

Abstract
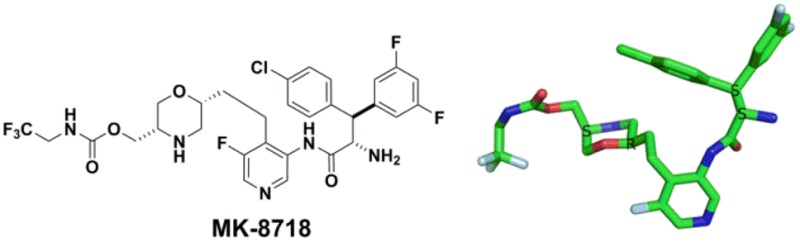
A novel HIV protease inhibitor was designed using a morpholine core as the aspartate binding group. Analysis of the crystal structure of the initial lead bound to HIV protease enabled optimization of enzyme potency and antiviral activity. This afforded a series of potent orally bioavailable inhibitors of which MK-8718 was identified as a compound with a favorable overall profile.
Keywords: MK-8718, HIV, protease, inhibitor
HIV protease is an aspartyl protease that catalyzes the proteolytic cleavage of polypeptide precursors into mature enzymes and structural proteins that are essential components of HIV.1 Inhibition of HIV protease prevents conversion of HIV particles into their mature infectious form and is an important approach for therapeutic intervention in HIV infection.2 HIV protease inhibitors (PIs) have played a crucial role as a therapy for the treatment of HIV.3,4 However, challenges still remain for these molecules in the form of strict dosing regimens, high pill burden, significant side effects, and the occurrence of resistant strains.5 In addition, HIV PIs have traditionally suffered from poor pharmacokinetic properties, including poor oral absorption and low metabolic stability.6,7
The first crystal structures of inhibitors bound to HIV protease were reported 25 years ago.8,9 Since then, structure based design has played a key role in the development of new inhibitors.10 A crucial structural feature of the majority of HIV PIs is the presence of a hydroxyl group that forms a hydrogen bond with the carboxylic acid functionalities of the catalytic Asp-25 and Asp-25′ residues in the enzyme active site.11 We were interested in pursuing analogues of inhibitors where an amine, instead of a hydroxyl group, forms the key interaction with the Asp-25 and Asp-25′ acidic residues of the enzyme.12 This type of inhibitor was attractive to us due to the potential improvement in physical properties offered by the polar amine functionality.
The starting point for the design of our compounds were the pyrrolidine-type inhibitors such as 1 (Figure 1) reported by Coburn et al.13 Although these molecules offered improved solubility over their hydroxyl counterparts, the pyrrolidine functionality introduced a number of undesirable off target activities, which may be attributed to the presence of a basic pyrrolidine nitrogen.14 This prompted us to consider designing a new core that would maintain the solubility enhancing amine functionality, while reducing the basicity of the nitrogen, with the goal of reducing off target activity. To this end, we felt a morpholine core offered a number of attractive properties. First, the inductively electron withdrawing effect of the oxygen lowers the pKa of the nitrogen; second the core provides access to a number of vectors, which could be used to append substituents to fill the remaining pockets of HIV protease.15 Our initial design was the truncated analogue 2 shown in Figure 1. It was envisioned that 2 would have sufficient affinity for the enzyme to enable a crystal structure of the inhibitor bound to the enzyme to be solved. This in turn could be used to design in the remaining functionality required to produce a potent inhibitor.
Figure 1.

Pyrrolidine based inhibitor 1 and proposed morpholine based inhibitor 2.
Synthesis of 2 began with commercially available aldehyde 3 as outlined in Scheme 1. Homologation and subsequent olefin reduction afforded aldehyde 4. Reductive amination followed by debenzylation afforded primary amine 5. Coupling of succinate ester 6(16) with amine 5 afforded amide 7, which was deprotected to yield desired product 2 as a mixture of diastereoisomers.
Scheme 1.

Reagents and conditions: (a) (i) Ph3P=CHCHO, THF, 50 °C; (ii) Pd/C, 30 psi H2, EtOAc, RT; (b) BnNH2, NaBH4, THF/MeOH, RT; (ii) Pd/C, 50 psi H2, MeOH, RT; (c) NaHCO3, THF/H2O, 0 °C; (d) TFA, Et3SiH, CH2Cl2, RT.
