

Abstract
Purpose
We employed a metabolomics-based approach with the goal to better understand the molecular signatures of glioblastoma (GBM) cells and tissues, with an aim towards identifying potential targetable biomarkers for developing more effective and novel therapies.
Experimental Design
We used liquid chromatography coupled with mass spectrometry (LC-MS/Q-TOF and LC-MS/QQQ) for the discovery and validation of metabolites from primary and established GBM cells, GBM tissues, and normal human astrocytes.
Results
We identified tryptophan, methionine, kynurenine, and 5-methylthioadenosine as differentially regulated metabolites (DRMs) in GBM cells compared to normal human astrocytes (NHAs). Unlike NHAs, GBM cells depend on dietary methionine for proliferation, colony formation, survival, and to maintain a deregulated methylome (SAM:SAH ratio). In methylthioadenosine phosphorylase (MTAP) deficient GBM cells, expression of MTAP transgene did not alter methionine dependency, but compromised tumor growth in vivo. We discovered that a lack of the kynurenine metabolizing enzymes kynurenine monooxygenase and/or kynureninase promotes the accumulation of kynurenine, which triggers immune evasion in GBM cells. Insilico analysis of the identified DRMs mapped the activation of key oncogenic kinases that promotes tumorigenesis in GBM. We validated this result by demonstrating that the exogenous addition of DRMs to GBM cells in vitro, results in oncogene activation as well as the simultaneous downregulation of Ser/Thr phosphatase PP2A.
Conclusion
We have connected a four-metabolite signature, implicated in the methionine and kynurenine pathways, to the promotion and maintenance of GBM. Together, our data suggest that these metabolites and their respective metabolic pathways serve as potential therapeutic targets for GBM.
Keywords: GBM, Metabolomics, Essential amino acid
Introduction
Glioblastoma (GBM), grade IV malignant glioma, is one of the most aggressive types of human cancer, with a median patient survival time of 12-15 months (1). Despite the deadliness of this disease, much is still unknown about the metabolic pathology of GBM because tumor heterogeneity and genetic alterations cause an altered metabolism and metabolic profile within the same tumor, making it difficult to correlate metabolic signatures. In recent years, metabolomic-based approaches have been recognized as an emerging tool to discover products of cellular biochemical reactions that fuel cell proliferation in a variety of malignancies (2-6). Furthermore, metabolomic profiling has led to the discovery and identification of numerous key cellular pathways (7, 8). Applications of metabolomics in clinical oncology have shown strong potential in the early detection, diagnosis, and prognosis of cancer as well as being a predictive/pharmacodynamic biomarker of drug efficacy (9, 10).
Utilizing LC-MS/MS, a technique extensively used for analyzing metabolites in a variety of in vitro cell culture systems (11-15), we report the differential regulation of tryptophan and methionine and their respective metabolites kynurenine and 5′-methylthioadenosine (MTA) in GBM compared to normal human astrocytes. After identifying these metabolites, the primary goal of this work was to elucidate the underlying biochemical mechanisms that determine their respective roles in promoting the tumorigenic phenotype of GBM. It was reported over a half a century ago that cancer cells rely on methionine for proliferation and survival (16-18). Since this time, the mechanism(s) underlying this methionine dependency have not been determined. Additionally, while previous studies have shown that the deregulation of the tryptophan pathway promotes immune evasion in GBM (19), there is a lack of understanding of the role of the tryptophan pathway in oncogenic activation.
In this study, we have investigated the mechanisms behind the differential regulation of these metabolites in GBM by evaluating the status of key enzymes in the methionine and tryptophan metabolic pathways. We have addressed the knowledge gaps that remain in the understanding of methionine dependency and the role of the tryptophan pathway in oncogenic activation. The findings reported here advance the understanding of how the methionine and tryptophan pathways contribute to the activation of oncogenic kinases and promote tumorigenesis in GBM.
Materials and Methods
Study approval
This study was conducted in accordance with OSU Intuitional Review Boards for IRB (2009C0065), IACUC (2009A0127) and IBC (2009R0169).
Cell culture
The 13 primary GBM cell lines used in this study were isolated from GBM patient tissues and authenticated by neuro-pathologist at The Ohio State University (OSU34, OSU35, OSU38, OSU53, OSU61, OSU68, OSU94, OSU96, MDNSC2, MDNSC20, MHG8, MGH74, and 146). The 4 commercially available cell lines (U118, U87, LN18, and LN229) were obtained from ATCC. GBM cells were maintained in DMEM (Life Technologies), supplemented with 10% FBS (Sigma-Aldrich), and 1% antibiotic-antimycotic (Life Technologies). Normal Human Astrocytes were obtained from Lonza and were maintained in Clonetics™ AGM™ Astrocyte Growth Medium (Lonza). Cells were cultured at 37° C under a gas phase of 95% air and 5% CO2. All studies were conducted within 5 passages. Before extracting intracellular and extracellular metabolites from Normal Human Astrocytes for metabolic profiling, the cells were grown in the same DMEM media as the GBM cell lines to maintain dietary consistency. In order to have a methionine free-DMEM for the methionine dependence studies, we used DMEM absent of glutamine, methionine, cystine, and pyruvate (Life Technologies) and supplemented with glutamine (4mM), cystine (0.2mM), and pyruvate (1mM). Methionine (20 μM) was added to the methionine free-DMEM for low methionine studies.
Gene expression analysis: RT-PCR
RNA was isolated and converted to cDNA from GBM cells and NHAs using the RNeasy® Mini Kit and Superscript® III First-Strand synthesis. Using quantitative RT-PCR, the expression level of GAPDH, IDO1, IDO2, TDO2, MTAP, AHCY, MAT1A, MAT2A, KMO, KYNU, and LAT1 were measured using the Taqman® gene expression assay. The gene expression profile data were generated using the C1000™ Thermal Cycler-CFX96™ Real-Time System and analyzed using the BIO-RAD CFX Manager. GAPDH was used as the control.
Statistical consideration
All results were confirmed in at least three independent experiments, and data from one representative experiment were shown. All quantitative data are presented as mean ± SD. The statistical analysis was performed using SAS 9.2 (SAS Institute) or Graph Pad Prism 5. Student's t-tests were used for comparisons of means of quantitative data between groups. Values of p<0.05 were considered significant.
Supplemental information
Detailed protocols for western analysis, H&E staining, cell culture and counting, metabolite extraction, global metabolic profiling using LC-MS (Q-TOF), data analysis, MTT proliferation and colony formation assays are provided as supplemental information.
