

Abstract
Chemotherapy for colorectal cancer is currently offered to patients based on the stage of their cancer, and there is evidence to show an overall survival benefit with 5-fluorouracil-based (5-FU) therapy for patients with lymph node metastasis who receive it. The pathogenesis of colorectal cancer involves genomic instability, with about 15% of tumors demonstrating a form of genomic instability called high-frequency microsatellite instability (MSI-H) and due to loss of DNA mismatch repair function, and the remainder of colorectal tumors lacking MSI-H with retained DNA mismatch repair function and called microsatellite stable (MSS), with a large proportion of these tumors demonstrating another form of genomic instability called chromosomal instability. There is now evidence to show that the form of genomic instability that is present in a patient’s colorectal cancer may predict a survival benefit from 5-FU. In particular, patients whose colorectal tumors have MSI-H do not gain a survival benefit with 5-FU as compared to patients with MSS tumors. In vitro evidence supports these findings, as MSI-H colon cancer cell lines are more resistant to 5-FU compared to MSS cell lines. More specifically, components of the DNA mismatch repair system have been shown to recognize and bind to 5-FU that becomes incorporated into DNA and which could be a trigger to induce cell death. The binding and subsequent cell death events would be absent in colorectal tumors with MSI-H, which have lost intact DNA mismatch repair function. These findings suggest that: (a) tumor cytotoxicity of 5-FU is mediated by DNA mechanisms in addition to well-known RNA mechanisms, and (b) patients whose tumors demonstrate MSI-H may not benefit from 5-FU therapy. Future studies should include a better understanding of the cellular mechanisms of the DNA recognition of 5-FU, multi-centered prospective trials investigating the survival benefit of 5-FU based on genomic instability, and the investigation of alternative chemotherapeutic regimens for patients with MSI-H tumors to improve survival.
Keywords: DNA mismatch repair, microsatellite instability, colorectal cancer, chemotherapy, treatment
1. Introduction
Approximately 147,000 cases will be diagnosed, and 57,000 individuals will die of colorectal cancer (CRC) in the US in 2005, ranking it second behind lung cancer as the deadliest cancer [1]. The most important prognostic indicator has traditionally been the stage of the tumor at diagnosis, with depth of invasion being the most critical factor [2]. However, there is considerable variation in the behavior of tumors even within the same pathologic stage, indicating that the molecular make-up of the tumor may be a more reliable predictor of its natural history and perhaps its biological response to chemotherapy. Among the most studied molecular markers correlated with survival in colorectal cancer is microsatellite instability (MSI). The finding of high frequency MSI (MSI-H) in CRC has been associated with a favorable patient prognosis when compared to patients with microsatellite stable (MSS) tumors [3,4], but paradoxically a poor response to 5-fluorouracil (5-FU)-based chemotherapy [5–7]. Here, we review the basic and clinical evidence for differences in chemotherapy responses for patients with MSI-H colorectal tumors, and the implications for future treatment of these patients.
2. Adjuvant chemotherapy with 5-fluorouracil
Treatment for advanced stage colorectal cancer includes surgical therapy and adjuvant chemotherapy. Most patients with advanced colorectal cancer that undergo surgical treatment alone will develop a recurrence of their disease. Adjuvant chemotherapy regimens have been tried with success to improve the outcome of patients with apparent residual disease after primary surgical resection [8,9]. Almost all adjuvant chemotherapy for advanced stage colorectal cancer involves the agent 5-fluorouracil (5-FU), typically in combination with levamisole or leukovorin [9]. Thus, 5-FU, introduced more than 40 years ago, remains the mainstay of chemotherapeutic treatment of colorectal cancer. In particular, 5-FU-based chemotherapy improves survival in patients with stage III colon cancer [10–12], and in patients with stage II and III rectal cancer [9]. Because colorectal cancer is so common, nearly every new chemotherapeutic agent has been tried. Other agents developed for colorectal cancer treatment, including irinotecan (camptothecin) and oxaliplatin, are not any more effective than 5-FU [13].
While chemotherapy with 5-FU is the best treatment option and has become the standard of care in advanced stage colorectal cancer, individual patient tumor response rates are still overall poor. In a meta-analysis of randomized trials, continuous infusion of 5-FU had a tumor response rate of 22% [14]. In the seven randomized trials included in this meta-analysis, the tumor response rates ranged between 20 and 30%. There is no current method to determine which patient will have a tumor response to 5-FU. There are likely several reasons why some colorectal tumors do not respond to treatment with 5-FU. Importantly, the loss of DNA mismatch repair within the patient’s tumor is associated with no survival benefit from 5-FU.
3. Loss of DNA mismatch repair and microsatellite instability
Colorectal cancers develop as a consequence of genomic instability. That is, during the transformation process from normal to tumor, the DNA of a colonocyte is damaged in a particular pattern that drives tumorigenesis. Approximately 15% of sporadic colorectal tumors display MSI-H, mostly as a result of epigenetic silencing by hypermethylation of the hMLH1 gene [15,16]. The remaining 85% of sporadic colorectal cancer can be termed microsatellite stable [MSS], with most tumors in this category following a molecular pathway characterized by chromosomal instability whereby sequential mutations and allelic loss of key regulatory genes culminate in the transformation of an adenoma into cancer [17,18].
The phenotype of MSI (particularly MSI-High, defined as ≥ 30% of DNA microsatellite markers mutated within a colorectal tumor) [19] is caused by a defective DNA MMR system. DNA MMR is an evolutionarily conserved system capable of repairing mispaired nucleotides and short mismatched insertion-deletion loops (IDLs) in DNA, presumably as a result of insertion mistakes made by DNA polymerase during replication. DNA MMR replaces the mispair on the newly synthesized daughter strand [20,21]. The mispair or IDL is recognized and bound by either hMutSα (a heterodimer of hMSH2 and hMSH6) or hMutSβ (a heterodimer of hMSH2 and hMSH3) in a highly orchestrated manner (Fig. 1). hMutSα is capable of binding to single base pair mismatches and single IDLs whereas hMutSα can only bind IDLs [22,23]. Recruitment of the hMutLα complex (heterodimer of hMLH1 and hPMS2) by hMutSα or hMutSβ subsequently targets the DNA to complete the repair process. Defects in the MMR genes hMSH2, hMLH1, and hMSH6 in humans have been clearly linked to Lynch syndrome (also known as hereditary nonpolyposis colorectal cancer, or HNPCC), an autosomal dominant condition in which one of the MMR genes is mutated in germline cells [24–30].