Although morpholine 2 was shown to be a modest inhibitor of HIV protease (11% inhibition at 1 μM), we decided to pursue a crystal structure of 2 bound to the enzyme. Pleasingly, a crystal structure was solved as depicted in Figure 2. It can be seen from this structure that the morpholine nitrogen of 2 indeed forms the desired key interactions with the Asp-25 and Asp-25′ acidic residues of the enzyme. In addition, the (R)-stereochemistry at the 2-position of the morpholine appeared to be preferred, and it could be seen that the two aryl groups occupy the S1 and S3 regions of the enzyme. With this valuable information in hand, we sought to improve the binding affinity by adding appropriate substituents to occupy additional key pockets of the enzyme.
Figure 2.

Overlay of the enzyme bound conformations of 2 (green) and 8 (magenta). The flaps have been cut away for optimum viewing. The 5-position is marked with an *.
The design of our next generation inhibitor was inspired by overlaying the enzyme bound conformation of our initial morpholine based inhibitor 2 with that of inhibitor 8, containing a P2 aryl substituent17 as shown in Figure 2. This analysis suggested that incorporation of an aryl P2 substituent into our morpholine based inhibitors could be beneficial for enzyme affinity. In addition, it was evident that a substituent at the 5-position of the morpholine could be used to reach out and occupy the S1′ pocket of the enzyme. This led us to pursue compound 9a.
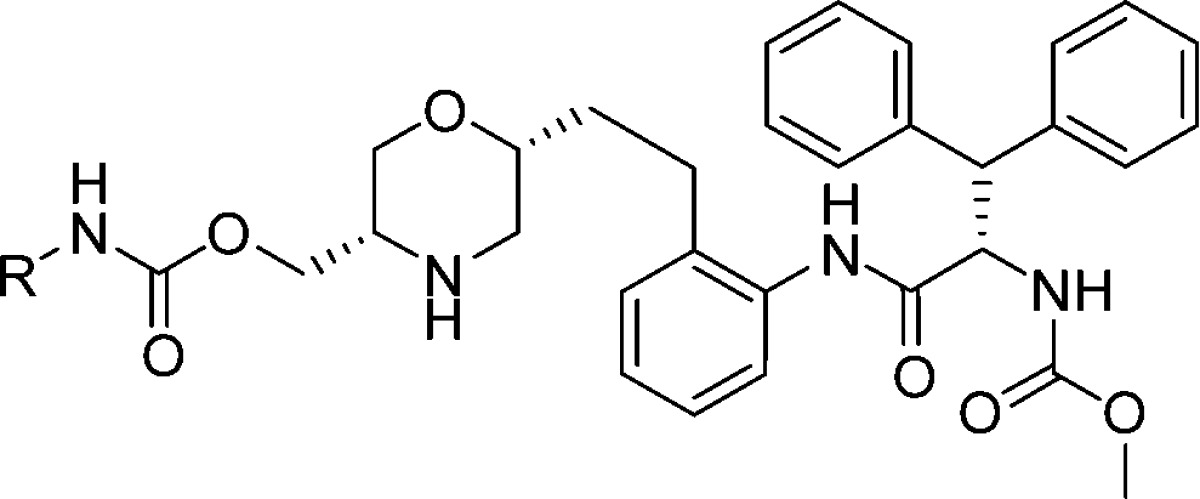
Synthesis of 9a is depicted in Scheme 2 and began by reaction of amino alcohol1810 with (R)-epichlorohydrin, using reaction conditions reported for the enantiomer,19 to give morpholine 11. Swern oxidation of 11 afforded aldehyde 12. A subsequent Wittig reaction, followed by reduction, afforded aniline 13. Coupling of acid 6 with aniline 13 afforded amide 14. Silyl deprotection to afford 15, followed by carbamate formation and morpholine deprotection afforded the desired compound 9a. Pleasingly, 9a was shown to be a potent inhibitor of HIV protease (IC50 = 6 nM). In addition, the compound showed antiviral activity in a cell based assay (IC95 = 202 nM). This promising activity encouraged us to benchmark the pharmacokinetic profile of 9a, shown in Table 1. The compound displayed moderate clearance in rat, coupled with a short half-life, and low bioavailability. With these results in hand and given our ability to introduce a P1′ substituent late in the synthesis, we decided to prepare analogues to explore the impact of this substituent on potency and pharmacokinetic properties. A subset of the analogues are shown in Table 1. A variety of substituents were tolerated; however, the trifluorethylcarbamate substituent present in 9i afforded a nice balance of potency, clearance, and oral bioavailability.