Results
Metabolic profiling discovers methionine, 5′-methylthioadenosine, tryptophan, and kynurenine as differentially regulated metabolites in GBM cells
The cellular metabolites in the intracellular compartment and extracellular milieu of 13 primary and 4 established GBM cell lines and Normal Human Astrocytes (NHAs) were analyzed using liquid chromatography (LC) coupled with Quadrupole Time of flight (Q-TOF) spectrometry in positive ion mode. The results showed a distinct difference in the total number of mapped metabolites between the intracellular compartment and extracellular milieu of the GBM cell lines and NHAs (Figs. S1A-B). Extrapolating the data to a principal component analysis plot, the metabolic profiles of the GBM cell lines and NHAs exhibited a clear separation based on their respective molecular mass, retention time, and ion abundance profiles (Figs. S1C-D). This extensive list of metabolites was condensed using multivariate statistical methods (20). Resultantly, using NHAs as the control, we identified four statistically significant differentially regulated metabolites (DRMs) in GBM cells. Kynurenine and 5′-methylthioadenosine (MTA) were identified as DRMs in the intracellular compartment and extracellular milieu, while methionine and tryptophan were discovered as DRMs in the intracellular compartment alone (Fig. 1). The total and extracted ion chromatograms for both the intracellular compartment and extracellular milieu have been provided in the Supplemental Figures 2 and 3. In addition to retention time and fragmentation pattern, the identification of these metabolites were confirmed by overlaying the molecular masses of the metabolites fragmentation patterns with the fragmentation patterns of their respective pure synthetic compounds (Figs. S4-7). Total ion chromatogram and multiple reactions monitoring method (MRM) were used to quantitatively validate each identified DRM (Figs. S8-9). The results confirmed the increased uptake of methionine and tryptophan in the intracellular compartment and the increased secretion of kynurenine and MTA to the extracellular milieu of GBM cells (Figs. 2A-D).
Figure 1. Qualitative analysis identified four differentially regulated metabolites (DRMs) in GBM.
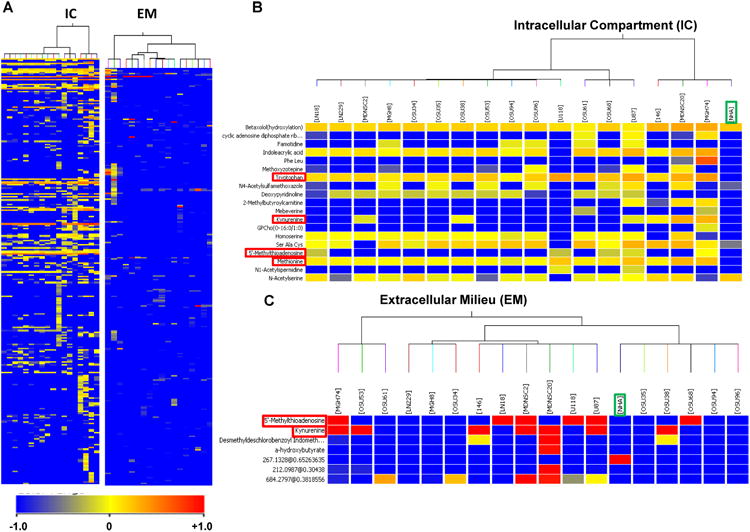
LC-MS/MS analysis was used to generate the intracellular compartment and extracellular milieumetabolomic profiles for commercially available and patient-derived GBM cell lines and NHAs. This data has been presented as heat maps, the identified DRMs are marked by red boxes, while the NHAs are marked by green boxes.
[A] The heat map of the intracellular compartment and extracellular milieu of the GBM cell lines and NHAs analyzed.
[B] In the intracellular compartment, four metabolites were discovered as upregulated DRMs: tryptophan, kynurenine, methionine, and MTA.
[C] In the extracellular milieu, two metabolites were discovered as DRMs: kynurenine and MTA.
Figure 2. Quantitative validation of DRMs and expression levels of LAT1 in GBM cells.
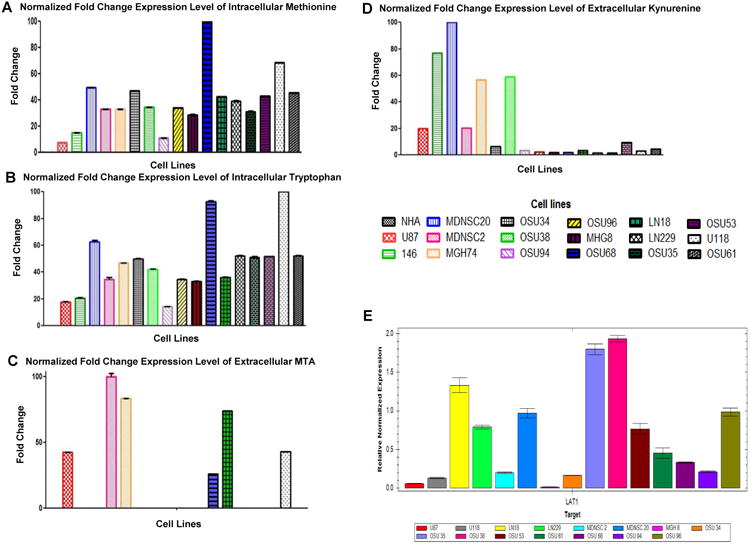
The identified DRMs were further validated and quantified through the multiple reactions monitoring method (MRM) by selecting the appropriate qualifier and quantifier, using Triple Quad LC-MS. The results are presented as fold changes normalized to their total cell number (intracellular compartment) or total volume of spent media (extracellular milieu).
[A-B] The quantitative analysis results showed significantly increased uptake of methionine and tryptophan in the intracellular compartment in all tested GBM cell lines.
[C] 6 of the 17 GBM cell lines tested showed increased secretion of MTA to the extracellular milieu.
[D] 9 of the 17 GBM cell lines examined showed increased secretion of kynurenine to the extracellular milieu.
[E] Using qRT-PCR, we measured the expression levels of LAT1 in GBM cells. Expression levels of LAT1 in GBM cells exhibit affinity for essential amino acids. However, there is no linear correlation between the uptake of methionine or tryptophan and LAT1 expression levels. Gapdh was used as a control.
Given the increased uptake of the L-type amino acids methionine and tryptophan, we investigated whether this is a result of the upregulation of the L-type amino acid transporter 1 (LAT1), which exhibits high affinity for essential amino acids. The expression levels of LAT1 (Fig. 2E) did not correlate with the uptake of methionine and tryptophan, which could be a result of not considering the uptake of other L-type amino acids. Furthermore, the results suggest that the increased uptake of methionine and tryptophan is driven by the altered regulation of other cellular processes.
GBM cells are dependent on dietary methionine for proliferation, colony formation, and survival
The intracellular concentration of methionine in the GBM cell lines ranged from 5 to 100-fold higher than NHAs (Fig. 2A). Due to the fact that methionine is an essential and multifunctional amino acid required for protein synthesis, polyamine production, DNA/RNA/protein methylation, and glutathione synthesis(21, 22), we hypothesized that GBM cells are dependent on dietary methionine to support their rapid proliferation, colony formation, and survival. We measured the effect of methionine on GBM cell growth and colony formation by conducting MTT and clonogenic survival assays in media with and without methionine (Figs. 3A-B). GBM cells grown in the absence of methionine showed a 40-60% inhibition of cell proliferation and a significantly decreased colony forming ability, confirming our hypothesis that GBM cells are dependent on environmental methionine for survival.
