

Summary
Background
Accumulating clinical evidence suggests that hyperhomocysteinemia (HHC) is correlated with Alzheimer’s disease (AD) and vascular dementia.
Objective
This study was carried out to elucidate the specific role of elevated homocysteine (HC) levels in AD pathophysiology.
Methods
Immunohistochemistry was used to examine amyloid-beta (Aβ) deposition along blood vessels, also known as cerebral amyloid angiopathy (CAA), fibrin(ogen) deposition, and their correlation to each other in the brains of AD patients with and without HHC. To study AD-HHC comorbidity in detail, an AD mouse model was administered a high methionine diet for several months. Parenchymal Aβ plaques, CAA-positive vessels, and fibrin deposits were then assessed by immunohistochemistry at different stages of AD progression. Memory deficits were evaluated with contextual fear conditioning and the Barnes maze. Additionally, the effect of HC and its metabolite, homocysteine thiolactone (HCTL), on the Aβ-fibrinogen interaction was analyzed by pull-down, ELISA, and fibrin clot formation and fibrinolysis assays in vitro.
Results
We found increased fibrin(ogen) levels and Aβ deposits in the blood vessels and brain parenchyma of AD patients with HHC. We demonstrate that HC and HCTL enhance the interaction between fibrinogen and Aβ, promote the formation of tighter fibrin clots, and delay clot fibrinolysis. Additionally, we show that diet-induced HHC in an AD mouse model leads to severe CAA and parenchymal Aβ deposition, as well as significant impairments in learning and memory.
Conclusions
These findings suggest that elevated levels of plasma HC/HCTL contribute to AD pathology via the Aβ-fibrin(ogen) interaction.
Keywords: Alzheimer disease, amyloid beta-peptides, cerebral amyloid angiopathy, fibrinogen, homocysteine
Introduction
There is increasing evidence that cerebrovascular dysfunction, such as altered cerebral blood flow, cerebral vasculature damage, and abnormal hemostasis, plays a critical role in Alzheimer’s disease (AD) pathogenesis[1–5]. Although the precise mechanism by which vascular dysfunction contributes to the development of AD remains unknown, it has been suggested that the β-amyloid peptide (Aβ) may act as a mediator for cerebrovascular impairment[2, 6, 7]. Aβ accumulates in the brain parenchyma and cerebral blood vessels in AD. The deposition of Aβ in cerebral blood vessels, known as cerebral amyloid angiopathy (CAA), has deleterious effects on the cerebrovasculature, promoting the degeneration of vessel wall components[1] while also reducing cerebral blood flow[7]. Several studies in AD mouse models demonstrate that a disrupted cerebrovasculature can induce cell damage and loss in CAA-positive vessels in early stages of AD[8, 9].
We and others have shown that elevated plasma fibrinogen levels could be a potential risk factor for AD[10–18]. Fibrinogen, a large plasma glycoprotein, is converted by thrombin into fibrin, which polymerizes into blood clots. We have recently shown that Aβ42 can directly interact with fibrinogen and induce its oligomerization in vitro[18]. Fibrin clots formed in the presence of Aβ42 have an abnormal structure and are resistant to degradation by fibrinolytic enzymes in vitro and in vivo[19, 20]. Fibrin(ogen) deposition leads to increased inflammation and disruption of the blood brain barrier (BBB) in AD patients and mouse models[13, 19, 21–23]. Furthermore, fibrin deposition parallels the severity of CAA in the brains of AD patients and mice[19, 23]. Moreover, pharmacological or genetic depletion of fibrinogen lessens CAA pathology[19], reduces cognitive impairment[19], suppresses microglial activation[13], and reduces BBB disruption[13] in AD mouse models. These studies support the possibility that Aβ can compromise cerebral blood flow through its interaction with fibrinogen, exacerbating AD pathology.
Elevated plasma levels of homocysteine (HC), a sulfur-containing amino acid derived from methionine[24], is known as hyperhomocysteinemia (HHC). HHC is associated with venous thromboembolism, atherosclerosis[25], and microvasculopathy[26]. Evidence suggests that HHC alters fibrin clot structure and stability ex vivo[27] by inducing fibrin accumulation, impairing perivascular fibrinolysis, and causing prothrombotic defects in vivo[28]. Interestingly, high levels of HC are consistently observed and correlated with cognitive decline in AD and vascular dementia patients[29, 30]. In addition, some studies have shown that HHC-inducing diets (high methionine or low folate) increase Aβ levels and/or deposition[31–33] and further impair cognitive decline[32, 34] in various AD mouse models. Despite these critical insights, the exact impact of elevated HC levels on vascular dysfunction in AD remains undefined. We hypothesized that concomitant HHC could aggravate AD pathogenesis via the Aβ-fibrinogen interaction. We examined the correlation between plasma HC concentration and fibrin(ogen)-containing CAA-positive vessels in AD patients’ brains. We also determined the effects of HC and its metabolite, homocysteine thiolactone (HCTL), on the Aβ-fibrinogen interaction, fibrin network structure, and fibrinolysis in vitro. Furthermore, we induced HHC in an AD transgenic mouse model to examine its effects on AD progression in vivo. Our studies suggest that HC and HCTL not only contribute to AD pathology, but they do so via the Aβ-fibrin(ogen) interaction.
Methods
Aβ42, homocysteinylated fibrinogen, and FragD
Aβ42 or biotinylated Aβ42 peptides (Anaspec) were reconstituted in 4% NH4OH in 50mM TBS, pH 7.4, and aliquots were stored at 80°C. Before use, aliquots were centrifuged for 10 min at 12,000×g to remove pre- aggregated material. Aβ42 concentration was determined by BCA assay.
To homocysteinylate fibrinogen or fragment D (FragD), human plasminogen-free fibrinogen (29.4 μM, Calbiochem) and purified FragD[18] (34μM) were incubated with various doses of HC (Santa Cruz Biotechnology ) or HCTL (Sigma) at 37°C for 18h. To remove unreacted HC and HCTL, HC/HCTL-incubated fibrinogen samples were gel-filtered with a Sephadex G-25 column (GE Healthcare). Control and homocysteinylated fibrinogen and FragD concentrations were adjusted to 2.94μM or 5.67μM and subjected to assays described below.