Scheme 2.

Reagents and conditions: (a) (R)-epichlorohydrin, LiClO4, NaOMe, toluene/MeOH, RT; (b) (i) Pd(OH)2, Boc2O, NEt3, 45 psi H2, RT; (ii) oxalyl chloride, NEt3, DMSO, CH2Cl2, −78 °C; (c) (i) K2CO3, 18-crown-6, (2-nitrobenzyl)triphenylphosphonium bromide, DME, RT; (ii) Pd/C, 50 psi H2, EtOH, RT; (d) (S)-2-((methoxycarbonyl)amino)-3,3-diphenylpropanoic acid, HATU, 2,6-lutidine, DMF, RT; (e) TBAF, THF, RT; (f) (i) benzylisocyanate, DCM, RT; (ii) TFA, DCM, 0 °C.
Table 1. Profiles of Compounds 9a–9j.

Assay for inhibition HIV protease as described in the Supporting Information (n = 2).
Assay for inhibition of viral infection as described in the Supporting Information (n = 2).
1 mpk IV (60% PEG 400/water), 5 mpk PO (0.5% methylcellulose). n = 2 rats.
Having identified an acceptable P1′ substituent, we decided to see if additional potency or pharmacokinetic improvements could be realized by optimizing the P2 substituent. A series of analogues were designed and the requisite anilines necessary for P2 SAR exploration were synthesized as shown in Scheme 3 and Scheme 4. For the fluorophenyl P2 precursor shown in Scheme 3, Wittig reaction of aldehyde 12 afforded olefin 16. Subsequent TBS deprotection gave alcohol 17. Carbamate formation followed by reduction (nitro and olefin) afforded aniline 18.
Scheme 3.

Reagents and conditions: (a) K2CO3, 18-crown-6, (2-fluoro-6-nitrobenzyl)triphenylphosphonium bromide,20 DME, RT; (b) HCl, MeOH, RT; (c) (i) 2,2,2-trifluoroethylamine, CDI, pyridine, RT; (ii) Pd(OH)2, 50 psi H2, CF3CH2OH, RT.
Scheme 4.

Reagents and conditions: (a) dimethyl (1-diazo-2-oxopropyl)phosphonate, K2CO3, MeOH, RT; (b) (i) 5-fluoro-4-iodopyridin-3-amine, 7% (PPh3)2PdCl2, 10% CuI, Et3N, CH3CN, 70 °C; (ii) 50% PtO2, 50 psi H2, CF3CF2OH, RT; (c) (i) Cbz-Cl, pyridine, 0 °C; (ii) TBAF, THF, RT; (d) (i) 2,2,2-trifluoroethylamine, CDI, pyridine, 60 °C; (ii) Pd/C, 1 atm H2, EtOH, RT.
For the pyridyl P2 precursors in Scheme 4, Seyferth–Gilbert homologation21 of aldehyde 12 afforded alkyne 19. Sonagashira coupling,22 followed by alkyne reduction gave anilines 20 and 21. Cbz protection followed by TBS deprotection afforded alcohols 22 and 23. Carbamate formation followed by Cbz-deprotection gave the desired anilines 24 and 25. As described in Scheme 7, anilines 18, 24, and 25 were coupled to commercially available (S)-2-((methoxycarbonyl)amino)-3,3-diphenylpropa-noic acid and (S)-2-((tert-butoxycarbonyl)amino)-3,3-diphenylpropanoic acid using POCl3. Boc-deprotection gave final compounds 9k to 9o shown in Table 2. Key discoveries from this set of compounds were as follows.