Fig. 1.
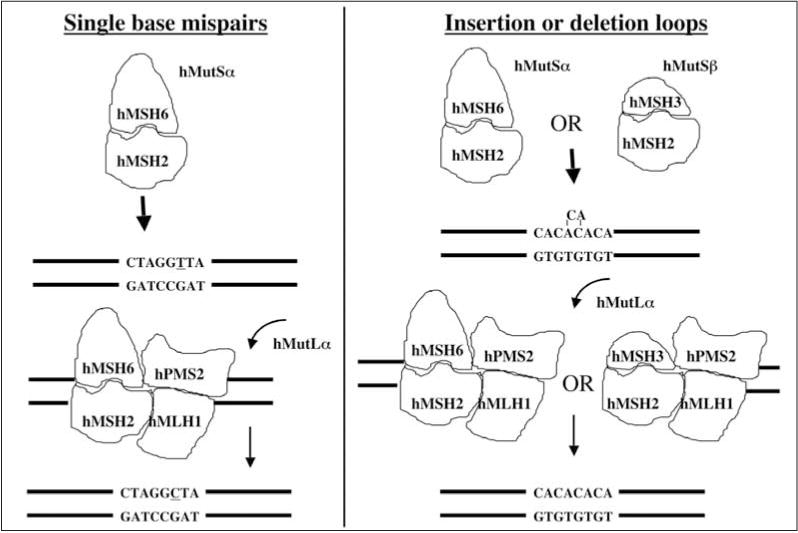
Schematic for DNA mismatch repair. The heterodimer hMutSα recognizes and binds single mispairs and small insertion-deletion loops (IDL), whereas hMutSβ only binds IDLs. The subsequent events to effect excision and re-synthesis of the DNA are identical between the two heterodimers. The heterodimer hMutSα has been shown to recognize 5-FU incorporated into DNA.
It has been noted that failure to correct post-replication mismatches results in a 10- to 100-fold increase in mutation rate [31,32]. Thus, absence of MMR results in a hypermutable environment with recognizable frameshift mutations in predominantly mono-, di-, and tri-nucleotide microsatellite tracts, as well as within genes that contain such sequences in their coding regions, such as TGFBR2, ACVR2, and BAX [33–38]. Mutation within these genes and the subsequent loss of protein function are thought to drive the pathogenesis of MSI-H colorectal tumors [39,40].
4. A more favorable prognosis for patients with MSI-H colorectal cancer (in the absence of adjuvant chemotherapy)
Clinically, tumors with MSI-H are correlated with the tumor’s location in the proximal colon, have a histological poor grade, and are inversely related to allelic loss [41–43]. Microsatellite unstable tumors also tend to be diploid, mucinous with signet cell features, and have a surrounding lymphoid reaction. This phenotype appears to be the result of rapid neoplastic progression in Lynch syndrome and sporadic tumors that exhibit MSI [43–45].
Patients with MSI-H tumors have been associated with a more favorable prognosis compared to patients with MSI-L or MSS tumors [3,4]. One study which examined the clinical outcome in 607 young patients (< 50 years of age) with colorectal cancer found that MSI-H tumors had a significant survival advantage (hazard ratio of 0.42, p < 0.0001) over MSS tumors [3]. In addition, MSI-H tumors had a significantly lower likelihood of metastasizing to regional lymph nodes (odds ratio 0.33, P < 0.001) and distant organs (odds ratio 0.49, P = 0.02). Indeed, the favorable prognosis of MSI-H CRC have been documented by several other studies [46,47], and a recent systematic review of pooled studies confirmed the relationship between tumor MSI-H and patient survival, with a combined hazard ratio for overall survival associated with MSI-H at 0.65 (95% CI 0.59 to 0.71) [4]. The biologic basis for the tumor MSI-H and patient survival association has not been established. It has been postulated that the lymphocyte infiltration seen in MSI-H tumors is a reflection of an enhanced host immune response invoked by the presence of numerous mutated products [44,48]. Several studies have correlated a lymphocytic infiltration in CRC with increased survival [49,50]. Others have also suggested that the enormous mutational burden resulting from loss of MMR activity may be self-limiting in that essential cell functions may be hindered [51]. However, these explanations remain speculative at this time, and the molecular mechanism underlying the relatively indolent behavior of MSI-H tumors remains elusive. The fact that patients with MSI-H tumors have a better prognosis over patients with MSS tumors originally confounded some studies examining 5-FU chemotherapy, with most lacking the appropriate control group for comparative purposes.
5. Response to 5-fluorouraci-based chemotherapy in patients with MSI-H tumors
Patients with tumors that exhibit high-frequency MSI (MSI-H) are typically treated the same as those without the MSI-H phenotype, with the stage of the tumor being the major determinant of which patients will benefit from adjuvant chemotherapy. However, recent evidence indicates that the MSI status of the tumor may also be important.