Pull-down assay
Unmodified/control fibrinogen (0.5nM) and FragD (25nM) or HC/HCTL- modified fibrinogen (0.5nM) and FragD (25nM) were incubated with or without biotinylated Aβ42 (200nM) for 1h at room temperature (RT) in binding buffer (50mM Tris·HCl, pH 7.4, 150mM NaCl, 0.01% NP-40, 0.1% BSA, and protease inhibitors). The samples were incubated with streptavidin-coupled magnetic beads (Invitrogen) for 1h and washed five times with binding buffer. Aβ-fibrinogen or Aβ-FragD complexes were eluted by heating to 80°C for 5 min in non-reducing sample buffer and subjected to Western blot analysis using an anti-fibrin(ogen) antibody (Dako). To compare the amounts of Aβ being pulled down, dot blots were performed (4G8, Covance).
ELISAs for Aβ-fibrinogen and Aβ-FragD interaction
Biotinylated Aβ42 was immobilized for 1h at RT on a Reacti-Bind streptavidin-coated plate (ThermoScientific). The plate was washed and incubated with either control fibrinogen (50nm) or FragD (100nM) or HC/HCTL-modified fibrinogen (50nM) or FragD (100nM) and then incubated with anti-fibrinogen antibody. The plate was washed and then incubated with HRP-conjugated secondary antibody and Ultra TMB solution (ThermoScientific). Absorbance was read at 405nm.
Fibrin clot formation
Control fibrinogen or HC/HCTL-modified fibrinogen (2.7μM) was mixed with Alexa Fluor-488 fibrinogen (0.3μM; Invitrogen) in HEPES buffer, mounted on glass-bottom microwell dishes (MatTek), and supplemented with 5mM CaCl2 and 0.5U/mL thrombin (Sigma) ± Aβ42 (3μM). Samples were then visualized by Zeiss LSM510 confocal laser scanning microscope with a 40-Axiovert 1.2/water objective. To analyze Aβ-influenced fibrin clot structure and size, 3–4 images were obtained as Z-stack slices taken every 0.5μm (11 slices/image) at 50μm above the glass surface. Images containing fibrin(ogen) clumps were projected two-dimensionally to produce the final image, equally thresholded, and then analyzed using NIH ImageJ software.
Fibrin clot turbidity assay
Fibrin clot formation and lysis assays were performed in triplicate[20]. To measure fibrin polymerization, control fibrinogen (1.5μM) and HC/HCTL- modified fibrinogen were mixed with thrombin and CaCl2. To measure clot formation and lysis, fibrinogen and HC/HCTL-incubated fibrinogen with or without Aβ42 (3μM) were mixed with plasminogen, thrombin, tPA (Genentech), and CaCl2 in HEPES buffer. Clot half-lysis time was calculated as the time between maximal and half-maximal turbidity. Plasminogen was purified from human plasma provided by NY Blood Center[19, 20].
Human brain immunohistochemistry
Human post-mortem frontal cortex and hippocampal samples from non-demented individuals and AD patients with or without HHC were obtained from The Thomas Willis Oxford Brain Collection at Oxford University Hospital. Paraffinized sections were prepared for immunohistochemical analysis[19]. Brain sections were incubated with rabbit anti-fibrinogen antibody (Dako), developed with deaminobenzidine, and counterstained with 1% Thioflavin S (ThioS; Sigma) in 70% ethanol or triple-immunostained using primary antibodies against fibrinogen, collagen IV (Fitzgerald), and Aβ (6E10, Covance), incubated with fluorescent secondary antibodies, and imaged using an inverted Zeiss Axiovert 200 microscope. To determine the total area of vascular fibrin(ogen) or CAA, 25–30 images from the frontal cortex or hippocampus were collected, thresholded using ImageJ, quantified, and normalized by area of fibrin(ogen)- or CAA-positive vessels in non-demented individuals for fibrin(ogen) staining or AD control patients for CAA staining.
Animals and diet treatment
TgCRND8 AD mice[35] and wild-type (WT) littermates (7–8 weeks-of-age) were fed standard control (CON), high methionine (MET), or high glycine (GLY) diet (Harlan) for 2 or 5 months. All animals had unrestricted access to food and water. Body weights and food consumption were monitored weekly. Mice were maintained and treated in accordance with Rockefeller’s IACUC.
Mouse brain immunohistochemistry
Following treatment, AD and WT mice were transcardially perfused with saline/heparin. Brains were removed and post-fixed overnight in 4% paraformaldehyde at 4°C, stored in 30% sucrose at 4°C until sinking, and sectioned with a sliding microtome into 30μm-thick sagittal sections. Brain sections were stained and imaged as described above. Olfactory bulb, hippocampus, and cortex of each mouse were analyzed for parenchymal Aβ deposition and CAA. Images were then thresholded using ImageJ and quantified.
Behavior
The Barnes maze was used to assess spatial learning and memory[36, 37]. Animals were subjected to two trials/day for five days with a 30 min intertrial interval. Trials were recorded and analyzed with Ethovision (Noldus). Fear conditioning was used to examine contextual memory[36].
Statistical analysis
All values are expressed as mean±SEM. Statistical significance was assessed as described in text. P<0.05 was considered statistically significant.
Results
Fibrin deposits and CAA are increased in AD patients with high plasma HC
To examine the effect of elevated HC on fibrin homeostasis and CAA pathology, we analyzed vascular fibrin and Aβ deposition in post-mortem brain tissue from AD patients and non-demented individuals with normal or elevated levels of plasma HC (Supplementary Table 1). Consistent with our previous reports[19, 23], fibrin and Aβ co-deposition along large (≥20μm) vessels was elevated in AD patients compared to controls (Fig 1A,B; Fig S1). Brain tissue from AD patients with HHC had more fibrin deposits (Fig 1B,E) and CAA (Fig 1C,F) in the frontal cortex and hippocampus compared to samples from AD patients with normal HC levels. Furthermore, a highly significant correlation between fibrin deposition and CAA was found in both the frontal cortex and hippocampus of AD patients (Fig S2A,B). Plasma HC levels did not influence fibrin or CAA deposition in non-demented individuals (not shown), but did so in the frontal cortex (Fig S2C,E) and hippocampus (Fig S2D,F) of AD patients. These data strongly suggest that high plasma HC levels contribute to fibrin accumulation and CAA in AD.
Figure 1. Increased fibrinogen deposition and CAA in the brains of AD patients with HHC.

