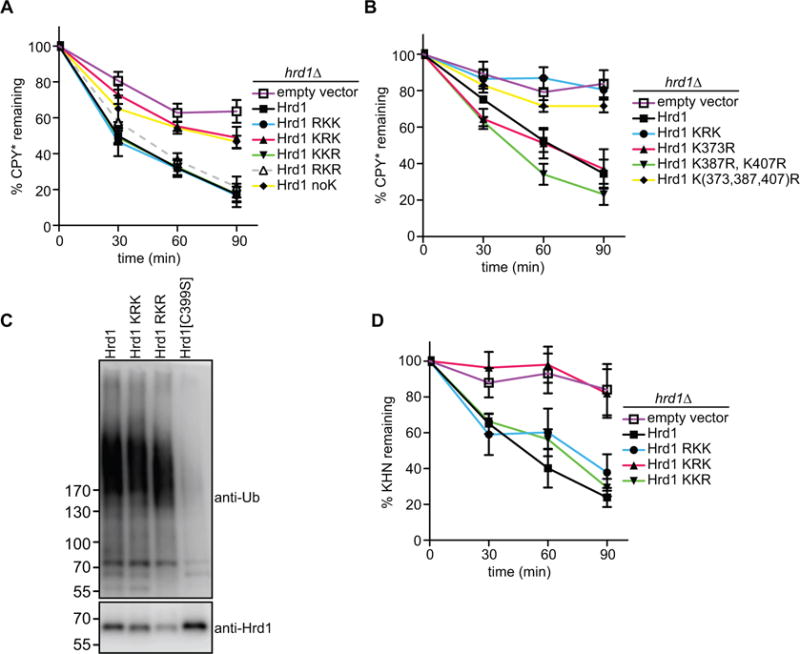
(A) The degradation of CPY* was followed in S. cerevisiae cells lacking Hrd1 (hrd1Δ), which contained either an empty vector or expressed Hrd1 mutants with lysine-to-arginine mutations from a single-copy plasmid. At each time point after cycloheximide addition, proteins were analyzed by SDS-PAGE and immunoblotting with HA antibodies (see also Figure S6A). Data are represented as mean +/− SEM from at least three experiments. Note that CPY* is not degraded in the KRK mutant.
(B) As in (A), but with Hrd1 mutants carrying the indicated lysine to arginine substitutions in the RING-finger domain. Note that a Hrd1 mutant carrying mutations in three lysines (373, 387, 407) is inactive in ERAD of CPY*.
(C) Wild-type Hrd1 or the indicated Hrd1 mutants with a His10-tag at their C-termini were expressed in hrd1Δ cells from a single-copy plasmid. The proteins were purified with IMAC beads under denaturing conditions, separated by SDS-PAGE, and subjected to immunoblotting with Hrd1 and ubiquitin antibodies. Note that the RKR mutant is polyubiquitinated.
(D) As in (A), but with the ERAD-L substrate KHN. (See also Figure S6G)
See also Figure S6.
To analyze which of the nine lysines in the RING-finger domain are important for ERAD, we systematically altered these lysines to arginines and tested the variants for CPY* degradation. The simultaneous mutation of three lysines (positions 373, 387, and 407; see Figure S6C) abolished all degradation activity (Figure 6B). Individually, these substitutions had no effect, indicating redundancy of the lysines. The lysine substitutions had a similar effect on the degradation of CPY*-TM and CPY*-TM2 (Figure S6D and S6E).
To assess whether Hrd1 is autoubiquitinated in S. cerevisiae cells containing all ERAD components, i.e. under conditions where Hrd1 is stable, we expressed a functional His-tagged version in hrd1Δ cells from a single-copy plasmid under its endogenous promoter (Figure S6F). Hrd1-His was purified under denaturing conditions and subjected to SDS-PAGE, followed by immunoblotting with ubiquitin and Hrd1 antibodies (Figure 6C). Polyubiquitinated Hrd1 was seen not only with the wild-type and KRK proteins, but also with the variant that contained lysines only in the RING-finger domain (RKR), although the levels of this protein were lower due to its reduced stability (Figure S6B). Ubiquitination was caused by Hrd1 itself, because no modification was observed with the ubiquitination-defective mutant Hrd1 C399S (Figure 6C). These results indicate that Hrd1 is autoubiquitinated in its RING domain in vivo.