Scheme 7.

Reagents and conditions: (a) (i) (S)-2-((methoxycarbonyl)amino)-3,3-diphenylpropa-noic acid or (S)-2-((tert-butoxycarbonyl)amino)-3,3-diphenylpropanoic acid, POCl3, pyridine, 0 °C; (ii) 4 M HCl, dioxane, RT; (b) (i) 30, 39, or 40, POCl3, pyridine, 0 °C; (ii) Pd/C, 1 atm H2, MeOH, RT, or PPh3, THF/H2O, reflux; (iii) 4 M HCl, dioxane, RT.
Table 2. Profile of Compounds 9i, 9k–9t, and Atazanavir.
| Rat PKc |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | X | R1 | R2 | R3 | R4 | R5 | R6 | Enzymea IC50 (nM) | Antiviralb IC95 (nM) | Cl (mL/min/kg) | %F |
| atazanavir | 0.04 | 17 | nd | nd | |||||||
| 9i | C | H | H | H | H | H | Moc | 27 | 120 | 36 | 19 |
| 9k | C | F | H | H | H | H | Moc | 12 | 95 | 36 | 37 |
| 9l | C | F | H | H | H | H | H | 3.1 | 230 | nd | nd |
| 9m | N | H | H | H | H | H | Moc | 14 | 330 | 81 | <5 |
| 9n | N | F | H | H | H | H | Moc | 3.5 | 94 | 11 | 15 |
| 9o | N | F | H | H | H | H | H | 9.9 | 150 | 13 | 59 |
| 9p | C | F | F | H | H | F | H | 6.4 | 75 | 45 | 30 |
| 9q | C | F | F | F | H | F | H | 8.2 | 61 | 22 | 14 |
| 9r | N | F | F | H | H | F | H | 2.4 | 66 | 16 | 37 |
| 9s | N | F | F | F | H | F | H | 2.4 | 130 | nd | nd |
| 9t (MK-8718) | N | F | Cl | F | H | F | H | 0.8 | 49 | 11 | 25 |
Assay for inhibition HIV protease as described in the Supporting Information (n = 2).
Assay for inhibition of viral infection as described in the Supporting Information (n = 2).
For compounds 9i–9n: 1 mpk IV (60% PEG 400/water), 5 mpk PO (0.5% methylcellulose). n = 2 rats. For compounds 9o–9t: 2 mpk IV (1:1 DMSO/PEG400), 10 mpk PO (10% tween 80/water). n = 2 rats.
A pyridyl P2 improved binding affinity (9i vs 9m) and a fluorophenyl P2 improved binding affinity and oral bioavailability (9i vs 9k). Combining these two findings afforded 9n, which offered a favorable combination of antiviral activity (IC95 = 94 nM) and rat clearance. Two primary amine derivatives were also prepared and offered improvements in binding affinity (9l vs 9k) and bioavailability (9o vs 9n).
At this point we turned our attention to optimizing the P1 and P3 phenyl substituents. It is evident from Figure 2 that the S1 and S3 pockets of the enzyme are not equivalent, even though they are occupied by identical P1 and P3 substituents. This inspired us to examine unsymmetrical P1 and P3 groups to further improve enzyme affinity. A series of designs were modeled and prioritized for synthesis. Synthesis of the requisite acid precursors was carried out as shown in Scheme 5. para-Fluorophenyl substituted acryloyl chloride 26(23) was treated with the anion of (S)-4-phenyloxazolidin-2-one to afford the desired acryloyloxazolidinone 27.24 Copper-catalyzed Grignard addition was used to introduce the desired P3 substituent in a highly stereoselective manner.25 Subsequent electrophilic azide transfer to the chiral enolate of 28 afforded the desired α-azidocarboximide 29. Hydrogen peroxide mediated hydrolysis afforded azido acid 30.26 In addition to a mono-fluorinated P3 substituent, we were also interested in a difluoro substituted P3. Unfortunately the 3,5-difluorophenyl Grignard reagent was insufficiently reactive to undergo copper catalyzed Grignard addition to 27. Accordingly, we used the route shown in Scheme 6 whereby the more reactive Grignard of the P1 substituent was utilized.