We performed immunohistochemical analysis for fibrin(ogen) (FBG, green), Aβ (blue), and collagen IV (COL4, red) in frontal cortex (A) and hippocampus (D) samples from non-demented control individuals (Sup Fig 1) and AD patients with normal (total HC concentration <15μmol/L) or high (HHC; total HC concentration ≥15μmol/L) plasma HC levels. Scale bars =100μm. Fibrin(ogen) in the frontal cortex (B) and hippocampus (E) was quantified and normalized to control individual samples. Results represent mean ± SEM. (*p<0.05, **p<0.01, ***p<0.001; One-way ANOVA with Neuman-Keuls post-hoc test). CAA-positive vessels were defined by Aβ- and COL4-positive signal larger than 20μm in diameter. CAA in the frontal cortex (C) and hippocampus (F) was normalized to AD control patient samples. Student’s t-test revealed a significant difference in the hippocampus between AD patients with HHC (AD-HHC) and those with normal plasma HC levels (AD); *p<0.05). One-way ANOVA with Tukey post-hoc test also revealed statistical significance. CON, non-demented control patients with normal plasma HC levels; HHC, non-demented patients with HHC; AD, AD patients with normal plasma HC levels; AD-HHC, AD patients with HHC.
HC and HCTL enhance the Aβ-fibrinogen interaction via homocysteinylation of the FragD region of fibrinogen
Given that there are significantly greater levels of fibrin deposits in AD patients with concomitant HHC, we examined the effects of HC/HCTL on fibrinogen in vitro. Consistent with previous reports[27, 38], HC/HCTL affected fibrin polymerization and degradation. We observed significant dose-dependent effects of HC/HCTL on the lag time to clot (Fig S3A,C) and maximal clot turbidity (Fig S3B,D); the higher the levels of HC/HCTL, the slower the fibrin clots formed and the lower their turbidity (Fig S3A–D). To visualize the effects of increased HC/HCTL on clotting, fibrinogen incubated with increasing amounts of HC or HCTL was analyzed by confocal microscopy (Fig S3E). These results suggest that high HC or HCTL levels may modify fibrinogen structurally thereby altering fibrin clot formation.
Since fibrinogen directly binds to Aβ42 and this interaction increases fibrinogen oligomerization in vitro[18], we investigated whether HC or HCTL affected fibrinogen oligomerization and its interaction with Aβ. We incubated biotinylated-Aβ42 with control, HC-, or HCTL-modified fibrinogen and performed pull-down assays. There was a stronger interaction between HC/HCTL-modified fibrinogen and Aβ42 than control fibrinogen (Fig 2A). At higher concentrations of HC or HCTL, fibrinogen oligomerized in the absence of Aβ42 (Fig S4). Consistent with these data, ELISAs showed increased binding absorbance of HC- or HCTL-modified fibrinogen with Aβ42, suggesting that modification of fibrinogen by HC/HCTL increases the interaction between fibrinogen and Aβ42 (Fig 2B).
Figure 2. Enhanced Aβ42-fibrinogen interaction via homocysteinylation of FragD/fibrinogen.
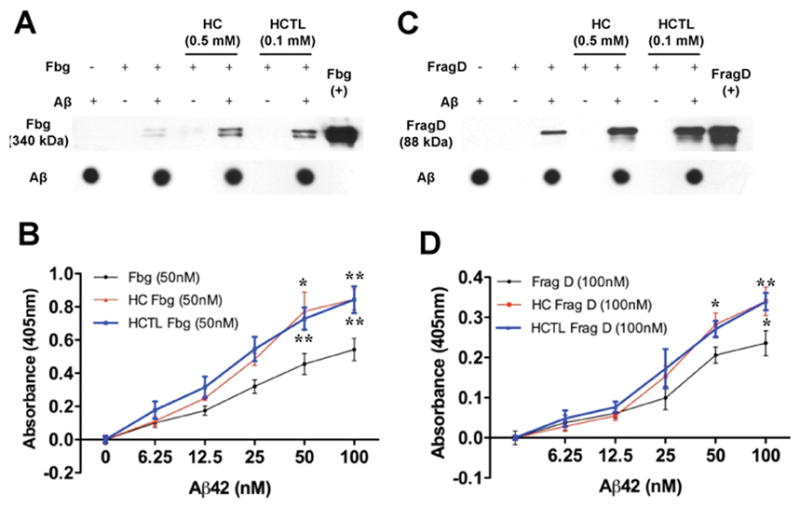
Control, HC- (0.5mM), or HCTL- (0.1mM) modified human fibrinogen (Fbg; 29.4μM) and FragD (34μM) were incubated in the presence or absence of biotinylated Aβ42. Unreacted HC/HCTL was removed as described in Methods. Pull down assays were performed using streptavidin-conjugated magnetic beads. More fibrinogen (A) and FragD (C) were pulled down when the incubation reactions contained HC or HCTL. Dot blots against Aβ42 showed that comparable amounts of Aβ42 were pulled down in all experiments. Pull down assays were performed 3–4 independent times, and representative blots are shown. (B, D) Binding curves for the interactions of fibrinogen (black in B; 50nM) and FragD (black in D; 100nM) with Aβ42 in the absence or presence of HC (red) or HCTL (blue) were determined by ELISA. The results are shown as mean ± SEM from four different experiments. Aβ42-fibrinogen interaction is significantly enhanced in the presence of HC or HCTL (*p<0.05, **p<0.01).
We have shown that fibrinogen binds to Aβ at its fragment D (FragD) region[18]. To examine whether the enhanced interaction between HC/HCTL-modified fibrinogen and Aβ42 is due to direct homocysteinylation of FragD, pull-down assay (Fig 2C) and ELISA (Fig 2D) were performed. HC and HCTL dramatically increased the FragD-Aβ42 interaction, suggesting that HC/HCTL positively influence the fibrinogen-Aβ42 interaction through homocysteinylation of FragD.
HC and HCTL affect fibrin clot formation and fibrinolysis
To further investigate how HC and HCTL affect fibrin clot structure, we induced clot formation using Alexa Fluor488- conjugated fibrinogen in the absence or presence of Aβ42 and HC/HCTL. Consistent with previous results[19, 20], Aβ42 induced the formation of non-homogeneous fibrin fibers while also significantly increasing fibrin aggregate size (p<0.001; Fig S5A-C). HC- or HCTL- modification of fibrinogen not only enhanced the formation of irregular clusters in the presence of Aβ (Fig S5A), it also significantly increased their size (Fig S5B,C). These data suggest that modification of fibrinogen by HC/HCTL promotes and enhances the formation of Aβ-induced irregular clusters within fibrin clots.
To investigate whether HC or HCTL affects fibrinolysis, in vitro degradation assays were performed with fibrinogen (control or HC/HCTL-modified) with or without Aβ42. Consistent with our previous data[19, 20], the dissolution of Aβ42-altered fibrin clots was significantly delayed (blue vs black, Fig S5D-G). However, modification of fibrinogen by HC/HCTL further increased the half-lysis time in the presence of Aβ42 (red vs blue, Fig S5D–G), but did not affect fibrinolysis in the absence of Aβ (gray vs black, Fig S5D–G). Collectively, our data suggest that HC/HCTL affect the structure of Aβ-altered fibrin clots, resulting in their delayed dissolution.