Figure 3. GBM cells are dependent on dietary methionine to maintain the methylome for hyper proliferation and survival.
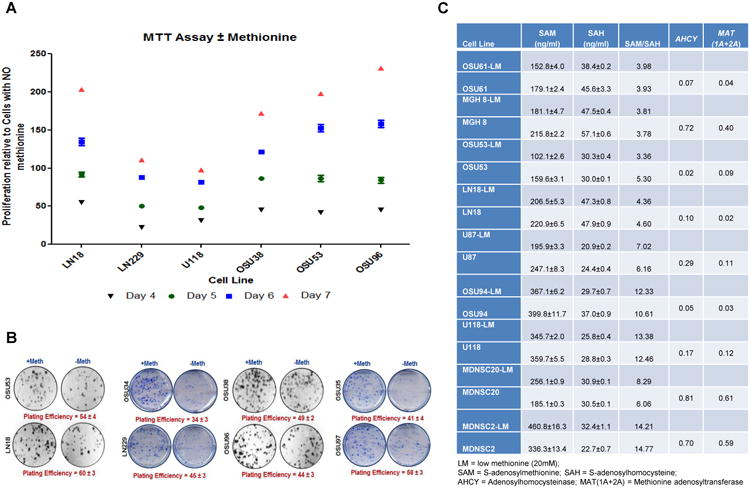
[A] A MTT proliferation assay was conducted using GBM cell lines in DMEM media with and without methionine. The cell lines grown in media without methionine underwent decreased proliferation over a 7 day period. Conversely, all the GBM cell lines exhibited a higher rate of proliferation in the presence of methionine. Thus, the availability of extracellular methionine correlates with the proliferative potential of GBM cells.
[B] Clonogenic assays were performed using GBM cell lines to measure the correlation between the availability of dietary methionine and survival. The results showed that the colony forming ability of the GBM cell lines tested was significantly decreased in the absence of methionine. Plating efficiencies varied from 35-60% depending on cell line.
[C] GBM cells were grown in DMEM media containing high (200 μM) and low (20 μM) concentrations of methionine. The concentration of SAM and SAH were then estimated using LC-MS (QQQ) spectrometry. Quantitative RT-PCR then measured the expression levels of AHCY and MAT (1A+2A) in the GBM cell lines.
Increased methionine consumption alters the SAM : SAH ratio and methylation landscape
When methionine enters the methionine metabolic cycle (Fig. S10), methionine adenosyl transferase (MAT1A), and its paralog MAT2A, catalyze the conversion of methionine to the primary methyl donor of the cell, S-adenosylmethionine (SAM). SAM is then partitioned between the de novo and salvage pathways. In the salvage pathway, SAM is utilized in polyamine synthesis, generating MTA as a by-product (21, 23). MTA is then phosphorylated by MTA phosphorylase (MTAP) producing 5′-methylthioribose-1-phosphate (MTR1P) (23-25). MTR1P then undergoes further processing, which produces methionine as an end product. Alternatively, in the de novo pathway, SAM synthesized from assimilated methionine donates methyl groups to DNA, RNA, and protein catalyzed by methyltransferases, during its conversion to S-adenosylhomocysteine (SAH) (26). Adenosylhomocysteinase (AHCY) converts SAH to homocysteine, which undergoes further processing and is converted back to methionine. Importantly, the SAM : SAH ratio, also defined as cellular methylation potential, directly regulates cellular methylation and gene transcription. Additionally, alterations to AHCY, an enzyme also thought to be important for transmethylation reactions (27), can affect the cellular levels of SAH and corresponding methylation index (SAM : SAH). Therefore, the regulation of the SAM : SAH ratio is of interest because aberrant DNA methylation patterns are known to promote oncogenic kinase activation, tumor suppressor inactivation, and induce widespread change in signaling cascades.
In order to quantitatively measure the change in the DNA methylation pattern based on changes in the level of dietary methionine, we used a LC-MS/QQQ Mass spectrometer to determine the SAM and SAH expression levels in GBM cell lines cultured in medium containing 200 μM or 20 μM of methionine (Fig. S11). In order to stay within the normal physiological conditions of human cells, we used 20 μM of methionine because this is the concentration found in human blood. As a group, GBM cells grown in media with a decreased methionine concentration expressed significantly decreased levels of SAM, however, showed no significant alteration in SAH expression level (Fig. 3C). This disparity suggests that the increased concentration of methionine in these GBM cells gets preferentially shunted to the methionine salvage pathway for protein synthesis.
Given that decreased dietary methionine significantly altered the SAM : SAH ratio in GBM cells, we hypothesized that altered expression levels of key enzymes that regulate SAM and SAH cellular concentrations, MAT1A, MAT2A, and AHCY, were responsible for the dysregulation of the SAM : SAH ratio. The results showed that MAT2A was expressed by all the GBM cell lines tested, while MAT1A was expressed by only one GBM cell line. Subsequently, only one-third of the GBM cells lines used showed significant expression levels of AHCY (Fig. 3C). The higher expression levels of MAT2A and AHCY in MDNSC2 and MDNSC20 present a correlation with the increased productions of SAM when grown in media with low methionine levels that result in a change in methylation potential. Due to the fact that SAH is a competitive inhibitor of SAM-dependent methyltransferase reactions, one possible explanation for this phenomenon may be that the increased expression levels of AHCY decrease competitive inhibition of SAM-dependent methylation, resulting in an increased SAM level. This trend is intriguing because an increase in the cellular methylation potential that regulates the reorganization of the GBM methylome may contribute to the overall aggressive tumorigenic phenotype of GBM.
Decreased expression or loss of MTAP alters methionine metabolism in GBM
We went on to investigate whether the altered expression of MTAP accounts for the increased secretion of MTA to the extracellular milieu by GBM cells. Using qRT-PCR, we found the MTAP expression levels in GBM cells were significantly lower than the expression level in NHAs (Fig. 4A). The cell lines LN18, LN229, U87, and U118 did not show any amplification of the MTAP transcript. Genomic sequencing data available for these cell lines confirmed the loss of MTAP due to LOH at 9q21. Additionally, the majority of the primary cell lines used did not express CDKN2A, which could be due to the co-deletion of CDKN2A and MTAP, as they are located in close proximity. Therefore, the decreased expression or LOH of MTAP results in an intracellular accumulation of MTA, likely explaining the increased secretion of MTA to the extracellular milieu by GBM cells in order to minimize MTA-induced cytotoxicity. Additionally, this loss of MTAP expression may, at least in part, account for the increased consumption of methionine, given this loss of expression would significantly decrease the ability of these cells to recycle methionine.