Scheme 5.

Reagents and conditions: (a) nBuLi, (S)-4-phenyloxazolidin-2-one, THF, −78 °C; (b) (3-Fluorophenyl)magnesium bromide, CuBr.SMe2, THF, −20 °C; (c) NaHMDS, trisyl azide, THF, −78 °C; (d) H2O2, LiOH, NaHCO3, THF/H2O, 0 °C.
Scheme 6.

Reagents and conditions: (a) nBuLi, (R)-4-phenyloxazolidin-2-one, THF, −78 °C; (b) (4-fluoro or chlorophenyl)magnesium bromide, CuBr·SMe2, THF, −20 °C; (c) H2O2, LiOH, NaHCO3, THF/H2O, 0 °C; (d) SOCl2, CH2Cl2, reflux; (d) nBuLi, (S)-4-phenyloxazolidin-2-one, THF, −78 °C; (d) NaHMDS, trisyl azide, THF, −78 °C; (d) H2O2, LiOH, NaHCO3, THF/H2O, 0 °C.
3,5-Difluoro substituted acryloyl chloride 31 was treated with the anion of (R)-4-phenyloxazolidin-2-one to afford the desired acrylooxazolidinone 32. Copper-catalyzed Grignard addition afforded acryloyloxazolidinones 33 and 34. Switching of the chiral auxiliaries afforded 35 and 36, which underwent electrophilic azide transfer to give α-azidocarboximides 37 and 38. Hydrogen peroxide mediated hydrolysis liberated azido acids 39 and 40. As depicted in Scheme 7 anilines 18 and 25 were coupled to azido acids 30, 39, and 40. Azide reduction, followed by Boc-removal, afforded the desired targets 9p–9t.
The relevant data for this set of compounds is shown in Table 2. The combination of para-fluorophenyl as the P1 substituent and a meta-fluorophenyl as the P3 substituent improved cell based antiviral activity (9p vs 9l). Adding a second fluorine to the P3 phenyl served to maintain cell based activity while lowering rat clearance (9p vs 9q). Incorporation of a P2 pyridyl substituent further improved rat clearance relative to the P2 phenyl, while maintaining cell based activity (9r vs 9p). Finally, replacing the P1 para-fluoro of 9r with a para-chloro substituent afforded 9t. This compound offered improvements in binding affinity, cell-based antiviral activity, rat clearance, and oral bioavailability. Additional preclinical evaluation of 9s–9t showed that 9t had the most favorable overall profile. Based on these desirable attributes 9t was designated as MK-8718 and was chosen for further studies to enable entry into the clinic for assessment of human pharmacokinetic properties.
In summary, a series of HIV protease inhibitors were designed containing a novel morpholine based aspartate binding group. Structure based optimization of the P1, P1′, P2, and P3 substituents was carried out to improve cell based antiviral activity and rat pharmacokinetic properties. This resulted in the identification of MK-8718, a potent HIV protease inhibitor with a favorable pharmacokinetic profile with potential for further development.
Acknowledgments
The authors wish to thank Dr. Anthony Roecker who assisted in proof-reading of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00135.
Synthetic experimental details for the synthesis of MK-8718 and 9b – 9j along with descriptions of primary biological assays. Experimental details for the remaining compounds can be found in the following patent: WO 2014043019 A1 (PDF)
Accession Codes
X-ray crystallographic data for 2, 8, 9a, and MK-8718 bound to HIV protease have been deposited in the RCSB protein data bank (pdb codes 5IVQ, 5IVR, 5IVS, and 5IVT).
The authors declare no competing financial interest.