Paradoxically, the favorable natural history of MSI-H CRC does not parallel its response to chemotherapy. Early reports appeared to indicate that 5-FU adjuvant chemotherapy appeared to be beneficial for patients with MSI-H CRC, but these studies were limited by small or non-randomized study population [52–55]. Recent studies powered by larger sample sizes and with appropriate control groups have demonstrated that patients with MSI-H tumors do not appear to derive any benefit from 5-FU-based adjuvant chemotherapy (Table 1), and there is evidence to suggest that chemotherapy may even be detrimental to patients with MSI-H CRC. In one study of 204 patients with sporadic stage II and III CRC, retrospective survival analysis failed to show a difference in survival among patients with MSI-H tumors irrespective of whether 5-FU was administered (p = 0.52) [5]. In contrast, there was a significant survival advantage with 5-FU chemotherapy among patients with MSI-L and MSS tumors (p = 0.0478). Similarly, another study of 505 stage II and III colorectal cancer patients failed to show a survival benefit with 5-FU in patients with MSI-H colorectal cancers (p = 0.4), while patients with MMR-competent MSI-L and MSS tumors benefited with improved survival with 5-FU (p = 0.0001) [7]. Another study examined 570 tissue specimens from patients with stage II and III CRC who had been enrolled in randomized trials of 5-FU-based chemotherapy [6]. Among 287 patients not receiving chemotherapy, the 5-year survival rate was higher among patients with MSI-H tumors compared to those with MSH-L or MSS tumors (88.0% vs. 68.4%, p = 0.004), a finding consistent with the more favorable prognosis of MSI-H tumors. However, among patients who received chemotherapy, those with MSI-H tumors were associated with a slightly lower 5-year survival rate compared to the MSH-L or MSS tumors (70.7% vs. 75.5%, p = 0.66). Furthermore, among patients with MSI-H tumors, chemotherapy with 5-FU was associated with a worse outcome (hazard ratio for death, 2.14, p = 0.11). In addition to the lack of 5-FU survival benefit in patients with sporadic MSI-H tumors, stage III patients with HNPCC do not demonstrate a 5-year survival benefit with 5-FU over untreated patients [56] (Table 1). Collectively, these results suggest that 5-FU-based chemotherapy does not prolong survival in patients with MSI-H CRC and may even be detrimental. On a molecular level, these findings suggest that 5-FU-mediated cytotoxicity may be dependent on intact DNA MMR gene function.
Table 1.
Studies of 5-FU treatment, survival, and MSI status
| Published Study (year) | Total Number of Patients | Number of 5-FU treated patients | Stage assessed for survival | MSI-H survival benefit with 5-FU? | MSS/MSI-L survival benefit with 5-FU? |
|---|---|---|---|---|---|
| Ribic et al. (2003) | 570 | 283 | II–III | NO | YES |
| Carethers et al. (2004) | 204 | 66 | II–III | NO | YES |
| deVos et al. (2004)* | 92 | 28 | III | NO | N/A |
| Jover et al. (2006)** | 754 | 260 | II–III | NO | YES |
HNPCC patients only. The other studies contained mostly sporadic patients.
Only 505 patients were stage II–III. Seven patients had a germline MMR gene mutation.
6. DNA mismatch repair: A mediator of chemotoxicity
In addition to its role in recognizing and directing repair of polymerase mistakes in DNA, the human MMR system is also capable of recognizing certain DNA adducts caused by exogenous alkylation damage [57–59]. The SN1 methylating agent N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) creates O6-methylguanine as its principal adduct. This adduct is recognized by the MMR system, and results in a G2/M cell cycle arrest and cell death [57]. Similarly, when 6-thioguanine (6-TG) is incorporated into the DNA of MMR-proficient cells, a G2/M cell cycle arrest is induced [58]. Both the MNNG adduct and 6-TG are thought to be recognized by the MMR system because of mispairing with T (or C) with the altered nucleotide on the newly synthesized DNA strand. Cell cycle arrest at the G2/M checkpoint prevents mutations (namely G to A transitions) in daughter cells. Adducts formed by cisplatin and carboplatin intercalate and distort DNA, which are also recognized by the MMR proteins [60–62] (Table 2). However, substituted amine analogues of cisplatin, namely oxaliplatin, tetraplatin, transplatin, JM335, and JM216, form different types of adducts and are not apparently recognized by the MMR system [60]. Similarly, 8-azaguanine (8-AG) is not recognized in MMR-intact cells [58]. Irinotecan, the topoisomerase I inhibitor, seems to induce its toxicity independent of the DNA MMR system [63–65]. Indeed, several compounds that induce toxicity upon colorectal cancer cells may work independently of DNA MMR, and some of these drugs will need further evaluation to exploit as independent treatments for patients with MSI-H colorectal cancer (Table 2).
Table 2.
Some chemotherapeutic compounds and relationship with MMR substrate recognition
| Compound | Does MMR recognition mediate toxicity? | References |
|---|---|---|
| N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) | YES | 57, 84 |
| 6-thioguanine | YES | 58 |
| cisplatin, carboplatin | YES | 60–62 |
| oxaliplatin, tetraplatin, transplatin, JM335, and JM216 | NO | 60 |
| 8-azaguanine | NO | 58 |
| 5-fluorouracil | YES | 68 |
| doxorubicin, epirubicin, mitoxantrone | YES | 64 |
| bleomycin | NO | 85 |
| dacarbazine | YES | 86 |
| irinotecan/camptothecin, topotecan | NO | 63–65 |
| docetaxel, paclitaxel | NO | 64 |
| mitomycin C | NO | 87 |
7. DNA mismatch repair and 5-FU recognition
5-FU is a fluoropyrimidine that is incorporated into RNA (mRNA, rRNA, and tRNA), is an inhibitor of thymidylate synthetase (which catalyzes the conversion of dUMP to dTMP), and has some incorporation into DNA [66–68] (Fig. 2). The cytotoxic effects of 5-FU have traditionally been attributed to its ability to inhibit thymidylate synthetase and its interference with RNA processing [66]. Under normal conditions as shown in Fig. 2, dUTPase prevents the incorporation of dUTP and FdUTP into DNA by dephosphorylating the nucleotides to dUMP and FdUMP, respectively [69,70] (enzyme #13, Fig. 2). However, since 5-FU can inhibit thymidylate synthetase, accumulation of dUMP and FdUMP occurs, which exhausts the ability of dUTPase to metabolize dUTP and FdUTP. As dUTP and FdUTP levels rise and those of TTP fall, dUTP and FdUTP replace TTP as substrates for DNA polymerases, and are incorporated into DNA [66] (enzyme #11, Fig. 2). Nonetheless, uracyl-N-glycosylase, an enzyme that removes uracil bases from DNA after the spontaneous deamination of deoxycytidine, will typically remove the incorporated uracil bases (enzyme #14, Fig. 2). In spite of this, TTP is not available and the DNA strand will be repaired using dUTP or FdUTP as a substrate. 5-FU incorporation into DNA had been previously observed [67,71,72], but the consequences of this phenomenon was not known until recently as there was no reported correlation between 5-FU incorporation into DNA and cytotoxicity [71–73]. The first demonstration of MMR-mediated 5-FU toxicity came from in vitro studies demonstrating that cell lines with intact MMR function were selectively killed with 5-FU treatment, while MSI-H cells were resistant to 5-FU [68]. These results were corroborated by a subsequent study demonstrating that colon cancer cells with biallelic hypermethylation of hMLH1 lost their resistance to 5-FU in the presence of a demethylating agent [74]. More recent studies have shown that hMutSα can recognize and bind DNA containing 5-FU [75,76], indicating that resistance to 5-FU in MMR-deficient cells may be attributable to the direct interaction between 5-FU and MMR proteins. Indeed, hMutSα, the heterodimer of hMSH2 and hMSH6, shows greater affinity for 5-FU incorporated into DNA than its natural substrate, a typical base mispair [75].