Concomitant HHC and AD enhances Aβ deposition in the mouse brain
We examined the effects of HHC on vascular and parenchymal Aβ deposition in TgCRND8 AD mice[35]. HHC was induced by feeding 7–8-week-old AD and WT mice a high methionine (MET) diet for 2 or 5 months. Standard (CON) and high glycine (GLY) diets were used as controls; GLY diet was used as a control for the increase in a single amino acid in the experimental diet (Sup Table 2). No gross changes in appearance or morbidity were observed, although MET-fed mice gained significantly less weight than control groups (Sup Table 3). Consistent with previous reports[35], we observed more Aβ plaques in the cortex, olfactory bulb, and hippocampus of MET-fed AD mice compared to CON- and GLY-fed mice (at 4 and 7 months-of-age) (Fig 3, S6; not shown). Compared to controls, MET diet caused a notable increase in the total Aβ-positive area in all regions examined at 4 months-of-age (Fig 3B) and in the cortex at 7 months-of-age (Fig 3A,C). Large vessel (≥20μm) CAA was detected at relatively low abundance in the brains of AD mice at 4 months-of-age but was dramatically increased by 7 months-of-age (Fig 3A,D,E; S6), suggesting that large vessel CAA develops after Aβ plaque formation in these mice. Although MET diet only led to an increase in large vessel CAA in the AD cortex at 4 months-of-age (Fig 3D), it markedly enhanced CAA-positive area in all brain regions examined by 7 months-of-age (Fig 3A,E; S6). Furthermore, CAA in capillaries (<20μm) was dramatically increased in MET-fed AD mice compared to control groups at both 4 and 7 months-of-age (not shown). Neither control nor MET diets affected the formation of Aβ deposits in the parenchyma or vessels of the WT mouse brains (not shown).
Figure 3. HHC increases parenchymal and vascular Aβ deposition in the brains of transgenic AD mice.

TgCRND8 AD mice and their WT littermates (7–8 weeks-of-age) were treated with CON, GLY, or MET diet for 2 or 5 months. (A) Immunohistochemical analysis was performed for fibrin(ogen) (FBG, green), Aβ (blue), and collagen IV (COL4, red) in the cortex of AD and WT mice. Aβ staining represents both parenchymal plaques and CAA. Arrows indicate representative large (≥20μm) COL4-positive vessels that were also fibrinogen- and Aβ-positive. As in humans, Aβ signal often localized with FBG. (B,C) Bar graphs depict the total Aβ-positive area (parenchymal and vascular Aβ deposits) throughout the olfactory bulb (OB), hippocampus (HP), and cortex (CTX) in AD mice treated for 2 months (B) or 5 months (C). There was significantly more Aβ deposition in brain regions of MET-fed AD mice compared to CON and GLY diet-fed AD mice (***p<0.001; One-way ANOVA with Newman-Keuls post hoc test). There was no Aβ-positive staining in any of the WT mouse groups (not shown). (D,E) Quantification of large CAA-positive vessels (via Aβ-positive staining aligned with COL4 signal) in each brain region. The number of CAA-positive vessels was significantly increased in the CTX of AD mice fed MET diet for 2 months compared to that of CON and GLY diet-fed mice. Mice treated with MET diet for 5 months showed a significant increase in the number of CAA-positive vessels in all brain regions examined compared to CON and GLY diets (*p<0.05, **p<0.01, ***p<0.001; One-way ANOVA with Newman-Keuls post hoc test). Statistical significance indicates a difference when compared with CON diet-treated AD mice. Parenchymal and vascular Aβ signal was quantified in 4–5 sections from 6–9 mice per experimental group. C, Control diet; G, high glycine diet; M, high methionine diet.
Concomitant HHC exacerbates cognitive deficits in AD mice
To examine whether the HHC-induced increase in CAA exacerbates cognitive deficits in AD mice, we performed the Barnes maze using 7-month-old mice that had been administered CON, GLY, or MET diet for 5 months. During acquisition training, two-way ANOVA revealed significant effects of genotype and diet on the time to reach the escape hole as well as a significant interaction between genotype and diet on time to enter the escape hole (Fig 4A). Furthermore, MET-fed AD mice took significantly longer to find and enter the escape hole compared to CON-treated AD mice on Days 3 and 4 (Fig 4A). When the latency to the target hole was quantified as area under the curve, CON-treated AD mice took significantly longer to reach the target hole compared with CON-treated WT mice (Fig 4B). Notably, MET-treated AD mice exhibited significantly longer entry latencies compared to control AD mouse groups (Fig 4B).
Figure 4. HHC exacerbates memory impairment in AD mice.

(A,B) Spatial learning and memory were measured using the Barnes maze, which consisted of 10 training trials over 5 days (2 trials/day) at 7 months-of-age (after 5 months of diet treatment, n=6–9/group). (A) Latency to reach the escape hole. There was a significant difference between MET diet-fed AD mice (blue) and CON diet-fed mice (black and red) in the latency to reach the target hole on the third and fourth days of training. (**p<0.01; Two-way ANOVA with Bonferroni post hoc test, [F(15,115)=2.152, p<0.05]). (B) Bar graph represents area under the curve derived from (A) (*p<0.05; mean ± SEM by student’s t-Test). (C) Contextual fear conditioning was used to measure learning and memory in AD and WT mice fed CON, GLY, or MET diet (n=7–10/group). Bar graph depicts percent freezing (immobility) during contextual testing (mean ± SEM) and shows a significant difference between CON diet-fed AD and WT mice and between CON diet- and MET diet-fed AD mice. (*p<0.05; Two-way ANOVA with Bonferroni post hoc test [F(2,41)=4.982, p<0.05].
We also performed contextual fear conditioning to assess memory impairment. Consistent with previous reports[39], control AD groups demonstrated a robust deficit in contextual fear conditioning at 7 months-of-age compared to the control WT groups (Fig 4C). Analysis by two-way ANOVA showed significant effects of genotype and diet as well as a significant interaction between genotype and diet. Compared to AD mice fed CON or GLY diet, MET diet induced an even more significant deterioration in their contextual memory (Fig 4C). There was no difference between genotype and diet in basal freezing activity (data not shown). These data suggest that HHC-induced cerebrovascular changes could exacerbate cognitive decline in AD.