Finally, we tested whether mutation of the lysines in the RING domain would generally affect the degradation of ERAD substrates in vivo. Indeed, the KRK variant was defective in the degradation of two established ERAD-L substrates (KHN and KWW; (Vashist et al., 2001; Vashist and Ng, 2004)), whereas the other lysine mutants had little effect (Figure 6D; Figure S6G, S6H, and S6I). Taken together, these results suggest that autoubiquitination of Hrd1 is required for the retrotranslocation of ERAD substrates.
DISCUSSION
In this study we provide direct evidence that Hrd1 mediates the retrotranslocation of a polypeptide across a membrane. Using reconstituted proteoliposomes containing Hrd1 and a membrane-bound substrate, we show that a luminal misfolded protein domain can cross the membrane. Because translocation requires an intact transmembrane domain of Hrd1, we propose that Hrd1 forms a conduit or channel for the polypeptide through the membrane. Polyubiquitination of Hrd1, rather than substrate modification, is necessary for retrotranslocation. Our in vivo data support the idea that autoubiquitination of Hrd1 in its RING-finger domain is the essential modification that allows retrotranslocation of ERAD substrates.
Taken together, these results lead to the following model for retrotranslocation across the ER membrane (Figure 7). The Hrd1 channel is initially closed (stage 1). Next, it interacts with a luminal segment of the substrate (stage 2). Upon autoubiquitination of Hrd1, a polypeptide segment moves to the cytosolic side of the membrane (stage 3). The polypeptide chain can probably slide in either direction through the open Hrd1 channel. Polyubiquitination of the substrate is not required for the initial emergence of a polypeptide segment on the cytosolic side of the membrane, but it could serve as a ratcheting mechanism to bias the movement of substrate towards the cytosol. The bulky polyubiquitin chain could directly prevent back-sliding of the polypeptide chain into the ER lumen (stage 4). In addition, the polyubiquitin chains could recruit the Cdc48 complex on the cytosolic side of the membrane, which would augment the ratcheting mechanism. The Cdc48 complex is not essential for initiation of retrotranslocation, but is required for the subsequent membrane extraction step (stage 5). The fundamental point in our model is that Hrd1 forms a ubiquitin-gated protein-conducting channel.
Figure 7. A model for Hrd1-mediated retrotranslocation.
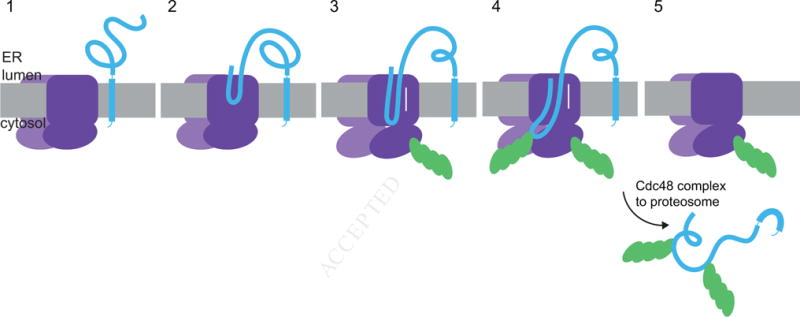
The scheme shows different stages of basic ERAD. Stage 1 shows the ubiquitin ligase Hrd1 and a single-spanning ERAD substrate containing a misfolded luminal domain. In stage 2, a segment of the misfolded domain interacts with the Hrd1 ligase. In stage 3, Hrd1 is autoubiquitinated, the substrate is inserted as a loop, and can slide back and forth inside the Hrd1 channel. In stage 4, the substrate is polyubiquitinated, which prevents back-sliding of the polypeptide into the ER lumen. In stage 5, the Cdc48 ATPase complex extracts the polyubiquitinated substrate from the ER membrane.
See also Table S1.