Fig. 2.
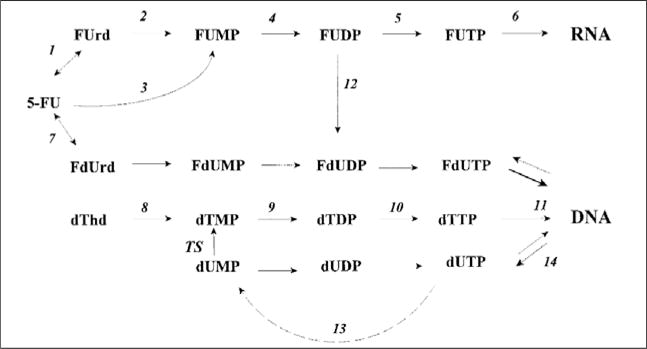
Intracellular metabolism of 5-FU. The numbers denote the following enzymes: (1) uridine phosphorylase, (2) uridine kinase, (3) orotate phsophoribosyltransferase, (4) and (9) pyrimidine kinase, (5) and (10) pyrimidine diphosphate kinase, (6) RNA polymerase, (7) thymidine phosphorylase, (8) thymidine kinase, (11) DNA polymerase, 12 ribonucleotide reductase, (13) deoxyuridine triphosphate pyrophosphatase, (14) uracil-DNA-glycosylase. TS = thymidylate synthase. 5-FU can be incorporated into RNA, and DNA due to its ability to block TS and exhaust the availability of dTTP, leaving only dUTP or FdUTP available for new DNA synthesis.
It is not clear how the MMR system recognizes 5-FU incorporated into DNA. Unlike bulky intercalating adducts such as cisplatin, or O6-methylguanine adducts formed by treatment with MNNG, 5-FU incorporation may not physically distort the DNA double helix because it does not interfere with interstrand hydrogen bonding (Fig. 3). How 5-FU might be recognized by DNA mismatch repair is speculation, but it may involve the highly charged fluorine atom that may deform the DNA double strand enough to be recognized by MMR proteins.
Fig. 3.
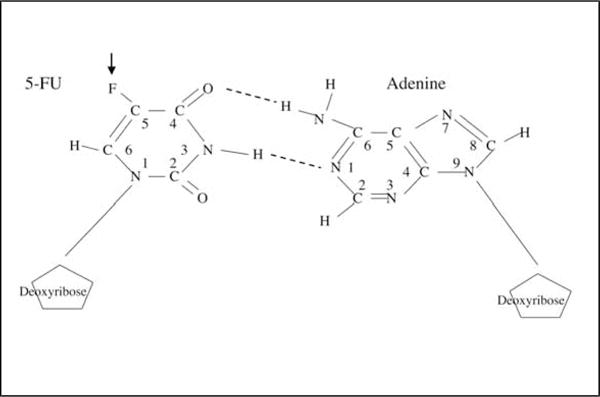
5-Fluorodeoxyuracil pairing with adenine in DNA. The fluorine molecule is at the 5 position (arrow), and does not interfere with hydrogen binding with adenine, but is recognized by DNA mismatch repair proteins.
The downstream signaling pathways triggered by MMR recognition of modified DNA have been partially elucidated for some chemotherapeutic agents but remains undefined for 5-FU. For example, introduction of O6-MeG into DNA results in a G2/M cell cycle arrest that is dependent on an intact MMR system and involves the ATM and Rad3-related (ATR) and CHK1 kinases [77]. p53, another downstream target of ATR [78], has also been shown to become phosphorylated during MMR-mediated repair of DNA damaged by O6-MeG [79]. Apoptosis induced by hMutSα recognition of O6-MeG lesions appears to involve mitochondrial signaling that activate both caspase-dependent and caspase-independent pathways [80]. The cellular response invoked by MMR in response to cisplatin involves activation of c-ABL, which promotes apoptosis through regulation of P73, a P53-related protein [81,82]. Much less is known about the signaling cascade induced by MMR recognition of 5-FU. Restoration of MMR function in MMR-deficient colon cancer cells results in restoration of G2/M cell cycle arrest after treatment with 5-FU [76,83], but it is not known whether the same pathways delineated for other agents are involved. Interestingly, apoptosis was noted to occur at low levels in both MMR-proficient and MMR-deficient cells after exposure to 5-FU and irradiation [83], which likely may represent the non-MMR-mediated toxicities of 5-FU.