Discussion
This study is the first to demonstrate significant increases in fibrin(ogen) deposition and CAA severity in the brains of AD patients with concomitant HHC compared to those with normal plasma HC levels. HHC, induced by a high methionine diet, also enhanced parenchymal and vascular Aβ deposition and aggravated cognitive decline in transgenic AD mice compared to AD mice administered control diets. Furthermore, our in vitro and in vivo data both suggest that high plasma HC has deleterious effects on vascular AD pathophysiology via the Aβ-fibrinogen interaction. These conclusions are based on two findings: 1) HC/HCTL affected fibrin clot formation and amplified the Aβ-fibrinogen interaction via modification of FragD, and 2) in the presence of Aβ, HC/HCTL-modified fibrinogen augmented clot size and delayed fibrin clot dissolution.
Recent clinical studies have shown that AD pathogenesis is associated with cerebrovascular dysfunction, such as reduced cerebral blood flow and altered hemostasis[3, 5], implying an association between vascular and neurological pathologies[2]. Several studies suggest that fibrinogen also plays a crucial role in these abnormalities[13, 14, 19]. Not only does fibrin(ogen) localize with CAA in the cerebral vessels and brain parenchyma of AD patients and mice[14, 19, 23], but fibrin(ogen) deposition increases with age and correlates with Aβ plaque levels in brains of AD mice and patients [10]. Furthermore, pharmacological or genetic depletion of fibrinogen attenuates CAA pathology and synaptic dysfunction in AD mice[10, 13, 19]. Consistently, we found that fibrin(ogen) deposition was enriched along CAA-containing cerebral vessels in AD patients and was associated with CAA severity in the frontal cortex and hippocampus of AD patients (Fig S2), indicating that the degree of fibrin(ogen) deposition may affect the development of CAA in the AD neurovasculature.
We also show that HHC results in increased fibrin(ogen) deposition and CAA in AD patients (Fig 1). Correlation analyses showed that plasma HC level was significantly associated with both fibrin accumulation and CAA severity in the frontal cortex and hippocampus of AD patients (Fig S2). Other studies have suggested that elevated plasma HC may contribute to vascular fibrin accumulation and reduced fibrinolysis in vascular diseases[40]. HHC is known to disturb hemostasis and shift hemostatic mechanisms, such as altered thrombosis and fibrinolysis [41]. Several studies demonstrated that HC-modified fibrinogen was more resistant to fibrinolysis[42] and formed fibrin clots with thinner fibers than normal fibrinogen in vitro[38]. We considered the possibility that HC/HCTL directly alters fibrin clot formation by modifying fibrinogen. We show by turbidity assay and confocal microscopy that abnormally high levels of HC or HCTL can modify fibrinogen, inducing its oligomerization and abnormal fibrin clot formation in vitro (Fig S3). These results indicate that homocysteinylation leads to atypical fibrin(ogen) properties.
The concentrations of HC/HCTL used in in vitro experiments were higher than physiological levels. We initially performed experiments with physiological concentrations of HC (5–100 μm) and HCTL (0.05–0.5 μm) (Sup Table 1), but these concentrations did not show a difference in lag time and maximal turbidity during clot formation. The higher concentrations used accelerated the reaction and allowed us to model this long-term disease in vitro in a reasonable time frame.
We also examined the possibility that HC/HCTL could increase Aβ42 fibrillization. Incubation of Aβ42 with various doses of HC (5–50mM) or HCTL (0.05–5mM) for up to 96 hours did not affect total β-sheet content via the Thioflavin T assay. Therefore, HC/HCTL-modified fibrinogen is crucial to the changes we observed in Aβ deposition.
It is known that Aβ can specifically interact with fibrinogen and subsequently alter fibrin clot structure[13, 18–20]. Aβ-influenced abnormal fibrin clots are resistant to fibrinolysis due to their tighter network of thinner fibers[18, 19]. The present studies confirm that HC/HCTL-modified fibrinogen more strongly interacts with Aβ42 (Fig 2A,B) and forms enlarged, irregular clots in the presence of Aβ42 (Fig S5A-C). Moreover, Aβ-influenced HC/HCTL-modified fibrin clots showed a delayed half-lysis time during fibrinolysis (Fig S5D-G), suggesting that HC/HCTL alters Aβ-influenced fibrin clot structure and causes delayed fibrin clot dissolution. Therefore, abnormally high levels of plasma HC/HCTL could be critically important in enhancing the deposition of fibrin and Aβ in the cerebral vasculature, via the Aβ-fibrinogen interaction.
HC/HCTL-modified FragD interacted more strongly with Aβ than control FragD in pull-down assays and ELISAs (Fig 2C,D). Importantly, Sauls and colleagues demonstrated that fibrinogen has 12 lysine residues that can be modified by HCTL, four of which are within FragD[38]. Since HC/HCTL-modified fibrinogen produced significantly larger aggregates during clot formation with Aβ but we previously showed that Aβ does not induce FragD oligomerization[18], our data suggest that HC/HCTL may homocysteinylate lysine residues in another region of fibrinogen in addition to FragD. Our results suggest that HCTL-modified residues within FragD account for the increased interaction with Aβ, supporting an important functional mechanism in AD/HHC.
In neuropathological studies, CAA is commonly observed in AD patients[7, 43, 44] and can induce neurovascular damage and disrupt blood flow in the brain, resulting in cognitive impairment[7, 45]. Studies have shown that amelioration of CAA in the brain improves memory deficits in AD mice[19, 46]. Recently, Li and Pratico reported that HHC induced by a diet deficient in folate and vitamins B6 and B12 increased CAA in AD transgenic mice[47]. Here, we report that HHC induced by a methionine-rich diet increased CAA severity and exacerbated spatial and contextual memory loss in AD mice (Figs 3, 4). These data strongly suggest that plasma HC can influence CAA pathology and cognitive impairment in AD. In the brains of AD mice, parenchymal Aβ can be cleared by way of the vasculature, but severe CAA hinders this clearance route[7]. Our studies showed that MET diet significantly increased both parenchymal and vascular Aβ deposition in AD mice compared to CON-fed AD mice (Fig 3). It is possible that the increased deposition of parenchymal Aβ could ultimately lead to impaired clearance through the vasculature, facilitating Aβ-induced abnormal fibrin clot formation. It is also possible that that high plasma HC/HCTL levels could modify fibrinogen and induce its oligomerization, leading to accumulation of Aβ-fibrinogen deposits. Given that homocysteinylation of fibrinogen induced its oligomerization (Fig S3) and strengthened its interaction with Aβ42 (Fig 2 and S4) in vitro, increased HC/HCTL levels may prompt more Aβ-fibrinogen accumulation within the cerebral vasculature. This hypothesis is supported by a report that fibrinogen and Aβ deposits are elevated in the brains of transgenic HHC mice[48]. Our results are the first to suggest that HC/HCTL may aggravate cognitive dysfunction and pathology in AD by way of the Aβ-fibrinogen interaction.