The proposal that Hrd1 forms a channel is consistent with the fact that the overexpression of Hrd1 in S. cerevisiae makes the other components of the Hrd1 complex dispensable, while all downstream cytosolic components, including the Cdc48 complex, are still required (Carvalho et al., 2010). The cells probably remain viable because Hrd1 alone can discriminate between folded and misfolded polypeptides (Stein et al., 2014), although other components increase the efficiency of substrate selection (Denic et al., 2006). The role for Hrd1 as a channel is supported by crosslinking experiments in S. cerevisiae, which showed that a retrotranslocating substrate interacts with Hrd1 inside the ER membrane (Carvalho et al., 2010). Hrd1 forms homo-oligomers (Carvalho et al., 2010; Horn et al., 2009; Bernardi et al., 2010), and could form a membrane path from at least 12 TM segments (each Hrd1 molecule has 6 TMs). Our data now show that Hrd1 alone can mediate the retrotranslocation of a polypeptide, providing the strongest evidence for it forming a retrotranslocon. However, this does not exclude the possibility that other membrane proteins, specifically Der1, contribute to the polypeptide conduit across the membrane (Mehnert et al., 2014).
Auto-modification of a ubiquitin ligase is often considered to be an in vitro artifact or a signal for degradation (for review, see (de Bie and Ciechanover, 2011)), although in the case of the RING1B ligase, autoubiquitination has been reported to enhance its activity (Ben-Saadon et al., 2006). In our system, autoubiquitination of Hrd1 serves to open the retrotranslocation channel. The best evidence for this model comes from our in vitro experiments, particularly from the demonstration that a misfolded protein domain in the lumen of reconstituted proteoliposomes cannot be modified by the soluble ubiquitin ligase Rsp5, but becomes accessible to modification by Hrd1 after Hrd1 autoubiquitination. Because only Hrd1 itself is available for modification at the beginning of the reaction, autoubiquitination is likely responsible for the initial movement of the misfolded domain across the membrane. Consistent with this conclusion, we show that preventing lysine polyubiquitination in the RING-finger domain abolishes Hrd1 function in vitro and in vivo, even though it remains active in ubiquitination. Hrd1 is polyubiquitinated in wild-type yeast cells, despite the fact that it is stable, further arguing that the modification is not just a signal for degradation.
Our model implies that autoubiquitination causes a conformational change in Hrd1. We found that ubiquitinated Hrd1 binds substrate with higher affinity than the unmodified ligase. The affinity change appears to be caused by the attachment of ubiquitin chains to one of three lysines in the RING-finger domain. Our results show that these lysines, only one of which is highly conserved, have redundant functions. Thus, perhaps it only matters that a bulky ubiquitin chain is attached to a certain region of the RING-finger.
Polyubiquitination of Hrd1 is probably tightly controlled in wild-type yeast cells to activate Hrd1 only when substrate arrives and to prevent untimely degradation of the ligase. Indeed, overexpression of Hrd1 or deletion of HRD3 lead to excessive autoubiquitination of Hrd1, which in turn causes degradation of the ligase (Gardner et al., 2000). In wild-type cells, Hrd3 binding to Hrd1 might serve as a “brake”, preventing autoubiquitination until substrate is bound to Hrd3 or Hrd1. Based on the stability of Hrd1, one probably has to invoke the function of a deubiquitinating enzyme that removes polyubiquitin chains from Hrd1. The interaction of Hrd1 with its binding partners in the membrane might also prevent the membrane extraction of transiently polyubiquitinated Hrd1 by the Cdc48 complex.
Although we indicate in the scheme in Figure 7 that substrate binds to Hrd1 before autoubiquitination, the order of events is currently unclear. In detergent, unmodified Hrd1 can bind substrate, although more weakly than after autoubiquitination. However, it is uncertain whether these results apply to the interaction in intact membranes. In any case, we consider it likely that the substrate inserts as a loop into the Hrd1 channel (Figure 7, stages 2 and 3). Although we cannot exclude that the N-terminus of our substrate inserts head on into Hrd1, loop insertion seems more plausible, because there are probably many more internal segments that can interact with the TM domain of Hrd1. A similar loop insertion model could apply to soluble, luminal ERAD substrates and is analogous to that established for the initial insertion of a secretory protein into the Sec61/SecY channel during translocation in the opposite direction (Shaw et al., 1988).