8. Conclusion
In summary, MSI represents a promising disease marker for CRC because of the favorable prognosis associated with MSI-H CRC. However, recent data suggests that it may also serve as a reliable marker for response to chemotherapy. An intact DNA MMR system appears to be necessary to mediate the cytotoxicity of several chemotherapeutic agents, including 5-FU. Both cell cycle arrest and cell death following exposure to 5-FU have been shown to be dependent on MMR proteins, and recognition of 5-FU incorporation into DNA by the MMR proteins appears to be a critical step in this process. Although the cellular mechanisms for the DNA recognition of 5-FU by MMR need to be elucidated, the discrimination of MMR recognition of incorporated 5-FU has important clinical implications in the treatment of CRC, since several studies have now shown that patients with MSI-H tumors do not derive any benefit from and may even be harmed by 5-FU-based adjuvant chemotherapy. There is currently an impetus for tailoring chemotherapeutic regimens based on the molecular profile of the tumor, and it is conceivable that MSI status may be a contraindication to 5-FU treatment in the future. However, prospective, randomized, and well-controlled studies are needed for absolute confirmation before any recommendations can be implemented. Furthermore, additional agents that might be beneficial towards patient survival regardless of the MSI status of the tumor need to be furthered explored.
Abbreviations
- 5-FU
5-fluorouracil
- MMR
DNA mismatch repair
- MSI-H
high-frequency microsatellite instability
- MSI-L
low frequency microsatellite instability
- MSS
microsatellite stable
- CRC
colorectal cancer
- TS
thymidylate synthase
- O6-MeG
O6-methylguanine
- 6-TG
6-thioguanine
- 8-AG
8-azoguanine
Footnotes
Supported by the US Public Health Service [CA90231 and DK067287], the California Department of Health Services Cancer Research Program, and the University of California Cancer Research Coordinating Committee.
References
- 1.Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, Feuer EJ, Thun MJ. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.O’Connell JB, Maggard MA, Ko CY. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. J Natl Cancer Inst. 2004;96:1420–1425. doi: 10.1093/jnci/djh275. [DOI] [PubMed] [Google Scholar]
- 3.Gryfe R, Kim H, Hsieh ET, Aronson MD, Holowaty EJ, Bull SB, Redston M, Gallinger S. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med. 2000;342:69–77. doi: 10.1056/NEJM200001133420201. [DOI] [PubMed] [Google Scholar]
- 4.Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–618. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 5.Carethers JM, Smith EJ, Behling CA, Nguyen L, Tajima A, Doctolero RT, Cabrera BL, Goel A, Arnold CA, Miyai K, Boland ANCCR. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology. 2004;126:394–401. doi: 10.1053/j.gastro.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 6.Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, Tu D, Redston M, Gallinger S. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jover R, Zapater P, Castells A, Llor X, Andreu M, Cubiella J, Pinol V, Xicola R, Bujanda L, Rene JM, Clofent J, Bessa X, Morillas JD, Nicolas-Perez D, Paya A, Alenda C. Mismatch repair status in the prediction of benefit from adjuvant fluorouracil chemotherapy in colorectal cancer. Gut. 2006 doi: 10.1136/gut.2005.073015. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boland CR. Diagnosis and management of primary and metastatic colorectal cancer. Seminars in Gastrointestinal Disease. 1992;3:33–45. [Google Scholar]
- 9.Boland CR, Sinicrope FA, Brenner DE, Carethers JM. Colorectal cancer prevention and treatment. Gastroenterology. 2000;118:S115–S128. doi: 10.1016/s0016-5085(00)70010-2. [DOI] [PubMed] [Google Scholar]
- 10.Laurie JA, Moertel CG, Fleming TR, Wieand HS, Leigh JE, Rubin J, McCormack GW, Gerstner JB, Krook JE, Malliard J, et al. Surgical adjuvant therapy of large-bowel carcinoma: an evaluation of levamisole and the combination of levamisole and fluorouracil. The North Central Cancer Treatment Group and the Mayo Clinic. J Clin Oncol. 1989;7:1447–1456. doi: 10.1200/JCO.1989.7.10.1447. [DOI] [PubMed] [Google Scholar]
- 11.Moertel CG, Fleming TR, Macdonald JS, Haller DG, Laurie JA, Goodman PJ, Ungerleider JS, Emerson WA, Tormey DC, Glick JH, et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N Engl J Med. 1990;322:352–358. doi: 10.1056/NEJM199002083220602. [DOI] [PubMed] [Google Scholar]
- 12.Moertel CG, Fleming TR, Macdonald JS, Haller DG, Laurie JA, Tangen CM, Ungerleider JS, Emerson WA, Tormey DC, Glick JH, Veeder MH, Mailliard JA. Fluorouracil plus levamisole as effective adjuvant therapy after resection of stage III colon carcinoma: a final report. Ann Intern Med. 1995;122:321–326. doi: 10.7326/0003-4819-122-5-199503010-00001. [DOI] [PubMed] [Google Scholar]
- 13.Rea DW, Nortier JW, Ten Bokkel Huinink WW, Falk S, Richel DJ, Maughan T, Groenewegen G, Smit JM, Steven N, Bakker JM, Semiond D, Kerr DJ, Punt CJ. A phase I/II and pharmacokinetic study of irinotecan in combination with capecitabine as first-line therapy for advanced colorectal cancer. Ann Oncol. 2005;16:1123–1132. doi: 10.1093/annonc/mdi227. [DOI] [PubMed] [Google Scholar]
- 14.Meta-analysis Group in Cancer. Efficacy of intravenous continuous infusion of fluorouracil compared with bolus administration in advance colorectal cancer. J Clin Oncology. 1998;16:301–308. doi: 10.1200/JCO.1998.16.1.301. [DOI] [PubMed] [Google Scholar]