Many clinical studies suggest comorbidity of AD and HHC, yet homocysteinylated-fibrinogen in the blood or brain of AD patients has not yet been reported. Interestingly, HHC patients deficient in cystathionine β-synthase (CBS), an enzyme responsible for converting homocysteine to cystathionine, have significantly elevated plasma levels of N-Hcy-fibrinogen[49, 50]. This fibrinogen N-homocysteinylation leads to resistance of fibrin clot lysis[27, 38] and increases risk of thrombosis[27, 51]. Moreover, experimental studies provide evidence that MET diet-induced HHC results in increased fibrin deposition in the vasculature and delayed fibrinolysis in the kidneys, lung, and liver[28], which complements the symptoms of CBS-deficient patients. Although we did not show evidence of fibrinogen N-homocysteinylation in our HHC mouse model, our study provides the first evidence that HC/HCTL-modified fibrinogen may be responsible for aggravating AD pathology by way of the Aβ-fibrinogen interaction.
Several studies have shown that AD mice have abnormal clotting abilities compared to controls[10, 19, 52]. We have also shown that AD patients have increased cerebral fibrin deposition, corresponding to their CAA severity[10, 53]. Moreover, our in vitro studies demonstrate a detailed mechanism as to how Aβ affects fibrin clot structure and interferes with clot lysis by preventing the binding of plasminogen to fibrin[18–20]. Altogether, these results could suggest that vessel obstruction results from the formation of persistent Aβ-laden fibrin clots, implying the existence of a close relationship between AD pathophysiology and stroke/microinfarcts. Many clinical studies have shown that asymptomatic spontaneous cerebral emboli[54] and microinfarcts[55] are more common in AD patients. Furthermore, these ischemic events lead to weakening of blood vessels, increasing the likelihood of subsequent microhemorrhage and hemorrhagic stroke, a process referred to as hemorrhagic transformation[56]. Thus, our findings that HC/HCTL-modified fibrinogen induced bigger Aβ-fibrin aggregates in clots and delayed fibrinolysis suggest that HHC could aggravate CAA pathology, including hemorrhage, in both AD and stroke.
Emerging evidence strongly indicates diverse roles of HHC on AD pathophysiology. Although several studies have explored the effect of HHC on Aβ plaque formation and tau phosphorylation in AD mice[57], there are still conflicting results about the role of HHC on AD pathophysiology. Here, we show that heightened plasma HC levels enhance the Aβ-fibrinogen interaction and exacerbate vascular pathogenesis and cognitive decline in AD. This finding and further studies that show HHC deteriorates the cerebrovascular environment through the Aβ-fibrinogen interaction will provide novel insights and may help develop therapeutics for AD.
Supplementary Material
Essentials.
Evidence suggests a comorbidity between hyperhomocysteinemia (HHC) and Alzheimer's disease (AD).
Homocysteine (HC) could affect the β-amyloid (Aβ)-fibrinogen interaction in AD pathology.
AD patients with concomitant HHC have increased fibrin and Aβ deposits in their brains.
HC contributes to AD pathology via the Aβ-fibrinogen interaction.
Acknowledgments
This work was supported by the National Institute of Health (NS050537), Sackler Center for Biomedicine and Nutrition Research and the Sackler Foundation, Litwin Foundation, Mellam Family Foundation, May and Samuel Rudin Family Foundation, Mr. John A. Herrmann, Jr, and the Blanchette Hooker Rockefeller Fund. The authors thank members of the Strickland Laboratory for scientific discussion. We are also grateful to the Thomas Willis Oxford Brain Collection at Oxford University Hospitals for providing the human samples for this study.
Footnotes
Addendum – Y. C. Chung performed the research, analyzed data, and wrote the manuscript; A. Kruyer performed experiments, analyzed data, and assisted in manuscript writing; Y. Yao assisted in manuscript writing; E. Feierman assisted in experiments; A. Richards assisted in manuscript editing; S. Strickland assisted in study design and data analysis; and E. H. Norris assisted in study design, data analysis, and writing and editing the manuscript.
Disclosure of Conflicts of Interest – The authors have no conflicts of interest.
References
- 1.de la Torre JC. Is Alzheimer's disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol. 2004;3:184–90. doi: 10.1016/S1474-4422(04)00683-0. [DOI] [PubMed] [Google Scholar]
- 2.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347–60. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 3.Luchsinger JA, Reitz C, Honig LS, Tang MX, Shea S, Mayeux R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology. 2005;65:545–51. doi: 10.1212/01.wnl.0000172914.08967.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sagare AP, Bell RD, Zlokovic BV. Neurovascular defects and faulty amyloid-beta vascular clearance in Alzheimer's disease. J Alzheimers Dis. 2013;33(Suppl 1):S87–100. doi: 10.3233/JAD-2012-129037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiesmann M, Kiliaan AJ, Claassen JA. Vascular aspects of cognitive impairment and dementia. J Cereb Blood Flow Metab. 2013;33:1696–706. doi: 10.1038/jcbfm.2013.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sweeney MD, Sagare AP, Zlokovic BV. Cerebrospinal fluid biomarkers of neurovascular dysfunction in mild dementia and Alzheimer's disease. J Cereb Blood Flow Metab. 2015;35:1055–68. doi: 10.1038/jcbfm.2015.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thal DR, Griffin WS, de Vos RA, Ghebremedhin E. Cerebral amyloid angiopathy and its relationship to Alzheimer's disease. Acta Neuropathol. 2008;115:599–609. doi: 10.1007/s00401-008-0366-2. [DOI] [PubMed] [Google Scholar]
- 8.Iadecola C, Gorelick PB. Converging pathogenic mechanisms in vascular and neurodegenerative dementia. Stroke. 2003;34:335–7. doi: 10.1161/01.str.0000054050.51530.76. [DOI] [PubMed] [Google Scholar]