In our in vitro assay, a substantial segment of the substrate crossed the membrane. However, it remains unclear whether the entire luminal substrate domain moved to the outside of the vesicles. Even the retrotranslocation of smaller segments might require the substrate to be unfolded or loosely folded. In our system, such a state may have been achieved by the treatment of substrates with urea and harsh detergent. In vivo, luminal chaperones are likely required to keep the substrate in a retrotranslocation-competent state (Nishikawa et al., 2001).
While directionality of translocation might normally be provided by a polyubiquitin-mediated ratcheting mechanism, likely enhanced by cytosolic binding factors, such as the Cdc48 complex (Bays et al., 2001; Jarosch et al., 2002; Rabinovich et al., 2002; Ye et al., 2001) and possibly the proteasome (Morris et al., 2014), substrate ubiquitination does not seem to be essential. In several cases, including the ERAD substrate used in this study, all lysines can be deleted without preventing degradation, while ubiquitination machinery is still required (Hassink et al., 2006; Wang et al., 2013; Yu and Kopito, 1999). Some of these lysine-free proteins are ubiquitinated at other amino acids. However, there are substrates, such as cholera toxin, that are not ubiquitinated (Rodighiero et al., 2002), providing strong evidence that the critical modification occurs on another component. In this case, directionality may be provided by polypeptide folding in the cytosol. In other cases, an unmodified substrate might interact with cytosolic factors, or be prevented from back-sliding by other mechanisms.
Although our data show that Hrd1 alone can mediate retrotranslocation of ERAD substrates, other components of the Hrd1 complex play an important role in vivo. Yos9 and Hrd3 are involved in substrate selection in the ER lumen and act upstream of Hrd1 (Denic et al., 2006; Gauss et al., 2006). Der1 probably helps to insert luminal substrates into the Hrd1 channel (Carvalho et al., 2010; Mehnert et al., 2014). Usa1 serves as a scaffold, providing a link between Hrd1 and Der1 and facilitating the oligomerization of Hrd1 (Carvalho et al., 2010; Horn et al., 2009). Specific details for how these proteins contribute to retrotranslocation remain to be clarified.
We suggest that other multi-spanning ubiquitin ligases of the ER function similarly to Hrd1. In both S. cerevisiae and mammals, several RING-finger ligases are known that are involved in the ubiquitination and degradation of different substrates (Table S1). All of these ligases contain several TMs and may form oligomers. We propose that they all form ubiquitin-gated protein-conducting channels.
EXPERIMENTAL PROCEDURES
Details of experimental procedures and protein purification are described in the Extended Experimental Procedures.
Yeast strains and plasmids
All deletion strains used in this study were purchased from GE Dharmacon and are derivatives of BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0). Plasmids were constructed using standard restriction cloning and Gibson assembly.
Protein purification and labeling
CPY*-TM, CPY*-TM2, CPY-TM, CPY-TM2 and Hrd1 were C-terminally labeled using a GGGC-coupled fluorophore (DyLight680, DyLight800, or Alexa488) and sortase (Popp et al., 2007; Stein et al., 2014). Where indicated, substrates were labeled with Alexa488-tetrafluorophenyl ester or DyLight800-N-hydroxysuccinimide ester on amines. The labeled proteins were purified by size-exclusion chromatography.
ERAD-substrate degradation
Cycloheximide chases were performed as described (Gardner et al., 1998), with variations described in the supplemental information.
Binding assays
Hrd1 binding assays were performed as described (Stein et al., 2014), with variations described in the supplemental information.
Ubiquitination assays
Ubiquitination assays were performed as described (Stein et al., 2014), with variations described in the supplemental information.