- 15.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, Kane MF, Kolodner RD, Vogelstein B, Kunkel TA, Baylin SB. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, Li GM, Drummond J, Modrich PL, Sedwick WD, Markowitz SD. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci USA. 1998;95:8698–8702. doi: 10.1073/pnas.95.15.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 18.Goel A, Arnold CN, Niedzwiecki D, Chang DK, Ricciardiello L, Carethers JM, Dowell JM, Wasserman L, Compton C, Mayer RJ, Bertagnolli MM, Boland CR. Characterization of sporadic colon cancer by patterns of genomic instability. Cancer Res. 2003;63:1608–1614. [PubMed] [Google Scholar]
- 19.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 20.Hare JT, Taylor JH. One role for DNA methylation in vertebrate cells is strand discrimination in mismatch repair. Proc Natl Acad Sci USA. 1985;82:7350–7354. doi: 10.1073/pnas.82.21.7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas DC, Roberts JD, Kunkel TA. Heteroduplex repair in extracts of human HeLa cells. J Biol Chem. 1990;266:3744–3751. [PubMed] [Google Scholar]
- 22.Acharya S, Wilson T, Gradia S, Kane MF, Guerrette S, Marsischky GT, Kolodner R, Fishel R. hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc Natl Acad Sci USA. 1996;93:13629–13634. doi: 10.1073/pnas.93.24.13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Genschel J, Littman SJ, Drummond JT, Modrich P. Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J Biol Chem. 1998;273:19895–19901. doi: 10.1074/jbc.273.31.19895. [DOI] [PubMed] [Google Scholar]
- 24.Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, Peltomaki P, Sistonen P, Aaltonen LA, Nystrom-Lahti M, Guan XY, Zhang J, Meltzer PS, Yu JW, Kao FT, Chen DJ, Cerosaletti KM, Fournier REK, Todd S, Lewis T, Leach RJ, Naylor SL, Weissenbach J, Mecklin JP, Jarvinen H, Petersen GM, Hamilton SR, Green J, Jass J, Watson P, Lynch HT, Trent JM, de la Chapelle A, Kinzler KW, Volgelstein B. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- 25.Fishel R, Lescoe MK, Rao MRS, Copeland NG, Jenkins NA, Garber J, Kane MR. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 26.Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM, Adams MD, Venter JC, Hamilton SR, Petersen GM, Watson P, Lynch HT, Peltomaki P, Mecklin JP, de la Chapelle A, Kinzler KW, Volgelstein B. Mutation of a mutL homolog in hereditary colon cancer. Science. 1994;263:1625–1629. doi: 10.1126/science.8128251. [DOI] [PubMed] [Google Scholar]
- 27.Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, Kane M, Earabino C, Lipford J, Lindblom A, Tannergard P, Bollag RJ, Godwin AR, Ward D, Nordenskjold M, Fishel R, Kolodner R, Liskay RM. Mutation in the DNA mismatch repair homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258–261. doi: 10.1038/368258a0. [DOI] [PubMed] [Google Scholar]
- 28.Akiyama Y, Sato H, Yamada T. Germ-line mutation of the hMSH6/GTBP gene in an atypical HNPCC kindred. Cancer Res. 1997;57:3920–3923. [PubMed] [Google Scholar]
- 29.Wijnen J, de Leeuw W, Vasen H, van der Klift H, Moller P, Stormorken A, Meijers-Heijboer H, Lindhout D, Menko F, Vossen S, Moslein G, Tops C, Brocker-Vriends A, Wu Y, Hofstra R, Sijmons R, Cornelisse C, Morreau H, Fodde R. Familial endometrial cancer in female carriers of MSH6 germline mutations. Nature Genet. 1999;23:142–144. doi: 10.1038/13773. [DOI] [PubMed] [Google Scholar]
- 30.Kolodner RD, Tytell JD, Schmeits J, Kane MF, Das Gupta R, Weger J, Wahlberg S, Fox EZ, Peel D, Ziogas A, Garber JE, Syngal S, Anton-Culver H, Li FP. Germ-line msh6 mutations in colorectal cancer families. Cancer Res. 1999;59:5068–5074. [PubMed] [Google Scholar]
- 31.Branch P, Hampson R, Karran P. DNA mismatch binding defects, DNA damage tolerance, and mutator phenotypes in human colorectal carcinoma cell lines. Cancer Res. 1995;55:2304–2309. [PubMed] [Google Scholar]
- 32.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Ann Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 33.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B, Brattain M, Willson JKV. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 34.Malkhosyan S, Rampino N, Yamamoto H, Perucho M. Frameshift mutator mutations. Nature. 1996;382:499–500. doi: 10.1038/382499a0. [DOI] [PubMed] [Google Scholar]
- 35.Mori Y, Yin J, Rashid A, Leggett BA, Young J, Simms L, Kuehl PM, Langenberg P, Meltzer SJ, Stine OC. Instabilotyping: comprehensive identification of frameshift mutations caused by coding region microsatellite instability. Cancer Res. 2001;61:6046–6049. [PubMed] [Google Scholar]
- 36.Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, Perucho M. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;75:967–969. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- 37.Jung B, Doctolero RT, Tajima A, Nguyen AK, Keku T, Sandler RS, Carethers JM. Loss of activin receptor type 2 protein expression in microsatellite unstable colon cancers. Gastroenterology. 2004;126:654–659. doi: 10.1053/j.gastro.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 38.Carethers JM, Pham T-TT. Mutations of transforming growth factor ?1 type II receptor, BAX, and insulin-like growth factor II receptor genes in microsatellite unstable cell lines. In Vivo. 2000;14:13–20. [PubMed] [Google Scholar]
- 39.Grady WM, Rajput A, Myeroff L, Liu DF, Kwon K, Willis J, Markowitz S. Mutation of the type II transforming growth factor-beta receptor is coincident with the transformation of human colon adenomas to malignant carcinomas. Cancer Res. 1998;58:3101–3104. [PubMed] [Google Scholar]