- 9.Kruyer A, Soplop N, Strickland S, Norris EH. Chronic Hypertension Leads to Neurodegeneration in the TgSwDI Mouse Model of Alzheimer's Disease. Hypertension. 2015;66:175–82. doi: 10.1161/HYPERTENSIONAHA.115.05524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cortes-Canteli M, Mattei L, Richards AT, Norris EH, Strickland S. Fibrin deposited in the Alzheimer's disease brain promotes neuronal degeneration. Neurobiol Aging. 2015;36:608–17. doi: 10.1016/j.neurobiolaging.2014.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jantaratnotai N, Schwab C, Ryu JK, McGeer PL, McLarnon JG. Converging perturbed microvasculature and microglial clusters characterize Alzheimer disease brain. Curr Alzheimer Res. 2010;7:625–36. doi: 10.2174/156720510793499039. [DOI] [PubMed] [Google Scholar]
- 12.Lominadze D, Dean WL, Tyagi SC, Roberts AM. Mechanisms of fibrinogen-induced microvascular dysfunction during cardiovascular disease. Acta Physiol (Oxf) 2010;198:1–13. doi: 10.1111/j.1748-1716.2009.02037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paul J, Strickland S, Melchor JP. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer's disease. J Exp Med. 2007;204:1999–2008. doi: 10.1084/jem.20070304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryu JK, McLarnon JG. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer's disease brain. J Cell Mol Med. 2009;13:2911–25. doi: 10.1111/j.1582-4934.2008.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu G, Zhang H, Zhang S, Fan X, Liu X. Plasma fibrinogen is associated with cognitive decline and risk for dementia in patients with mild cognitive impairment. Int J Clin Pract. 2008;62:1070–5. doi: 10.1111/j.1742-1241.2007.01268.x. [DOI] [PubMed] [Google Scholar]
- 16.van Oijen M, Witteman JC, Hofman A, Koudstaal PJ, Breteler MM. Fibrinogen is associated with an increased risk of Alzheimer disease and vascular dementia. Stroke. 2005;36:2637–41. doi: 10.1161/01.STR.0000189721.31432.26. [DOI] [PubMed] [Google Scholar]
- 17.Thambisetty M, Simmons A, Hye A, Campbell J, Westman E, Zhang Y, Wahlund LO, Kinsey A, Causevic M, Killick R, Kloszewska I, Mecocci P, Soininen H, Tsolaki M, Vellas B, Spenger C, Lovestone S, AddNeuroMed C. Plasma biomarkers of brain atrophy in Alzheimer's disease. PLoS One. 2011;6:e28527. doi: 10.1371/journal.pone.0028527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahn HJ, Zamolodchikov D, Cortes-Canteli M, Norris EH, Glickman JF, Strickland S. Alzheimer's disease peptide beta-amyloid interacts with fibrinogen and induces its oligomerization. Proc Natl Acad Sci U S A. 2010;107:21812–7. doi: 10.1073/pnas.1010373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cortes-Canteli M, Paul J, Norris EH, Bronstein R, Ahn HJ, Zamolodchikov D, Bhuvanendran S, Fenz KM, Strickland S. Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer's disease. Neuron. 2010;66:695–709. doi: 10.1016/j.neuron.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zamolodchikov D, Strickland S. Abeta delays fibrin clot lysis by altering fibrin structure and attenuating plasminogen binding to fibrin. Blood. 2012;119:3342–51. doi: 10.1182/blood-2011-11-389668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rybarczyk BJ, Lawrence SO, Simpson-Haidaris PJ. Matrix-fibrinogen enhances wound closure by increasing both cell proliferation and migration. Blood. 2003;102:4035–43. doi: 10.1182/blood-2003-03-0822. [DOI] [PubMed] [Google Scholar]
- 22.Vidal B, Serrano AL, Tjwa M, Suelves M, Ardite E, De Mori R, Baeza-Raja B, Martinez de Lagran M, Lafuste P, Ruiz-Bonilla V, Jardi M, Gherardi R, Christov C, Dierssen M, Carmeliet P, Degen JL, Dewerchin M, Munoz-Canoves P. Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev. 2008;22:1747–52. doi: 10.1101/gad.465908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hultman K, Strickland S, Norris EH. The APOE epsilon4/epsilon4 genotype potentiates vascular fibrin(ogen) deposition in amyloid-laden vessels in the brains of Alzheimer's disease patients. J Cereb Blood Flow Metab. 2013;33:1251–8. doi: 10.1038/jcbfm.2013.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hajjar KA. Homocysteine: a sulph'rous fire. J Clin Invest. 2001;107:663–4. doi: 10.1172/JCI12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Humphrey LL, Fu R, Rogers K, Freeman M, Helfand M. Homocysteine level and coronary heart disease incidence: a systematic review and meta-analysis. Mayo Clin Proc. 2008;83:1203–12. doi: 10.4065/83.11.1203. [DOI] [PubMed] [Google Scholar]
- 26.Markus HS. Genes, endothelial function and cerebral small vessel disease in man. Exp Physiol. 2008;93:121–7. doi: 10.1113/expphysiol.2007.038752. [DOI] [PubMed] [Google Scholar]
- 27.Sauls DL, Wolberg AS, Hoffman M. Elevated plasma homocysteine leads to alterations in fibrin clot structure and stability: implications for the mechanism of thrombosis in hyperhomocysteinemia. J Thromb Haemost. 2003;1:300–6. doi: 10.1046/j.1538-7836.2003.00053.x. [DOI] [PubMed] [Google Scholar]
- 28.Jacovina AT, Deora AB, Ling Q, Broekman MJ, Almeida D, Greenberg CB, Marcus AJ, Smith JD, Hajjar KA. Homocysteine inhibits neoangiogenesis in mice through blockade of annexin A2-dependent fibrinolysis. J Clin Invest. 2009;119:3384–94. doi: 10.1172/JCI39591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCaddon A, Davies G, Hudson P. Nutritionally independent B12 deficiency and Alzheimer disease. Arch Neurol. 2000;57:607–8. doi: 10.1001/archneur.57.4.607. [DOI] [PubMed] [Google Scholar]
- 30.Ravaglia G, Forti P, Maioli F, Martelli M, Servadei L, Brunetti N, Porcellini E, Licastro F. Homocysteine and folate as risk factors for dementia and Alzheimer disease. Am J Clin Nutr. 2005;82:636–43. doi: 10.1093/ajcn.82.3.636. [DOI] [PubMed] [Google Scholar]