Figure 4. MTAP rescue inhibits tumor formation but does not reverse methionine dependency.
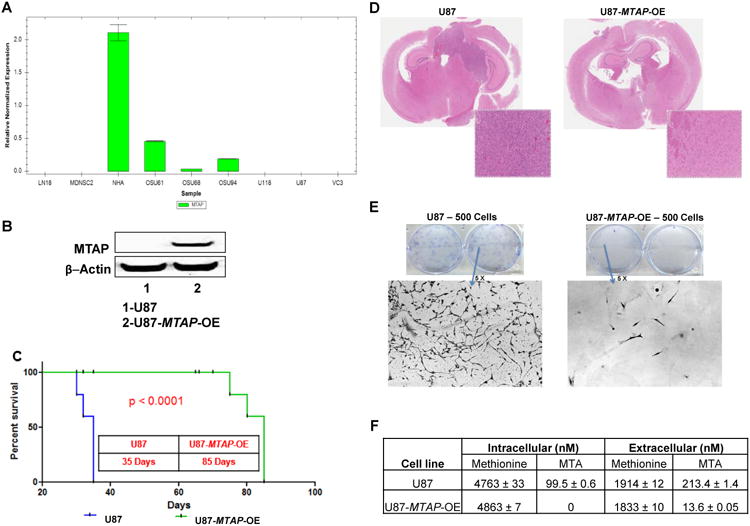
[A] Quantitative RT-PCR was used to determine the expression levels of MTAP in GBM cell lines and NHAs. The results showed that in comparison to NHAs, the GBM cell lines tested had significantly decreased or no expression of MTAP. GAPDH was used as the control.
[B] Lentiviral transfection of U87 cells (inherently MTAP-deficient) was used to create an isogenic U87 cell line that overexpresses MTAP (U87-MTAP-OE). Western blot analysis was then used to confirm the overexpression of MTAP in U87-MTAP-OE cells.
[C] NOD-SCID mice were injected with 100,000 cells from the U87 or U87-MTAP-OE cell lines. The Kaplan-Meier (KPM) plot shows the increased survival benefit of NOD-SCID mice injected with the U87-MTAP-OE cell line, as compared to the U87 cell line. The median survival benefit of mice injected with the transfected U87-MTAP-OE cell line was approximately 50 days.
[D] Hematoxylin-Eosin (H&E) stained coronal sections depict an aggressive tumor in mice bearing U87 cells and a residual tumor in mice bearing U87-MTAP-OE cells. The staining clearly shows the lack of tumor forming ability when MTAP is expressed. A higher magnification of the remaining tumors have also been included.
[E] 500 cells from the U87 and U87-MTAP-OE cell lines were seeded on 6-well plates. After 2 weeks, the plates were stained. The U87-MTAP-OE cells showed significantly decreased tumor forming ability in relation to the U87 cells. Higher magnification (5×) pictures of each plate have also been included.
[F] Mass spectral quantitative analysis using the MRM method shows that the uptake of methionine is similar between the U87 and U87-MTAP-OE cell lines. However, there is a significant decrease of MTA in both the intracellular compartment and extracellular milieu of U87-MTAP-OE cells.
MTAP rescue compromises tumor forming potential of GBM in vivo without altering methionine uptake
To assess the effect that a MTAP loss has on the tumor forming ability and methionine uptake of GBM cells, MTAP transgene was expressed in MTAP-deficient U87 cells (U87-MTAP-OE) (Fig. 4B). To estimate the relative tumor forming ability, NOD-SCID mice were injected intracranially with the U87 and U87-MTAP-OE cell lines (28). The mice injected with the U87-MTAP-OE cell line showed an increase in survival of approximately 50 days and a significant decrease in tumor size in relation to the mice injected with the U87 cells (Fig. 4C-D). Clonogenic assays with the U87-MTAP-OE cell line demonstrated significantly decreased colony forming ability (Fig. 4E). Quantifying the concentration of methionine and MTA in the intracellular compartment and extracellular milieu of the U87 and U87-MTAP-OE cell lines, there was no change in the uptake of methionine. However, the levels of conversion of methionine to MTA and MTA secretion to the extracellular milieu (15-fold decrease) were markedly compromised in the U87-MTAP-OE cells (Fig. 4F). This suggests that the methionine dependence phenotype of MTAP-deficient U87 cells cannot be reverted by the introduction of a functional MTAP gene. Our data suggests that methionine uptake is independent of MTAP gene status and intracellular concentration of MTA, rather, MTAP overexpression deregulates other genes involved in the methionine salvage pathway that contribute to the methionine dependence phenotype of GBM cells.
Methionine as a tracer for GBM
Methionine is an essential neutral amino acid that can readily cross the blood-brain barrier through neutral amino acid transporters and accumulate in the area of an active tumor. The uptake of methionine in a normal brain is relatively low as compared to those with gliomas, hence providing a potential advantage over 2-deoxy-2-18Fluoro-D-glucose (FDG) (29). Previous studies have shown specificity and sensitivity of MET-PET over FDG (30-32). Our metabolic data shows that accumulation of methionine is specific to GBM cells. To confirm the clinical relevance of methionine as a tracer for GBM, we have determined the methionine concentration in fresh patient GBM biopsy tissue (Suppl. Materials, Table S2). This is the first study which forms the foundation and rationale to use methionine as a tracer for PET imaging GBM tumors.
Tryptophan 2,3-dioxygenase-2 (TDO2) is primarily responsible for the conversion of tryptophan to kynurenine in GBM cells
Under normal conditions, kynurenine formed from tryptophan is converted to quinolinic acid and NAD+. However, during adaptive immune response, there is an increase in pro-inflammatory cytokines that increase the activity of the indoleamine 2,3-dioxygenase-1/2 (IDO1/IDO2) and tryptophan 2,3-dioxygenase-2 (TDO2) enzymes (33). These enzymes are responsible for the conversion of tryptophan to kynurenine and are proven immunosuppressant agents, as kynurenine suppresses anti-tumor immune responses in many cancer types, including GBM (34). The increased uptake of tryptophan coupled with the increased secretion of kynurenine to the extracellular milieu by GBM cells discovered in our metabolic profiling suggested the activation of the IDO/TDO enzymes. To investigate this activity, we measured the mRNA and protein expression levels of IDO1, IDO2, and TDO2 in GBMs and NHAs (Figs. 5A-C, S12). Our results showed a significant upregulation of TDO2 in GBM cells. This is in accordance with previous reports that have shown TDO2 is primarily responsible for catabolizing tryptophan to kynurenine in gliomas (19).
Figure 5. Expression levels of key enzymes in the tryptophan pathway and insilico analysis of the DRMs in GBM cells.