Supplementary Material
References
- Erickson-Viitanen S.; Manfredi J.; Viitanen P.; Tribe D. E.; Tritch R.; Hutchison C. A. 3rd; Loeb D. D.; Swanstrom R. Cleavage of HIV-1 gag polyprotein synthesized in vitro: sequential cleavage by the viral protease. AIDS Res. Hum. Retroviruses 1989, 5 (6), 577–91. 10.1089/aid.1989.5.577. [DOI] [PubMed] [Google Scholar]
- Kohl N. E.; Emini E. A.; Schleif W. A.; Davis L. J.; Heimbach J. C.; Dixon R. A. F.; Scolnick E. M.; Sigal I. S. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. U. S. A. 1988, 85 (13), 4686–90. 10.1073/pnas.85.13.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensing A. M. J.; van Maarseveen N. M.; Nijhuis M. Fifteen years of HIV Protease Inhibitors: raising the barrier to resistance. Antiviral Res. 2010, 85 (1), 59–74. 10.1016/j.antiviral.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Osswald H. L.; Prato G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J. Med. Chem. 2016, 10.1021/acs.jmedchem.5b01697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensing A. M.; Calvez V.; Gunthard H. F.; Johnson V. A.; Paredes R.; Pillay D.; Shafer R. W.; Richman D. D. 2014 Update of the drug resistance mutations in HIV-1. Top Antivir Med. 2014, 22 (3), 642–50. [PMC free article] [PubMed] [Google Scholar]
- Kempf D. J. Progress in the discovery of orally bioavailable inhibitors of HIV protease. Perspect. Drug Discovery Des. 1995, 2 (3), 427–36. 10.1007/BF02172035. [DOI] [Google Scholar]
- Boffito M.; Maitland D.; Samarasinghe Y.; Pozniak A. The pharmacokinetics of HIV protease inhibitor combinations. Curr. Opin. Infect. Dis. 2005, 18 (1), 1–7. 10.1097/00001432-200502000-00002. [DOI] [PubMed] [Google Scholar]
- Miller M.; Jaskolski M.; Rao J. K. M.; Leis J.; Wlodawer A. Crystal structure of a retroviral protease proves relationship to aspartic protease family. Nature (London, U. K.) 1989, 337 (6207), 576–9. 10.1038/337576a0. [DOI] [PubMed] [Google Scholar]
- Swain A. L.; Miller M. M.; Green J.; Rich D. H.; Schneider J.; Kent S. B. H.; Wlodawer A. X-ray crystallographic structure of a complex between a synthetic protease of human immunodeficiency virus 1 and a substrate-based hydroxyethylamine inhibitor. Proc. Natl. Acad. Sci. U. S. A. 1990, 87 (22), 8805–9. 10.1073/pnas.87.22.8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti F.; Frecer V.; Miertus S. Inhibitors of HIV-Protease from Computational Design. A History of Theory and Synthesis Still to be Fully Appreciated. Curr. Pharm. Des. 2014, 20 (21), 3398–3411. 10.2174/13816128113199990628. [DOI] [PubMed] [Google Scholar]
- Jaskolski M.; Tomasselli A. G.; Sawyer T. K.; Staples D. G.; Heinrikson R. L.; Schneider J.; Kent S. B.; Wlodawer A. Structure at 2.5-A resolution of chemically synthesized human immunodeficiency virus type 1 protease complexed with a hydroxyethylene-based inhibitor. Biochemistry 1991, 30 (6), 1600–9. 10.1021/bi00220a023. [DOI] [PubMed] [Google Scholar]
- Wood J. M.; Maibaum J.; Rahuel J.; Gruetter M. G.; Cohen N.-C.; Rasetti V.; Rueger H.; Goeschke R.; Stutz S.; Fuhrer W.; Schilling W.; Rigollier P.; Yamaguchi Y.; Cumin F.; Baum H.-P.; Schnell C. R.; Herold P.; Mah R.; Jensen C.; O’Brien E.; Stanton A.; Bedigian M. P. Structure-based design of aliskiren, a novel orally effective renin inhibitor. Biochem. Biophys. Res. Commun. 2003, 308 (4), 698–705. 10.1016/S0006-291X(03)01451-7. [DOI] [PubMed] [Google Scholar]
- Coburn C. A.; Holloway M. K.; Vacca J. P.. Preparation of phenylsulfonylpyrrolidinylaminoethyl diphenylalaninamides as HIV protease inhibitors. WO2010138338A1, 2010.