- 40.Jung B, Smith EJ, Doctolero RT, Gervaz P, Alonso JC, Keku T, Sandler RS, Carethers JM. Influence of target gene mutation on survival, stage, and histology in sporadic microsatellite unstable colon cancers. International Journal of Cancer. 2006 doi: 10.1002/ijc.21710. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ionov Y, Peinado MA, Malkhosyan S, et al. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–561. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- 42.Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993;260:816–819. doi: 10.1126/science.8484122. [DOI] [PubMed] [Google Scholar]
- 43.Aaltonen LA, Peltomaki P, Leach FS, Sistonen P, Pylkkanen L, Mecklin JP, Jarvinen H, Powell SM, Jen J, Hamilton SR, Petersen GM, Kinzler KW, Vogelstein B, de la Chapelle A. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812–816. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 44.Smyrk TC, Watson P, Kaul K, Lynch HT. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer. 2001;91:2417–2422. [PubMed] [Google Scholar]
- 45.Tsao JL, Yatabe Y, Salovaara R, et al. Genetic reconstruction of individual colorectal tumor histories. Proc Natl Acad Sci USA. 2000;97:1236–1241. doi: 10.1073/pnas.97.3.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guidoboni M, Gafa R, Viel A, Doglioni C, Russo A, Santini A, Del Tin L, Macri E, Lanza G, Boiocchi M, Dolcetti R. Microsatellite instability and high content of activated cytotoxic lymphocytes identify colon cancer patients with a favorable prognosis. Am J Pathol. 2001;159:297–304. doi: 10.1016/S0002-9440(10)61695-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Samowitz WS, Curtin K, Ma KN, Schaffer D, Coleman LW, Leppert M, Slattery ML. Microsatellite instability in sporadic colon cancer is associated with an improved prognosis at the population level. Cancer Epidemiol Biomarkers Prev. 2001;10:917–923. [PubMed] [Google Scholar]
- 48.Bodmer W, Bishop T, Karran P. Genetic steps in colorectal cancer. Nat Genet. 1994;6:217–219. doi: 10.1038/ng0394-217. [DOI] [PubMed] [Google Scholar]
- 49.Jass JR. Lymphocytic infiltration and survival in rectal cancer. J Clin Pathol. 1986;39:585–589. doi: 10.1136/jcp.39.6.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Canna K, McArdle PA, McMillan DC, McNicol AM, Smith GW, McKee RF, McArdle CS. The relationship between tumour T-lymphocyte infiltration, the systemic inflammatory response and survival in patients undergoing curative resection for colorectal cancer. Br J Cancer. 2005;92:651–654. doi: 10.1038/sj.bjc.6602419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shibata D, Peinado MA, Ionov Y, Malkhosyan S, Perucho M. Genomic instability in repeated sequences is an early somatic event in colorectal tumorigenesis that persists after transformation. Nat Genet. 1994;6:273–281. doi: 10.1038/ng0394-273. [DOI] [PubMed] [Google Scholar]
- 52.Hemminki A, Mecklin JP, Jarvinen H, Aaltonen LA, Joensuu H. Microsatellite instability is a favorable prognostic indicator in patients with colorectal cancer receiving chemotherapy. Gastroenterology. 2000;119:921–928. doi: 10.1053/gast.2000.18161. [DOI] [PubMed] [Google Scholar]
- 53.Lukish JR, Muro K, DeNobile J, Katz R, Williams J, Cruess DF, Drucker W, Kirsch I, Hamilton SR. Prognostic significance of DNA replication errors in young patients with colorectal cancer. Ann Surg. 1998;227:51–56. doi: 10.1097/00000658-199801000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elsaleh H, Powell B, Soontrapornchai P, Joseph D, Goria F, Spry N, Iacopetta B. p53 gene mutation, microsatellite instability and adjuvant chemotherapy: impact on survival of 388 patients with Dukes’ C colon carcinoma. Oncology. 2000;58:52–59. doi: 10.1159/000012079. [DOI] [PubMed] [Google Scholar]
- 55.Elsaleh H, Joseph D, Grieu F, Zeps N, Spry N, Iacopetta B. Association of tumour site and sex with survival benefit from adjuvant chemotherapy in colorectal cancer. Lancet. 2000;355:1745–1750. doi: 10.1016/S0140-6736(00)02261-3. [DOI] [PubMed] [Google Scholar]
- 56.de Vos tot Nedervee Cappel WH, Neulenbeld HJ, Keibeuker JH, Nagengast FM, Menko FH, Griffioen G, Cats A, Morreau H, Gelderblom H, Vasen HF. Survival after adjuvant 5-FU treatment for stage III coon cancer in hereditary nonpolyposis colorectal cancer. In J Cancer. 2004;109:468–471. doi: 10.1002/ijc.11712. [DOI] [PubMed] [Google Scholar]
- 57.Carethers JM, Hawn MT, Chauhan DP, Luce MC, Marra G, Koi M, Boland CR. Competency in mismatch repair prohibits clonal expansion of cancer cells treated with N-methyl-N′-nitro-N-nitrosoguanidine. J Clin Invest. 1996;98:199–206. doi: 10.1172/JCI118767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hawn MT, Umar A, Carethers JM, Marra G, Kunkel TA, Boland CR, Koi M. Evidence for a connection between the mismatch repair system and the G2 cell cycle checkpoint. Cancer Res. 1995;55:3721–3725. [PubMed] [Google Scholar]
- 59.Aebi S, Kurdi-Haidar B, Gordon R, Cenni B, Zheng H, Fink D, Christen RD, Boland CR, Koi M, Fishel R, Howell SB. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res. 1996;56:3087–3090. [PubMed] [Google Scholar]
- 60.Fink D, Nebel S, Aebi S, Zheng H, Cenni B, Nehme A, Christen RD, Howell SB. The role of DNA mismatch repair in platinum drug resistance. Cancer Res. 1996;56:4881–4886. [PubMed] [Google Scholar]
- 61.Papouli E, Cejka P, Jiricny J. Dependence of the cytotoxicity of DNA-damaging agents on the mismatch repair status of human cells. Cancer Res. 2004;64:3391–3394. doi: 10.1158/0008-5472.CAN-04-0513. [DOI] [PubMed] [Google Scholar]
- 62.Li GM. DNA mismatch repair and cancer. Front Biosci. 2003;8:997–1017. doi: 10.2741/1121. [DOI] [PubMed] [Google Scholar]