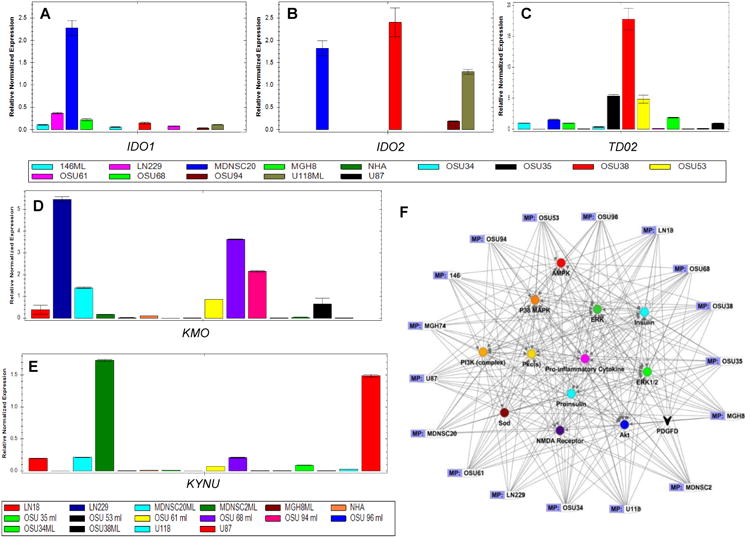
We used qRT-PCR to evaluate the expression levels of the key enzymes in the tryptophan metabolic pathway: [A] IDO1, [B] IDO2, [C] TDO2, [D] KMO, and [E] KYNU in GBM cells and NHAs.
[F] Additionally, the identified DRMs were put into IPA analysis to explore deregulated signaling networks. The analysis results from each GBM cell line were overlaid to identify the common core pathway members. Next, we conducted insilico mapping of the metabolome and proteome using 6 GBM cell lines (Fig. S13). Overlapping the insilico analysis from each GBM cell line, we identified the activation of signaling networks known to mediate treatment resistance. Of note, the oncogenic enzymes Akt, PI3K, and pro-inflammatory cytokines all showed high levels of activation as a result of increased uptake of methionine and kynurenine.
Lack of KMO and KYNU accumulates kynurenine in the extracellular milieu, a key mediator of immune evasion
Under normal physiological conditions, tryptophan is converted to the neurotransmitter serotonin. However, under pathophysiological conditions, the rate of conversion from tryptophan to kynurenine is increased due to the upregulation of TDO/IDO enzymes. The increase in IDO/TDO activity in GBM cell lines tested suggests tryptophan is shunted away from the normal neurotransmitter (serotonin) pathway and into the kynurenine pathway. In NHAs, kynurenine is almost exclusively converted to kynurenic acid, a neuroprotective compound. Conversely, kynurenine-3-monooxygenase (KMO) catalyzes the hydroxylation of kynurenine to form 3-hydroxykynurenine. Kynureninase (KYNU), a pyridoxal-5′-phosphate dependent enzyme, then catalyzes the cleavage of kynurenine and 3-hydroxykynurenine into anthranilic and 3-hydroxyanthranilic acids, respectively. These products are then converted into quinolinic acid, a neurotoxic NMDA receptor antagonist (Fig. S12). Kynurenine that is not degraded by KMO or KYNU may bind to the aryl hydrocarbon receptor that recruits immune-suppressive T cells, leading to immune evasion.
We hypothesized that the increased amount of kynurenine secreted to the extracellular milieu by GBM cell lines tested, was a result of a change in the expression levels of KMO and KYNU. Further, qRT-PCR showed a lack of expression of these enzymes in GBM cells (Fig. 5D-E). GBM cells expressing KMO or KYNU had decreased amounts of kynurenine accumulation due to further metabolic conversions downstream. Therefore, an interesting correlation exists between KMO/KYNU expression and extracellular milieu kynurenine concentrations, which suggests that the lack of KMO or KYNU expression is one of the factors influencing the rate-limiting step for the utilization of kynurenine.
The differentially regulated metabolites activate key oncogenic kinases
To discover connections between the DRMs and key oncogenic enzymes/proteins in GBM cell lines, we used Ingenuity Pathway Analysis (IPA), and identified amino acid metabolism, molecular transport at the cellular level, and nervous system development as predicted functions with the highest scores (Figs. 5F, S13). This analysis revealed that the identified DRMs were linked to a number of key oncogenic signaling proteins including: PI3K, Akt, ERK, p38-MAPK, PKC, and others, that partly mediate the highly proliferative, angiogenic, invasive, and treatment resistant nature of GBM cells.
To validate the findings from our insilico analysis, we measured the activation of oncogenic protein kinases following the exogenous addition of methionine and kynurenine to GBM and NHA cells (Fig. 6A). Sharp increases in the phosphorylation of Akt, PI3K, ERK1/2, and IGF1R were observed with peak intensity ranging from 5-30 minutes depending on the cell line; no detectable increase in phosphorylation levels of these proteins was observed in NHAs (Fig. S14). Additionally, the spikes in the phosphorylation levels of these kinases were found to be associated with a decrease in the expression of PP2A subunits (Fig. 6B). While further investigation is required to uncover the mechanistic basis behind the connection of these DRMs to the activation of key oncogenic proteins, this discovery is intriguing as it presents the potential for novel therapeutic targets for GBM.
Figure 6. Exogenous addition of methionine or kynurenine decreases PP2A expression, upregulating the phospho-proteins responsible for the activation of oncogenic kinases and graphical abstract.

[A] In the U87 and LN18 cell lines, exogenous addition of methionine or kynurenine leads to an immediate activation of oncogenic kinases. The expression levels of various kinases (both total and phosphorylated protein) were analyzed on western blots at 5, 15, and 30 minutes after the addition of each metabolites. The densitometric calculations were carried out and plotted (Fig. S14). At varying time points all of the oncogenic kinases showed an increase in expression across the cell lines. β-actin and the total protein extracts were used as loading controls.
[B] The activation of PP2A subunits were measured using western blot analysis at 5,15, and 30 minutes after the exogenous addition of kynurenine or methionine to U87 and LN18 cells. The densitometric calculations are provided below each lane. A depletion of the phosphatase subunits PP2A-A, PP2A-B, and PP2A-C were observed corresponding to the increased activation of oncogenic kinases.β-actin was used as the control.
[C] The graphical abstract provided describes how the four-metabolite signature discovered in this study progresses the tumorigenic phenotype of GBM.
Discussion
In GBM, there is an urgent need to identify biomarkers to improve diagnosis, prognosis, and treatment efficacy. In these endeavors, applications of metabolomics hold a promising potential in complementing existing functional genomics approaches. Here, we have utilized a metabolomics-based technique and discovered four differential regulated metabolites and accessed their involvement in promoting a tumorigenic phenotype in GBM.