- Cavalli A.; Poluzzi E.; De Ponti F.; Recanatini M. Toward a pharmacophore for drugs inducing the long QT syndrome: insights from a CoMFA study of HERG K+ channel blockers. J. Med. Chem. 2002, 45 (18), 3844–3853. 10.1021/jm0208875. [DOI] [PubMed] [Google Scholar]
- Hall H. K. Jr. Correlation of the base strengths of amines. J. Am. Chem. Soc. 1957, 79, 5441–4. 10.1021/ja01577a030. [DOI] [Google Scholar]
- Milot G.; Branchaud S.; Stranix B. R.. Lysme-based prodrugs of aspartyl protease inhibitors and processes for their preparation. WO2007062526A1, 2007.
- Moradei O. M.; Crane S.; McKay D. J.; Lebrun M.-E.; Truong V. L.. Preparation of amino acid amides containing a sulfonamide bond as HIV protease inhibitors. WO2012055034A1.
- Martinez M. M.; Hoppe D. Total Synthesis of (−)-α-Kainic Acid by (−)-Sparteine-Mediated Asymmetric Deprotonation-Cycloalkylation. Org. Lett. 2004, 6 (21), 3743–3746. 10.1021/ol0485666. [DOI] [PubMed] [Google Scholar]
- De Man A. P. A.; Sterrenburg J.-G.; Raaijmakers H. C. A.; Kaptein A.; Oubrie A.; Rewinkel J. B. M.; Jans C. G. J. M.; Wijkmans J. C. H. M.; Barf T. A.; Cooper A. B.; Kim R. M.; Boga S. B.; Zhu H. Y.; Gao X.; Yao X.; Anand R.; Wu H.; Liu S.; Yang C.; Alhassan A.-B.; Wang J.; Yu Y.; Liu J.; Vaccaro H. M.. Bicyclic heterocycles as BTK inhibitors and their preparation. WO2013010380A1.
- Carmosin R. J.; Carson J. R.; Pitis P. M.. Preparation of octahydropyrrolo-[3,4-c]carbazoles useful as analgesic agents. WO9965911A1.
- Gilbert J. C.; Weerasooriya U. Diazoethenes: their attempted synthesis from aldehydes and aromatic ketones by way of the Horner-Emmons modification of the Wittig reaction. A facile synthesis of alkynes. J. Org. Chem. 1982, 47 (10), 1837–45. 10.1021/jo00349a007. [DOI] [Google Scholar]
- Sonogashira K. Development of Pd-Cu catalyzed cross-coupling of terminal acetylenes with sp2-carbon halides. J. Organomet. Chem. 2002, 653 (1–2), 46–49. 10.1016/S0022-328X(02)01158-0. [DOI] [Google Scholar]
- Li H.; Wang C.; Sanchez T.; Tan Y.; Jiang C.; Neamati N.; Zhao G. Amide-containing diketoacids as HIV-1 integrase inhibitors: Synthesis, structure-activity relationship analysis, and biological activity. Bioorg. Med. Chem. 2009, 17 (7), 2913–2919. 10.1016/j.bmc.2009.01.077. [DOI] [PubMed] [Google Scholar]
- Eldrup A. B.; Soleymanzadeh F.; Taylor S. J.; Muegge I.; Farrow N. A.; Joseph D.; McKellop K.; Man C. C.; Kukulka A.; De Lombaert S. Structure-Based Optimization of Arylamides as Inhibitors of Soluble Epoxide Hydrolase. J. Med. Chem. 2009, 52 (19), 5880–5895. 10.1021/jm9005302. [DOI] [PubMed] [Google Scholar]
- Nicolas E.; Russell K. C.; Hruby V. J. Asymmetric 1,4-addition of organocuprates to chiral α,β-unsaturated N-acyl-4-phenyl-2-oxazolidinones: a new approach to the synthesis of chiral β-branched carboxylic acids. J. Org. Chem. 1993, 58 (3), 766–70. 10.1021/jo00055a039. [DOI] [Google Scholar]
- Evans D. A.; Britton T. C. Electrophilic azide transfer to chiral enolates. A general approach to the asymmetric synthesis of α-amino acids. J. Am. Chem. Soc. 1987, 109 (22), 6881–3. 10.1021/ja00256a069. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.