- 63.Magrini R, Bhonde MR, Hanski ML, Notter M, Scherubl H, Boland CR, Zeitz M, Hanski C. Cellular effects of CPT-11 on colon carcinoma cells: dependence on p53 and hMLH1 status. Int J Cancer. 2002;101:23–31. doi: 10.1002/ijc.10565. [DOI] [PubMed] [Google Scholar]
- 64.Fedier A, Schwarz VA, Walt H, Carpini RD, Haller U, Fink D. Resistance to topoisomerase poisons due to loss of DNA mismatch repair. Int J Cancer. 2001;93:571–576. doi: 10.1002/ijc.1356. [DOI] [PubMed] [Google Scholar]
- 65.Fallik D, Borrini F, Boige V, Viguier J, Jacob S, Miquel C, Sabourin JC, Ducreaax M, Praz F. Microsatellite instability is a predictive factor of the tumor response to irinotecan in patients with advanced colorectal cancer. Cancer Res. 2003;63:5738–5744. [PubMed] [Google Scholar]
- 66.Parker WB, Cheng YC. Metabolism and mechanism of action of 5-fluorouracil. Pharmacol Ther. 1990;48:381–395. doi: 10.1016/0163-7258(90)90056-8. [DOI] [PubMed] [Google Scholar]
- 67.Major PP, Egan E, Herrick D, Kufe DW. 5-Fluorouracil incorporation in DNA of human breast carcinoma cells. Cancer Res. 1982;42:3005–3009. [PubMed] [Google Scholar]
- 68.Carethers JM, Chauhan DP, Fink D, Nebel S, Bresalier RS, Howell SB, Boland CR. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology. 1999;117:123–131. doi: 10.1016/s0016-5085(99)70558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Caradonna SJ, Cheng YC. The role of deoxyuridine triphosphate nucleotidohydrolase, uracil-DNA glycosylase, and DNA polymerase alpha in the metabolism of FUdR in human tumor cells. Molec Pharmac. 1980;18:513–520. [PubMed] [Google Scholar]
- 70.Ingraham HA, Tseng BY, Goulian M. Mechanism for exclusion of 5-fluorouracil from DNA. Cancer Res. 1980;40:998–1001. [PubMed] [Google Scholar]
- 71.Lonn U, Lonn S. DNA lesions in human neoplastic cells and cytotoxicity of 5-fluoropyrimidines. Cancer Res. 1986;46:3866–3870. [PubMed] [Google Scholar]
- 72.Parker WB, Kennedy KA, Klubes P. Dissociation of 5-fluorouracil-induced DNA fragmentation from either its incorporation into DNA or its cytotoxicity in murine T-lymphoma (S-49) cells. Cancer Res. 1987;47:979–982. [PubMed] [Google Scholar]
- 73.Danenberg PV, Lockshin A. Fluorinated pyrimidines as tight-binding inhibitors of thymidylate synthetase. Pharmac Ther. 1981;13:69–90. doi: 10.1016/0163-7258(81)90068-1. [DOI] [PubMed] [Google Scholar]
- 74.Arnold CN, Goel A, Boland CR. Role of hMLH1 promoter hypermethylation in drug resistance to 5-fluorouracil in colorectal cancer cell lines. Int J Cancer. 2003;106:66–73. doi: 10.1002/ijc.11176. [DOI] [PubMed] [Google Scholar]
- 75.Tajima A, Hess MT, Cabrera BL, Kolodner RD, Carethers JM. The mismatch repair complex hMutS alpha recognizes 5-fluorouracil-modified DNA: implications for chemosensitivity and resistance. Gastroenterology. 2004;127:1678–1684. doi: 10.1053/j.gastro.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 76.Meyers M, Wagner MW, Mazurek A, Schmutte C, Fishel R, Boothman DA. DNA mismatch repair-dependent response to fluoropyrimidine-generated damage. J Biol Chem. 2005;280:5516–5526. doi: 10.1074/jbc.M412105200. [DOI] [PubMed] [Google Scholar]
- 77.Stojic L, Mojas N, Cejka P, Di Pietro M, Ferrari S, Marra G, Jiricny J. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 2004;18:1331–1344. doi: 10.1101/gad.294404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Duckett DR, Bronstein SM, Taya Y, Modrich P. hMutSalpha- and hMutLalpha-dependent phosphorylation of p53 in response to DNA methylator damage. Proc Natl Acad Sci USA. 1999;96:12384–12388. doi: 10.1073/pnas.96.22.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hickman MJ, Samson LD. Apoptotic signaling in response to a single type of DNA lesion, O(6)-methylguanine. Mol Cell. 2004;14:105–116. doi: 10.1016/s1097-2765(04)00162-5. [DOI] [PubMed] [Google Scholar]
- 81.Nehme A, Baskaran R, Aebi S, Fink D, Nebel S, Cenni B, Wang JY, Howell SB, Christen RD. Differential induction of c-Jun NH2-terminal kinase and c-Abl kinase in DNA mismatch repair-proficient and -deficient cells exposed to cisplatin. Cancer Res. 1997;57:3253–3257. [PubMed] [Google Scholar]
- 82.Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG, Jr, Levrero M, Wang JY. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399:806–809. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- 83.Meyers M, Wagner MW, Hwang HS, Kinsella TJ, Boothman DA. Role of the hMLH1 DNA mismatch repair protein in fluoropyrimidine-mediated cell death and cell cycle responses. Cancer Res. 2001;61:5193–5201. [PubMed] [Google Scholar]
- 84.Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, Boland CR. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N′-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 1994;54:4308–4312. [PubMed] [Google Scholar]
- 85.Li HR, Shagisultanova EI, Yamashita K, Piao Z, Perucho M, Malkhosyan SR. Hypersensitivity of tumor cell lines with microsatellite instability to DNA double strand break producing chemotherapeutic agent bleomycin. Cancer Res. 2004;64:4760–4767. doi: 10.1158/0008-5472.CAN-04-0975. [DOI] [PubMed] [Google Scholar]
- 86.Sanada M, Takagi Y, Ito R, Sekiguchi M. Killing and mutagenic actions of dacarbazine, a chemotherapeutic alkylating agent, on human and mouse cells: effects of Mgmt and Mlh1 mutations. DNA Repair. 2004;3:413–420. doi: 10.1016/j.dnarep.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 87.Pors K, Patterson LH. DNA mismatch repair deficiency, resistance to cancer chemotherapy and the development of hypersensitive agents. Curr Top Med Chem. 2005;5:1133–1149. doi: 10.2174/156802605774370883. [DOI] [PubMed] [Google Scholar]