We have shown that GBM cells are dependent on dietary methionine for proliferation, colony formation, and survival. This is in accordance with a previous report that increased levels of methionine uptake in cancer cells are driven by methionine dependency(16). In addition, two previous studies have shown methionine depletion over 10-12 days decreases ATP and glutathione pools, sensitizing refractory tumors to the cytotoxic anticancer drugs cisplatin, doxorubicin, carmustine, N,N′-bis(2-chloroethyl)-N-nitrosourea, and temozolomide by inducing mitotic and cell cycle arrest, apoptotic death, and widespread necrosis in tumors(17, 18). Therefore, dietary methionine and the methionine metabolic cycle may present promising therapeutic targets for combating GBM.
The increased uptake of methionine by GBM cells alters the SAM : SAH ratio. This is significant because changes in this ratio change the total amount of methylation of DNA, RNA, and protein, resulting in the reorganization of the cellular methylation pattern and regulation of gene transcription. Aberrant DNA methylation patterns, typically tumor suppressor genes are silenced by promoter hypermethylation and oncogenes are activated by promoter hypomethylation, have been associated with a large number of human malignancies. Therefore, this change in cellular methylation may activate oncogenic kinases and change cellular regulation of gene transcription that enables increased proliferation. This result reinforces the potential use of a dietary methionine restriction strategy to improve treatment outcome.
The premier biochemical and genetic etiology of methionine dependency in cancer cells is not fully understood. While deletions, polymorphisms, and altered expression of key enzymes in the methionine metabolic cycle have been implicated, there has been no report that directly links these enzymes to methionine dependency. To this end, we observed that MTAP expression is significantly decreased or undergoes a loss of MTAP due to LOH at human chromosome 9p21 in a number of tumorigenic GBM cell lines. This deletion or lower expression level of MTAP coincides with previously reported data that showed in a number of cancer types, including GBM, that the MTAP gene is frequently co-deleted with p16INK4a (located 100kb telomeric at locus 9p21) (22, 35, 36). In MTAP-deficient U87 GBM cells, the expression of MTAP transgene was sufficient to compromise the tumor forming capacity of these cells. Importantly, this suggests that MTAP may function as an independent tumor suppressor gene. This notion is further supported by previous work demonstrating that the MTAP locus can be deleted independently of p16INK4a (37). While this indicates that MTAP rescue may provide a means of increasing the survival of GBM patients, the overexpression of MTAP did not alter the methionine dependency of U87 cells. A similar observation was made in MTAP-deficient MCF-7 breast cancer cells, in which overexpression of MTAP transgene reduced the tumor growth, but failed to reverse the methionine dependency (38, 39). Thus, it appears that there are mutations in other gene(s) involved in the methionine metabolic pathway responsible for rendering cells methionine dependent in GBM. Moving forward, further studies are necessary to determine the underlying molecular mechanism(s) responsible for methionine dependency in GBM.
The current standard of care for newly diagnosed GBM is surgical resection, followed by adjuvant chemotherapy and radiotherapy (1, 40). The prolific growth of sophisticated and accurate radiation therapy techniques such as stereotactic radiotherapy, radiosurgery, intensity-modulated radiotherapy, proton therapy, etc., would allow improved survival rates to be achieved. This increased survival benefit is possible because using high-dose irradiation on a limited target volume could eradicate tumor cells while minimizing radiation exposure to adjacent normal tissues. This study forms a foundation to incorporate MET-PET imaging into the clinic and will have significant implications in the treatment strategy for GBM patients for both surgical resection and radiation treatment planning. A variety of cancer cells express immunogenic antigens, but manage to escape the immune response by employing a number of immune evading mechanisms (41). One of the immune escaping mechanisms that GBM cells adapt is the conversion of tryptophan to kynurenine instead of serotonin (19, 34, 42) and this reaction is fueled by activated TDO/IDO enzymes in GBM. Here, we found that the intracellular level of tryptophan was higher in GBM cells relative to NHAs and that GBM cells produced kynurenine primarily through the catalytic action of TDO2. The lack of KMO expression and corresponding accumulation of kynurenine in the extracellular milieu suggests this may be the rate-limiting step for the utilization of kynurenine. Previous studies have shown that kynurenine, but not other tryptophan catabolic products downstream of kynurenine, directly activate aryl hydrocarbon receptors, resulting in the activation of immunosuppressant agents (43). Taken together these results suggest that reinforcing KMO or KYNU activity may result in the utilization of kynurenine, decreasing the excess amount of kynurenine in the extracellular milieu that triggers this immune evasion. A result that could restore immune surveillance. Additionally, a recent report has revealed that interferon-gamma stimulation significantly potentiates the expression of the kynurenine pathway enzymes IDO1, IDO2, KYNU, and KMO in cultured human glioma cells (44). It has been shown that both tryptophan starvation and tryptophan catabolites (kynurenine, 3-hydroxykynurenine, 3-hydroxyanthranilic acid, and quinolinic acid) contribute to an effector mechanism of immune tolerance (45). This also supports our data suggesting that reinstating KMO or KYNU would generate kynurenine pathway catabolites that could contribute to the alteration of the regulatory T cell environment in GBM to combat the immune response. This treatment aim involving KMO or KYNU may not only restore anti-tumor immunity but also may reduce the capacity for malignant cells to activate oncogenic kinases. These data provides further evidence for a potential role of the tryptophan-kynurenine metabolic pathway products as novel and attractive therapeutic targets for the treatment of GBM.
Interestingly, we observed that when kynurenine was added to GBM cells in vitro, it induced the activation of PI3K and Akt, which correlated with the downregulation of protein phosphatase PP2A subunits responsibe for reducing the phospho-Ser/Thr levels in many signaling proteins (46, 47). Previous studies have shown that inhibition of PP2A by a variety of inhibitors enhances cell growth and proliferation, which is partially attributed to the activation of the RTK-RAS-PI3K pathway (48, 49). Furthermore, it has been shown that pharmacologic inhibition of PP2A may be a general method for enhancing the effectiveness of cancer treatments that damage DNA or disrupt components of cell replication (50). Thus, our data have unfolded a new function of kynurenine in GBM cells. Additionally, we note that exogenous methionine also induced activation of these kinases and reduced the expression of the PP2A subunits. These data show a direct link between the increased uptake of methionine and kynurenine and the upregulation of key oncogenic signaling proteins. This result further supports the notion of targeting these two metabolites for the development of new GBM therapies.
In sum, we have identified a four-metabolite signature that aids in the immune evasion, activation of oncogenic kinases, proliferation, survival, and development of treatment resistance in GBM cells. A schematic of the metabolic alterations that induce the passage to a more progressive cancer state driven by increased tryptophan and methionine uptake is presented in the graphical abstract (Fig. 6C). While additional investigation is required to further understand the molecular mechanisms that connect these four DRMs to driving tumorigenesis, our results present exciting novel drug targets to combat GBM.
Supplementary Material
Translational Relevance.