- 31.Fuso A, Nicolia V, Cavallaro RA, Ricceri L, D'Anselmi F, Coluccia P, Calamandrei G, Scarpa S. B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances PS1 and BACE expression and amyloid-beta deposition in mice. Mol Cell Neurosci. 2008;37:731–46. doi: 10.1016/j.mcn.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 32.Zhuo JM, Portugal GS, Kruger WD, Wang H, Gould TJ, Pratico D. Diet-induced hyperhomocysteinemia increases amyloid-beta formation and deposition in a mouse model of Alzheimer's disease. Curr Alzheimer Res. 2010;7:140–9. doi: 10.2174/156720510790691326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhuo JM, Pratico D. Acceleration of brain amyloidosis in an Alzheimer's disease mouse model by a folate, vitamin B6 and B12-deficient diet. Exp Gerontol. 2010;45:195–201. doi: 10.1016/j.exger.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernardo A, McCord M, Troen AM, Allison JD, McDonald MP. Impaired spatial memory in APP-overexpressing mice on a homocysteinemia-inducing diet. Neurobiol Aging. 2007;28:1195–205. doi: 10.1016/j.neurobiolaging.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 35.Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–70. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- 36.Ahn HJ, Glickman JF, Poon KL, Zamolodchikov D, Jno-Charles OC, Norris EH, Strickland S. A novel Abeta-fibrinogen interaction inhibitor rescues altered thrombosis and cognitive decline in Alzheimer's disease mice. J Exp Med. 2014;211:1049–62. doi: 10.1084/jem.20131751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walker JM, Fowler SW, Miller DK, Sun AY, Weisman GA, Wood WG, Sun GY, Simonyi A, Schachtman TR. Spatial learning and memory impairment and increased locomotion in a transgenic amyloid precursor protein mouse model of Alzheimer's disease. Behav Brain Res. 2011;222:169–75. doi: 10.1016/j.bbr.2011.03.049. [DOI] [PubMed] [Google Scholar]
- 38.Sauls DL, Lockhart E, Warren ME, Lenkowski A, Wilhelm SE, Hoffman M. Modification of fibrinogen by homocysteine thiolactone increases resistance to fibrinolysis: a potential mechanism of the thrombotic tendency in hyperhomocysteinemia. Biochemistry. 2006;45:2480–7. doi: 10.1021/bi052076j. [DOI] [PubMed] [Google Scholar]
- 39.Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, Pawlik M, Peterhoff CM, Yang AJ, Wilson DA, St George-Hyslop P, Westaway D, Mathews PM, Levy E, Cuervo AM, Nixon RA. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–77. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Di Minno MN, Tremoli E, Coppola A, Lupoli R, Di Minno G. Homocysteine and arterial thrombosis: Challenge and opportunity. Thrombosis and haemostasis. 2010;103:942–61. doi: 10.1160/TH09-06-0393. [DOI] [PubMed] [Google Scholar]
- 41.Malinowska J, Kolodziejczyk J, Olas B. The disturbance of hemostasis induced by hyperhomocysteinemia; the role of antioxidants. Acta Biochim Pol. 2012;59:185–94. [PubMed] [Google Scholar]
- 42.Undas A, Williams EB, Butenas S, Orfeo T, Mann KG. Homocysteine inhibits inactivation of factor Va by activated protein C. J Biol Chem. 2001;276:4389–97. doi: 10.1074/jbc.M004124200. [DOI] [PubMed] [Google Scholar]
- 43.Attems J. Sporadic cerebral amyloid angiopathy: pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol. 2005;110:345–59. doi: 10.1007/s00401-005-1074-9. [DOI] [PubMed] [Google Scholar]
- 44.Burgermeister P, Calhoun ME, Winkler DT, Jucker M. Mechanisms of cerebrovascular amyloid deposition. Lessons from mouse models. Ann N Y Acad Sci. 2000;903:307–16. doi: 10.1111/j.1749-6632.2000.tb06381.x. [DOI] [PubMed] [Google Scholar]
- 45.Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ. Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology. 2002;58:1629–34. doi: 10.1212/wnl.58.11.1629. [DOI] [PubMed] [Google Scholar]
- 46.Park L, Zhou J, Zhou P, Pistick R, El Jamal S, Younkin L, Pierce J, Arreguin A, Anrather J, Younkin SG, Carlson GA, McEwen BS, Iadecola C. Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc Natl Acad Sci U S A. 2013;110:3089–94. doi: 10.1073/pnas.1300021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li JG, Pratico D. High levels of homocysteine results in cerebral amyloid angiopathy in mice. J Alzheimers Dis. 2015;43:29–35. doi: 10.3233/JAD-141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muradashvili N, Tyagi R, Metreveli N, Tyagi SC, Lominadze D. Ablation of MMP9 gene ameliorates paracellular permeability and fibrinogen-amyloid beta complex formation during hyperhomocysteinemia. J Cereb Blood Flow Metab. 2014;34:1472–82. doi: 10.1038/jcbfm.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jakubowski H, Boers GH, Strauss KA. Mutations in cystathionine beta-synthase or methylenetetrahydrofolate reductase gene increase N-homocysteinylated protein levels in humans. FASEB J. 2008;22:4071–6. doi: 10.1096/fj.08-112086. [DOI] [PubMed] [Google Scholar]
- 50.Sikora M, Marczak L, Kubalska J, Graban A, Jakubowski H. Identification of N–homocysteinylation sites in plasma proteins. Amino Acids. 2014;46:235–44. doi: 10.1007/s00726-013-1617-7. [DOI] [PubMed] [Google Scholar]
- 51.Splaver A, Lamas GA, Hennekens CH. Homocysteine and cardiovascular disease: biological mechanisms, observational epidemiology, and the need for randomized trials. Am Heart J. 2004;148:34–40. doi: 10.1016/j.ahj.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 52.Klohs J, Baltes C, Princz-Kranz F, Ratering D, Nitsch RM, Knuesel I, Rudin M. Contrast-enhanced magnetic resonance microangiography reveals remodeling of the cerebral microvasculature in transgenic ArcAbeta mice. J Neurosci. 2012;32:1705–13. doi: 10.1523/JNEUROSCI.5626-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hultman K, Strickland S, Norris EH. The APOE varepsilon4/varepsilon4 genotype potentiates vascular fibrin(ogen) deposition in amyloid-laden vessels in the brains of Alzheimer's disease patients. J Cereb Blood Flow Metab. 2013;33:1251–58. doi: 10.1038/jcbfm.2013.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Purandare N, Burns A. Cerebral emboli in the genesis of dementia. J Neurol Sci. 2009;283:17–20. doi: 10.1016/j.jns.2009.02.306. [DOI] [PubMed] [Google Scholar]
- 55.Brundel M, de Bresser J, van Dillen JJ, Kappelle LJ, Biessels GJ. Cerebral microinfarcts: a systematic review of neuropathological studies. J Cereb Blood Flow Metab. 2012;32:425–36. doi: 10.1038/jcbfm.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang J, Yang Y, Sun H, Xing Y. Hemorrhagic transformation after cerebral infarction: current concepts and challenges. Ann Transl Med. 2014;2:81. doi: 10.3978/j.issn.2305-5839.2014.08.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhuo JM, Wang H, Pratico D. Is hyperhomocysteinemia an Alzheimer's disease (AD) risk factor, an AD marker, or neither? Trends Pharmacol Sci. 2011;32:562–71. doi: 10.1016/j.tips.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.