Reconstitution into proteoliposomes
1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) (Avanti Polar Lipids) and CPY*-TM substrate were completely solubilized in Triton X-100 (Anatrace) and the detergent was removed with Bio-Beads SM2 (Bio-Rad). Decyl maltose neopentyl glycol (DMNG) was added to partially solublize the liposomes before addition of Hrd1. After Hrd1 incorporation, DMNG was removed using detergent-removal spin columns (Pierce). For color switching assays, prior to Hrd1 incorporation the proteoliposomes were incubated with sortase and GGGC-coupled-fluorophore. The vesicles were purified by flotation in a glycerol gradient.
Fluorescence-quenching retrotranslocation assay
Proteoliposomes were generated with unlabeled Hrd1 and amine-coupled Alexa488 CPY*-TM. In the proteoliposomes, CPY*-TM was in two orientations; each population was labeled with different dyes at the C-terminus (DyLight680 and DyLight800). The vesicles were incubated with Alexa488-antibodies, floated in a glycerol gradient, and incubated with ubiquitination machinery in the absence of ATP. After addition of ATP, the fluorescence of Alexa488 coupled to CPY*-TM was followed in a plate reader.
Denaturing pull-downs
Hrd1-His10 was expressed from the HRD1 endogenous promoter on a centromeric plasmid. Cells were collected in mid-log phase and Hrd1 was purified on His-tag Dynabeads (Life Technologies).
Supplementary Material
Acknowledgments
We thank Nicholas Bodnar, Stefan Schoebel, and Alex Stein for help with some in vitro experiments, and Neil Blok, Stefan Schoebel, Alex Stein, and Xudong Wu for their critical reading of the manuscript. We also thank the ICCB-Longwood Screening Facility at Harvard Medical School for access to the M5 plate reader. This work is supported by the NIH/NIGMS Award R01GM052586. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. R.D.B. is the Fraternal Order of Eagles Fellow of the Damon Runyon Cancer Research Foundation, DRG-2184-14. T.A.R. is a Howard Hughes Medical Institute Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
R.D.B. and T.A.R. conceived and designed the experiments. R.D.B performed the experiments. R.D.B. and T.A.R. wrote the manuscript.
References
- Bays NW, Wilhovsky SK, Goradia A, Hodgkiss-Harlow K, Hampton RY. HRD4/NPL4 Is Required for the Proteasomal Processing of Ubiquitinated ER Proteins. Mol Biol Cell. 2001;12:4114–4128. doi: 10.1091/mbc.12.12.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Saadon R, Zaaroor D, Ziv T, Ciechanover A. The polycomb protein Ring1B generates self atypical mixed ubiquitin chains required for its in vitro histone H2A ligase activity. Mol Cell. 2006;24:701–711. doi: 10.1016/j.molcel.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Bernardi KM, Williams JM, Kikkert M, van Voorden S, Wiertz EJ, Ye Y, Tsai B. The E3 ubiquitin ligases Hrd1 and gp78 bind to and promote cholera toxin retro-translocation. Mol Biol Cell. 21:140–151. doi: 10.1091/mbc.E09-07-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordallo J, Wolf DH. A RING-H2 finger motif is essential for the function of Der3/Hrd1 in endoplasmic reticulum associated protein degradation in the yeast Saccharomyces cerevisiae. FEBS Lett. 1999;448:244–248. doi: 10.1016/s0014-5793(99)00362-2. [DOI] [PubMed] [Google Scholar]
- Cadwell K, Coscoy L. Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science. 2005;309:127–130. doi: 10.1126/science.1110340. [DOI] [PubMed] [Google Scholar]
- Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Carvalho P, Stanley AM, Rapoport TA. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell. 2010;143:579–591. doi: 10.1016/j.cell.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Ye Y. Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struc Mol Biol. 2014;21:325–335. doi: 10.1038/nsmb.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinson I, Corey RA, Allen WJ. Channel crossing: how are proteins shipped across the bacterial plasma membrane? Philos Trans R Soc Lond B Biol Sci. 2015;370 doi: 10.1098/rstb.2015.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bie P, Ciechanover A. Ubiquitination of E3 ligases: self-regulation of the ubiquitin system via proteolytic and non-proteolytic mechanisms. Cell Death Differ. 2011;18:1393–1402. doi: 10.1038/cdd.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- Finger A, Knop M, Wolf DH. Analysis of two mutated vacuolar proteins reveals a degradation pathway in the endoplasmic reticulum or a related compartment of yeast. Eur J Biochem. 1993;218:565–574. doi: 10.1111/j.1432-1033.1993.tb18410.x. [DOI] [PubMed] [Google Scholar]