We identified tryptophan and methionine as differentially regulated metabolites (DRMs) in the intracellular compartment of glioblastoma cell lines and tissue samples as compared to normal human astrocytes. This altered regulation results from a lack of key enzymes in the tryptophan and methionine metabolic pathways. The expression of MTAP, responsible for recycling methionine, is significantly decreased or deleted, resulting in increased intracellular concentrations of methionine. MTAP rescue studies suggest MTAP is a tumor suppressor. Since methionine is an essential amino acid, methionine positron emission tomography can be efficiently used to delineate normal/tumor volume leading to precise surgical resection and higher radiation dose treatment planning. In the tryptophan pathway, activation of IDO/TDO enzymes accumulates kynurenine leading to immune evasion. Reinforcing kynurenine catabolic enzymes KYNU/KMO could antagonize this effect and retain immune surveillance. Insilico analysis, mapped the DRMs to the activation of oncogenic kinases which was confirmed in vitro.
Acknowledgments
We thank Dr. Sean Lawler, Brigham and Women's Hospital, Harvard Medical School, Boston, MA for sharing with us brain tumor tissues which were used for isolating primary cells, Cell lines MDNSC2 and MDNSC20 were provided by Dr. Howard Colman, The University of Utah School of Medicine, Salt Lake City, Utah, which were grown as adherent monolayers for our study. We thank Dr. S. Jaharul Haque for editing the manuscript and Ananya Kamalakannan for her help with graphical abstract.
Funding: NIH/NCI awards R01CA108633, RC2CA148190, The Brain Tumor Funders Collaborative Group, and The Arthur G. James Comprehensive Cancer Center
NIH/NCI 1RC2CA148190, 1RO1CA169368, 1RO1CA11522358, The Ohio State University Comprehensive Cancer Center and Radiation Therapeutic Oncology Group.
Footnotes
Authors' Contributions: Conceived and designed the experiments: KP, AC; Analyzed the data: TK, KP; Performed the experiments: TK, KP, NG, SK, RS, JRJ, NS, KTL; Contributed reagents/materials/analysis tools: DP, MO, EB, BR, ARC, TL, SF, AC; Wrote the paper: TK, KP.
Disclosure of Potential Conflicts of Interest: The authors declare no potential conflicts of interest.
References
- 1.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 2.Bowen BP, Northen TR. Dealing with the unknown: metabolomics and metabolite atlases. Journal of the American Society for Mass Spectrometry. 2010;21:1471–6. doi: 10.1016/j.jasms.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Mayr M. Metabolomics: ready for the prime time? Circulation Cardiovascular genetics. 2008;1:58–65. doi: 10.1161/CIRCGENETICS.108.808329. [DOI] [PubMed] [Google Scholar]
- 4.Ryan D, Robards K. Metabolomics: The greatest omics of them all? Analytical chemistry. 2006;78:7954–8. doi: 10.1021/ac0614341. [DOI] [PubMed] [Google Scholar]
- 5.Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457:910–4. doi: 10.1038/nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 6.Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–4. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weckwerth W, Fiehn O. Can we discover novel pathways using metabolomic analysis? Current opinion in biotechnology. 2002;13:156–60. doi: 10.1016/s0958-1669(02)00299-9. [DOI] [PubMed] [Google Scholar]
- 8.Zhang GF, Sadhukhan S, Tochtrop GP, Brunengraber H. Metabolomics, pathway regulation, and pathway discovery. The Journal of biological chemistry. 2011;286:23631–5. doi: 10.1074/jbc.R110.171405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldsmith P, Fenton H, Morris-Stiff G, Ahmad N, Fisher J, Prasad KR. Metabonomics: a useful tool for the future surgeon. The Journal of surgical research. 2010;160:122–32. doi: 10.1016/j.jss.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 10.Spratlin JL, Serkova NJ, Eckhardt SG. Clinical applications of metabolomics in oncology: a review. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:431–40. doi: 10.1158/1078-0432.CCR-08-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuperlovic-Culf M, Barnett DA, Culf AS, Chute I. Cell culture metabolomics: applications and future directions. Drug discovery today. 2010;15:610–21. doi: 10.1016/j.drudis.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 12.Griffin JL, Shockcor JP. Metabolic profiles of cancer cells. Nature reviews Cancer. 2004;4:551–61. doi: 10.1038/nrc1390. [DOI] [PubMed] [Google Scholar]
- 13.Khoo SHG, Al-Rubeai M. Metabolomics as a complementary tool in cell culture. Biotechnol Appl Bioc. 2007;47:71–84. doi: 10.1042/BA20060221. [DOI] [PubMed] [Google Scholar]
- 14.Ramanathan A, Wang C, Schreiber SL. Perturbational profiling of a cell-line model of tumorigenesis by using metabolic measurements. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5992–7. doi: 10.1073/pnas.0502267102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Want EJ, Nordstrom A, Morita H, Siuzdak G. From exogenous to endogenous: the inevitable imprint of mass spectrometry in metabolomics. Journal of proteome research. 2007;6:459–68. doi: 10.1021/pr060505+. [DOI] [PubMed] [Google Scholar]
- 16.Sugimura T, Birnbaum SM, Winitz M, Greenstein JP. Quantitative nutritional studies with water-soluble, chemically defined diets. VIII. The forced feeding of diets each lacking in one essential amino acid. Archives of biochemistry and biophysics. 1959;81:448–55. doi: 10.1016/0003-9861(59)90225-5. [DOI] [PubMed] [Google Scholar]
- 17.Poirson-Bichat F, Goncalves RA, Miccoli L, Dutrillaux B, Poupon MF. Methionine depletion enhances the antitumoral efficacy of cytotoxic agents in drug-resistant human tumor xenografts. Clinical cancer research : an official journal of the American Association for Cancer Research. 2000;6:643–53. [PubMed] [Google Scholar]
- 18.Kokkinakis DM, Hoffman RM, Frenkel EP, Wick JB, Han Q, Xu M, et al. Synergy between methionine stress and chemotherapy in the treatment of brain tumor xenografts in athymic mice. Cancer research. 2001;61:4017–23. [PubMed] [Google Scholar]
- 19.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 20.Broadhurst DI, DB K. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics. 2007;2:171–96. [Google Scholar]
- 21.Finkelstein JD. Methionine metabolism in mammals. The Journal of nutritional biochemistry. 1990;1:228–37. doi: 10.1016/0955-2863(90)90070-2. [DOI] [PubMed] [Google Scholar]
- 22.Cavuoto P, Fenech MF. A review of methionine dependency and the role of methionine restriction in cancer growth control and life-span extension. Cancer treatment reviews. 2012;38:726–36. doi: 10.1016/j.ctrv.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Albers E. Metabolic characteristics and importance of the universal methionine salvage pathway recycling methionine from 5′-methylthioadenosine. IUBMB life. 2009;61:1132–42. doi: 10.1002/iub.278. [DOI] [PubMed] [Google Scholar]