- Foresti O, Rodriguez-Vaello V, Funaya C, Carvalho P. Quality control of inner nuclear membrane proteins by the Asi complex. Science. 2014;346:751–755. doi: 10.1126/science.1255638. [DOI] [PubMed] [Google Scholar]
- Gardner R, Cronin S, Leader B, Rine J, Hampton R. Sequence determinants for regulated degradation of yeast 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell. 1998;9:2611–2626. doi: 10.1091/mbc.9.9.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, Swarbrick GM, Bays NW, Cronin SR, Wilhovsky S, Seelig L, Kim C, Hampton RY. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol. 2000;151:69–82. doi: 10.1083/jcb.151.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauss R, Jarosch E, Sommer T, Hirsch C. A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nat Cell Biol. 2006;8:849–854. doi: 10.1038/ncb1445. [DOI] [PubMed] [Google Scholar]
- Hassink G, Kikkert M, van Voorden S, Lee SJ, Spaapen R, van Laar T, Coleman CS, Bartee E, Fruh K, Chau V, et al. TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem J. 2005;388:647–655. doi: 10.1042/BJ20041241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassink GC, Barel MT, Van Voorden SB, Kikkert M, Wiertz EJ. Ubiquitination of MHC class I heavy chains is essential for dislocation by human cytomegalovirus-encoded US2 but not US11. J Biol Chem. 2006;281:30063–30071. doi: 10.1074/jbc.M602248200. [DOI] [PubMed] [Google Scholar]
- Horn SC, Hanna J, Hirsch C, Volkwein C, Schutz A, Heinemann U, Sommer T, Jarosch E. Usa1 functions as a scaffold of the HRD-ubiquitin ligase. Mol Cell. 2009;36:782–793. doi: 10.1016/j.molcel.2009.10.015. [DOI] [PubMed] [Google Scholar]
- Huyer G, Piluek WF, Fansler Z, Kreft SG, Hochstrasser M, Brodsky JL, Michaelis S. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J Biol Chem. 2004;279:38369–38378. doi: 10.1074/jbc.M402468200. [DOI] [PubMed] [Google Scholar]
- Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, Sommer T. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol. 2002;4:134–139. doi: 10.1038/ncb746. [DOI] [PubMed] [Google Scholar]
- Khmelinskii A, Blaszczak E, Pantazopoulou M, Fischer B, Omnus DJ, Le Dez G, Brossard A, Gunnarsson A, Barry JD, Meurer M, et al. Protein quality control at the inner nuclear membrane. Nature. 2014;516:410–413. doi: 10.1038/nature14096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Finger A, Braun T, Hellmuth K, Wolf DH. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 1996;15:753–763. [PMC free article] [PubMed] [Google Scholar]
- Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- Mehnert M, Sommer T, Jarosch E. Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat Cell Biol. 2014;16:77–86. doi: 10.1038/ncb2882. [DOI] [PubMed] [Google Scholar]
- Morris LL, Hartman IZ, Jun DJ, Seemann J, DeBose-Boyd RA. Sequential actions of the AAA-ATPase valosin-containing protein (VCP)/p97 and the proteasome 19 S regulatory particle in sterol-accelerated, endoplasmic reticulum (ER)-associated degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem. 2014;289:19053–66. doi: 10.1074/jbc.M114.576652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa S, Fewell SW, Kato Y, Brodsky JL, Endo T. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J Cell Biol. 2001;153:1061–70. doi: 10.1083/jcb.153.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Rapoport TA. Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Ann Rev Biophys. 2012;41:21–40. doi: 10.1146/annurev-biophys-050511-102312. [DOI] [PubMed] [Google Scholar]