- 24.Carson DA, Nobori T, Kajander EO, Carrera CJ, Kubota M, Yamanaka H. Methylthioadenosine (MeSAdo) phosphorylase deficiency in malignancy. Advances in experimental medicine and biology. 1988;250:179–85. doi: 10.1007/978-1-4684-5637-0_16. [DOI] [PubMed] [Google Scholar]
- 25.Lee JE, Settembre EC, Cornell KA, Riscoe MK, Sufrin JR, Ealick SE, et al. Structural comparison of MTA phosphorylase and MTA/AdoHcy nucleosidase explains substrate preferences and identifies regions exploitable for inhibitor design. Biochemistry. 2004;43:5159–69. doi: 10.1021/bi035492h. [DOI] [PubMed] [Google Scholar]
- 26.Zingg JM, Jones PA. Genetic and epigenetic aspects of DNA methylation on genome expression, evolution, mutation and carcinogenesis. Carcinogenesis. 1997;18:869–82. doi: 10.1093/carcin/18.5.869. [DOI] [PubMed] [Google Scholar]
- 27.Shu S, Mahadeo DC, Liu X, Liu W, Parent CA, Korn ED. S-adenosylhomocysteine hydrolase is localized at the front of chemotaxing cells, suggesting a role for transmethylation during migration. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:19788–93. doi: 10.1073/pnas.0609385103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palanichamy K, Acus K, Jacob RJ, Chakravarti A. Clinically relevant brain tumor model and device development for experimental therapeutics. Journal of Analytical Oncology. 2015;4:5–12. [Google Scholar]
- 29.Singhal T, Narayanan TK, Jacobs MP, Bal C, Mantil JC. 11C-methionine PET for grading and prognostication in gliomas: a comparison study with 18F-FDG PET and contrast enhancement on MRI. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2012;53:1709–15. doi: 10.2967/jnumed.111.102533. [DOI] [PubMed] [Google Scholar]
- 30.Herholz K, Holzer T, Bauer B, Schroder R, Voges J, Ernestus RI, et al. 11C-methionine PET for differential diagnosis of low-grade gliomas. Neurology. 1998;50:1316–22. doi: 10.1212/wnl.50.5.1316. [DOI] [PubMed] [Google Scholar]
- 31.Grosu AL, Weber WA, Riedel E, Jeremic B, Nieder C, Franz M, et al. L-(methyl-11C) methionine positron emission tomography for target delineation in resected high-grade gliomas before radiotherapy. International journal of radiation oncology, biology, physics. 2005;63:64–74. doi: 10.1016/j.ijrobp.2005.01.045. [DOI] [PubMed] [Google Scholar]
- 32.Matsuo M, Miwa K, Tanaka O, Shinoda J, Nishibori H, Tsuge Y, et al. Impact of [11C]methionine positron emission tomography for target definition of glioblastoma multiforme in radiation therapy planning. International journal of radiation oncology, biology, physics. 2012;82:83–9. doi: 10.1016/j.ijrobp.2010.09.020. [DOI] [PubMed] [Google Scholar]
- 33.Platten M, von Knebel Doeberitz N, Oezen I, Wick W, Ochs K. Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors. Front Immunol. 2014;5:673. doi: 10.3389/fimmu.2014.00673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer research. 2012;72:5435–40. doi: 10.1158/0008-5472.CAN-12-0569. [DOI] [PubMed] [Google Scholar]
- 35.Nobori T, Karras JG, Della Ragione F, Waltz TA, Chen PP, Carson DA. Absence of methylthioadenosine phosphorylase in human gliomas. Cancer research. 1991;51:3193–7. [PubMed] [Google Scholar]
- 36.Nobori T, Takabayashi K, Tran P, Orvis L, Batova A, Yu AL, et al. Genomic cloning of methylthioadenosine phosphorylase: a purine metabolic enzyme deficient in multiple different cancers. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:6203–8. doi: 10.1073/pnas.93.12.6203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brat DJ, James CD, Jedlicka AE, Connolly DC, Chang E, Castellani RJ, et al. Molecular genetic alterations in radiation-induced astrocytomas. The American journal of pathology. 1999;154:1431–8. doi: 10.1016/S0002-9440(10)65397-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christopher SA, Diegelman P, Porter CW, Kruger WD. Methylthioadenosine phosphorylase, a gene frequently codeleted with p16(cdkN2a/ARF), acts as a tumor suppressor in a breast cancer cell line. Cancer research. 2002;62:6639–44. [PubMed] [Google Scholar]
- 39.Tang B, Li YN, Kruger WD. Defects in methylthioadenosine phosphorylase are associated with but not responsible for methionine-dependent tumor cell growth. Cancer research. 2000;60:5543–7. [PubMed] [Google Scholar]
- 40.Palanichamy K, Chakravarti A. Combining drugs and radiotherapy: from the bench to the bedside. Curr Opin Neurol. 2009;22:625–32. doi: 10.1097/WCO.0b013e3283327d33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 42.Adams S, Braidy N, Bessede A, Brew BJ, Grant R, Teo C, et al. The kynurenine pathway in brain tumor pathogenesis. Cancer research. 2012;72:5649–57. doi: 10.1158/0008-5472.CAN-12-0549. [DOI] [PubMed] [Google Scholar]
- 43.Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185:3190–8. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adams S, Teo C, McDonald KL, Zinger A, Bustamante S, Lim CK, et al. Involvement of the kynurenine pathway in human glioma pathophysiology. PloS one. 2014;9:e112945. doi: 10.1371/journal.pone.0112945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol. 2006;176:6752–61. doi: 10.4049/jimmunol.176.11.6752. [DOI] [PubMed] [Google Scholar]
- 46.Mumby M. PP2A: unveiling a reluctant tumor suppressor. Cell. 2007;130:21–4. doi: 10.1016/j.cell.2007.06.034. [DOI] [PubMed] [Google Scholar]
- 47.Seshacharyulu P, Pandey P, Datta K, Batra SK. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett. 2013;335:9–18. doi: 10.1016/j.canlet.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Casillas AM, Amaral K, Chegini-Farahani S, Nel AE. Okadaic acid activates p42 mitogen-activated protein kinase (MAP kinase; ERK-2) in B-lymphocytes but inhibits rather than augments cellular proliferation: contrast with phorbol 12-myristate 13-acetate. The Biochemical journal. 1993;290(Pt 2):545–50. doi: 10.1042/bj2900545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Puustinen P, Junttila MR, Vanhatupa S, Sablina AA, Hector ME, Teittinen K, et al. PME-1 protects extracellular signal-regulated kinase pathway activity from protein phosphatase 2A-mediated inactivation in human malignant glioma. Cancer research. 2009;69:2870–7. doi: 10.1158/0008-5472.CAN-08-2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu J, Kovach JS, Johnson F, Chiang J, Hodes R, Lonser R, et al. Inhibition of serine/threonine phosphatase PP2A enhances cancer chemotherapy by blocking DNA damage induced defense mechanisms. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:11697–702. doi: 10.1073/pnas.0905930106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.