- Pilon M, Schekman R, Romisch K. Sec61p mediates export of a misfolded secretory protein from the endoplasmic reticulum to the cytosol for degradation. EMBO J. 1997;16:4540–4548. doi: 10.1093/emboj/16.15.4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp MW, Antos JM, Grotenbreg GM, Spooner E, Ploegh HL. Sortagging: a versatile method for protein labeling. Nat Chem Biol. 2007;3:707–708. doi: 10.1038/nchembio.2007.31. [DOI] [PubMed] [Google Scholar]
- Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S. AAA-ATPase p97/Cdc48p, a Cytosolic Chaperone Required for Endoplasmic Reticulum-Associated Protein Degradation. Mol Cell Biol. 2002;22:626–634. doi: 10.1128/MCB.22.2.626-634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodighiero C, Tsai B, Rapoport TA, Lencer WI. Role of ubiquitination in retro-translocation of cholera toxin and escape of cytosolic degradation. EMBO Rep. 2002;3:1222–1227. doi: 10.1093/embo-reports/kvf239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer A, Wolf DH. Sec61p is part of the endoplasmic reticulum-associated degradation machinery. EMBO J. 2009;28:2874–2884. doi: 10.1038/emboj.2009.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AS, Rottier PJ, Rose JK. Evidence for the loop model of signal-sequence insertion into the endoplasmic reticulum. Proc Natl Acad Sci USA. 1988;85:7592–7596. doi: 10.1073/pnas.85.20.7592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, Okuda-Shimizu Y, Hendershot LM. Ubiquitylation of an ERAD substrate occurs on multiple types of amino acids. Mol Cell. 2010;40:917–926. doi: 10.1016/j.molcel.2010.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A, Ruggiano A, Carvalho P, Rapoport TA. Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell. 2014;158:1375–1388. doi: 10.1016/j.cell.2014.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taxis C, Hitt R, Park SH, Deak PM, Kostova Z, Wolf DH. Use of modular substrates demonstrates mechanistic diversity and reveals differences in chaperone requirement of ERAD. J Biol Chem. 2003;278:35903–35913. doi: 10.1074/jbc.M301080200. [DOI] [PubMed] [Google Scholar]
- Vashist S, Kim W, Belden WJ, Spear ED, Barlowe C, Ng DT. Distinct retrieval and retention mechanisms are required for the quality control of endoplasmic reticulum protein folding. J Cell Biol. 2001;155:355–368. doi: 10.1083/jcb.200106123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashist S, Ng DTW. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Yang J, Huibregtse JM. Functional domains of the Rsp5 ubiquitin-protein ligase. Mol Cell Biol. 1999;19:342–352. doi: 10.1128/mcb.19.1.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Herr RA, Chua WJ, Lybarger L, Wiertz EJ, Hansen TH. Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J Cell Biol. 2007;177:613–624. doi: 10.1083/jcb.200611063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yu YY, Myers N, Hansen TH. Decoupling the role of ubiquitination for the dislocation versus degradation of major histocompatibility complex (MHC) class I proteins during endoplasmic reticulum-associated degradation (ERAD) J Biol Chem. 2013;288:23295–23306. doi: 10.1074/jbc.M113.482018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner W, Schekman R. Protein translocation across biological membranes. Science. 2005;310:1452–1456. doi: 10.1126/science.1113752. [DOI] [PubMed] [Google Scholar]
- Wiertz EJHJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA, Ploegh HL. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 1996;384:432–438. doi: 10.1038/384432a0. [DOI] [PubMed] [Google Scholar]
- Willer M, Forte GM, Stirling CJ. Sec61p is required for ERAD-L: genetic dissection of the translocation and ERAD-L functions of Sec61P using novel derivatives of CPY. J Biol Chem. 2008;283:33883–33888. doi: 10.1074/jbc.M803054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- Yu H, Kopito RR. The role of multiubiquitination in dislocation and degradation of the alpha subunit of the T cell antigen receptor. J Biol Chem. 1999;274:36852–36858. doi: 10.1074/jbc.274.52.36852. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.